

Abstract
Ca2+‐sensor proteins are generally implicated in insulin release through SNARE interactions. Here, secretagogin, whose expression in human pancreatic islets correlates with their insulin content and the incidence of type 2 diabetes, is shown to orchestrate an unexpectedly distinct mechanism. Single‐cell RNA‐seq reveals retained expression of the TRP family members in β‐cells from diabetic donors. Amongst these, pharmacological probing identifies Ca2+‐permeable transient receptor potential vanilloid type 1 channels (TRPV1) as potent inducers of secretagogin expression through recruitment of Sp1 transcription factors. Accordingly, agonist stimulation of TRPV1s fails to rescue insulin release from pancreatic islets of glucose intolerant secretagogin knock‐out(−/−) mice. However, instead of merely impinging on the SNARE machinery, reduced insulin availability in secretagogin−/− mice is due to β‐cell loss, which is underpinned by the collapse of protein folding and deregulation of secretagogin‐dependent USP9X deubiquitinase activity. Therefore, and considering the desensitization of TRPV1s in diabetic pancreata, a TRPV1‐to‐secretagogin regulatory axis seems critical to maintain the structural integrity and signal competence of β‐cells.
Keywords: Ca2+ signalling, β‐cell, diabetes, endocannabinoid, exocytosis
Subject Categories: Molecular Biology of Disease, Signal Transduction
Introduction
Life‐long maintenance of the cellular integrity of Langerhans islets is critical for biphasic hormone release and therefore adequate responses to environmental and metabolic demands. β‐cells produce insulin, and it is their progressive demise that is the causative basis of both type 1 and 2 diabetes (Deng et al, 2004; Ize‐Ludlow et al, 2011). Despite intense research on restorative strategies to facilitate cell turnover or the expansion of the remnant β‐cell pool, available solutions amenable to diabetic conditions remain limited. The lack of breakthroughs is particularly concerning when considering the manifold receptor‐dependent signalling events modulating β‐cell survival.
Changes in intracellular Ca2+ levels regulate many cellular processes within pancreatic β‐cells, ranging from transcription (German et al, 1990), protein folding (Creutz et al, 1994; Lievremont et al, 1997) and insulin exocytosis (Henquin et al, 2002) to the control of cell fate (Skrzypski et al, 2015). Translation of Ca2+ signals into physiologically meaningful output relies, in part, on Ca2+‐sensor proteins, which upon Ca2+ binding undergo conformational changes and can phasically activate or inactivate their specific target partners via protein–protein interactions (Nelson & Chazin, 1998; Shen et al, 2008). To be available in sufficient amounts to decode Ca2+ signals, the intracellular turnover of Ca2+‐sensors themselves may be Ca2+ regulated (van Der Luit et al, 1999), thus allowing precise control over their activity. Therefore, the coupling of receptor‐mediated Ca2+ signalling to transcriptional programmes regulating Ca2+‐sensor availability in β‐cells represents an appealing yet modestly explored niche to integrate β‐cell survival and functionality in type 2 diabetes.
Secretagogin is an EF‐hand Ca2+‐sensor protein, which is amongst the most abundant proteins in pancreatic β‐cells (Wagner et al, 2000). Primarily, secretagogin is thought to interact with cytoskeletal proteins (Maj et al, 2010; Yang et al, 2016), soluble N‐ethylmaleimide‐sensitive‐factor attachment receptor (SNARE) components (e.g. SNAP‐25, synaptobrevins; Rogstam et al, 2007; Romanov et al, 2015) and cargo proteins (Bauer et al, 2011) to modulate regulated insulin exocytosis. Accordingly, secretagogin loss‐of‐function studies in vitro show negative correlation with insulin release (Wagner et al, 2000; Hasegawa et al, 2013; Yang et al, 2016). Secondly, secretagogin has been implicated in the regulation of β‐cell proliferation (Wagner et al, 2000). Therefore, and even if causality between secretagogin and insulin expression and release exists, the SNARE interactome of secretagogin alone is likely insufficient to chiefly account for the progressive loss of pancreatic β‐cells in diabetes. Instead, the analysis of alternative regulatory pathways seems warranted, particularly to map receptor‐to‐gene transcription axes impacting the Ca2+‐dependent regulation of cell turnover.
Here, we combined bulk mRNA expression profiling of pancreatic islets obtained from a human cohort and single‐cell RNA‐seq from diabetic donors to establish an inverse correlation between secretagogin expression and the incidence of type 2 diabetes. To determine the identity of Ca2+‐permeable channels regulating secretagogin function in β‐cells, we focused on TRP family members that can fine‐tune Ca2+‐dependent transcriptional elements. Pharmacological probing of TRP channels revealed that transient receptor potential type 1 vanilloid channels (TRPV1) are particularly poised to induce secretagogin mRNA and protein expression. We then combined in silico models, secretagogin promoter assays, biochemistry and mouse genetics to establish that Sp1‐dependent promoter activation is causal to TRPV1‐mediated secretagogin expression. Next, we generated secretagogin−/− mice that by 6 weeks of age display glucose intolerance, which coincides with the progressive loss of their β‐cell mass. By using global proteomics and histochemical tools, we find that Ca2+‐bound secretagogin can coordinate protein folding and turnover. Accordingly, secretagogin−/− mice exhibit disrupted protein chaperonin activity and significant endoplasmic reticulum (ER) stress. Likewise, secretagogin mRNA levels in pancreatic islets from human diabetic donors negatively correlate with activating transcription factor 4 (ATF4) and CCAAT‐enhancer‐binding protein homologous protein (CHOP) expression. Cellular models and acute genetic manipulations reveal that secretagogin deletion renders its interacting partners, ubiquitin carboxyl‐terminal hydrolases (USP9X and USP7) inactive, thus shifting the balance of β‐cell proliferation/death towards increased apoptosis. Therefore, a ubiquitously Ca2+‐driven TRPV1‐to‐secretagogin transcriptional axis is suggested as a signal transduction pathway whose deregulation can curtail the life‐long structural integrity and functional competence of pancreatic β‐cells.
Results
Secretagogin is a molecular predictor of type 2 diabetes
Secretagogin is a Ca2+‐sensor protein, and when silenced in vitro negatively couples to insulin secretion (Wagner et al, 2000; Hasegawa et al, 2013; Yang et al, 2016). Secretagogin mRNA and protein expression are preferentially associated with endocrine cell identities, including hormone or neuropeptide‐producing cells in brain, pituitary, gastrointestinal tract and pancreas in both human and mouse (Fig EV1A and B; reviewed in: Alpar et al, 2012). More specifically, secretagogin immunoreactivity was seen in glucagon+/α‐cells, insulin+/β‐cells, somatostatin+/δ‐cells and pancreatic polypeptide+/PP‐cells in the islets of Langerhans in mouse (Fig EV1C). Thus, secretagogin is a ubiquitous cellular mark in the endocrine pancreas. Nevertheless, this study only focuses on dissecting secretagogin function in differentiated β‐cells.
Figure EV1. Body‐wide secretagogin distribution in human and mouse.
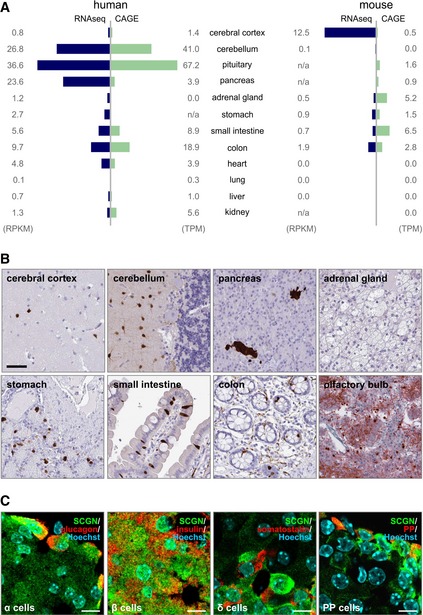
- Secretagogin expression in 12 human and mouse tissue types based on correlated RNA‐seq and cap analysis of gene expression (CAGE) data. Data are expressed either as reads per kilobase of transcript per million mapped reads (RPKM) or tags per million (TPM). In human, secretagogin is mainly expressed in the CNS, endocrine glands (pancreas and pituitary) and the gastrointestinal tract. A similar expression pattern is observed in mouse with the exception of the cerebellum, which lacks secretagogin expression.
- Immunohistochemistry reveals secretagogin expression in a subset of (inter)neuron‐like cells in the cerebral cortex and molecular layer of the cerebellum, endocrine pancreas, enteroendocrine cells of the stomach, small intestine and colon, as well as olfactory bulb of human subjects. Scale bars = 100 μm.
- Secretagogin was detected in glucagon+ α‐cells, insulin+ β‐cells, somatostatin (SST)+ δ‐cells and some pancreatic polypeptide+ cells in adult mouse pancreas. Scale bars = 5 μm.
Despite in vitro probing of its function in exocytosis earlier, the effects of secretagogin deficiency on glucose homoeostasis—particularly in humans—remain unknown. Therefore, we established whether secretagogin could partake in the development of a diabetic phenotype by analysing gene expression data from pancreatic islets of n = 32 subjects diagnosed with type 2 diabetes and n = 170 healthy controls (Fadista et al, 2014). We found a significant negative correlation between secretagogin mRNA expression and the incidence of diabetes (log2FC = −0.42, P = 0.0066; Fig 1A). Higher secretagogin mRNA expression also predicted lower glycated haemoglobin in blood (HbA1c; R 2 = 0.059, P = 0.0097; Fig 1A1). Moreover, a significant positive correlation between secretagogin and insulin mRNA expression (R 2 = 0.2, P = 2.7e−11; Fig 1A2) suggested a role for secretagogin in the hormonal control of glucose metabolism in vivo.
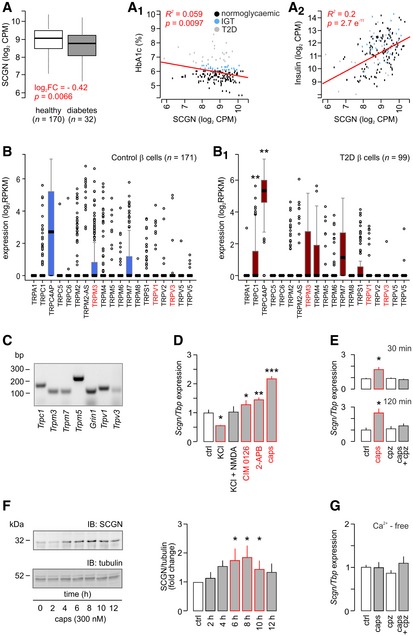
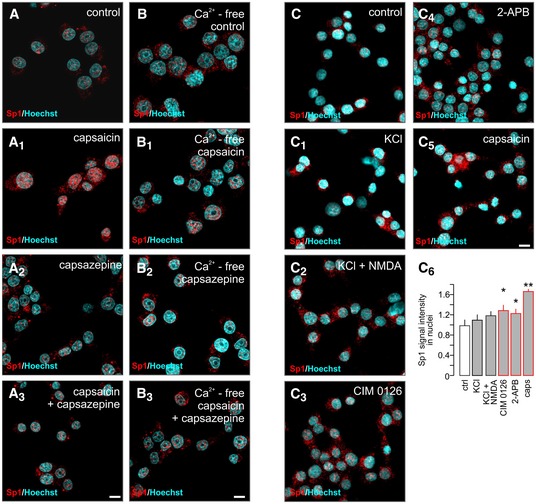
Figure 1. TRP family channels induce secretagogin expression.
-
A–A2Transcriptome analysis (bulk) of human (n = 202) pancreatic islets reveals decreased secretagogin (SCGN) expression in patients with type 2 diabetes (T2D; A). A negative correlation between secretagogin expression and the level of glycated haemoglobin (HbA1c) is shown (A1). Conversely, secretagogin mRNA levels positively correlate with insulin expression (A2). Data were presented as log2 counts per million (CPM). IGT, impaired glucose tolerance. Boxes represent 25th percentiles ± 90th percentiles, horizontal lines represent median values.
-
B, B1Single‐cell RNA‐seq analysis of TRP family members in β‐cells from healthy (n = 6) and type 2 diabetic (n = 4) donors reveals retained TRP expression in β‐cells from diabetic subjects. Data were expressed as log2 reads per kilobase of transcript per million mapped reads (RPKM). Boxes represent 25th percentiles ± 90th percentiles, horizontal lines represent mean values.
-
CReverse‐transcription PCR products of select TRP channels and Grin1 (encoding NMDA receptor subunit 1) in INS‐1E cells.
-
DAcute agonist stimulation (30 min) of TRPM3 (CIM 0126; 1 μM), TRPV3 (2‐APB; 25 μM) and TRPV1 (capsaicin; caps; 300 nM) promotes secretagogin expression in INS‐1E cells with the most pronounced effect evoked by capsaicin. INS‐1E cell depolarization with KCl (30 mM, 30 min) decreases secretagogin expression, which was reversed by NMDA.
-
ECapsazepine, a TRPV1 antagonist (10 μM), occluded capsaicin‐induced secretagogin expression at both 30 and 120 min.
-
FLong‐term (2–12 h) stimulation of INS‐1E cells with capsaicin increases secretagogin protein content. Quantitative data reflect fold changes in SCGN signal intensity normalized to tubulin.
-
GCapsaicin fails to increase secretagogin mRNA expression in Ca2+‐free media. Representative immunoblots are shown.
TRP channels regulate secretagogin mRNA expression
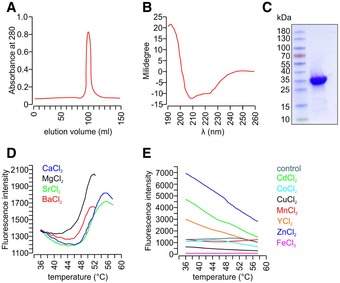
Ca2+‐sensors are regulated by ionic conductances through Ca2+ permeable channels, thus allowing their efficient translation of environmental stimuli into adequately‐tuned physiological cellular responses (Celio, 1990; Schwaller, 2010). Considering that ion binding of any sensor protein is not restricted to a single cation, we first tested secretagogin's metal ion specificity (the basis for being a “Ca2+‐sensor”) using recombinant secretagogin (Fig EV2). We demonstrate seceretagogin's preference for bivalent members of the Beryllium group (Be, Ca, Mg, Sr) for protein‐fold stabilization using the Thermofluor technique (Ericsson et al, 2006), which particularly favours Ca2+ over Mg2+ under quasi‐physiological ion concentrations considering its ~100‐fold higher affinity for Ca2+ (Rogstam et al, 2007).
Figure EV2. Recombinant His6‐tagged secretagogin only binds bivalent ions—calcium and its analogues.
-
ASize exclusion chromatography confirming the purity of recombinant His6‐tagged secretagogin.
-
BProtein folding integrity confirmed by circular dichroism spectroscopy returned the spectrum of a typical all‐α fold as expected based on the known high‐resolution structure of the zebrafish homologue (Bitto et al, 2009).
-
CCoomassie blue staining of purified His6‐tagged secretagogin.
-
D, EMetal ion stabilization by secretagogin was investigated using the Thermofluor method (Ericsson et al, 2006). Typical thermal denaturation was observed in the presence of Ba2+, Ca2+, Mg2+, Sr2+ (all Ca2+‐analogues; D), while the presence of other metal ions (E) resulted in curves where temperature‐dependent unfolding could not be detected. All measurements were done in triplicate; representative results are shown.
We hypothesized that Ca2+ entry could serve as an essential feedback for the expression of this Ca2+‐sensor protein. Therefore, we searched for Ca2+ permeable channels that (i) can work independently or in tandem with glucose‐regulated voltage‐gated Ca2+ channels, which are lowly expressed in β‐cells (Rorsman & Trube, 1986), (ii) their phasic activity can fine‐tune β‐cell Ca2+ signalling, (iii) their ligand levels are changing in type 2 diabetes and (iv) can affect cell proliferation (tissue size) during pancreas development or in pancreatic tumours since secretagogin is re‐expressed upon tumorigenic transformation (Wagner et al, 2000; Birkenkamp‐Demtroder et al, 2005; Lai et al, 2006). An appealing candidate satisfying the above criteria is the transient receptor potential (TRP) family of channels, which are evolutionarily conserved in the endocrine lineage, Ca2+ permeable, and their genetic ablation is implicated in tissue size control (Zhao et al, 2013; Riera et al, 2014; Malenczyk et al, 2015). When screening single‐cell RNA‐seq data in 270 β‐cells from healthy and type 2 diabetic donors (Segerstolpe et al, 2016), we find that 17 TRP family members are expressed (> 0.001 RPKM; Fig 1B) with TRPC1 and TRPC4AP levels significantly increased in diabetes. Expression of most other channels was retained in β‐cells from diabetic donors. We validated mRNA expression for Trpc1, Trpm3, Trpm5, Trpm7, Trpv1 and Trpv3 as representatives of the different TRP subfamilies (Clapham et al, 2001) as well as for Grin1 (Marquard et al, 2015) in INS‐1E cells (Fig 1C). Next, we determined the effect of agonist stimulation of NMDA receptor, TRPM3, TRPV1 and TRPV3 with exogenous ligands (NMDA (20 μM), CIM 0126 (1 μM; Held et al, 2015), capsaicin (300 nM) and 2‐APB (25 μM; Hu et al, 2009), respectively) on secretagogin mRNA expression. We applied 30 mM KCl to induce depolarization‐dependent Ca2+ influx as control. After 30 min, CIM 0126 (P < 0.05), 2‐APB (P < 0.01) and capsaicin (P < 0.001) increased secretagogin mRNA levels with capsaicin producing the most pronounced change (Fig 1D). In turn, KCl‐induced passive Ca2+ overload reduced secretagogin expression, which was modulated by NMDA co‐application (Fig 1D). These data show that temporally confined, receptor‐mediated signalling events produce meaningful signals to induce secretagogin transcription.
TRPV1‐induced secretagogin accumulation in β‐cells is Ca2+ dependent
TRPV1s are particularly interesting since they are rapidly activated by anandamide, other endovanilloids and, for example, capsaicin (Di Marzo & De Petrocellis, 2012). In pancreas, both α‐ and β‐cells express rate‐limiting enzymes of anandamide metabolism and TRPV1s (Akiba et al, 2004; Malenczyk et al, 2013, 2015), which have been functionally implicated in the control of the intracellular Ca2+ level, insulin secretion and β‐cell turnover (Akiba et al, 2004; De Petrocellis et al, 2007; Riera et al, 2014; Malenczyk et al, 2015). Anandamide is present in circulating blood, and its levels significantly increase in obesity and type 2 diabetes (Matias et al, 2006). In juvenile mouse plasma, we measured anandamide concentrations to be 20.4 ± 8.2 nM, superseding pKi for anandamide to activate TRPV1s (5.78 nM; Ross et al, 2001).
We verified TRPV1 involvement in capsaicin‐induced secretagogin mRNA expression (30 and 120 min time‐points are shown; P < 0.05; Fig 1E) by demonstrating its sensitivity to capsazepine (TRPV1 antagonist, 10 μM) pre‐treatment. We could also see the stimulatory effect of capsaicin on protein translation in INS‐1E cells (2–12 h; Fig 1F). Finally, we confirmed the role of Ca2+ in the transcriptional regulation of secretagogin expression by showing that removal of extracellular Ca2+ precluded the TRPV1‐dependent transcription of secretagogin (Fig 1G). Thus, a Ca2+‐dependent mechanism might operate downstream from TRPV1 channels to control secretagogin function in β‐cells.
TRPV1s recruit Sp1 transcription factors to regulate secretagogin transcription
We then analysed the mouse and human secretagogin promoter in silico to predict binding sites for transcription factors potentially involved in its expressional control (Tables EV1 and EV2). Since Ca2+ influx seems indispensable to increase secretagogin mRNA content in INS‐1E cells, we focused on the Ca2+‐dependent transcription factor Sp1 because its consensus binding sites were detected in the putative secretagogin promoter (−1,400 to +1) in both species (Fig 2A and A1). Notably, Sp1 activity has previously been linked to transmembrane channels permeable for bivalent cations, including TRPV1 (Moon et al, 2012).
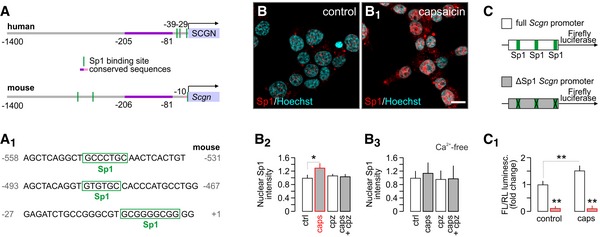
Figure 2. TRPV1 agonism promotes the Sp1‐dependent activity of the predicted secretagogin promoter.
-
A, A1In silico prediction of Sp1 transcription factor binding sites within the human and murine secretagogin promoters (up to −1,400 bps). (A1) Consensus recognition sequences exported from (A).
-
B–B3Capsaicin (caps; TRPV1 agonist; 300 nM for 30 min) induces Sp1 translocation to the nucleus (determined as increased Sp1 immunoreactivity) in INS‐1E cells. Representative images are shown. Hoechst 33342 was used as nuclear counterstain. Scale bar = 5 μm. (B2) This capsaicin effect is blocked by capsazepine (cpz; TRPV1 antagonist, 10 μM). Capsaicin is also ineffective in the absence of extracellular Ca2+ (B3). Representative images for quantitative data are shown in Fig EV3. Data were expressed as means ± s.d. from triplicate experiments with n ≥ 100 cells/group quantified for (B2, B3).
-
C, C1Deletion of predicted Sp1 binding sites in the Scgn promoter (C) abrogates basal and capsaicin‐induced (300 nM for 30 min) promoter activity defined as a ratio of firefly to Renilla luciferase chemiluminescence (3.5 h after stimulation; C1). Data were expressed as means ± s.d. from triplicate experiments.
Acute capsaicin stimulation (300 nM; 30 min) of TRPV1 receptors in INS‐1E cells induced the nuclear translocation of Sp1. This response was capsazepine sensitive, confirming TRPV1 involvement (P < 0.05 vs. control; Figs 2B–B2 and EV3A–A3). Similarly, activation of TRPM3 and TRPV3 led to Sp1 translocation (Fig EV3C–C6) at magnitudes lesser than for TRPV1, corroborating our observations on secretagogin mRNA expression (Fig 1C). Likewise, TRPV1 stimulation under Ca2+‐free conditions occluded Sp1 translocation (Figs 2B3 and EV3B–B3).
Figure EV3. Activation of TRP channels promotes the nuclear translocation of Sp1.
-
A–A3Capsaicin (caps; TRPV1 agonist; 300 nM, 30 min) induces Sp1 translocation to the nuclei of INS‐1E cells (determined as an increased level of Sp1 immunoreactivity). This capsaicin effect is blocked by capsazepine (cpz; TRPV1 antagonist, 10 μM). Scale bar = 5 μm.
-
B–B3Capsaicin is ineffective in the absence of extracellular Ca2+. Scale bar = 5 μm.
-
C–C6CIM 0126 (TRPM3 agonist; 1 μM, 30 min), 2‐APB (TRPV3 agonist; 25 μM, 30 min) and capsaicin promote nuclear Sp1 import, whereas KCl (30 mM, 30 min) alone or in combination with NMDA (NMDA receptor agonist; 20 μM, 30 min) remains ineffective. Representative images are shown. Hoechst 33342 was used as a nuclear counterstain. Scale bar = 5 μm. Data are expressed as means ± s.d.; n ≥ 100 cells/group from triplicate experiments, **P < 0.01, *P < 0.05; pairwise comparisons/one‐way ANOVA.
To define the requirement of Sp1 binding to the enhancement of secretagogin transcription, we generated a construct with the full mouse secretagogin promoter (Scgn‐full, its 1,400 bps 5′ flanking region) driving firefly luciferase in comparison to a construct in which predicted Sp1 binding sites were deleted (Scgn‐ΔSp1; Fig 2C). Capsaicin (300 nM; 30 min) significantly increased luciferase‐induced chemiluminescence in the Scgn‐full construct (P < 0.01 vs. control; Fig 2C1), measured as the ratio of firefly to Renilla luciferase activity. In contrast, Scgn‐ΔSp1 resulted in a substantial decrease in both basal and capsaicin‐stimulated luciferase expression and activity (P < 0.01 vs. Scgn‐full; Fig 2C1), demonstrating the critical contribution of Sp1 transcription factors to regulating secretagogin expression.
Secretagogin links TRPV1 activity to regulated insulin exocytosis
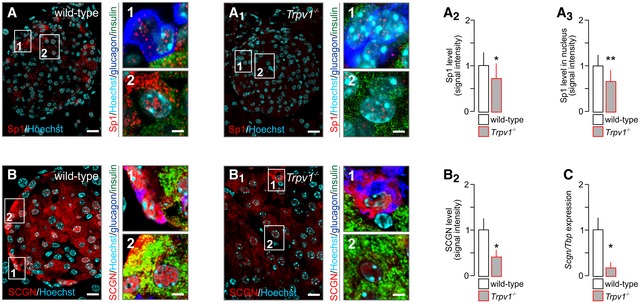
We used Trpv1 −/− mice to confirm Sp1 function downstream from TRPV1s in β‐cells in vivo. We show significantly reduced total and nuclear Sp1 immunoreactivity (P < 0.01 and P < 0.01, respectively; Fig 3A–A3) in Trpv1 −/− mice relative to wild‐type littermates. Coincidently, secretagogin protein (P < 0.05; Fig 3B–B2) and mRNA levels (P < 0.05; Fig 3C) were significantly reduced on a Trpv1 −/− background. Cumulatively, the above data show that endovanilloid signalling in β‐cells promoting Ca2+ influx via TRPV1s can modulate secretagogin expression through an Sp1‐dependent transcriptional mechanism.
Figure 3. TRPV1 deletion in vivo downregulates secretagogin expression.
-
A–A3Reduced immunoreactivity for total (A2) and nuclear (A3) Sp1 in pancreata of Trpv1 −/− mice as compared to wild‐type controls.
-
B–B2Reduced secretagogin (SCGN) immunoreactivity in pancreata from Trpv1 −/− mice. Representative images are shown. Open boxes reflect the general location of numbered insets. Hoechst 33342 was used as nuclear counterstain.
-
CLikewise, Scgn mRNA expression in pancreatic islets isolated from Trpv1 −/− mice is significantly decreased.
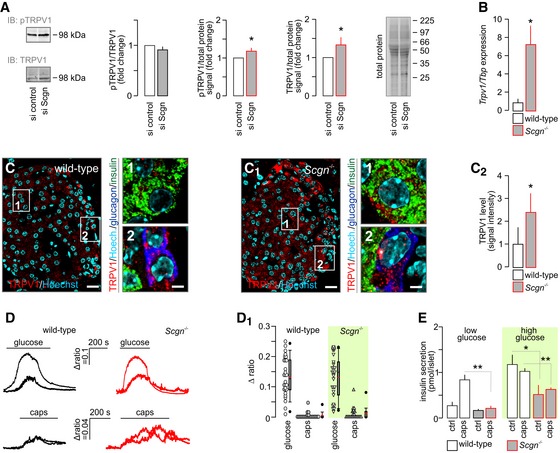
Considering that circulating levels of anandamide, the chief endogenous TRPV1 agonist, are increased in type 2 diabetes (Matias et al, 2006; Starowicz et al, 2008) and lead to TRPV1 desensitization (Zsombok et al, 2011; Sanz‐Salvador et al, 2012; Gao et al, 2012), we asked whether reduced secretagogin levels in Trpv1 −/− mice represent an adaptive response or instead the loss of secretagogin itself can per se precipitate diabetes and modulate TRPV1 signalling. In cultured INS‐1E cells, we found that acute siRNA silencing of secretagogin increased TRPV1 protein levels (P < 0.05; Figs 4A and EV4A and B). Nevertheless, TRPV1 phosphorylation at its Ser800 residue did not change (as the ratio of total TRPV1; Fig 4A) suggesting that excess TRPV1s brought about by secretagogin knock‐down do not confer gain‐of‐function. Next, we generated secretagogin−/− mice (for histological validation of protein loss see: Fig EV4C–E). Notably, secretagogin−/− mice present > sixfold increase in Trpv1 mRNA expression (P < 0.05; Fig 4B), as well as elevated TRPV1 protein levels in pancreatic islets (P < 0.05), particularly in β‐cells (Fig 4C–C2). Therefore, we used Ca2+ imaging to establish whether surplus TRPV1s are functionally competent or, alike our in vitro observations (Fig 4A), are signal deficient. Ca2+ imaging in dissociated pancreatic islets showed no difference in either glucose (16 mM) or capsaicin (300 nM)‐stimulated Ca2+ influx between secretagogin−/− and wild‐type β‐cells (Fig 4D and D1). Furthermore, and despite the upregulation of TRPV1s, capsaicin, (300 nM) stimulation of pancreatic islets isolated from secretagogin−/− mice was insufficient to potentiate basal (2.75 mM glucose) or rescue glucose‐induced insulin release (16.5 mM glucose; both P < 0.01 vs. wild type; Fig 4E). Cumulatively, our data suggest that secretagogin acting downstream of Ca2+ influx is an essential effector linking TRPV1 activity and glucose metabolism to regulated insulin exocytosis in β‐cells, and whose loss can stem insulin release even in the presence of excess TRPV1s.
Figure 4. Compensatory upregulation of TRPV1 expression is insufficient to rescue insulin secretion in secretagogin knock‐out mice.
-
AScgn silencing in INS1‐E cells does not affect TRPV1 phosphorylation (expressed as pTRPV1/TRPV1) despite significantly increasing total TRPV1. Total protein dye‐labelling was used as control. A representative immunoblot is shown. Quantitative data were expressed as fold changes in signal intensity vs. control and normalized to total protein. Data were expressed as means ± s.d. from triplicate experiments.
-
BTrpv1 mRNA expression in secretagogin knock‐out (Scgn −/−) mice. Data were expressed as means ± s.d. from triplicate experiments.
-
C–C2Histochemical detection of TRPV1s in pancreatic islets of Scgn −/− mice confirmed their increased amounts relative to wild‐type littermates (C2). Representative images are shown. Hoechst 33342 was used as nuclear counterstain. Scale bars = 15 μm and 2 μm (numbered inserts). Quantitative results were expressed as means ± s.d. from triplicate experiments; n > 10 islets (C–C2).
-
D, D1In dissociated islets from Scgn −/− and wild‐type mice, β‐cells were identified by their Ca2+ responses to 16 mM glucose (upper traces). Subsequent bath application of capsaicin (caps; 300 nM) barely mobilized intracellular Ca2+ in β‐cells from either genotype (bottom traces). (D1) Quantitative analysis of glucose‐ and capsaicin‐induced Ca2+ responses in wild‐type and Scgn −/− β‐cells are shown as dot density plots combined with box plots representing medians and 10th, 25th, 70th and 90th percentiles. Red lines within box plots represent mean and black median values. Peak values during the first 100 s (for capsaicin) or 300 s (for glucose) stimulation were plotted. Mann–Whitney rank‐sum test failed to reveal statistically significant differences between wild‐type and Scgn −/− samples for either stimulation.
-
ECapsaicin (300 nM) is ineffective in potentiating basal (low glucose) or glucose‐stimulated insulin secretion from pancreatic islets isolated from Scgn −/− mice. Data were expressed as means ± s.d. from n = 30 islets (E) from at least three mice/genotype.
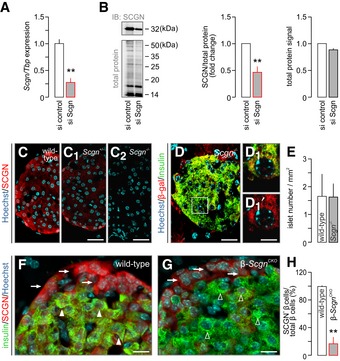
Figure EV4. Validation of secretagogin silencing by siRNA and gene knock‐out strategies.
-
ASignificantly decreased Scgn mRNA expression was observed 48 h after transfection of INS‐1E cells with a targeting Scgn siRNA. TATA‐binding protein (Tbp), a housekeeping gene, was used to normalize gene expression. Data were expressed as means ± s.d. from triplicate experiments; **P < 0.01 (Student's t‐test).
-
BWestern blotting confirmed successful secretagogin silencing by siRNA. Cy5‐labelling of the total protein load was shown to normalize secretagogin data. Representative immunoblot is shown. Quantitative analysis of immunoblots depicts fold changes in signal intensity vs. control and is normalized to the total protein level. Data were expressed as means ± s.d. from triplicate experiments; **P < 0.01 (Student's t‐test).
-
C–C2Decreased level of secretagogin (SCGN) immunoreactivity in pancreatic islets of Scgn +/− mice and the complete lack of secretagogin in pancreatic islets from Scgn −/− mice confirm (i) an allelic dosage effect on protein content and (ii) the specificity of antibodies. Scale bars = 25 μm.
-
D, D1β‐galactosidase (β‐gal) immunosignal in a pancreatic islet of Scgn −/− mice confirm the successful insertion of the LacZ/Δgeo cassette used to disrupt the Scgn gene. Hoechst 33342 was used as a nuclear counterstain. Scale bars = 25 μm and 3 μm in inset.
-
EQuantitative analysis of the pancreas in Scgn −/− mice shows no change in islet number relative to wild‐type controls. Islet numbers were normalized to tissue area. Data were expressed as means ± s.d. from triplicate experiments.
-
F, GLack of secretagogin immunosignals in insulin+ β‐cells (open and closed arrowheads) confirms successful tamoxifen‐induced secretagogin ablation. In contrast, secretagogin expression in α‐cells is retained (arrows) β‐Scgn CKO mice. Scale bars = 10 μm.
-
HQuantitative analysis of the ratio of secretagogin+ β‐cells amongst all insulin+ β‐cells in wild‐type and β‐Scgn CKO mice. Data were expressed as means ± s.d. (percentages) from > 3 mice/genotype; **P < 0.001 (Student's t‐test).
Source data are available online for this figure.
Secretagogin−/− mice develop progressing glucose intolerance
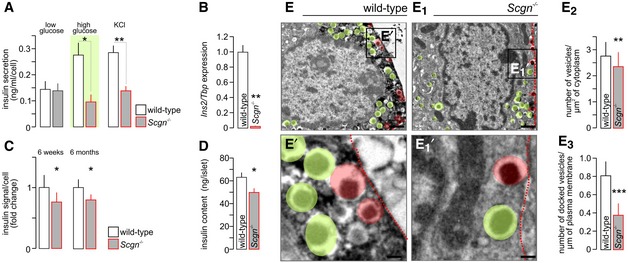
We went on to test whether secretagogin loss‐of‐function may be causal to a diabetic phenotype in vivo. Indeed, secretagogin−/− mice at both 6 weeks and 6 months of age were glucose intolerant as shown by their attenuated blood glucose clearance (Fig 5A). Moreover, secretagogin−/− mice at 6 months of age presented significantly elevated fasting blood glucose levels (P < 0.01; Fig 5A). Notwithstanding, secretagogin−/− mice presented no change in their body weight relative to their wild‐type littermates (17.6 ± 1.5 g [secretagogin−/−] vs. 17.3 ± 1.7 g [wild type] at 6 weeks and 28.7 ± 3.5 g [secretagogin−/−] vs. 28.5 ± 3.7 g [wild type] at 6 months). We then generated β‐cell‐specific secretagogin−/− mice (Ins1‐CreERT::secretagoginfloxed/floxed; “β‐Scgn CKO”) to confirm that the detrimental effect of secretagogin loss on glucose metabolism relies specifically on β‐cell dysfunction. β‐Scgn CKO mice lacked detectable immunoreactivity for secretagogin in > 80% of β‐cells 1 week after daily tamoxifen administration (P < 0.001; Fig EV4F–H) and presented both elevated fasting blood glucose levels (P < 0.01; Fig 5B) and significantly attenuated blood glucose clearance (Fig 5B). Next, we found that pancreatic islets from secretagogin−/− mice, unlike those of their wild‐type littermates, were insensitive to glucose (P < 0.05 vs. wild type) or KCl stimulation (P < 0.01 vs. wild type) ex vivo (Fig EV5A). These data were supported by decreased insulin mRNA expression (P < 0.01; Fig EV5B), decreased intensity of insulin immunoreactivity in individual β‐cells (at both 6 weeks and 6 months, P < 0.05; Fig EV5C) and reduced total insulin content in pancreatic islets (P < 0.05; Fig EV5D) in secretagogin−/− mice. Disrupted insulin production in secretagogin−/− mice was also suggested by the reduced number of insulin granules in β‐cells (P < 0.01; Fig EV5E–E2) with a particularly notable effect on the number of docked vesicles (in direct contact with the plasma membrane of β‐cells; P < 0.001; Fig EV5E3). These data together with previous in vitro interaction analyses of secretagogin and cytoskeletal proteins (tubulin and molecular motors of axonal traffic; Maj et al, 2010; Bauer et al, 2011), as well as components of the exocytosis machinery (Rogstam et al, 2007; Romanov et al, 2015) unequivocally place secretagogin within the SNARE complex.
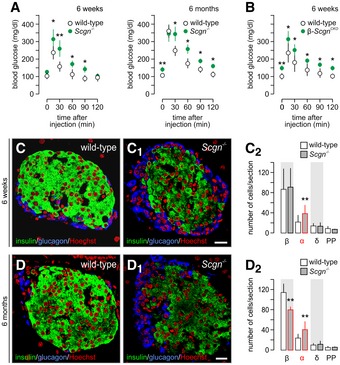
Figure 5. Secretagogin−/− mice develop progressive glucose intolerance and loss of β‐cells.
-
AGlucose intolerance in secretagogin−/− (Scgn −/−) mice at 6 weeks of age (left). Six‐month‐old Scgn −/− mice have a pre‐diabetic profile of glucose utilization (right), including significantly elevated fasting blood glucose levels. n = 6 animals/genotypes/age was used.
-
BElevated fasting blood glucose levels and impaired glucose clearance in β‐Scgn CKO was observed at 6 weeks of age. n = 6 mice/genotype.
-
C–C2At the age of 6 weeks, pancreatic islets of Scgn −/− mice contain an increased number of α‐cells in conjunction with unchanged β‐cell numbers (C2).
-
D–D2At the age of 6 months, however, a significant decrease in β‐cells together with persistent α‐cell hyperplasia was seen in Scgn −/− mice relative to wild‐type littermates. Representative images are shown. Hoechst 33342 was used as nuclear counterstain (pseudo‐coloured in red).
Figure EV5. Decreased insulin content and impaired vesicle docking in β‐cells lead to disrupted insulin secretion in secretagogin−/− mice.
-
APancreatic islets isolated from Scgn −/− mice fail to secrete insulin in response to high glucose (16.75 mM) or KCl (30 mM) stimulation.
-
B–DPancreatic islets isolated from Scgn −/− animals show decreased expression of insulin (Ins2) mRNA (B), reduced intensity of insulin immunoreactivity per β‐cell both at 6 weeks and 6 months of age (C), and decreased insulin content (D).
-
E–E3Electron microscopy of pancreatic β‐cells in glucose challenged mice (30 min post‐injection) reveals a reduction in the total number of insulin granules (E2), including fewer docked vesicles (E3) in β‐cells of Scgn −/− mice (vs. wild‐type controls). Dashed red lines mark the plasmalemma. Green shading highlights insulin‐containing secretory granules. Semitransparent red shading marks docked vesicles (in direct contact with the plasmalemma). Representative ultramicrographs are shown. Scale bars = 1 μm (E, E1) and 150 nm (E′, E1′).
When assaying the number of cell types within pancreatic islets of both 6‐week and 6‐month‐old secretagogin−/− mice, we found an age‐dependent β‐cell loss, which was accompanied with α‐cell hyperplasia (Fig 5C and D). The number of δ‐ and PP‐cells remained unchanged at either time point. Since α‐to‐β‐cell imbalance is a principal cause of progressive glucose intolerance and diabetes (Deng et al, 2004), a phenotype secretagogin−/− mice exhibit by 6 months of age (P < 0.01 vs. wild type at 6 months of age, Fig 5C–D2), we tested whether β‐cell loss is a surrogate of secretagogin loss‐of‐function or instead, secretagogin directly controls β‐cell survival and turnover.
Secretagogin modulates protein folding and degradation through protein–protein interactions
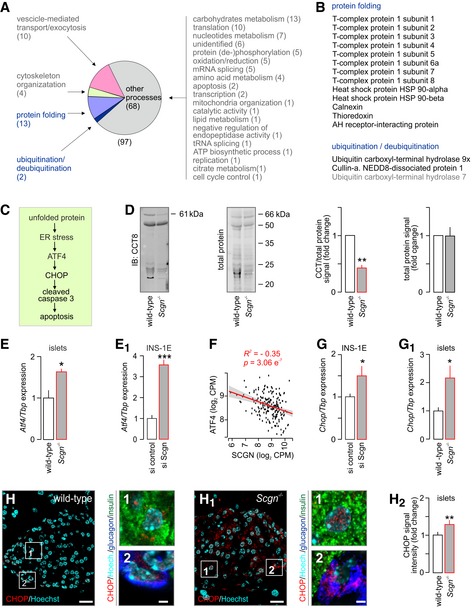
Considering the abundance of secretagogin on, for example, ER membranes (Romanov et al, 2015; Hanics et al, 2017), we screened possible protein–protein interactions unrelated to either the cytoskeleton or SNARE assembly. By using His6‐tagged recombinant secretagogin in the presence of Ca2+ (100 μM), we precipitated putative interacting partners from INS‐1E cells and identified their amino acid sequences by mass spectrometry. We retained canonical interacting proteins participating in vesicle‐mediated transport, exocytosis and cytoskeletal organization as positive controls (Fig 6A, Table EV3). Here, we focused on captured proteins controlling protein folding and (de‐)ubiquitination (Fig 6B) since disrupted protein maturation is particularly detrimental for secretory cells and could lead to ER stress and eventual cell death (Fig 6C). When annotating secretagogin's interacting partners functionally grouped as those with “protein folding” involvement, we find members of the chaperonin‐containing complex (CCT; Fig 6B, Table EV3), known to assist cytoplasmic proteins including actin, tubulin, p53 and c‐Myc to acquire their native and functionally active conformations (Grantham et al, 2002; Koch et al, 2007; Lin et al, 2012; Trinidad et al, 2013). As such, we then probed T‐complex protein 1 subunit 8 (CCT8) in pancreatic islets of secretagogin−/− mice and verified its levels to be significantly reduced (Fig 6D).
Figure 6. Secretagogin interacts with members of the protein folding machinery to control endoplasmic reticulum homoeostasis.
-
AProteins identified as molecular interactors when using purified His6‐tagged secretagogin as bait include (by GO function determination) those for protein folding, vesicle‐mediated transport, exocytosis as well as protein (de‐)ubiquitination. Bracketed numbers refer to the number of proteins per GO cluster. “Other processes” refer to those, which were not analysed in detail. Nevertheless, the identity, discovery rate and peptide fragmentation data for all interacting proteins from our LC‐MS/MS analyses are listed in Table EV3.
-
BList of interacting partners involved in protein folding and protein ubiquitination/deubiquitination.
-
CSchema of predicted steps triggering β‐cell death upon unfolded protein response when genetically ablating secretagogin. Decreased protein folding in β‐cells is posited to induce endoplasmic reticulum (ER) stress, expression of Atf4 transcription factor and the pro‐apoptotic protein CHOP, leading to the activation of caspase 3 and, ultimately, apoptotic cell death.
-
DThe level of T‐complex protein 1 subunit 8 (CCT8) is significantly decreased in pancreatic islets isolated from Scgn −/− mice, as compared to wild‐type controls. Left: Representative immunoblot with adjacent Cy5‐labelled total protein control are shown. Right: Fold change in CCT8 and total protein in Scgn −/− pancreata vs. wild‐type littermates. Data are expressed as means ± s.d. from n = 3 mice/genotype.
-
E, E1Atf4 expression is significantly increased in pancreatic islets isolated from Scgn −/− mice, as compared to wild‐type controls (E) and upon silencing of secretagogin expression (si Scgn) in INS‐1E cells (E1). Data represent means ± s.d. from triplicate experiments.
-
FSecretagogin mRNA levels are negatively correlated with ATF4 expression in human pancreatic islets. Means ± s.d. log2 counts per million (CPM) are shown.
-
G, G1mRNA expression of the pro‐apoptotic protein Chop is significantly increased in INS‐1E cells upon secretagogin silencing (G) and in pancreatic islets isolated from Scgn −/− mice (G1). Data were normalized to those in control/wild type in triplicate experiments (G) or n = 3 mice/genotype (G1) and expressed as means ± s.d.
-
H–H2Increased CHOP immunoreactivity in pancreatic islets of Scgn −/− mice as compared to wild‐type controls. Representative images are shown. Hoechst 33342 was used as a nuclear counterstain. Scale bars = 15 μm, and 2 μm in numbered insets. (H2) Quantitative data are expressed as means ± s.d. n = 20 islets from 3 mice/genotype were analyzed.
Additionally, mRNA expression of Atf4, a typical marker of ER stress (Harding et al, 2003), was significantly increased in islets from secretagogin−/− mice relative to wild‐type littermate controls (P < 0.05; Fig 6E). We validated the above data as being predictive, rather than auxiliary to progressive diabetes in our secretagogin−/− model, by acute siRNA silencing of secretagogin expression in INS‐1E cells (Fig EV4A and B). These experiments (48 h survival after siRNA exposure) confirmed significantly increased Atf4 RNA expression (P < 0.001; Fig 6E1). If reduced secretagogin expression induces ER stress then one can hypothesize a negative correlation between secretagogin and ATF4 mRNA levels in human diabetic subjects. Indeed, we found a negative correlation in human pancreatic islets (R 2 = −0.35, P < 0.001; Fig 6F) emphasizing a likely mechanistic link between secretagogin loss‐of‐function and ensuing ER stress.
Unresolved ER stress and the decreased stability of proteins affecting the cell cycle may lead to apoptosis (Suzuki et al, 2001; Yan et al, 2014). Since the number of β‐cells in pancreatic islets of secretagogin−/− mice is significantly reduced when compared to wild‐type littermates (Fig 5D–D2), we analysed the effect of secretagogin deficiency on the level of CHOP (Fig 6C), which mediates apoptosis (Matsumoto et al, 1996). Chop mRNA expression was significantly upregulated in both INS‐1E cells upon secretagogin knock‐down (P < 0.05; Fig 6G) and in secretagogin−/− islets (P < 0.05; Fig 6G1) relative to their respective controls. These data were extended by showing significantly increased CHOP immunoreactivity in pancreatic islets from secretagogin−/− mice (P < 0.01; Fig 6H–H2) to suggest that ER stress‐related cell death is the likely cause of β‐cell loss in secretagogin−/− mice.
Secretagogin interacts with USP9X in β‐cells to control cell survival
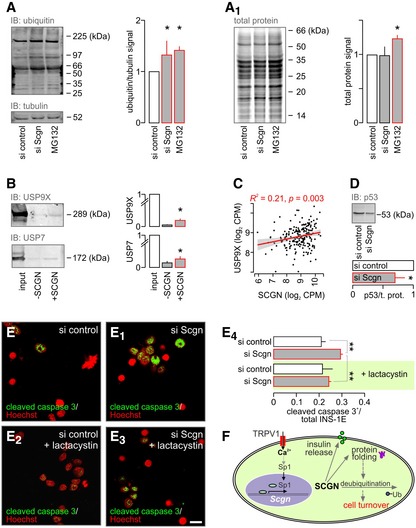
Impaired protein folding, the primary cause of ER stress in β‐cells, is resolved by increased protein degradation through ubiquitination (Allen et al, 2004). Acute silencing of secretagogin expression increased the level of ubiquitinated proteins (total) in INS‐1E cells by ~50%, equivalent to that of the proteasome inhibitor MG132 (10 μM, 6 h; Fig 7A). However, and contrasting the effect of MG132, secretagogin knock‐down did not increase the total protein content of INS‐1E cells (Fig 7A1), suggesting either a reduced rate of protein translation or accelerated proteasomal degradation. Our interactome profiling (Fig 6A and B) suggested that Ca2+‐bound secretagogin could selectively interact with the ubiquitin carboxyl‐terminal hydrolase USP9X, and possibly USP7. Secretagogin's interaction with both USP9X and USP7 was first validated by pull‐down with His6‐tagged secretagogin followed by immunoblot detection (Fig 7B). Secondly, we found a significant correlation between secretagogin and USP9X mRNA expression in our human cohort (R 2 = 0.21, P < 0.001; Fig 7C) lending further support to functional interactions. Subsequently, we hypothesized that secretagogin's interaction with ubiquitin carboxyl‐terminal hydrolases could prime their deubiquitinating/protein stabilizing activity (with enzymatic activity being reduced on null backgrounds). We tested this concept by detecting known USP9X and USP7 substrates involved in cell cycle control: p53, Mdm2, Mcl‐1 (Li et al, 2002; Schwickart et al, 2010). As such, secretagogin knock‐down produced a pronounced decrease in p53 levels in INS‐1E cells (Figs 7D and EV6A), validating that secretagogin could interact with USP9X, and thus modulate its activity.
Figure 7. Secretagogin regulates β‐cell turnover through modulating protein deubiquitination.
-
A, A1(A) Accumulation of ubiqitinated proteins in INS‐1E cells with siRNA‐mediated secretagogin knock‐down. MG132 (10 μM, 6 h), a proteasome inhibitor, was used as a positive control. (A1) In contrast to MG132, secretagogin silencing does not alter the total protein level.
-
BUbiquitin carboxyl‐terminal hydrolases USP9X and USP7 remained in the eluted fraction upon pull‐down with His6‐tagged secretagogin, suggesting protein–protein interaction.
-
CUSP9X mRNA expression positively correlates with the level of secretagogin mRNA in human pancreatic islets.
- D
-
E–E4Proteasome inhibition by lactacystin (5 μM, 6 h) with concomitant siRNA‐mediated secretagogin knock‐down significantly reduces the number of cleaved caspase 3+ (apoptotic) INS‐1E cells. Representative images are shown. Hoechst 33342 was used as a nuclear counterstain (pseudo‐coloured in red). Scale bar = 5 μm.
-
FProposed mechanism of the upstream regulation of secretagogin (SCGN) expression to determine β‐cell survival. TRPV1‐mediated increase in intracellular Ca2+ promotes the translocation of Sp1 to the nucleus to induce Scgn transcription. SCGN not only participates in insulin secretion but also controls protein folding and cell turnover by interacting with ubiquitin carboxyl‐terminal hydrolases.
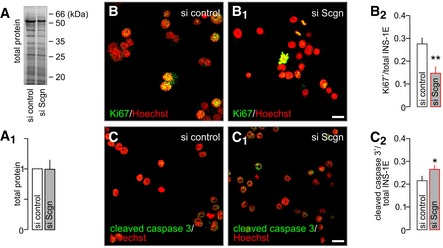
Figure EV6. Secretagogin knock‐down decreases the rate of INS‐1E cell proliferation and promotes apoptosis.
-
A, A1Representative immunoblot showing Cy5‐labelled proteins (“total load”) complementing the analysis of p53 in Fig 7D and allowing quantitation by expression of fold changes vs. control.
-
B–B2siRNA‐mediated silencing of secretagogin expression results in the decreased number of proliferating (Ki67+) INS‐1E cells. Hoechst 33342 was used as a nuclear counterstain (pseudo‐coloured in red).
-
C–C2Simultaneously, secretagogin knock‐down increases the number of INS‐1E cells undergoing apoptosis (cleaved caspase 3+).
To test the direct output of these changes on cell survival, we assayed Ki67 and cleaved caspase 3, markers of cell proliferation and apoptotic cell death, respectively, in INS‐1E cells after secretagogin knock‐down. Secretagogin silencing significantly decreased the number of Ki67+ INS‐1E cells (P < 0.01; Fig EV6B–B2). Simultaneously, the number of cleaved caspase 3+ INS‐1E cells significantly increased (P < 0.05; Fig EV6C–C2). Next, we asked whether inhibition of protein degradation could rescue the detrimental effect of secretagogin knock‐down on INS‐1E cell survival. We found that lactacystin (5 μM, 6 h), a proteasome inhibitor, significantly decreased the number of apoptotic INS‐1E cells which first underwent secretagogin silencing (P < 0.01 vs. siRNA without lactacystin; Fig 7E–E4), which confirmed secretagogin's role in the USP9X‐mediated modulation of protein turnover to impact β‐cell survival (Fig 7F).
Discussion
Our study identifies TRPV1 signalling converging onto secretagogin expression as an important regulatory axis for both β‐cell secretory function and survival. Combining single‐cell RNA‐seq in human β‐cells, studies in the INS‐1E cell line in vitro, Trpv1 and secretagogin knock‐out mouse models and the bulk analysis of gene expression in islets of a human cohort, we show that impairment of this signalling pathway precipitates both molecular and pathophysiological hallmarks of type 2 diabetes, that is impaired insulin secretion (Pfeifer et al, 1981), ER stress (Araki et al, 2003) and progressive loss of β‐cell mass (Butler et al, 2003). We specifically demonstrate that TRPV1 signals through Sp1 to enhance secretagogin promoter activity with resultant increases in secretagogin levels providing a molecular check‐point for protein folding and deubiquitination. Thus, we identify a hierarchical signalling cascade that can operate in parallel with secretagogin modulation of the SNARE (exocytosis) machinery, and ensure fundamentally different cellular consequences.
Ca2+‐sensor proteins are known to assist in protein folding, maintain ER homoeostasis and control cell fate (Heit et al, 2006; Bollo et al, 2010). Calcineurin has been shown to modulate β‐cell proliferation and survival in humans and rodents (Heit et al, 2006; Soleimanpour et al, 2010; Goodyer et al, 2012). Mice with β‐cell‐specific deletion of calcineurin are reported to develop age‐dependent type 2 diabetes due to a decrease in insulin production and loss of β‐cell mass (Heit et al, 2006). The mechanism of calcineurin's action in the control of β‐cell fate depends on the transcriptional regulation of insulin receptor substrate (Irs2; Shirakawa et al, 2013) and is clearly different from the action of secretagogin shown here. Despite calmodulin‐dependent signalling being essential for both proliferation and apoptosis in many cellular niches (Rasmusen & Means, 1990; Yu et al, 2002), its overexpression in β‐cells has been linked to impaired insulin secretion, β‐cell loss and diabetes (Yu et al, 2002). Opposing roles of secretagogin and calmodulin in the control of β‐cell turnover are in line with opposite changes in their protein levels seen in diabetic animal models (Bazwinsky‐Wutschke et al, 2010). Moreover, increased levels of calmodulin observed in diabetes could itself impair secretagogin expression, since Ca2+‐dependent interaction between calmodulin and the C‐terminus of TRPV1 has been shown to promote TRPV1 desensitization and decrease ligand‐induced Ca2+ currents (Numazaki et al, 2003). Thus, secretagogin can be viewed as a “master switch” amongst Ca2+‐sensors that ultimately determines cell fate irrespective of the molecular identity of upstream or secondarily interacting Ca2+‐sensor and/or Ca2+ buffer proteins.
Under metabolic stress, the proper function of protein chaperones and efficient unfolded protein response signalling are essential coping mechanisms that protect β‐cells from apoptosis (Allen et al, 2004). Secretagogin's interaction with the chaperonin‐containing complex, and a decreased CCT8 protein level in pancreatic islets of secretagogin−/− mice implicate secretagogin in participating in the regulation of protein folding. Importantly for this study, the activity of chaperonins is essential for many cytoskeletal proteins including α‐ and β‐ actin, α‐, β‐ and γ‐tubulin, and cell cycle regulators, that is p53 to acquire their functional conformation (Dekker et al, 2008; Trinidad et al, 2013). Considering a crucial role of cytoskeletal components in insulin transport and exocytosis in β‐cells (Henquin et al, 2012), misfolding of such proteins may be a key factor behind impaired insulin secretion in secretagogin−/− pancreatic islets.
Protein ubiquitination/deubiquitination and the proteasome machinery are crucial to control numerous functions in β‐cells including insulin secretion and the cell cycle (Ardestani et al, 2014; Sugawara et al, 2014; Suriben et al, 2015). Protein degradation can directly (e.g. by the control of Mcl‐1, p53, cyclins or caspases; Glotzer et al, 1991; Suzuki et al, 2001; Li et al, 2002; Yan et al, 2014) or indirectly (e.g. by removing misfolded proteins and decreasing ER overload; Sugawara et al, 2014) regulate cell fate. Impairment in the ubiquitin‐proteasome system in pancreatic islets has been directly linked to β‐cell dysfunction and the incidence of type 2 diabetes in humans (Bugliani et al, 2013). Here, we show secretagogin mRNA levels to closely correlate with ubiquitin carboxyl‐terminal hydrolase USP9X expression in humans. In experimental models, Ca2+‐bound secretagogin interacts with both USP9X and USP7, promoting their deubiquitinating activity. Notably, USP7 and USP9X chiefly control the levels of proteins determining cell cycle progression and apoptosis (e.g. p53, Mcl‐1 PCNA, Chk1; Li et al, 2002; Yan et al, 2014; Kashiwaba et al, 2015; Lecona et al, 2016; McGarry et al, 2016). Consequently, the decreased enzymatic activity of ubiquitin carboxyl‐terminal hydrolases results in increased ubiquitination and degradation of proteins essential for cell survival. These facts link secretagogin deficiency to β‐cell apoptosis, particularly since acute inhibition of the proteasome is sufficient to recover β‐cell viability upon silencing of secretagogin expression in vitro.
Pancreatic β‐ and α‐cells express key enzymes for anandamide metabolism (Akiba et al, 2004; Malenczyk et al, 2013, 2015). Moreover, substantial levels of circulating anandamide are found in blood (Matias et al, 2006). Taking these data together, both autocrine and paracrine activation of TRPV1s present on β‐cells could be hypothesized to regulate secretagogin expression. In fact, autocrine events or lateral ligand diffusion in β‐cell membranes might be favoured when considering that anandamide uses an intracellular site to bind the TRPV1 receptor (van der Stelt et al, 2005). Notably, endocannabinoids and endovanilloids acting on TRP channels have been shown to regulate intracellular Ca2+ level (De Petrocellis et al, 2007), pancreas development (Malenczyk et al, 2015), β‐cell turnover (Skrzypski et al, 2015) and insulin secretion (Akiba et al, 2004). Despite increased endocannabinoid levels, including anandamide, in obesity and diabetes (Matias et al, 2006; Starowicz et al, 2008), secretagogin expression in these conditions is downregulated (Hasegawa et al, 2013). As shown in Trpv1 −/− mice, chronically increased anandamide levels can lead to desensitization and/or intracellular delocalization and degradation of TRPV1s (Rossi et al, 2011; Sanz‐Salvador et al, 2012; Chen et al, 2015) and consequently disrupt secretagogin expression, thus priming β‐cell death. Importantly, physiological concentrations of anandamide are expected to serve as modulatory tonic stimuli for TRPV1‐dependent Ca2+ influx and promote “on‐demand” expression. It is noteworthy that Trpv1 −/− mice have been shown to preserve healthy glucose metabolism (Riera et al, 2014) unless subjected to high fat diet (Lee et al, 2015). We found that the number of pancreatic islet in Trpv1 −/− mice is unchanged compared to their wild‐type littermates (data not shown). Yet, as we previously showed, the average size of islet in Trpv1 −/− mice is increased due to a higher β‐cell number (Malenczyk et al, 2015). These changes could be seen as compensatory to the chronic reduction of secretagogin levels in Trpv1 −/− β‐cells. Thus, the results presented here also provide an alternative and equally beneficial secondary target for capsaicin patches clinically indicated for pain relief associated with diabetic neuropathy.
Undoubtedly, Ca2+ influx in β‐cells does not exclusively depend on the activity of TRPVs. Instead, the activity of other receptors and channels such as TRPCs (Qian et al, 2002), TRPMs (Cheng et al, 2007), NMDA receptors (Inagaki et al, 1995) and M3 muscarinic receptors (Molina et al, 2014) that move Ca2+ across the plasmalemma, likely jointly regulates secretagogin expression. Glucose metabolism‐dependent activity of voltage‐gated L‐, P/Q‐ and T‐type Ca2+ channels is considered the primary factor affecting Ca2+ homoeostasis in pancreatic β‐cells (Braun et al, 2008). Interestingly, a physiological glucose concentration (~5 mM) has been shown to most significantly, yet moderately stimulate the activity of the secretagogin promoter in insulinoma cells (Skovhus et al, 2006). This finding, particularly considering essential ramifications of the continued presence of secretagogin in β‐cells is compatible with a persistent demand on secretagogin expression. Thus, if one places secretagogin as a “master regulator” of β‐cell function and survival, then manifold signalling pathways likely converge on this Ca2+‐sensor to maintain its steady‐state levels, particularly under pathophysiological conditions of hypo‐ or hyperglycaemia. The concept that the expressional upregulation of secretagogin transcription is sensitive to rapid and phasic elevations in intracellular Ca2+ is supported by the finding that KCl inhibits, while any Ca2+‐permeable receptor we tested augments this regulatory step. Therefore, we view secretagogin as a key intracellular regulator that can respond to regionalized and transient Ca2+ elevation, which arises upon glucose‐independent signalling events to superposition onto glucose metabolism‐driven signal transduction cascades. Overall, our findings of a TRPV1‐to‐secretagogin signalling axis in genetic models and humans offers a novel and tangible therapeutic target for restoring β‐cell mass and function in diabetes.
Materials and Methods
Cell lines
INS‐1E cells (Merglen et al, 2004) were cultured at 37°C in RPMI‐1640 medium supplemented with glutamine (2 mM), glucose (11 mM), HEPES (10 mM), heat‐inactivated foetal bovine serum (FBS; 5%), sodium pyruvate (1 mM), β‐mercaptoethanol (50 μM), penicillin (50 μg/ml) and streptomycin (100 μg/ml). Cells were routinely sub‐cultured in 24‐well plates up to passage 120, and allowed to reach ~80% confluence. Knock‐down of secretagogin expression was achieved by the co‐application of secretagogin siRNA (50 nM; Santa Cruz) and jetPRIME transfection reagent (polyplus‐transfection SA) for 48 h. Scrambled siRNA (50 nM, Santa Cruz) was used as control. Ca2+‐free conditions were established by addition of EGTA (1 mM) to modified Krebs‐Ringer bicarbonate HEPES buffer containing (in mM): 135 NaCl, 3.6 KCl, 0.5 MgCl2, 1.5 NaH2PO4, 5 NaHCO3, 10 HEPES at pH 7.4.
Generation of secretagogin−/− and β‐Scgn CKO mice
Secretagogin−/− (Scgn −/−) mice (Hanics et al, 2017) were custom‐generated at MMRRC (University of California, Davis, Mouse Biology Program) using the “two‐in‐one” targeting strategy (Skarnes et al, 2011), which generates full knock‐outs by expressing a termination signal after exon 3 of the secretagogin gene. The ensuing truncated protein terminates before the first EF‐hand domain, excluding Ca2+‐binding activity. To generate β‐cell‐specific secretagogin knock‐out mice (β‐Scgn CKO), Scgn floxed/floxed mice were bred with Ins1‐CreERT mice (Jackson #024709). β‐Scgn CKO and wild‐type mice (4‐week old) were intraperitoneally injected with tamoxifen daily over a period of 5 days (200 mg/kg b.w. freshly dissolved in corn oil, both from Sigma; Tamarina et al, 2014). Secretagogin deletion was confirmed a week after the last tamoxifen injection (Fig EV4F–H). Animals (including Trpv1 −/− mice, Jackson #3770) were kept under standard housing conditions with a 12‐h/12‐h light/dark cycle and food and water available ad libitum. Experiments on live animals conformed to the European Communities Council Directive (86/609/EEC) were approved by regional ethical committees and regulated by applicable local laws (Tierversuchsgesetz 2012, BGBI, Nr. 114/2012, Austria). Particular effort was directed towards minimizing the number of animals used and their suffering during experiments.
Isolation of pancreatic islets
Islets of wild‐type and secretagogin−/− mice were obtained after perfusion of their pancreata with Hank's balanced salt solution (HBSS; Invitrogen/Gibco) containing collagenase (type I, 0.33 mg/ml; Sigma) and HEPES (25 mM) followed by purification on Histopaque 1077 gradients (Sigma). After repeated washes in HBSS containing 10% FBS, isolated islets were maintained in RPMI‐1640 medium supplemented as above in humidified atmosphere (5% CO2) at 37°C overnight prior to the commencement of hormone release measurements, assessment of gene expression or protein levels.
Calcium imaging
Single cells were prepared by isolation of pancreatic islets using the papain dissociation system (Worthington) with some modifications: 300 islets were incubated in 500 μl papain solution in EBSS supplemented with 0.005% DNase at 37°C for 20 min. Then, single cells were plated on 12‐mm coverslips coated with matrigel (Corning) in complete RPMI‐1640 (1:2 v/v ratio) and subsequently cultured in RPMI‐1640 medium supplemented as above in humidified atmosphere (5% CO2) at 37°C. Twenty‐four hours later, Ca2+ responses from isolated pancreatic cells were recorded using the Ca2+ indicator Fura‐2AM (Invitrogen). Ratiometric imaging was performed by using a VisiChrome monochromator (Visitron Systems) and CoolSnap HQ2 back‐cooled camera (Photometrics) mounted onto a Zeiss Axiovert microscope equipped with a water‐immersion 40×/differential interference contrast objective (Plan‐Apochromat/N.A. 1.0). Ca2+ responses were recorded in the VisiView software (Visitron Systems) and statistically analysed in SigmaPlot 13.0 (Systat Software Inc.). Krebs‐Ringer solution with low glucose was chosen as basic external solution and contained (in mM): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 1.5 CaCl2 and 1.5 MgCl2, 20 HEPES, 3 glucose (pH 7.4). Experiments were performed at 25°C.
Molecular pharmacology
The effect of the following drugs, alone or in combination, on the subcellular localization and/or protein levels of Sp1, secretagogin, ubiquitinated proteins, apoptotic (e.g. cleaved caspase 3), proliferation (e.g. Ki67) and cell cycle (e.g. p53) markers as well as insulin secretion in INS‐1E cells or pancreatic islets was assessed: 2‐aminoethoxydiphenylborane (2‐APB; non‐selective TRPV3 agonist, 25 μM), (E)‐N‐[(4‐hydroxy‐3‐methoxyphenyl)methyl]‐8‐methyl‐6‐nonenamide (capsaicin; TRPV1 agonist, 300 nM), N‐[2‐(4‐chlorophenyl)ethyl]‐1,3,4,5‐tetrahydro‐7,8‐dihydroxy‐2H‐2‐benzazepine‐2‐carbothioamide capsazepine; TRPV1 antagonist, 10 μM), 3,4‐dihydro‐N‐(5‐methyl‐3‐isoxazolyl)‐α‐phenyl‐1(2H)‐quinolineacetamide (CIM 0126; TRPM3 agonist, 1 μM), (2R,3S,4R)‐3‐hydroxy‐2‐[(1S)‐1‐hydroxy‐2‐methylpropyl]‐4‐methyl‐5‐oxo‐2‐pyrrolidinecarboxy‐N‐acetyl‐L‐cysteine thioester (lactacystin; proteasome inhibitor, 5 μM), N‐[(phenylmethoxy)carbonyl]‐L‐leucyl‐N‐[(1S)‐1‐formyl‐3‐methylbutyl]‐L‐leucinamide (MG132; proteasome inhibitor, 5 μM) and N‐methyl‐D‐aspartate (NMDA; NMDA receptor agonist 20 μM).
Production and purification of His6‐human secretagogin
Expression constructs for human secretagogin (UniProt ID: O76038) included amino acids 1–276. The coding sequence was amplified by PCR using appropriate primers, Pfu‐Turbo polymerase (Stratagene) and cDNA as template and cloned by a ligation independent method into a pNIC28Bsa4 expression vector (GenBank accession no. EF198106) providing a cleavable N‐terminal His6‐tag (H2N‐MHHHHHHSSGVDLGTENLYFQ*S). Protein was expressed in E. coli BL21 (DE3) at 21°C in Luria–Bertani broth (LB) supplemented with 30 μg/ml kanamycin. Protein expression was induced by the addition of 0.1 mM isopropyl β‐D‐1‐thiogalactopyranoside at OD600 of 0.5–0.6. Cells were harvested 20 h post‐induction, washed in 0.3 M NaCl and 10 mM Tris–HCl (pH 8.0) and disrupted by lysozyme (40 μg/ml) and DNaseI (2 μg/ml) treatment followed by sonication. Recombinant His6‐hSGCN was purified by affinity chromatography using His‐Pur™ Ni‐NTA resin (Thermo Scientific) and eluted with an increasing imidazole gradient. Using a desalting column (HiPrep‐26/10; Pharmacia Biotech), the elution buffer was exchanged to 0.15 M NaCl and 25 mM Tris–HCl (pH 8.0). Size exclusion chromatography was then performed (Fig EV2A and C) on a Superdex‐200 column (Pharmacia Biotech) equilibrated with 0.15 M NaCl and 25 mM Tris–HCl (pH 8.0). Pure protein samples were concentrated using a Centriprep YM‐10 centrifugal device (Millipore) to 21.2 mg/ml. Protein concentration was determined according to Bradford using Pierce‐Thermo reagents and bovine serum albumin (BSA) as standard. Samples were analysed by SDS–polyacrylamide gel electrophoresis, ESI‐MS mass spectrometry and their folding integrity was confirmed by circular dichroism spectroscopy (Fig EV2B). Protein samples were aliquoted in a buffer containing 0.15 M NaCl and 25 mM Tris–HCl (pH 8.0), flash‐frozen in liquid N2 and stored at −80°C.
In silico transcription factor binding site prediction and firefly luciferase assay
A 1,400‐bp fragment (−1,400 to +1) of the 5′ flanking region of both the murine (NC_000079.6) and human (NC_000006.12) secretagogin genes was analysed for potential transcription factor binding sites using the open‐label PROMO software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_6.4). For activity determination of the murine secretagogin promoter, its 1,400‐bp 5′ flanking region (−1,400 to +1) referred to as full Scgn promoter or a deletion mutant lacking predicted Sp1 binding sites at bps −548 to −542, −485 to −480 and −11 to −3 (Fig 2C) were custom‐synthesized (Eurofins Genomics) by cloning between XhoI and NcoI restriction sites into a pGL3‐Basic vector (Promega) also containing a firefly luciferase cassette. Subsequently, INS‐1E cells cultured in 96‐well plates (50,000/well) were co‐transfected with either construct and a pCMV‐green Renilla luciferase vector (Thermo Fisher; as control) using jetPRIME transfection reagent (Polyplus‐transfection SA). Eighteen h later, cells were stimulated with capsaicin (300 nM) for 30 min. After an additional 3.5 h, firefly and Renilla luciferase lumiensence was measured using the Dual‐Luciferase® Reporter Assay System (Promega) on a GloMax® luminometer (Promega). Data were analysed as a ratio of firefly to Renilla luciferase lumiensence intensity and normalized to values from unstimulated control cells.
RNA isolation and gene expression analysis
Total RNA from INS‐1E cells and pancreatic islets was isolated using the RNeasy Mini Kit (Qiagen) followed by DNase digestion and verifying RNA integrity on 2% agarose gels (500 ng RNA). cDNA was prepared by reverse transcription with random primers using the High‐capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed using 2× Master mix (Promega) and specific primer pairs (Table EV4). PCR products were resolved on 2% agarose gels and imaged on a ChemiDoc XRS+ system (Bio‐Rad). Quantitative PCR analysis (CFX Connect, Bio‐Rad) was performed using 50 ng of cDNA template, iTaq Universal SYBR Green Supermix (Bio‐Rad) and specific primer pairs (Table EV4).
Western blotting
Proteins were extracted from INS‐1E cells or isolated pancreatic islets using a radioimmunoprecipitation assay buffer containing (in mM): 50 Tris (pH 7.4), 150 NaCl, 10 NaF, 5 EDTA (pH 8.0), 1 Na3VO4, 1 PMSF, 1% TX‐100, pepstatin A (5 μg/μl), leupeptin (10 μg/μl) and aprotinine (2 μg/μl), optionally supplemented with MG132 (Tocris; 10 μM; to inhibit protein deubiquitination). Protein lysates were centrifuged at 12,000 g at 4°C for 10 min. For the analysis of total protein levels, lysates were pre‐incubated with carbocyanine (Cy) 5 labelling buffer (GE Healthcare) for 10 min. Samples were denatured in loading buffer (GE Healthcare) and boiled at 95°C for 5 min. Proteins were probed by loading 20 μg aliquots under denaturing conditions on SDS–PAGE (13.5% or 8–18% gradient gels), followed by wet transfer onto PVDF membranes. Primary antibodies were listed in Table EV5. After exposure to Cy3‐ or Cy5‐conjugated secondary antibodies (GE Healthcare, 1:2,500; 1 h), target proteins were visualized using an automated Amersham WB system (GE Healthcare).
Tissue preparation, immunocytochemistry and immunohistochemistry
For immunocytochemistry, cells were plated on 12‐mm coverslips coated with poly‐D‐lysine (0.001%), washed in 0.1 M phosphate buffer (PB, pH 7.4) and immersion fixed in 4% paraformaldehyde (PFA) for 20 min. Mice were transcardially perfused with ice‐cold 0.1 M PB, followed by 4% PFA in PB (100 ml at 3 ml/min flow speed). Pancreata were rapidly dissected and post‐fixed in 4% PFA overnight. After equilibrating in 30% sucrose for 48–72 h, tissues were cryosectioned at a thickness of 14 μm (Leica CM1850) and thaw‐mounted onto SuperFrost+ glass slides. After rinsing in 0.1 M PB, specimens were exposed to a blocking solution composed of 0.1 M PB, 10% normal donkey serum, 5% BSA and 0.3% TX‐100 for 3 h followed by overnight incubation with select combinations of primary antibodies (Table EV5). Cy2, 3 or 5‐conjugated secondary antibodies (1:300, Jackson) were applied in 0.1 M PB supplemented with 2% BSA (20–22°C, 2 h). Nuclei were routinely counterstained with Hoechst 33342 (1:20,000; Sigma). Cells and pancreas sections were imaged on a Zeiss LSM880 laser‐scanning microscope equipped with a Plan‐Apochromat 40×/1.3 oil immersion objective at 1.0–2.5× optical zoom. Images were acquired in the ZEN2010 software package. Quantitative analysis was performed using ImageJ 1.45 with appropriate plug‐ins. Multipanel images were assembled in CorelDraw X7 (Corel Corp.).
Electron microscopy
Male mice (n = 3/group) were fasted overnight (~15 h) with water available ad libitum. Thirty min after intraperitoneal glucose challenge (2 g/kg body weight), mice were euthanized by cervical dislocation and transcardially perfused with 0.9% NaCl, followed by fixative (4% PFA, 0.1% glutaraldehyde, 15% picric acid) in PB (3 ml/min flow speed). Pancreata were then dissected out and post‐fixed overnight in the same fixative. After vigorous washing in ice‐cold PB, vibratome sections were cut (40–60 μm) containing pancreatic islets. Sections were repeatedly washed in PB, cryoprotected in 30% sucrose and subsequently freeze–thawed three times in liquid N2. After extensive washing in PB, slices were incubated with H2O2 (1%, 20–22°C, 20 min) to block endogenous peroxidase activity. After washing again with PB, sections were incubated with rabbit anti‐insulin antibody (1:400; 4°C, 48 h). Sections were extensively washed, incubated with biotinylated horse anti‐mouse secondary antibody (1:250; Vector Labs., 20–22°C, 2 h), rinsed again and then exposed to pre‐formed avidin biotin‐peroxidase complexes (1:200; Vector, 2 h) and developed with 3,3′‐diaminobenzidine (5 mg/ml, 0.03% H2O2 as substrate). Sections were then osmificated (1% OsO4 in PB, 15 min), dehydrated in increasing ethanol concentrations followed by flat embedding in Durcupan (FLUKA, ACM). Ultrathin sections (60 nm) were cut on a Leica ultramicrotome, collected on Formvar‐coated single‐slot grids, and analysed with a Tecnai 10 electron microscope (FEI). Investigators were blinded to the experimental groups during the entire procedure. Quantitative analysis (n = 30 cells/group) was performed using ImageJ 1.45 with appropriate plug‐ins. The total number of insulin‐containing secretory vesicles was normalized to the cytoplasm surface. Insulin granules directly contacting the plasmalemma (Fig EV5E′ and E1′) were considered as docked, and their number was normalized to the length of the plasma membrane (expressed as vesicle/μm).
Pull‐down assay
Sixty micro gram of His6‐tagged secretagogin and INS‐1E cell lysate (400 μg of protein) was mixed in binding buffer (pH 7.4) containing (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2, 10 imidazole to obtain a 100‐μl total sample volume, and incubated together with 40 μl of pre‐equilibrated Ni‐NTA beads in Spin‐X Centrifuge Tube Filter 0.22 μm cellulose acetate columns (Costar) at 20–22°C for 30 min. In control experiments, His6‐tagged secretagogin was replaced with binding buffer alone to obtain 100 μl of total sample. Flow‐through fractions were removed after spinning at 1,500 g for 30 s. Beads were washed (3×) with 100 μl buffer (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2, 50 imidazole. Proteins were eluted with 70 μl elution buffer composed of (in mM): 25 Tris–HCl, 300 NaCl, 0.1 CaCl2 and 300 imidazole. Samples were stored at −80°C until processing for mass spectrometry.
LC‐MS/MS analysis of protein complexes including statistics
Eluted proteins (17.5 μg) of His6‐secretagogin (n = 3) and control pull‐downs (n = 3) were digested with trypsin via filter‐aided sample preparation (Wisniewski et al, 2009) with minor modifications (Aradska et al, 2015). Tryptic digests were desalted and concentrated on customized reversed‐phase C18 stage tips (Rappsilber et al, 2007). Purified peptides were reconstituted in 5% formic acid (FA), and 2 × 1 μg of each sample (technical replicates) was analysed by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). Peptides were separated on an Ultimate 3000 nanoRSLC system (Thermo Fisher) equipped with a PM100‐C18 pre‐column (3 μm, 75 μm × 20 mm) and a PepMap‐C18 analytical column (2 μm, 75 μm × 500 mm). Ten μl of each sample was loaded onto the trap column for 5 min using 0.1% FA in water at a flow rate of 5 μl/min. Afterwards, peptides were forward‐flushed to and separated on the analytical column at a flow rate of 300 nl/min using a 55‐min gradient ranging from 5 to 37.5% solvent B, followed by a 5‐min gradient from 37.5 to 50% solvent B and finally, to 90% solvent B for 5 min before re‐equilibration to 5% solvent B (solvent A: 0.1% FA in water; solvent B: 0.1% FA in 80% acetonitrile). Eluted peptides were in‐line analysed on a Thermo Q‐Exactive Plus mass spectrometer (Thermo Fisher) in positive ion mode. MS scans were performed in a range of m/z 380–1,800 at a resolution of 70,000. In a data‐dependent acquisition mode, the 20 most intense precursor ions were selected and fragmented via higher energy collisional dissociation at 27% normalized collision energy with a fixed first mass of 100 m/z. Fragment ions were detected at a resolution of 17,500. Dynamic exclusion of selected peptides was enabled for 60 s, and maximal accumulation times were set to 100 and 50 ms in MS and MSn modes, respectively. To describe the secretagogin‐specific interactome, all MS raw data files were analysed by the open‐source software MaxQuant 1.5.3.30 (Cox & Mann, 2008) as previously described (Smidak et al, 2016). Proteins were identified against the Swiss‐Prot rat (Rattus norvegicus) reference proteome database (as of November 2015; 31,457 entries) permitting a mass tolerance of 5 ppm and 20 ppm for MS and MS2 spectra, respectively. Maximum two missed cleavages were allowed and minimum two peptide identifications per protein were required. Carbamidomethylation of cysteines was set as fixed modification while methionine oxidation and N‐terminal protein acetylation were chosen as variable modifications. For both peptide and protein identification, the false discovery rate (FDR) was set to 0.01. MaxQuant results were further processed using the Perseus statistical package (version 1.5.4.1) to identify interactors from label‐free data (LFQ intensities). Proteins were filtered for reversed sequences, contaminants, and if they were only identified by site. Intensity values were log2‐transformed and proteins were further filtered for having LFQ values in minimum three samples at least in one group (either in the target or in the control). Zero intensities were input and replaced by normal distribution, and secretagogin pull‐downs were compared to control samples via two‐sided Student's t‐test with P < 0.05 (applying permutation‐based FDR for truncation). Proteins with a minimum twofold increase of their LFQ intensities in the target pull‐down were considered as specific for secretagogin if they were identified by at least two unique peptides.
LC‐MS/MS analysis of endocannabinoid contents
Extraction, purification and quantification of anandamide from the blood of embryos at embryonic day 18.5 followed published protocols (De Marchi et al, 2003; Matias et al, 2006). After lipid extraction and pre‐purification on silica gel columns, anandamide levels from three animals were analysed by isotope dilution using liquid chromatography‐atmospheric pressure chemical ionization‐mass spectrometry (Shimadzu LCMS‐2020).
Insulin secretion and total insulin content ELISA assay
Pancreatic islets were incubated in drug‐free modified Krebs‐Ringer bicarbonate HEPES buffer (KRBHB) containing (in mM): 135 NaCl, 3.6 KCl, 1.5 CaCl2, 0.5 MgCl2, 1.5 NaH2PO4, 5 NaHCO3, 10 HEPES (pH 7.4) and BSA (0.1%) supplemented with 2.75 mM glucose for 1 h. Subsequently, samples were transferred to microcentrifuge tubes at a density of 10 islets/tube. Islets were incubated in modified KRBHB supplemented with 2.75 mM glucose with or without 30 mM KCl or 16.5 mM glucose for 30 min. Alternatively, after overnight culturing, islets (30 islets/group) were homogenized in water using 27G needles. Homogenates were mixed with 0.18 M HCl in 96% EtOH (1:3 v/v ratio), incubated at 4°C for 16 h and centrifuged at 1,500 g at 4°C for 15 min. Supernatants were stored at −80°C until processing. Insulin levels were measured by a rat/mouse insulin ELISA Kit (Millipore) with insulin concentrations normalized to either the number of islets or β‐cells.
Glucose tolerance test
Male mice (n = 6/group) were fasted overnight (~15 h) with water available ad libitum. Blood glucose level was measured from tail blood using a FreeStyle Lite glucose meter (Abbott Diabetes Care) before (baseline, 0 min) as well as 15, 30, 60, 90 and 120 min after intraperitoneal glucose challenge (2 g/kg body weight). Data were expressed as mg/dl blood glucose.
RNA‐seq and histochemistry‐based body‐wide mapping of secretagogin distribution
Secretagogin expression in nervous and peripheral tissue in both human and mouse was determined based on (i) open‐source RNA‐seq data and (ii) cap analysis of gene expression (CAGE). RNA‐seq data in human was directly imported from the Genotype‐Tissue Expression Portal (GTEx; GTEx Consortium et al, 2013) while mouse RNA‐seq data were obtained from the ENCODE project repository (Kundaje et al, 2015; Fig EV1A, Table EV6). Secretagogin transcription start sides were quantified based on FANTOM CAGE data (Carninci et al, 2005) using the ZENBU genome browser (http://fantom.gsc.riken.jp/zenbu/). Antibody‐based protein distribution maps in human and mouse tissues were extracted from the Human Protein Atlas project (www.proteinatlas.org) based on antibodies raised against the C‐terminal domain of secretagogin complemented by immunohistochemistry of particular tissues (Fig EV1B, Table EV6) using the same antibody (Mulder et al, 2010).
Gene expression in human subjects
Islets from 202 cadaver donors of European ancestry were provided by the Nordic Islet Transplantation Programme (http://www.nordicislets.org). All procedures were approved by the ethics committee at Lund University and performed as previously described (Fadista et al, 2014). Briefly, purity of islets was assessed by dithizone followed by islets culture in CMRL 1066 (ICN Biomedicals) supplemented with 10 mM HEPES, 2 mM l‐glutamine, 50 μg/ml gentamicin, 0.25 μg/ml Fungizone (GIBCO), 20 μg/ml ciprofloxacin (Bayer Healthcare) and 10 mM nicotinamide at 37°C (5% CO2) for 1–9 days prior to RNA preparation. Total RNA was isolated with the AllPrep DNA/RNA Mini Kit following the manufacturer's instructions (Qiagen). RNA quality and concentration were measured using an Agilent 2100 bioanalyzer (Bio‐Rad) and a Nanodrop ND‐1000 (NanoDrop Technologies). Samples were stratified based upon glucose tolerance estimated from HbA1c, that is donors with normal glucose tolerance (HbA1c < 6%, n = 123), impaired glucose tolerance (IGT, 6% ≤ HbA1c < 6.5%, n = 47), and type 2 diabetes (HbA1c ≥ 6.5%, n = 32). A linear model adjusting for age and sex as implemented in the R Matrix eQTL package was used to determine the expression of genes associated with glucose tolerance status. Raw data were base called and de‐multiplexed using CASAVA 1.8.2 (Illumina) before alignment to hg19 with STAR. To count the number of reads aligned to specific transcripts feature Counts (v 1.4.4, http://bioinf.wehi.edu.au/featureCounts/) was used. Raw data were normalized using trimmed mean of M‐values (TMM) and transformed into log2 counts per million (log2CPM) using voom (limma R, Bioconductor) before linear modelling. After TMM‐normalization, batch effects were removed using the Combat function (sva package).
Single‐cell RNA‐seq of pancreatic islets
Analysis was performed based on a publicly available expression matrix (Segerstolpe et al, 2016) from 270 human β‐cells from six healthy (n = 171 cells) and four type 2 diabetic donors (n = 99 cells). Briefly, human tissue and primary islets were purchased from Prodo Laboratories Inc., providing islets isolated from donor pancreata obtained from deceased individuals with research consent from Organ Procurement Organizations. Human islet samples (85–95% pure) were cultured for 4 days in complete Prodo Islet Media Standard PIM(S) after arrival. Islets were dissociated and distributed by FACS into 384‐well plates. Single‐cell RNA‐seq libraries were produced with the Smart‐seq2 protocol (Segerstolpe et al, 2016). Sequencing was carried out on an Illumina HiSeq 2000 generating 43‐bp single‐end reads. Sequence reads were aligned towards the human genome (hg19 assembly) using STAR (v2.3.0e), and uniquely aligned reads within RefSeq gene annotations were used to quantify gene expression as RPKMs using the “rpkmforgenes” routine (sandberg.cmb.ki.se/media/data/rnaseq/).
Statistics
All experiments were performed in triplicate unless stated otherwise. Data from pharmacology experiments were analysed using one‐way ANOVA followed by pairwise comparisons where appropriate. Student's t‐test (independent group design) was used to statistically evaluate data on pancreatic islet morphology, INS‐1E proliferation and apoptosis and gene expression. Data from Ca2+‐imaging and RNA‐seq were analysed using the Mann–Whitney rank‐sum test. Data are expressed as means ± s.d. unless specified. Fold changes represent the percentage change from the untreated (control) value in individual experiments. A P < 0.05 value was considered statistically significant.
Accession numbers
Raw data on single‐cell RNA‐seq of pancreatic beta cells and human islets sequenced in bulk were deposited in ArrayExpress (E‐MTAB‐5061) and Gene Expression Omnibus (GSE50398), respectively. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD006544.
Author contributions
THa and KM conceived the study design; THa, LG, THö, GL, SK, RS and VDM procured funding for experimental work; KM, FG, ES, PS, ÅS, GT, RS, JM, RAR, EB and FP performed experiments. LW, RS, GS and SK provided unique reagents and databases; KM and THa wrote the manuscript, which was proofread and approved by all authors.
Conflict of interest
V.D.M. and T.Ha. declare support from GW Pharmaceuticals on projects unrelated to the focus of this report. All other authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 7
Acknowledgements
The authors thank Dr. J. Attems (Newcastle University, UK) for providing histochemical images on secretagogin in olfactory bulb and Ms. J. Schachendorfer for contributing to the initial characterization of β‐Scgn CKO mice. This work was supported by the Swedish Research Council (T.Ha., T.Hö., L.G.), Hjärnfonden (T.Ha.), the Novo Nordisk Foundation (K.M., T.Ha., T.Hö.), the European Research Council (Secret‐Cells 2015‐AdG‐695136; T.Ha.) and intramural funds of the Medical University of Vienna (T.Ha.). S.K. was supported for next‐generation sequencing by JDRF grants 3‐SRA‐2015‐20‐Q‐R, 17‐2011‐258. R.A.R. was supported by the European Molecular Biology Organization (long‐term fellowship, ALTF reference 586‐2014) and EU FP7 (Marie Curie Actions, EMBOCOFUND2012, grant GA‐2012‐600394).
The EMBO Journal (2017) 36: 2107–2125
References
- Akiba Y, Kato S, Katsube K, Nakamura M, Takeuchi K, Ishii H, Hibi T (2004) Transient receptor potential vanilloid subfamily 1 expressed in pancreatic islet beta cells modulates insulin secretion in rats. Biochem Biophys Res Commun 321: 219–225 [DOI] [PubMed] [Google Scholar]
- Allen JR, Nguyen LX, Sargent KE, Lipson KL, Hackett A, Urano F (2004) High ER stress in beta‐cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun 324: 166–170 [DOI] [PubMed] [Google Scholar]
- Alpar A, Attems J, Mulder J, Hokfelt T, Harkany T (2012) The renaissance of Ca2+‐binding proteins in the nervous system: secretagogin takes center stage. Cell Signal 24: 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aradska J, Bulat T, Sialana FJ, Birner‐Gruenberger R, Erich B, Lubec G (2015) Gel‐free mass spectrometry analysis of Drosophila melanogaster heads. Proteomics 15: 3356–3360 [DOI] [PubMed] [Google Scholar]
- Araki E, Oyadomari S, Mori M (2003) Impact of endoplasmic reticulum stress pathway on pancreatic beta‐cells and diabetes mellitus. Exp Biol Med (Maywood) 228: 1213–1217 [DOI] [PubMed] [Google Scholar]
- Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, Frogne T, Tao W, Oberholzer J, Pattou F, Kerr CJ, Maedler K (2014) MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med 20: 385–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MC, O'Connell DJ, Maj M, Wagner L, Cahill DJ, Linse S (2011) Identification of a high‐affinity network of secretagogin‐binding proteins involved in vesicle secretion. Mol BioSyst 7: 2196–2204 [DOI] [PubMed] [Google Scholar]
- Bazwinsky‐Wutschke I, Wolgast S, Muhlbauer E, Peschke E (2010) Distribution patterns of calcium‐binding proteins in pancreatic tissue of non‐diabetic as well as type 2 diabetic rats and in rat insulinoma beta‐cells (INS‐1). Histochem Cell Biol 134: 115–127 [DOI] [PubMed] [Google Scholar]
- Birkenkamp‐Demtroder K, Wagner L, Brandt SF, Bording AL, Gartner W, Scherubl H, Heine B, Christiansen P, Orntoft TF (2005) Secretagogin is a novel marker for neuroendocrine differentiation. Neuroendocrinology 82: 121–138 [DOI] [PubMed] [Google Scholar]
- Bitto E, Bingman CA, Bittova L, Frederick RO, Fox BG, Phillips GN Jr (2009) X‐ray structure of Danio rerio secretagogin: a hexa‐EF‐hand calcium sensor. Proteins 76: 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollo M, Paredes RM, Holstein D, Zheleznova N, Camacho P, Lechleiter JD (2010) Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PLoS ONE 5: e11925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P (2008) Voltage‐gated ion channels in human pancreatic beta‐cells: electrophysiological characterization and role in insulin secretion. Diabetes 57: 1618–1628 [DOI] [PubMed] [Google Scholar]
- Bugliani M, Liechti R, Cheon H, Suleiman M, Marselli L, Kirkpatrick C, Filipponi F, Boggi U, Xenarios I, Syed F, Ladriere L, Wollheim C, Lee MS, Marchetti P (2013) Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin‐proteasome system in pancreatic beta cell dysfunction. Mol Cell Endocrinol 367: 1–10 [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner‐Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta‐cell deficit and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110 [DOI] [PubMed] [Google Scholar]
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, et al (2005) The transcriptional landscape of the mammalian genome. Science 309: 1559–1563 [DOI] [PubMed] [Google Scholar]
- Celio MR (1990) Calbindin‐D‐28k and parvalbumin in the rat nervous system. Neuroscience 35: 375–475 [DOI] [PubMed] [Google Scholar]
- Chen J, Li L, Li Y, Liang X, Sun Q, Yu H, Zhong J, Ni Y, Chen J, Zhao Z, Gao P, Wang B, Liu D, Zhu Z, Yan Z (2015) Activation of TRPV1 channel by dietary capsaicin improves visceral fat remodeling through connexin43‐mediated Ca2+ influx. Cardiovasc Diabetol 14: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, Kinet JP, Fleig A, Penner R (2007) TRPM4 controls insulin secretion in pancreatic beta‐cells. Cell Calcium 41: 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Runnels LW, Strubing C (2001) The TRP ion channel family. Nat Rev Neurosci 2: 387–396 [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Creutz CE, Liou A, Snyder SL, Brownawell A, Willison K (1994) Identification of the major chromaffin granule‐binding protein, chromobindin A, as the cytosolic chaperonin CCT (chaperonin containing TCP‐1). J Biol Chem 269: 32035–32038 [PubMed] [Google Scholar]
- De Marchi N, De Petrocellis L, Orlando P, Daniele F, Fezza F, Di Marzo V (2003) Endocannabinoid signalling in the blood of patients with schizophrenia. Lipids Health Dis 2: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Marini P, Matias I, Moriello AS, Starowicz K, Cristino L, Nigam S, Di Marzo V (2007) Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta‐cells. Exp Cell Res 313: 2993–3004 [DOI] [PubMed] [Google Scholar]
- Dekker C, Stirling PC, McCormack EA, Filmore H, Paul A, Brost RL, Costanzo M, Boone C, Leroux MR, Willison KR (2008) The interaction network of the chaperonin CCT. EMBO J 27: 1827–1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng S, Vatamaniuk M, Huang X, Doliba N, Lian MM, Frank A, Velidedeoglu E, Desai NM, Koeberlein B, Wolf B, Barker CF, Naji A, Matschinsky FM, Markmann JF (2004) Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 53: 624–632 [DOI] [PubMed] [Google Scholar]
- van Der Luit AH, Olivari C, Haley A, Knight MR, Trewavas AJ (1999) Distinct calcium signaling pathways regulate calmodulin gene expression in tobacco. Plant Physiol 121: 705–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L (2012) Why do cannabinoid receptors have more than one endogenous ligand? Philos Trans R Soc Lond B Biol Sci 367: 3216–3228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P (2006) Thermofluor‐based high‐throughput stability optimization of proteins for structural studies. Anal Biochem 357: 289–298 [DOI] [PubMed] [Google Scholar]
- Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, Storm P, Osmark P, Ladenvall C, Prasad RB, Hansson KB, Finotello F, Uvebrant K, Ofori JK, Di Camillo B, Krus U, Cilio CM, Hansson O, Eliasson L, Rosengren AH, et al (2014) Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci USA 111: 13924–13929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Miyata K, Bhaskaran MD, Derbenev AV, Zsombok A (2012) Transient receptor potential vanilloid type 1‐dependent regulation of liver‐related neurons in the paraventricular nucleus of the hypothalamus diminished in the type 1 diabetic mouse. Diabetes 61: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MS, Moss LG, Rutter WJ (1990) Regulation of insulin gene expression by glucose and calcium in transfected primary islet cultures. J Biol Chem 265: 22063–22066 [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349: 132–138 [DOI] [PubMed] [Google Scholar]
- Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK (2012) Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell 23: 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham J, Ruddock LW, Roobol A, Carden MJ (2002) Eukaryotic chaperonin containing T‐complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro . Cell Stress Chaperones 7: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium , Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Walters G, Walters G, Walters G, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J, Fleming J, et al (2013) The genotype‐tissue expression (GTEx) project. Nat Genet, 45: 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanics J, Szodorai E, Tortoriello G, Malenczyk K, Keimpema E, Lubec G, Hevesi Z, Lutz MI, Kozsurek M, Puskar Z, Toth ZE, Wagner L, Kovacs GG, Hokfelt TG, Harkany T, Alpar A (2017) Secretagogin‐dependent matrix metalloprotease‐2 release from neurons regulates neuroblast migration. Proc Natl Acad Sci USA 114: E2006–E2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Kimoto M, Minakuchi H, Fujimura K, Hosoya K, Komatsu M, Kaneko Y, Kanda T, Tokuyama H, Hayashi K, Itoh H (2013) The hydrolase DDAH2 enhances pancreatic insulin secretion by transcriptional regulation of secretagogin through a Sirt1‐dependent mechanism in mice. FASEB J 27: 2301–2315 [DOI] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK (2006) Calcineurin/NFAT signalling regulates pancreatic beta‐cell growth and function. Nature 443: 345–349 [DOI] [PubMed] [Google Scholar]
- Held K, Kichko T, De CK, Klaassen H, Van BR, Vanherck JC, Marchand A, Reeh PW, Chaltin P, Voets T, Vriens J (2015) Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc Natl Acad Sci USA 112: E1363–E1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Ishiyama N, Nenquin M, Ravier MA, Jonas JC (2002) Signals and pools underlying biphasic insulin secretion. Diabetes 51(Suppl. 1): S60–S67 [DOI] [PubMed] [Google Scholar]
- Henquin JC, Mourad NI, Nenquin M (2012) Disruption and stabilization of beta‐cell actin microfilaments differently influence insulin secretion triggered by intracellular Ca2+ mobilization or store‐operated Ca2+ entry. FEBS Lett 586: 89–95 [DOI] [PubMed] [Google Scholar]
- Hu H, Grandl J, Bandell M, Petrus M, Patapoutian A (2009) Two amino acid residues determine 2‐APB sensitivity of the ion channels TRPV3 and TRPV4. Proc Natl Acad Sci USA 106: 1626–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Kuromi H, Gonoi T, Okamoto Y, Ishida H, Seino Y, Kaneko T, Iwanaga T, Seino S (1995) Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J 9: 686–691 [PubMed] [Google Scholar]
- Ize‐Ludlow D, Lightfoot YL, Parker M, Xue S, Wasserfall C, Haller MJ, Schatz D, Becker DJ, Atkinson MA, Mathews CE (2011) Progressive erosion of beta‐cell function precedes the onset of hyperglycemia in the NOD mouse model of type 1 diabetes. Diabetes 60: 2086–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwaba S, Kanao R, Masuda Y, Kusumoto‐Matsuo R, Hanaoka F, Masutani C (2015) USP7 is a suppressor of PCNA ubiquitination and oxidative‐stress‐induced mutagenesis in human cells. Cell Rep 13: 2072–2080 [DOI] [PubMed] [Google Scholar]
- Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR III, Menssen A, Hermeking H (2007) Large‐scale identification of c‐MYC‐associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6: 205–217 [DOI] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi‐Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Lu B, Xing X, Xu E, Ren G, Huang Q (2006) Secretagogin, a novel neuroendocrine marker, has a distinct expression pattern from chromogranin A. Virchows Arch 449: 402–409 [DOI] [PubMed] [Google Scholar]
- Lecona E, Rodriguez‐Acebes S, Specks J, Lopez‐Contreras AJ, Ruppen I, Murga M, Munoz J, Mendez J, Fernandez‐Capetillo O (2016) USP7 is a SUMO deubiquitinase essential for DNA replication. Nat Struct Mol Biol 23: 270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Jung DY, Kim JH, Patel PR, Hu X, Lee Y, Azuma Y, Wang HF, Tsitsilianos N, Shafiq U, Kwon JY, Lee HJ, Lee KW, Kim JK (2015) Transient receptor potential vanilloid type‐1 channel regulates diet‐induced obesity, insulin resistance, and leptin resistance. FASEB J 29: 3182–3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416: 648–653 [DOI] [PubMed] [Google Scholar]
- Lievremont JP, Rizzuto R, Hendershot L, Meldolesi J (1997) BiP, a major chaperone protein of the endoplasmic reticulum lumen, plays a direct and important role in the storage of the rapidly exchanging pool of Ca2+ . J Biol Chem 272: 30873–30879 [DOI] [PubMed] [Google Scholar]
- Lin YF, Lee YF, Liang PH (2012) Targeting beta‐tubulin:CCT‐beta complexes incurs Hsp90‐ and VCP‐related protein degradation and induces ER stress‐associated apoptosis by triggering capacitative Ca2+ entry, mitochondrial perturbation and caspase overactivation. Cell Death Dis 3: e434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj M, Gartner W, Ilhan A, Neziri D, Attems J, Wagner L (2010) Expression of TAU in insulin‐secreting cells and its interaction with the calcium‐binding protein secretagogin. J Endocrinol 205: 25–36 [DOI] [PubMed] [Google Scholar]
- Malenczyk K, Jazurek M, Keimpema E, Silvestri C, Janikiewicz J, Mackie K, Di Marzo V, Redowicz MJ, Harkany T, Dobrzyn A (2013) CB1 cannabinoid receptors couple to focal adhesion kinase to control insulin release. J Biol Chem 288: 32685–32699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenczyk K, Keimpema E, Piscitelli F, Calvigioni D, Bjorklund P, Mackie K, Di Marzo V, Hokfelt TG, Dobrzyn A, Harkany T (2015) Fetal endocannabinoids orchestrate the organization of pancreatic islet microarchitecture. Proc Natl Acad Sci USA 112: E6185–E6194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquard J, Otter S, Welters A, Stirban A, Fischer A, Eglinger J, Herebian D, Kletke O, Klemen MS, Stozer A, Wnendt S, Piemonti L, Kohler M, Ferrer J, Thorens B, Schliess F, Rupnik MS, Heise T, Berggren PO, Klocker N, et al (2015) Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat Med 21: 363–372 [DOI] [PubMed] [Google Scholar]
- Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, Petrosino S, Hoareau L, Festy F, Pasquali R, Roche R, Maj M, Pagotto U, Monteleone P, Di Marzo V (2006) Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta‐pancreatic cells and in obesity and hyperglycemia. J Clin Endocrinol Metab 91: 3171–3180 [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S (1996) Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett 395: 143–147 [DOI] [PubMed] [Google Scholar]
- McGarry E, Gaboriau D, Rainey MD, Restuccia U, Bachi A, Santocanale C (2016) The deubiquitinase USP9X maintains DNA replication fork stability and DNA damage checkpoint responses by regulating CLASPIN during S‐phase. Cancer Res 76: 2384–2393 [DOI] [PubMed] [Google Scholar]
- Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P (2004) Glucose sensitivity and metabolism‐secretion coupling studied during two‐year continuous culture in INS‐1E insulinoma cells. Endocrinology 145: 667–678 [DOI] [PubMed] [Google Scholar]
- Molina J, Rodriguez‐Diaz R, Fachado A, Jacques‐Silva MC, Berggren PO, Caicedo A (2014) Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes 63: 2714–2726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DO, Kang CH, Kang SH, Choi YH, Hyun JW, Chang WY, Kang HK, Koh YS, Maeng YH, Kim YR, Kim GY (2012) Capsaicin sensitizes TRAIL‐induced apoptosis through Sp1‐mediated DR5 up‐regulation: involvement of Ca(2+) influx. Toxicol Appl Pharmacol 259: 87–95 [DOI] [PubMed] [Google Scholar]
- Mulder J, Spence L, Tortoriello G, Dinieri JA, Uhlen M, Shui B, Kotlikoff MI, Yanagawa Y, Aujard F, Hokfelt T, Hurd YL, Harkany T (2010) Secretagogin is a Ca2+‐binding protein identifying prospective extended amygdala neurons in the developing mammalian telencephalon. Eur J Neurosci 31: 2166–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Chazin WJ (1998) An interaction‐based analysis of calcium‐induced conformational changes in Ca2+ sensor proteins. Protein Sci 7: 270–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M (2003) Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci USA 100: 8002–8006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer MA, Halter JB, Porte D Jr (1981) Insulin secretion in diabetes mellitus. Am J Med 70: 579–588 [DOI] [PubMed] [Google Scholar]
- Qian F, Huang P, Ma L, Kuznetsov A, Tamarina N, Philipson LH (2002) TRP genes: candidates for nonselective cation channels and store‐operated channels in insulin‐secreting cells. Diabetes 51(Suppl. 1): S183–S189 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- Rasmusen CD, Means AR (1990) Effects of changes in calmodulin levels on cell proliferation. Environ Health Perspect 84: 31–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera CE, Huising MO, Follett P, Leblanc M, Halloran J, Van AR, de Magalhaes Filho CD, Merkwirth C, Dillin A (2014) TRPV1 pain receptors regulate longevity and metabolism by neuropeptide signaling. Cell 157: 1023–1036 [DOI] [PubMed] [Google Scholar]
- Rogstam A, Linse S, Lindqvist A, James P, Wagner L, Berggard T (2007) Binding of calcium ions and SNAP‐25 to the hexa EF‐hand protein secretagogin. Biochem J 401: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov RA, Alpar A, Zhang MD, Zeisel A, Calas A, Landry M, Fuszard M, Shirran SL, Schnell R, Dobolyi A, Olah M, Spence L, Mulder J, Martens H, Palkovits M, Uhlen M, Sitte HH, Botting CH, Wagner L, Linnarsson S, et al (2015) A secretagogin locus of the mammalian hypothalamus controls stress hormone release. EMBO J 34: 36–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Trube G (1986) Calcium and delayed potassium currents in mouse pancreatic beta‐cells under voltage‐clamp conditions. J Physiol 374: 531–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Gibson TM, Brockie HC, Leslie M, Pashmi G, Craib SJ, Di Marzo V, Pertwee RG (2001) Structure‐activity relationship for the endogenous cannabinoid, anandamide, and certain of its analogues at vanilloid receptors in transfected cells and vas deferens. Br J Pharmacol 132: 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Bellini G, Luongo L, Torella M, Mancusi S, De Petrocellis L, Petrosino S, Siniscalco D, Orlando P, Scafuro M, Colacurci N, Perrotta S, Nobili B, Di Marzo V, Maione S (2011) The endovanilloid/endocannabinoid system: a new potential target for osteoporosis therapy. Bone 48: 997–1007 [DOI] [PubMed] [Google Scholar]
- Sanz‐Salvador L, ndres‐Borderia A, Ferrer‐Montiel A, Planells‐Cases R (2012) Agonist‐ and Ca2+‐dependent desensitization of TRPV1 channel targets the receptor to lysosomes for degradation. J Biol Chem, 287: 19462–19471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B (2010) Cytosolic Ca2+ buffers. Cold Spring Harb Perspect Biol 2: a004051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham‐Anderson J, Yue P, Dornan D, Huang DC, Dixit VM (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463: 103–107 [DOI] [PubMed] [Google Scholar]
- Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, Smith DM, Kasper M, Ammala C, Sandberg R (2016) Single‐cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Li H, Ou Y, Tao W, Dong A, Kong J, Ji C, Yu S (2008) The secondary structure of calcineurin regulatory region and conformational change induced by calcium/calmodulin binding. J Biol Chem 283: 11407–11413 [DOI] [PubMed] [Google Scholar]
- Shirakawa J, Togashi Y, Sakamoto E, Kaji M, Tajima K, Orime K, Inoue H, Kubota N, Kadowaki T, Terauchi Y (2013) Glucokinase activation ameliorates ER stress‐induced apoptosis in pancreatic beta‐cells. Diabetes 62: 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (2011) A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 474: 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovhus KV, Bergholdt R, Erichsen C, Sparre T, Nerup J, Karlsen AE, Pociot F (2006) Identification and characterization of secretagogin promoter activity. Scand J Immunol 64: 639–645 [DOI] [PubMed] [Google Scholar]
- Skrzypski M, Khajavi N, Mergler S, Szczepankiewicz D, Kolodziejski PA, Metzke D, Wojciechowicz T, Billert M, Nowak KW, Strowski MZ (2015) TRPV6 channel modulates proliferation of insulin secreting INS‐1E beta cell line. Biochim Biophys Acta 1853: 3202–3210 [DOI] [PubMed] [Google Scholar]
- Smidak R, Mayer RL, Bileck A, Gerner C, Mechtcheriakova D, Stork O, Lubec G, Li L (2016) Quantitative proteomics reveals protein kinases and phosphatases in the individual phases of contextual fear conditioning in the C57BL/6J mouse. Behav Brain Res 303: 208–217 [DOI] [PubMed] [Google Scholar]
- Soleimanpour SA, Crutchlow MF, Ferrari AM, Raum JC, Groff DN, Rankin MM, Liu C, De Leon DD, Naji A, Kushner JA, Stoffers DA (2010) Calcineurin signaling regulates human islet {beta}‐cell survival. J Biol Chem 285: 40050–40059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz KM, Cristino L, Matias I, Capasso R, Racioppi A, Izzo AA, Di Marzo V (2008) Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high‐fat diet. Obesity (Silver Spring) 16: 553–565 [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano MA, Campi B, McNaughton P, Geppetti P, Di Marzo V (2005) Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J 24: 3026–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Kano F, Murata M (2014) Rab2A is a pivotal switch protein that promotes either secretion or ER‐associated degradation of (pro)insulin in insulin‐secreting cells. Sci Rep 4: 6952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suriben R, Kaihara KA, Paolino M, Reichelt M, Kummerfeld SK, Modrusan Z, Dugger DL, Newton K, Sagolla M, Webster JD, Liu J, Hebrok M, Dixit VM (2015) beta‐cell insulin secretion requires the ubiquitin ligase COP1. Cell 163: 1457–1467 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Takahashi R (2001) Ubiquitin‐protein ligase activity of X‐linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase‐3 and enhances its anti‐apoptotic effect in Fas‐induced cell death. Proc Natl Acad Sci USA 98: 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarina NA, Roe MW, Philipson L (2014) Characterization of mice expressing Ins1 gene promoter driven CreERT recombinase for conditional gene deletion in pancreatic beta‐cells. Islets 6: e27685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad AG, Muller PA, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, Vousden KH (2013) Interaction of p53 with the CCT complex promotes protein folding and wild‐type p53 activity. Mol Cell 50: 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner L, Oliyarnyk O, Gartner W, Nowotny P, Groeger M, Kaserer K, Waldhausl W, Pasternack MS (2000) Cloning and expression of secretagogin, a novel neuroendocrine‐ and pancreatic islet of Langerhans‐specific Ca2+‐binding protein. J Biol Chem 275: 24740–24751 [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- Yan J, Zhong N, Liu G, Chen K, Liu X, Su L, Singhal S (2014) Usp9x‐ and Noxa‐mediated Mcl‐1 downregulation contributes to pemetrexed‐induced apoptosis in human non‐small‐cell lung cancer cells. Cell Death Dis 5: e1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SY, Lee JJ, Lee JH, Lee K, Oh SH, Lim YM, Lee MS, Lee KJ (2016) Secretagogin impacts insulin secretion in pancreatic beta cells by regulating actin dynamics and focal adhesion. Biochem J 437: 1791–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Niwa T, Miura Y, Horio F, Teradaira S, Ribar TJ, Means AR, Hasegawa Y, Senda T, Niki I (2002) Calmodulin overexpression causes Ca(2+)‐dependent apoptosis of pancreatic beta cells, which can be prevented by inhibition of nitric oxide synthase. Lab Invest 82: 1229–1239 [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang X, Kuang H, Wu J, Guo Y, Ma L (2013) Effect of TRPV1 channel on the proliferation and apoptosis in asthmatic rat airway smooth muscle cells. Exp Lung Res 39: 283–294 [DOI] [PubMed] [Google Scholar]
- Zsombok A, Bhaskaran MD, Gao H, Derbenev AV, Smith BN (2011) Functional plasticity of central TRPV1 receptors in brainstem dorsal vagal complex circuits of streptozotocin‐treated hyperglycemic mice. J Neurosci 31: 14024–14031 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 4
Source Data for Figure 7