

Abstract
The GTP/GDP nucleotide switch in Ras superfamily GTPases generally involves differential affinity toward downstream effectors, with the GTP-bound state having a higher affinity for effector than the GDP-bound state. We have developed a quantitative model of allosteric regulation of the Wiskott–Aldrich syndrome protein (WASP) by the Rho GTPase Cdc42 to better understand how GTPase binding is coupled to effector activation. The model accurately predicts WASP affinity for Cdc42, activity toward Arp2/3 complex, and activation by Cdc42 as functions of a two-state allosteric equilibrium in WASP. The ratio of GTPase affinities for the inactive and active states of WASP is appreciably larger for Cdc42–GTP than for Cdc42–GDP. The greater ability to distinguish between the two states of WASP makes Cdc42–GTP a full WASP agonist, whereas Cdc42–GDP is only a partial agonist. Thus, the nucleotide switch controls not only the affinity of Cdc42 for its effector but also the efficiency of coupling between the Cdc42-binding and allosteric equilibria in WASP. This effect can ensure high fidelity and specificity in Cdc42 signaling in crowded membrane environments.
Keywords: allostery, signal transduction
Small GTPases in the Ras superfamily function as molecular switches in vivo, cycling between an inactive GDP-bound state and an active GTP-bound state. The biochemical basis of switching generally involves a difference in affinity for downstream effector proteins. That is, the GTP-bound form of the GTPase has appreciably higher affinity for effectors than does the GDP-bound form. This concept has arisen from studies of interactions between GTPases and the minimal GTPase-binding domains (GBDs) of effectors (1–6). The affinity of a GTPase for a GBD typically switches 500- to 1,000-fold with a change in nucleotide. In many full-length GTPase effectors, the GBD makes autoinhibitory contacts with other activity-bearing regions of the protein. Structural and thermodynamic coupling (anticooperativity) between these intramolecular interactions and intermolecular interactions with the GTPase are believed to be central to GTPase-mediated activation of effector signaling. In most cases, we lack a quantitative description of the biochemistry of interactions between full-length effectors and their cognate GTPases. Thus, it is not clear whether, in cases of autoinhibited effectors, a simple change in affinity is sufficient to account for the change in GTPase-mediated activity triggered by the nucleotide switch.
The Rho subfamily of Ras GTPases, including Cdc42, Rac, and Rho, regulate a diverse range of cellular function, including signaling pathways controlling cell growth, motility, and cytoskeletal architecture (7). Cdc42 is an important regulator of actin cytoskeletal structure and dynamics. Cdc42 can communicate to the cytoskeleton through binding the Cdc42/Rac interactive binding (CRIB) motif located in the GBD of its autoinhibited effector, the Wiskott–Aldrich syndrome protein (WASP) (Fig. 1) (8). Activated WASP, in turn, activates the Arp2/3 complex, a nucleator of new actin filaments, through intermolecular interactions with its C-terminal verprolin homology/central domain/acidic domain (VCA) to generate cellular structures such as filopodia and lamellipodia.
Fig. 1.

Domain organization of WASP constructs used in this study. EVH1, Ena/Vasp homology 1; B, basic region; P, proline-rich region; V, verprolin homology region; C, central hydrophobic domain; A, acidic region; GGSGGS, linker. Color coding matches that of the autoinhibited WASP structure in Fig. 3.
We demonstrate here that the nucleotide switch controls the affinity between Cdc42 and WASP and the coupling between the Cdc42–WASP binding equilibrium and the allosteric equilibrium of WASP. Specifically, we find that Cdc42–GTP has a higher affinity for WASP than does Cdc42–GDP and that it is more effective at differentiating between the inactive and active states of WASP. The latter property imparts to the saturated Cdc42–GTP–WASP complex greater activity toward the Arp2/3 complex than to the saturated Cdc42–GDP–WASP complex. Thus, an important component of the nucleotide switch is modulation of the differential binding of Cdc42 for the two states of WASP, changing Cdc42 from a partial agonist in the GDP-bound form to a full agonist in the GTP-bound form.
Methods
Constructs and Proteins. Preparation and purification of WASP constructs and Cdc42 (residues 1–179) loaded with guanosine 5′-[β,γ-imido]triphosphate (GMPPNP) have been described elsewhere (9–11). Arp2/3 complex was isolated from bovine thymus (12). Actin was purified from rabbit skeletal muscle (13).
Chemical Denaturation. CD measurements (222 nm) were obtained on a Aviv Associates (Lakewood, NJ) 62DS spectrometer at 25°C by using 10 μM protein samples and urea or guanidine hydrochloride as denaturant. Chemical denaturation experiments were performed in 25 mM phosphate, pH 7/150 mM NaCl/2 mM MgCl2/2 mM DTT. Experimental data were fit to a six-parameter equation (14) to obtain ΔG of folding and m. Where performed, identical ΔG values were obtained for urea and guanidine hydrochloride denaturation.
Fluorescence Binding Assays. Binding affinity of WASP constructs for Cdc42 loaded with a fluorescent nonhydrolyzable GTP analog, mantGMPPNP, was measured on a Horiba Jobin Yvon Fluorolog 3-11 fluorimeter. Binding was followed by monitoring the quenching of mant fluorescence at 432 nm (λex = 350) upon addition of WASP protein. Experiments were performed in 25 mM phosphate, pH 7/150 mM NaCl/2 mM MgCl2/2 mM DTT. Equilibrium dissociation constants (KD) were obtained by fitting the data to a quadratic equation for a single-site binding isotherm (15).
Binding affinities of Arp2/3 for I290Q and basic region GBD (BGBD)–VCA were measured by using a rhodamine anisotropy fluorescence assay. A C-terminal cysteine residue was introduced by mutagenesis to I290Q and BGBD–VCA to facilitate labeling with tetramethylrhodamine 5′-malemide. Rhodamine fluorescence was monitored at 574 nm (λex = 552 nm). Rhodamine-labeled I290Q and BGBD–VCA displayed higher florescence anisotropy upon addition of Arp2/3 complex in the presence of 4 μM actin-latrunculin. Experiments and analysis were performed in the same buffers as above.
Because mant fluorescence can interfere with pyrene fluorescence in pyrene-actin polymerization assays and because mant labeling decreases the affinity of Cdc42 for WASP (16), we also measured affinity of Cdc42–GMPPNP and Cdc42–GDP for WASP proteins by using a previously described FRET-based assay as a prelude to the actin polymerization measurements below (17). Briefly, WASP proteins were fused to cyan-fluorescent protein (CFP) and yellow fluorescent protein at their N and C termini, respectively. Binding of Cdc42 was measured through changes in the ratio of yellow fluorescent protein emission (526 nm) to CFP emission (476 nm) after CFP excitation (λex = 433 nm). Experiments were performed in 20 mM Hepes, pH 7.3/150 mM KCl/2 mM MgCl2/2 mM DTT. Data were fit to a quadratic equation for a single-site binding isotherm (17).
Pyrene Actin Polymerization Assays. Actin polymerization assays were performed as described in ref. 12. Pyrene fluorescence at 407 nm (λex = 365 nm) was monitored over time. Polymerization assays were performed in 10 mM imidazole, pH 7/50 mM KCl/1 mM MgCl2/10 mM EGTA and contained 4 μM actin (6% pyrene), 10 nM Arp2/3, and 500 nM WASP proteins. Fractional activity is determined from the concentration of filament barbed ends calculated for each WASP construct relative to the isolated VCA domain as described in ref. 12.
NMR Acquisition. NMR data were collected on 500- or 600-MHz Varian Innova spectrometers at 25°C. To probe the structural details of WASP in complexes with Cdc42, 1H/15N transverse relaxation-optimized spectroscopy (TROSY) heteronuclear sequential quantum correlation (HSQC) spectra were acquired on 100 μM 2H/15N GBD–VCA protein in 10 mM imidazole, pH 7/50 mM KCl/1 mM MgCl2/10 mM EGTA in the presence of either 500 μM Cdc42–GMPPNP or 750 μM Cdc42–GDP. To determine the binding interface between Cdc42–GDP and GBD–VCA, 1H/15N TROSY HSQC spectra were acquired on a 100 μM sample of 2H/15N Cdc42–GDP in 20 mM Tris, pH 7/100 mM NaCl/2 mM MgCl2/5 mM DTT in the presence of increasing concentrations of GBD–VCA (up to 300 μM). Backbone 1HN, 15N, 13Cα, 13Cβ, and 13CO assignments were obtained for the Cdc42–GDP domain through the following 2H-decoupled, triple resonance spectra appliedtoa 2H/15N/13C-labeled sample: CT-HNCa, CT-HN(CO)Ca, HN(Ca)Cb, and HN(COCa)Cb (18).
All data sets were processed identically with nmrpipe (19) and analyzed with nmrview (20).
Formulation of the Two-State Allosteric Model. The development of a two-state allosteric model for WASP has been described in detail in ref. 10. Briefly, the fraction of WASP proteins in the active state is described by
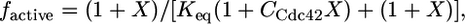 |
[1] |
where
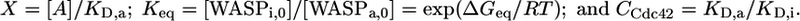 |
[2] |
[A] is the concentration of activator (Cdc42); [WASPi,0] and [WASPa,0] are the concentrations of inactive and active WASP in the absence of activator; KD,a and KD,i are the binding affinities of WASPa and WASPi for activator, respectively; ΔGeq is the free energy difference between the inactive and active states of WASP, which we approximate by the folding stability of the autoinhibited domain; and R and T are the ideal gas constant and temperature. In the absence of any activator, Eq. 1 simplifies to
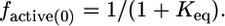 |
[3] |
In the presence of infinite activator, Eq. 1 can be described by
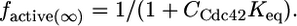 |
[4] |
The measured binding affinities reflect an equilibrium mixture of inactive and active WASP states and can be described by the following equation (10):
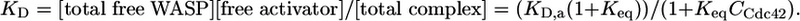 |
[5] |
Results
Development of a Two-State Model for Allostery in WASP. To understand the activation of WASP by Cdc42, we have developed a simple two-state model of WASP based on classical descriptions of allostery (10, 21, 22) (Fig. 2). In such models, the state of the system can be quantitatively described by only three parameters: Keq, the equilibrium constant between the inactive and active states of the free protein (WASP); C, the ratio of affinities of activator (Cdc42) for the active and inactive states of WASP (KD,a/KD,i); and A, the concentration of activator. Binding is coupled to activation through the product Keq*C, which is the equilibrium constant between the inactive and active states of the saturated protein–activator complex. Thus, activator binding shifts the equilibrium by the amount C. Briefly, our model proposes, based on a large body of biophysical data, that the allosteric equilibrium in WASP can be quantitatively represented by the folding equilibrium of the regulatory domain formed by intramolecular interactions between the GBD and the activity-bearing (toward the Arp2/3 complex) VCA elements of the protein. Additionally, the parameter C can be determined from the affinities of Cdc42 for engineered WASP proteins that are significantly stabilized and significantly destabilized relative to wild type. Previous work showed that the model quantitatively predicts the hydrogen exchange behavior and Cdc42–GTP binding affinity of WASP proteins over a range of folding stabilities (Keq values) (10). To extend this work across a wider range of Keq values and, more importantly, to examine the activity of different WASP proteins toward Arp2/3 complex, we generated a series of WASP constructs by introducing mutations in the GBD outside of the CRIB motif (Figs. 1 and 3). Proteins in this series have folding stabilities ranging from –5.8 to more than –1.5 kcal mol–1 (1 cal = 4.18 J) (Keq = 17,000 to <12), including M307A, which has a folding stability comparable to the protein most representative of full-length WASP (GBDPVCA) (Table 1) and two mutants, I294A and I290Q, whose stabilities against chemical denaturation were too low to measure accurately.
Fig. 2.

A two-state allosteric model for WASP. WASP exists in equilibrium between an inactive, folded autoinhibited state and an active, predominantly unfolded state. Cdc42 biases this equilibrium toward activation because of its higher affinity for the active state. WASPi and WASPa represent inactive and active states of WASP, respectively.
Fig. 3.

Mutations generated in this study. The minimal autoinhibited WASP structure (Protein Data Bank ID code 1EJ5) (9) consists of the GBD (residues 242–310; yellow and cyan) connected to the C domain helix (residues 461–492; red) by a GGSGGS linker. Elements of the GBD that contact Cdc42 directly in the Cdc42–GBD structure (Fig. 8A) (11) are yellow. Side chains of residues mutated in the background of the GBD–VCA construct are purple and are located in the GBD outside of the Cdc42 binding interface.
Table 1. Measured values of ΔG, Keq, and KD.
| WASP | ΔG, kcal·mol-1 | Keq | KD, μM |
|---|---|---|---|
| BGBD-C | -5.5 | 11,000 | 98 ± 13 |
| GBD—VCA | -4.2 | 1,200 | 114 ± 8 |
| M307A (GBD—VCA) | -3.4 | 310 | 71 ± 5 |
| GBDPVCA | -3.3 | 260 | n.d. |
| M307K (GBD—VCA) | -2.4 | 58 | 13 ± 1 |
| 1290A (GBD—VCA) | -1.5 | 13 | 4.0 ± 0.1 |
| 1294A (GBD—VCA) | -0.4† | 2 | 0.50 ± 0.05 |
| 1290Q (GBD—VCA) | n.d. | n.d. | 0.20 ± 0.04 |
| M307A (BGBD—VCA) | -3.9 | 730 | n.d. |
| M307K (BGBD—VCA) | -2.8 | 110 | n.d. |
| 1290A (BGBD—VCA) | -2.5 | 68 | n.d. |
| 1294A (BGBD—VCA) | -0.9† | 5 | n.d. |
n.d., not determined.
This measurement is only approximate and may not represent the unfolding of tertiary structure.
Cdc42-GTP Affinity for WASP Constructs. The binding affinity of each WASP construct for active Cdc42 was determined by monitoring the fluorescence quenching of mantGMPPNP-loaded Cdc42 in a titration (Fig. 4). The measured binding affinities displayed an inverse correlation with protein folding stability. For example, the most stable BGBD-C protein, which approximates the inactive state of WASP, has ≈500-fold lower affinity for Cdc42 than the least stable protein I290Q, which emulates the fully active state of WASP. Using the measured values of KD,a (I290Q) and KD,i (BGBD-C) to estimate CCdc42,GTP yields: CCdc42,GTP = KD,a/KD,i = 1.73 × 10–3. The greatest changes in KD occur in the transition region between stabilities of –4 and –1 kcal mol–1. Outside of this region, changes in KD are very small. The data can be fit well to the equation relating KD to ΔG (r2 = 0.97), yielding a CCdc42,GTP value of 2.35 × 10–3, similar to that determined from the endpoints alone, 1.73 × 10–3, and to the value of 3.4 × 10–3 previously measured under slightly different conditions (10).
Fig. 4.

Affinity of WASP proteins as a function of stability. KD values for Cdc42–GMPPNP are shown as black circles. The black line indicates the log fit of the data to Eq. 5.
Activation of Arp2/3 Complex by WASP Constructs. We next examined WASP activity toward Arp2/3 complex as a function of Keq by using a pyrene-actin assembly assay. As in Cdc42 binding, the WASP mutants have a range of Arp2/3-stimulatory activities (Fig. 5 A and C), which correlate inversely with folding stability. Highly destabilized proteins, such as I290A, display higher levels of activity (>0.5 relative to VCA), whereas more stable proteins, such as M307A, have significantly lower activity (<0.1). In addition, a second series of WASP constructs containing the basic sequence of amino acids [basic region (BR)] immediately N-terminal to the GBD (Fig. 1) was generated because this element has been shown to contribute to autoinhibition in the WASP homolog, N-WASP (23, 24). For all mutants, we find that addition of the BR increases the folding stability of WASP by ≈0.5 kcal·mol–1 when compared with non-BR WASP constructs (Table 1). This increase in stability causes a concomitant decrease in activity toward the Arp2/3 complex, such that BR and non-BR proteins fall on essentially the same fitted curve (see below) (Figs. 5 B and C). This result suggests that the BR decreases WASP activity through modulating the interaction between GBD and VCA rather than through an independent mechanism involving direct interaction with the Arp2/3 complex (23). Thus, WASP activity toward the Arp2/3 complex is limited by the folding stability of the autoinhibited domain and reflects the equilibrium between inactive and active states.
Fig. 5.

Activity of WASP proteins as a function of stability. (A) Pyrene-actin polymerization assays of 500 nM WASP proteins in the presence of 10 nM Arp2/3 complex (except “actin only” curve) and 4 μM actin (6% pyrene-labeled). (B) Actin polymerization assays comparing GBD–VCA mutant constructs with BGBD–VCA mutant constructs (indicated by a B-prefix). (C) Fractional activity (relative to VCA) of WASP proteins in the pyrene-actin polymerization assay. GBD–VCA and BGBD–VCA (wild-type and mutant proteins) are indicated by circles and triangles, respectively. The fractional activities predicted by the model in the absence of activator (Eq. 11 in Supporting Methods), in the presence of Cdc42–GDP (Eq. 12 in Supporting Methods), and in the presence of Cdc42–GTP are shown as black solid, green dashed, and blue dotted lines, respectively. The C values used for Cdc42–GMPPNP and Cdc42–GDP are 1.1 × 10–2 and 5.4 × 10–2, respectively.
To evaluate these data in terms of our two-state model, we first tested whether the Arp2/3 complex itself could bias the WASP allosteric equilibrium (i.e., whether CArp2/3 ≠ 1) (Supporting Methods, which is published as supporting information on the PNAS web site). By using a fluorescence binding assay, we measured the Arp2/3 complex affinity of rhodamine-labeled I290Q and BGBD–VCA, the active WASP proteins with the lowest and highest folding stabilities. Affinity was measured in the presence of 4 μM actin-latrunculin to approximate the conditions of the assembly assay. These measurements determined that CArp2/3 = 0.053. By using this value, the data in the assembly assay fit well (r2 = 0.97) (Fig. 5C) to the equation describing the fraction of the activated Arp2/3 complex and actin bound to active WASP, yielding a binding affinity of active WASP for Arp2/3 and actin (KD(Arp2/3),a = 21 nM) similar to that measured in a titration of VCA activity toward the Arp2/3 complex (data not shown) (KD(Arp2/3),a = 85 nM). Thus, the two-state model also reasonably describes WASP activity toward the Arp2/3 complex as a function of the allosteric equilibrium.
The Cdc42 Nucleotide Switch Modulates Coupling Between the GTPase-Binding and Allosteric Equilibria in WASP. Our model predicts that activators will shift the WASP allosteric equilibrium toward the activated state according to their C parameters (Fig. 5C). The magnitude of C dictates the maximum increase in activity that can be achieved by a single agonist. Fig. 6A demonstrates that for several mutants, activity toward the Arp2/3 complex is a linear function of the percent saturation of WASP by Cdc42, as expected. Highly destabilized WASP proteins, such as I294A, are predominantly unfolded so that the addition of Cdc42–GMPPNP has little or no further effect on the equilibrium or protein activity (Fig. 6A). However, addition of increasing amounts of Cdc42–GMPPNP can achieve full activation in even the most stabilized WASP proteins, such as GBD–VCA and M307A. Thus, C42,GTP, when coupled with CArp2/3, provides sufficient driving force to fully overcome WASP autoinhibition for Keq ≤ 1,200.
Fig. 6.

WASP activity in the presence of activators. (A) Fractional activity of different WASP constructs with increasing fractional saturation by Cdc42–GMPPNP. Lines show linear fits of the data for each protein. (B) Actin polymerizations assays of M307A were performed with increasing saturation by Cdc42–GMPPNP (blue) or Cdc42–GDP (green). Data points were fit to a linear slope and extrapolated to 100% saturation.
To examine the biophysical basis of the Cdc42 nucleotide switch, we applied the allosteric model to the GDP state of the GTPase. We measured the affinity of Cdc42–GDP for the most stable (50 μM) and least stable (2.7 μM) mutants, by using a FRET-based fluorescence assay (17), yielding a value of C42,GDP = 5.4 × 10–2. This is 5-fold less than the value measured for Cdc42–GMPPNP using the same assay (KD,i = 2.6 μM; KD,a = 29 nM; C42,GTP = 1.1 × 10–2). This result suggested that the GDP form of Cdc42 should be a poorer activator of WASP than the GTP form of Cdc42 at comparable levels of WASP saturation. Consistent with this prediction, addition of Cdc42–GDP to polymerization assays did increase the rates of actin polymerization achieved by each WASP mutant but to lower levels than achieved by Cdc42–GMPPNP (data not shown). Parallel titrations of Cdc42–GMPPNP or Cdc42–GDP to M307A show that Cdc42–GMPPNP can stimulate maximum WASP activity at saturation whereas Cdc42–GDP can only partially activate WASP even at saturation (Fig. 6B and Fig. 9, which is published as supporting information on the PNAS web site). Thus, the coupling of the GTPase binding equilibrium to the allosteric equilibrium in WASP is less efficient for Cdc42–GDP than for Cdc42–GMPPNP.
To better understand the structural details of WASP interaction with Cdc42, we acquired 1H/15N TROSY HSQC spectra of 2H/15N GBD–VCA free and in the presence of Cdc42–GMPPNP and Cdc42–GDP (Fig. 7). In free GBD–VCA, peaks corresponding to the folded autoinhibited core are well dispersed in the 2D plane (Fig. 7A). Peaks representing unstructured elements, including the CRIB motif and the V and A regions of the VCA domain, are clustered in the center of the spectrum with high intensity. Addition of Cdc42–GMPPNP to 99% saturation (Fig. 7B) causes substantial broadening of the resonances representing the folded core, including the upfield-shifted indole ring HN resonance of Trp-252, which is a good indicator of the structured autoinhibited domain (9, 25). Severe global broadening of the WASP resonances is consistent with a conformational equilibrium between folded and unfolded states on the surface of the GTPase. We also observe the appearance of downfield-shifted peaks characteristic of the bound CRIB motif (10, 11), suggesting that this element anchors the protein to the GTPase. In contrast, in the presence of 90% saturating Cdc42–GDP (limited by Cdc42 solubility) (Fig. 7C), peaks representing the autoinhibited core are virtually unaffected, indicating that the folded structure is heavily populated on the surface of the GTPase. Resonances from the CRIB motif disappear from the center of the spectrum, and some new downfield shifted peaks are now observed. In the reverse titration of unlabeled GBD–VCA into 2H/15N Cdc42–GDP, significant line-broadening at substoichiometric ligand is observed primarily for peaks corresponding to the Cdc42 α1-helix, β2/β3-hairpin, and α5-helix (Fig. 8B). These elements are all close to the bound CRIB motif in the Cdc42–GTP–GBD complex structure (Fig. 8A), suggesting that this element is similarly bound in the Cdc42–GDP–GBD–VCA complex. Most resonances corresponding to the switch 1 and switch 2 regions of Cdc42–GDP are not observed for either the free protein or the GBD–VCA complex because of exchange broadening. Thus, it is unclear how extensively the bound GBD–VCA contacts these elements. These results suggest that the CRIB motif interacts with the GTPase in a similar manner in the Cdc42–GTP and Cdc42–GDP complexes. In both cases, the autoinhibited domain is likely in equilibrium on the Cdc42 surface between the folded and unfolded states, with the latter probably bound as observed in the Cdc42–GTP–GBD structure (Fig. 8A). In both complexes, the folding equilibrium of WASP is shifted toward the unfolded state relative to the free protein. But because of their different C parameters, the two nucleotides lead to different proportions of the folded and unfolded states, with GDP favoring the folded state and GTP favoring the unfolded state. Thus, Cdc42–GTP is a full agonist, whereas Cdc42–GDP is only a partial agonist.
Fig. 7.

1H/15N TROSY HSQC spectra of 2H/15N GBD–VCA. (A) GBD–VCA (100 μM). (B) GBD–VCA (100 μM) in the presence of 500 μM Cdc42–GMPPNP (99% saturation). (C) GBD–VCA (100 μM) in the presence of 750 μM Cdc42–GDP (90% saturation). Peaks identified in A (red squares) and superimposed on C are GBD–VCA residues, including the CRIB motif, which broaden severely upon binding to Cdc42–GDP. Additional peaks identified in B and C (blue circles) are new downfield shifted peaks that appear upon binding to Cdc42–GTP (11) and Cdc42–GDP, respectively.
Fig. 8.

Binding of WASP to Cdc42–GTP and Cdc42–GDP. (A) Structure of a truncated WASP GBD (residues 230–277; yellow) bound to Cdc42–GTP (residues 1–178; purple) (Protein Data Bank ID code 1CEE) (11). The WASP CRIB motif is highlighted in orange. (B) The structure of Cdc42–GDP (purple; Protein Data Bank ID code 1DOA) (32). Residues whose 1H/15N TROSY HSQC resonances decrease in intensity by >60% or shift by >0.033 ppm when 50 μM GBD–VCA is added to 100 μM 2H/15N Cdc42–GDP are colored yellow. Differences in 1H and 15N chemical shifts were determined by using the equation Δδ = [(Δ1H)2 + (Δ15N*0.15)2]1/2. (C) Normalized ratios of 1H/15N Cdc42–GDP peak intensities in the presence of GBD–VCA relative to 1H/15N Cdc42–GDP peak intensities alone. Cdc42–GDP resonances that were absent from the spectra are indicated by an intensity ratio of 0. The secondary structural elements of Cdc42–GDP are indicated below.
Discussion
Our data here and reported previously (10) indicate that the activity and Cdc42-regulation of WASP can be explained accurately and quantitatively by a two-state model. Predictions of the model that WASP's affinity for Cdc42, activity toward Arp2/3 complex, and activation by Cdc42 are functions of the allosteric equilibrium in WASP are borne out experimentally. The nucleotide switch in Cdc42 modulates not only the absolute affinity of the GTPase for WASP, but also the ability of the GTPase to distinguish between the two states of WASP. That is, the C parameter of the allosteric model is nucleotide-dependent and smaller for Cdc42–GTP than Cdc42–GDP. This property leads to the unexpected result that Cdc42–GDP can only partially activate WASP, even at saturation, whereas Cdc42–GTP can activate fully. With the truncated WASP protein used in these studies, we observe a 2-fold difference in activity between the two nucleotide states of Cdc42, as predicted by our model (Fig. 5C). The model also predicts that this difference will increase for more stable WASP proteins or when WASP is stabilized through interactions involving the BR or perhaps other binding partners, such as WIP (26, 27), with a maximum differential of C42,GDP/C42,GTP = 5-fold (Eq. 12 in Supporting Methods).
The biophysical basis of the nucleotide switch is generally thought to be simply a change in GTPase affinity for effectors. Our data here point toward a more complex mechanism involving modulation of the coupling (or linkage) between two equilibria, the Cdc42–WASP binding equilibrium, and a conformational equilibrium in WASP. What is the physical basis for the stronger coupling of Cdc42–GTP relative to Cdc42–GDP? Hatley et al. (28) recently attempted to identify the critical residue interactions that mediate communication between the nucleotide and effector binding sites in the G protein family by using an approach termed Statistical Coupling Analysis. This method identifies coevolving sequence positions in large alignments of conserved domains. The authors argue that such coevolution is driven by energetic coupling of residues in 3D structures of the domain and, thus, that clusters of mutually coevolving sites represent energetically cooperative units. In the case of G proteins, they find a large cluster of coevolving residues that surround the nucleotide-binding pocket and effector-binding elements. In the active GTP-bound structures of several GTPases, including Ras, these residues form a continuously connected core, whereas, in the GDP-bound structures, the connectivity of the network is broken and disorganized. Hatley et al. (28) argue that an essential component of the switch many thus be formation of this larger, energetically cooperative unit. Assuming similar behavior in Cdc42, it is possible that only active WASP, which interacts strongly with switch 1 and switch 2 (11, 16), can make contacts that propagate energetically through this large cooperative network, affording the GTP state significant differential binding to the two conformations of WASP. Because the network is predicted to be disorganized in the GDP state, such larger cooperativity may not be accessed, and this state may discriminate more poorly between the two conformations of WASP. Certainly further physical studies are needed to explore this issue.
Activation of WASP by Cdc42 plays an important role in activation of T cells during engagement by antigen-presenting cells (29, 30). During this process, WASP and Cdc42–GTP are independently concentrated at the T cell–antigen presenting cell interface (31). Locally high concentrations of the two molecules result in spatially restricted WASP activation. We have postulated that the independence of WASP and Cdc42–GTP recruitment plays an important role in mediating the choice of this particular effector (among many known effectors) by the GTPase during T cell activation (31). Thus, Cdc42–GTP selects the effectors that are locally concentrated at the site of its own activation by guanine nucleotide exchange factor, resulting in a specific signaling event. A nucleotide switch based solely on a change in affinity could be overridden in such a scenario. That is, a high local concentration of WASP could overcome a weak GTPase affinity, enabling its inappropriate activation by Cdc42–GDP, degrading the fidelity of Cdc42 signaling. The properties of WASP and Cdc42 that we have discovered here would mitigate this effect by ensuring that the Cdc42–GDP complex with WASP has a weak activity toward the Arp2/3 complex. Thus, a nucleotide switch based on both a change in affinity and a change in the efficiency of coupling between the binding and allosteric equilibria of effectors ensures both high specificity and high fidelity in GTPase (including Cdc42) signaling pathways. Given the preponderance of autoinhibited GTPase effectors, and regulatory balances between partial and full agonists throughout biology, we predict that such behaviors will be widely observed in GTPase signaling pathways.
Supplementary Material
Acknowledgments
We thank N. Abdul-Manan (Vertex Pharmaceuticals, Cambridge, MA) for providing backbone chemical shifts of Cdc42–GDP; M. Buck for discussion; G. Amarasinghe for critical reading of the manuscript; C. Amezcua, S. C. Panchal, and P. Li for assistance with NMR data acquisition and analysis; and A. Seth (University of Texas Southwestern Medical Center) for the generous gift of FRET sensor proteins. This work was supported by National Institutes of Health Grant GM56322 and Welch Foundation Grant I-1544.
Author contributions: D.W.L. and M.K.R. designed research; D.W.L. performed research; D.W.L. and M.K.R. analyzed data; and D.W.L. and M.K.R. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: WASP, Wiskott–Aldrich syndrome protein; GBD, GTPase-binding domain; BGBD, basic region GBD; CRIB, Cdc42/Rac interactive binding; VCA, verprolin homology/central domain/acidic domain; GMPPNP, guanosine 5′-[β,γ-imido]triphosphate; TROSY, transverse relaxation-optimized spectroscopy; HSQC, heteronuclear sequential quantum correlation; BR, basic region.
References
- 1.Bourne, H. R. (1995) Philos. Trans. R. Soc. London B 349, 283–289. [DOI] [PubMed] [Google Scholar]
- 2.Buchwald, G., Hostinova, E., Rudolph, M. G., Kraemer, A., Sickmann, A., Meyer, H. E., Scheffzek, K. & Wittinghofer, A. (2001) Mol. Cell. Biol. 21, 5179–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buck, M., Xu, W. & Rosen, M. K. (2001) Biochemistry 40, 14115–14122. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman, G. R. & Cerione, R. A. (2000) Cell 102, 403–406. [DOI] [PubMed] [Google Scholar]
- 5.Vetter, I. R. & Wittinghofer, A. (2001) Science 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- 6.Lei, M., Lu, W., Meng, W., Parrini, M. C., Eck, M. J., Mayer, B. J. & Harrison, S. C. (2000) Cell 102, 387–397. [DOI] [PubMed] [Google Scholar]
- 7.Etienne-Manneville, S. & Hall, A. (2002) Nature 420, 629–635. [DOI] [PubMed] [Google Scholar]
- 8.Higgs, H. N. & Pollard, T. D. (2001) Annu. Rev. Biochem. 70, 649–676. [DOI] [PubMed] [Google Scholar]
- 9.Kim, A. S., Kakalis, L. T., Abdul-Manan, N., Liu, G. A. & Rosen, M. K. (2000) Nature 404, 151–158. [DOI] [PubMed] [Google Scholar]
- 10.Buck, M., Xu, W. & Rosen, M. K. (2004) J. Mol. Biol. 338, 271–285. [DOI] [PubMed] [Google Scholar]
- 11.Abdul-Manan, N., Aghazadeh, B., Liu, G. A., Majumdar, A., Ouerfelli, O., Siminovitch, K. A. & Rosen, M. K. (1999) Nature 399, 379–383. [DOI] [PubMed] [Google Scholar]
- 12.Higgs, H. N., Blanchoin, L. & Pollard, T. D. (1999) Biochemistry 38, 15212–15222. [DOI] [PubMed] [Google Scholar]
- 13.Pardee, J. D. & Spudich, J. A. (1982) Methods Enzymol. 85, 164–181. [DOI] [PubMed] [Google Scholar]
- 14.Pace, C. N. (1990) Trends Biotechnol. 8, 93–98. [DOI] [PubMed] [Google Scholar]
- 15.Marchand, J. B., Kaiser, D. A., Pollard, T. D. & Higgs, H. N. (2001) Nat. Cell Biol. 3, 76–82. [DOI] [PubMed] [Google Scholar]
- 16.Rudolph, M. G., Linnemann, T., Grunewald, P., Wittinghofer, A., Vetter, I. R. & Herrmann, C. (2001) J. Biol. Chem. 276, 23914–23921. [DOI] [PubMed] [Google Scholar]
- 17.Seth, A., Otomo, T., Yin, H. L. & Rosen, M. K. (2003) Biochemistry 42, 3997–4008. [DOI] [PubMed] [Google Scholar]
- 18.Muhandiram, D. R. & Kay, L. E. (1994) J. Magn. Reson. Ser. B 103, 203–216. [Google Scholar]
- 19.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- 20.Johnson, B. A. & Blevins, R. A. (1994) J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- 21.Monod, J., Wyman, J. & Changeux, J. P. (1965) J. Mol. Biol. 12, 88–118. [DOI] [PubMed] [Google Scholar]
- 22.Fersht, A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 23.Prehoda, K. E., Scott, J. A., Mullins, R. D. & Lim, W. A. (2000) Science 290, 801–806. [DOI] [PubMed] [Google Scholar]
- 24.Rohatgi, R., Ho, H. Y. & Kirschner, M. W. (2000) J. Cell Biol. 150, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson, J. R., Bickford, L. C., Morgan, D., Kim, A. S., Ouerfelli, O., Kirschner, M. W. & Rosen, M. K. (2004) Nat. Struct. Mol. Biol. 11, 747–755. [DOI] [PubMed] [Google Scholar]
- 26.Ho, H. Y., Rohatgi, R., Lebensohn, A. M., Le, M., Li, J., Gygi, S. P. & Kirschner, M. W. (2004) Cell 118, 203–216. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Quiles, N., Rohatgi, R., Anton, I. M., Medina, M., Saville, S. P., Miki, H., Yamaguchi, H., Takenawa, T., Hartwig, J. H., Geha, R. S. & Ramesh, N. (2001) Nat. Cell Biol. 3, 484–491. [DOI] [PubMed] [Google Scholar]
- 28.Hatley, M. E., Lockless, S. W., Gibson, S. K., Gilman, A. G. & Ranganathan, R. (2003) Proc. Natl. Acad. Sci. USA 100, 14445–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labno, C. M., Lewis, C. M., You, D., Leung, D. W., Takesono, A., Kamberos, N., Seth, A., Finkelstein, L. D., Rosen, M. K., Schwartzberg, P. L. & Burkhardt, J. K. (2003) Curr. Biol. 13, 1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cannon, J. L. & Burkhardt, J. K. (2002) Immunol. Rev. 186, 90–99. [DOI] [PubMed] [Google Scholar]
- 31.Cannon, J. L., Labno, C. M., Bosco, G., Seth, A., McGavin, M. H., Siminovitch, K. A., Rosen, M. K. & Burkhardt, J. K. (2001) Immunity 15, 249–259. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman, G. R., Nassar, N. & Cerione, R. A. (2000) Cell 100, 345–356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.