

Abstract
Mammalian target of rapamycin (mTOR) signaling is involved in a variety of kidney diseases. Clinical trials administering mTOR inhibitors to patients with FSGS, a prototypic podocyte disease, led to conflicting results, ranging from remission to deterioration of kidney function. Here, we combined complex genetic titration of mTOR complex 1 (mTORC1) levels in murine glomerular disease models, pharmacologic studies, and human studies to precisely delineate the role of mTOR in FSGS. mTORC1 target genes were significantly induced in microdissected glomeruli from both patients with FSGS and a murine FSGS model. Furthermore, a mouse model with constitutive mTORC1 activation closely recapitulated human FSGS. Notably, the complete knockout of mTORC1 by induced deletion of both Raptor alleles accelerated the progression of murine FSGS models. However, lowering mTORC1 signaling by deleting just one Raptor allele ameliorated the progression of glomerulosclerosis. Similarly, low-dose treatment with the mTORC1 inhibitor rapamycin efficiently diminished disease progression. Mechanistically, complete pharmacologic inhibition of mTOR in immortalized podocytes shifted the cellular energy metabolism toward reduced rates of oxidative phosphorylation and anaerobic glycolysis, which correlated with increased production of reactive oxygen species. Together, these data suggest that podocyte injury and loss is commonly followed by adaptive mTOR activation. Prolonged mTOR activation, however, results in a metabolic podocyte reprogramming leading to increased cellular stress and dedifferentiation, thus offering a treatment rationale for incomplete mTOR inhibition.
Keywords: focal segmental glomerulosclerosis, mTOR, Pathophysiology of Renal Disease and Progression, mitochondrial function, rapamycin, raptor
FSGS represents a major cause of nephrotic syndrome and it is the most common primary glomerular disorder causing ESRD in the United States.1 The histologic hallmark of initial FSGS is mediated by various known (secondary) or yet unknown (primary) insults directed to or inherent within the podocyte.2 Primary (idiopathic) FSGS is attributed to circulating permeability factors. Evidence for such circulating plasma factors came from the observation that proteinuria can be ameliorated in some patients with FSGS by plasmapheresis or immunoadsorption and that FSGS can recur rapidly after renal transplantation.3–5 Secondary FSGS can be due to familial and sporadic podocyte gene mutations, APOL1 gene variants, viral infections, and drug-induced podocyte injury.2 The fact that most gene mutations causing proteinuria and FSGS directly affect the podocyte led to the definition of FSGS as a podocytopathy. Interestingly, the genes causing FSGS encode for diverse podocyte proteins localizing to the cell membrane, actin cytoskeleton, mitochondria, lysosomes, nucleus, or the slit diaphragm.6 Adaptive FSGS is another distinct form of secondary FSGS, which appears to result from structural-functional adaptation to reduced renal mass and/or renal vasodilation followed by glomerular hypertrophy, glomerular hyperfiltration, and eventual progressive glomerular scarring.7 Clinically, the distinction of primary (idiopathic) and secondary FSGS is important because the treatment of secondary FSGS usually only consists of supportive measures but not immunosuppressive therapy.8,9 Despite the heterogeneity of primary and secondary causes leading to initial podocyte injury and loss, the progressing phase of FSGS seems in all cases to be underlined by a mismatch of podocyte numbers trying to cover the surface of the glomerular basement membrane.10 Subsequently, podocyte hypertrophy with eventual podocyte dedifferentiation and further podocyte loss appear to be self-sufficient driving factors of both primary and secondary FSGS progression. Until now, we are still lacking a precise understanding of the molecular signaling events underlying FSGS progression, hampering the development of an effective therapeutic approach to prevent progression to ESRD.11
Recent work, however, evidenced that podocyte size control is centrally regulated by mammalian target of rapamycin (mTOR)12,13 and that mTOR activation can contribute to glomerular disease progression.12–14 In agreement with these findings, caloric restriction in aging rats,10 which is known to significantly reduce mTOR activity, prevented the development of glomerular enlargement, proteinuria, and glomerulosclerosis.
In general, the mTOR pathway controls cellular growth, survival, and metabolism. The serine/threonine kinase mTOR is the catalytic subunit of two distinct complexes, mTOR complex 1 and 2 (mTORC1 and mTORC2), that can be distinguished by their unique composition and different substrates.15 Rapamycin-sensitive adaptor protein of mTOR (RAPTOR) is, among other proteins, an essential component of mTORC1.16 The activation of mTORC1 predominantly results in direct phosphorylation of the ribosomal S6 kinase (S6K) and the eukaryotic translation initiation factor 4E–binding protein (4E-BP), which stimulate ribosome biogenesis and protein translation to increase cell mass.17,18 The use of rapamycin, a selective mTORC1 inhibitor, in steroid-resistant FSGS and FSGS recurrence after kidney transplantation, has been reported both successful and deleterious, indicating an incomplete understanding of the role of the mTOR pathway in this glomerular disease.19–25 Here, we present a translational approach encompassing transgenic animal models, pharmacologic animal studies, and human data to precisely delineate the role of mTOR in FSGS.
Results
FSGS Is Associated with mTORC1 Activation in Humans and Mice
To study the potential association of mTORC1 activation and podocyte injury in human FSGS, we analyzed glomerular gene expression of microdissected glomeruli from patients with primary FSGS or minimal change disease (MCD) in comparison to healthy controls (living donors). Increased expression of major mTORC1 target genes responsible for mitochondrial biogenesis, fatty acid synthesis, and angiogenesis as well as mTOR mRNA itself indicated the activation of mTORC1 in primary FSGS, whereas transcription levels were not influenced in MCD (Figure 1A). Activation of mTORC1 could be confirmed on protein level, as phosphorylation of glomerular ribosomal protein S6 (pS6), a marker of mTORC1- dependent S6K activation, was significantly enhanced in glomeruli and especially podocytes of patients with FSGS compared with glomeruli of patients with MCD (Figure 1B). To further dissect the role of mTORC1 in the development of FSGS lesions, we investigated a commonly used FSGS mouse model, where the primary podocyte injury is induced by the injection of Adriamycin (ADR) in susceptible mouse strains. ADR injection resulted in significant phosphorylation of S6, highlighting mTORC1 activation as a common feature of human- and animal-associated FSGS-like lesions (Figure 1, C and D). Increasing proteinuria (Figure 1E) and glomerulosclerosis (Figure 1F) correlated well with the progression of podocyte loss (Figure 1, G–I).
Figure 1.

FSGS is associated with mTORC1 activation in humans and mice. (A) Gene expression of major mTORC1 target genes was significantly upregulated in microdissected glomeruli from patients with primary FSGS in comparison with MCD or healthy living donor controls (LD) (FSGS n=10, MCD n=5, LD n=18) (B) Immunohistochemistry staining of human kidney biopsy samples demonstrated enhanced mTORC1-dependent phosphorylation of S6 in glomeruli of patients with FSGS compared with patients with MCD (scale bars, 50 µm; representative images at high magnification; n=3 patients each). (C) mTORC1-dependent phosphorylation of S6 was enhanced in podocytes (arrows) of CD1 wild-type mice in response to injury after ADR injection (scale bars, 5 µm). (D) FSGS-like glomerular lesions in CD1 wild-type mice in ADR nephropathy (arrows). (E) Proteinuria increased over time as a marker for progressive glomerular damage after ADR injection (n=5). (F) Quantification of glomerulosclerosis. (G and H) There is a loss of podocytes in ADR nephropathy demonstrated as reduced WT1-positive nuclei per glomerulus in absolute numbers as well as after normalization for glomerular area. (I) Glomerular tuft volume is unchanged in ADR nephropathy. *, significant; n.s., not significant.
Interestingly, however, plasma from patients with recurrent primary (idiopathic) FSGS after transplantation was not associated with activation of mTORC1 (Supplemental Figure 1). Cultured human podocytes were treated for 24 hours with media containing 5% serum from patients with recurrent FSGS. Compared with treatment with serum from healthy controls, phosphorylation of S6 was not significantly altered in recurrent FSGS serum–treated podocytes. This suggests that mTORC1 is not the primary target of FSGS permeability factors, but rather might represent a chronic, potentially maladaptive response after a primary podocyte injury.
Constitutive mTORC1 Activation Causes FSGS-Like Lesions
Chronic mTORC1 activation in the podocyte was previously described to lead to podocyte hypertrophy and progressive glomerular disease.12 Because we identified mTORC1 activation as a hallmark of FSGS, we used this model to study the time course of FSGS development in response to mTOR hyperactivation. Therefore, we generated a podocyte-specific knockout of Tsc1 (Supplemental Figure 2A). Loss of Tsc1 results in constitutive hyperactivation of mTORC1 (Supplemental Figure 2D). In fact, Tsc1Δpodocyte mice already developed FSGS-like histologic lesions and nephrotic-range proteinuria at 3 weeks of age, confirming mTORC1 activation as a pathogenic factor driving the development of FSGS (Supplemental Figure 2, B, C, and E). At the cellular level, podocyte numbers were significantly reduced but the glomerular tuft volume was increased compared with controls, suggesting a hypertrophy of the remaining podocytes (Supplemental Figure 2, F–H).
Complete Genetic Abrogation of mTORC1 Activity Facilitates FSGS-Like Disease
Next, we tested the phenotypic consequences of suppressing ADR-induced mTOR activation by the genetic deletion of mTORC1 activity. Previously, we showed that deleting Raptor can efficiently diminish the function of mTORC1 in podocytes. The constitutive podocyte-specific deletion of Raptor during embryonic development (Raptor flox/flox; Nphs2.Cre mice) resulted in progressive proteinuria associated with growth retardation and increased mortality.13 Raptor deletion induced in adult mice (doxycycline-treated Raptor flox/flox; Nphs2.rtTA; tetO.Cre mice), however, caused neither proteinuria nor alteration of glomerular histology.13 Therefore, we combined the ADR-induced FSGS model with a timed inducible deletion of Raptor (Figure 2, A and B). Unexpectedly, mTORC1 abrogation did not ameliorate the course of FSGS-like lesions, but resulted in a dramatically increased vulnerability toward ADR-induced FSGS-like lesions. Compared with control mice, mice with induced homozygous Raptor deletion developed massively increased proteinuria (Figure 2C), showed much more severe signs of glomerulosclerosis (Figure 2, D and E), and exhibited a significantly increased loss of podocytes (Figure 2, F and G). The total glomerular tuft volume was not different between Raptor-deficient and controls (Figure 2H).
Figure 2.

Complete genetic abrogation of mTORC1 activity facilitates FSGS-like disease. (A) Doxycycline-inducible Raptor knock-out model (Raptorflox/flox; NPHS2.rtTA; tetO.Cre) in mice of mixed (C57BL/6J/ICR) genetic background for inducible deletion of mTORC1. (B) Western blot analysis of isolated glomeruli confirmed Raptor deletion as well as reduced mTORC1-dependent phosphorylation of S6. (C) Deletion of Raptor in adult mice (inducible RaptorΔpodocyte) resulted in significantly increased proteinuria and (D and E) accelerated glomerulosclerosis in a model of ADR-induced FSGS (white arrows, areas with focal sclerosis; bars, 50 µm; glomerulosclerosis index after el Nahas et al. at 8 weeks of experiment; n=3). (F and G) Enhanced loss of podocytes in inducible RaptorΔpodocyte is demonstrated by reduced WT1-positive nuclei per glomerulus in absolute numbers as well as after normalization for glomerular area, whereas (H) glomerular tuft volume is not different in both groups. (I) Subtotal nephrectomy (Nx) after inducible deletion of Raptor in podocytes of adult mice (RaptorΔpodocyte) versus Raptor+/+ mice resulted in severe proteinuria (week 5 of experiment, n=7, respectively) and (J) progressive decline of renal function (serum urea measured after 8 weeks of follow up; n=5 and n=8, respectively). (K) Histologic and transmission electron microscopy images of Nx mice confirm severe glomerular lesions and foot process effacement in Raptor-deficient mice. (L–N) Nx in WT animals induces glomerular hypertrophy and podocyte loss, demonstrated as reduced WT1-positive nuclei per glomerulus in absolute numbers as well as after normalization for glomerular area. In contrast, glomerular hypertrophy is blocked in RaptorΔpodocyte, whereas WT1-positive nuclei differ only in absolute values but not after normalization for glomerular area. *, significant; n.s., not significant; WT, wild type.
To corroborate these surprising findings, we performed a similar series of experiments by combining a subtotal nephrectomy (Nx) model—known to cause FSGS—with induced deletion of Raptor in adult animals (doxycycline-treated Raptor flox/flox; Nphs2.rtTA; tetO.Cre mice). Again, compared with Nx wild-type mice, induced Raptor-deficient Nx mice developed more severe proteinuria, renal failure, and uremia (Figure 2, I and J). Histologic examination and transmission electron microscopy documented increased glomerular lesions, podocyte foot process effacement, and tubular casts of Nx Raptor-deficient versus control mice (Figure 2K, Supplemental Figure 3). In agreement with the ADR model with an induced homozygous Raptor deletion, a significant loss of podocytes was detected after Nx whereas no glomerular hypertrophy was seen, underlining the need of mTOR activation for podocyte and subsequent glomerular hypertrophy (Figure 2, L–N).
Partial Genetic Reduction of mTORC1 Activity Ameliorates FSGS-Like Disease
The fact that constitutive mTORC1 activation by itself can cause FSGS-like lesions, but complete loss of mTORC1 activity aggravates underlying FSGS models, suggested a nonlinear role of mTOR in FSGS-related glomerular disease. Although some mTOR activation is likely required for a compensatory adaptive hypertrophy of remaining podocytes, persistent mTOR hyperactivation presumably propagates podocyte dedifferentiation and progressive podocyte loss. Conceptually, this model suggested a modest mTOR inhibition as a novel therapeutic strategy. To genetically test this hypothesis, we generated mice lacking just one Raptor allele in podocytes (RaptorHet podocyte, Figure 3A). Reduced RAPTOR expression was documented by western blot analysis (Figure 3B) and was associated with significantly reduced mTOR activation in response to ADR-induced FSGS (Figure 3C). Strikingly, progression of proteinuria in ADR nephropathy was substantially ameliorated in RaptorHet podocyte mice (Figure 3D). Furthermore, quantification of glomerular lesions demonstrated unambiguously less glomerulosclerosis in RaptorHet podocyte compared with control mice (Figure 3, E and G). In-depth quantitative stereologic analyses using the dissector principle 3 weeks after ADR injection allowed us to directly determine podocyte cell volumes and revealed that podocyte-specific lowering of mTORC1 activity in ADR nephropathy completely prevented podocyte hypertrophy, whereas podocyte number and glomerular tuft volume were not different (Figure 3, F and H). In summary, these data demonstrate that modest genetic mTOR inhibition can ameliorate FSGS progression.
Figure 3.

Partial genetic reduction of mTORC1 activity ameliorates FSGS-like disease. (A) Heterozygous deletion of Raptor in podocytes (RaptorHet podocyte) in mice of ICR genetic background (B) reduces RAPTOR protein level in glomerular lysates, and (C) reduces activity of the mTORC1 pathway in ADR FSGS model, expressed as percentage of glomerular area with phosphorylated S6 in immunofluorescence (mean of 20 glomeruli per mouse; n=3 per column). (D) Proteinuria was reduced after heterozygous deletion of Raptor in ADR-adaptive FSGS model (n=7 and n=8, respectively). (E and G) RaptorHet podocyte prevented glomerulosclerosis (glomerulosclerosis index after el Nahas et al. at 8 weeks of experiment; n=4 per column; and PAS staining, white arrows). (F and H) Quantitative stereologic analyses showed reduced podocyte volume in in RaptorHet podocyte but not in wild-type animals (n=3 and n=3, respectively; bars=SD); silver methenamine stainings (white arrows; bar, 20 µm). *, significant; n.s., not significant.
Metabolic Podocyte Reprogramming by mTOR Signaling
To gain more insights into the molecular consequences of mTOR signaling in podocytes, we analyzed the mTOR-dependent metabolic profile in immortalized human podocyte cells. Key features of cellular metabolism are reflected by mitochondrial respiration and ATP generation. Interestingly, mTOR signaling exhibited a dose-dependent effect on mitochondrial function. High-dose treatment with Torin 1 massively reduced mitochondrial respiration and ATP synthesis due to lower levels of the oxygen consumption rate, whereas low-dose treatment did not affect the oxidative phosphorylation (Figure 4, A and B) or the total ATP content (Figure 4C). In agreement with these results, high-dose treatment with Torin 1 lowered the expression levels of SDHB, a key component of the respiratory chain (Figure 4D, Supplemental Figure 4A), thereby directly linking mTOR downstream signaling events to oxidative ATP production. In addition, functional mitochondrial impairment in response to high-dose treatment with Torin 1 led to high levels of oxidative stress (Figure 4E). ADR itself had no effect on the mTOR signaling pathway in immortalized human podocytes (Figure 4F), but mitochondrial respiration and oxidative phosphorylation were impaired (Figure 4, G and H). Low-dose treatment with Torin 1, however, ameliorated ADR-induced mitochondrial dysfunction (Figure 4, I and J) and maintained the capacity for mitochondrial respiration and oxidative ATP synthesis. In contrast, high-dose Torin 1 treatment aggravates ADR-induced mitochondrial dysfunction (Figure 4, K and L).
Figure 4.

Metabolic podocyte reprogramming by mTOR signaling. (A) Mitochondrial function of immortalized cultured podocytes treated with different doses of the mTOR inhibitor Torin 1 for 14 days. Oxygen consumption rate (OCR) was measured at basal level and after the sequential addition of oligomycin (1 μM), FCCP (0.5 μM), and rotenone (Rot; 1.0 μM) + antimycin A (Ant; 1.0 μM; n=3, technical replicates). (B) Combined results underline the role of mTOR for mitochondrial respiratory function (n=3, biologic replicates; NMR, nonmitochondrial respiration). (C) Although total cellular ATP production remains unchanged after low-dose mTOR inhibition with Torin 1, it is suppressed in response to high-dose treatment (n=3, biologic replicates). (D) Protein levels of key components of the respiratory chain in immortalized cultured podocytes treated with different doses of Torin 1 for 14 days. (E) Assessment of oxidative stress by CellROX live cell fluorescence staining and FACS analysis of primary podocytes, displaying significantly induced ROS production (FL4-H fluorescence intensity) after high-dose mTOR inhibition compared with control and low-dose inhibition. (F) Immortalized cultured podocytes treated with ADR 3 µg/ml for indicated time show no changes in pS6. Treatment with mTOR inhibitor Torin 1 100 nM for 6 hours serves as a control. (G) Mitochondrial function of immortalized cultured podocytes treated with ADR 3 µg/ml for 24 hours or vehicle. OCR was measured at basal level and after the sequential addition of oligomycin (1 μM), FCCP (0.5 μM), and rotenone (Rot; 1.0 μM) + antimycin A (Ant; 1.0 μM; n=3, technical replicates). (H) Combined results demonstrate the effect of ADR for mitochondrial respiratory function (n=3, biologic replicates). (I) Mitochondrial function of immortalized cultured podocytes treated with ADR 3 µg/ml or ADR 3 µg/ml + Torin 1 10 nM for 24 hours. OCR was measured at basal level and after the sequential addition of oligomycin (1 μM), FCCP (0.5 μM), and rotenone (Rot; 1.0 μM) + antimycin A (Ant; 1.0 μM; n=3, technical replicates). (J) Combined results demonstrate the beneficial effect of Torin 1 on ADR-induced mitochondrial dysfunction (n=3, biologic replicates). (K) Mitochondrial function of immortalized cultured podocytes treated with ADR 3 µg/ml or ADR + Torin 1 100 nM for 24 hours. OCR was measured at basal level and after the sequential addition of oligomycin (1 μM), FCCP (0.5 μM), and rotenone (Rot; 1.0 μM) + antimycin A (Ant; 1.0 μM; n=3, technical replicates). (L) Combined results demonstrate the additive effect of high-dose Torin 1 treatment on ADR-induced mitochondrial dysfunction (n=3, biologic replicates).
These data are in line with the clinical observations in mice and unravel that complete inhibition of mTOR causes dramatic metabolic changes in podocytes, being associated with mitochondrial dysfunction and increased ROS production. Strikingly, however, incomplete mTOR inhibition preserves mitochondrial function even in the presence of an experimental FSGS model.
Rapamycin Treatment Prevents FSGS-Like Lesions in ADR Nephropathy
To test whether the genetic and metabolic data can be transferred to a new therapeutic rationale, we employed a rapamycin treatment protocol on mice with ADR-induced FSGS. Because a partial reduction of mTORC1 activity should be sufficient, we chose a low-dose protocol (0.5 µg/g mouse body wt three times weekly intraperitoneally) to avoid adverse effects of high-dose rapamycin. Low-dose rapamycin (first dose of rapamycin together with ADR) reduced mTORC1-dependent phosphorylation of S6 (Figure 5A) and significantly ameliorated proteinuria (Figure 5B). In agreement, podocyte numbers were preserved in rapamycin-treated animals (Figure 5, D and E), whereas glomerular volume did not differ significantly between the groups (Figure 5F). Histologic analysis evidenced that low-dose rapamycin prevents progressive glomerulosclerosis (Figure 5, C and G). Strikingly, the beneficial effect of pharmacologic mTORC1 inhibition in ADR nephropathy was fully preserved even when rapamycin treatment was started 2 weeks after ADR injection, suggesting that prolonged mTOR activation after a podocyte injury is the critical step for FSGS disease progression (Supplemental Figure 5A).
Figure 5.

Rapamycin treatment prevents FSGS-like lesions in ADR nephropathy. (A) pS6-stained glomerular area (mean of 20 glomeruli per mouse, n=3 per group). (B) Rapamycin ameliorated proteinuria in ADR nephropathy of mice (n=10 per group), and (C and G) preserved glomerular architecture (glomerulosclerosis index after el Nahas et al. at 8 weeks of experiment; n=3; PAS staining, white arrows). (D and E) Reduced loss of podocytes in CD1 wild-type mice due to rapamycin treatment after ADR injection was demonstrated by a reduced number of WT1-positive nuclei per glomerulus in absolute numbers as well as after normalization for glomerular area. (F) Glomerular tuft volume is not influenced by rapamycin treatment. (G) Low dose Rapamycin treatment prevented glomerulosclerosis. WT, wild type.
High-dose rapamycin (4 µg/g rapamycin three times weekly) protocols did not differ in their efficiency to prevent FSGS-like lesions from low-dose rapamycin applications in ADR nephropathy (Supplemental Figure 5, A and B). We did not observe any statistically significant effects of either low- or high-dose rapamycin on serum markers of glomerular function (Supplemental Figure 5B), indicating that our high-dose therapy protocol did not reach the same levels of mTOR inhibition as caused by genetic deletion of both Raptor alleles.
Discussion
ESRD represents a major burden for patients, societies, and worldwide health care systems. Glomerular diseases including diabetic nephropathy are the leading cause of ESRD. Strikingly, podocyte loss is a major predictor of glomerular disease progression26 and therefore podocytopathies like FSGS offer the unique opportunity to study common pathways leading to chronic renal disease progression. In models of targeted toxic podocyte ablation, the degree of podocyte depletion directly correlated with a threshold-dependent severity of FSGS-like disease and proteinuria.27 Interestingly, podocytes were also continuously lost after termination of toxin exposure, suggesting a secondary autonomous phase of podocyte loss.28 However, a comprehensive molecular pathogenic model for progressive podocyte loss and glomerulosclerosis has not been established.
mTORC1 Activation Propagates FSGS Progression
Here, we identified mTORC1 activation as a common response to primary or secondary podocyte loss in human FSGS as well as in FSGS-like animal models. Genetically induced constitutive mTORC1 activation proved that persistent mTORC1 activation is self-sufficient to cause progressive glomerulosclerosis. Recently, the critical role of mTORC1 for glomerular development has been demonstrated and it appears to be a general theme that podocyte injury leads to the reactivation of developmental programs such as Notch, Wnt, and mTOR.11,29 At the cellular level it was shown that activation of mTORC1 in diabetic animal models resulted in podocyte hypertrophy, dedifferentiation, and redistribution of slit diaphragm proteins, ultimately leading to podocyte loss and glomerulosclerosis.12,13 Furthermore, it was revealed that mTORC1 activation results in increased levels of Nox4, Nox1, and NADPH oxidase activity in podocytes,30 resulting in mTORC1-dependent ROS and ER stress signaling pathways known to instigate glomerular disease progression.12 Thus, persistent mTORC1 activation presents a cellular stress pathway in podocytes that specifically propagates glomerular disease and FSGS progression (Figure 6).
Figure 6.

Role of mTORC1 signaling in progressive glomerulosclerosis. (A) Schematic illustration of mTORC1 activity in FSGS. Initial podocyte injury is mediated by various known (secondary) or as yet unknown (primary) insults. Subsequently, mTORC1 is part of an adaptive response to podocyte loss. However, persistent mTOR activation drives podocyte dedifferentiation and further podocyte loss in self-sustained progression of FSGS. (B) Concept for individual and timed dosage effects of mTOR inhibition.
Adaptive Functions of mTORC1 Provide a Model To Understand Rapamycin-Induced Proteinuria
Although we identified mTORC1 activation as an underlying cause of glomerular disease progression, a timed inducible knockout of the mTORC1 complex did not prevent, but dramatically facilitated, glomerular disease with FSGS-like lesions. These data clearly point to a nonlinear dual role of mTORC1 function for glomerular maintenance versus disease progression. Recent findings indicated mTOR as a regulator of podocyte size, allowing podocytes to adjust for podocyte loss or an expanding glomerular surface.10–13,31,32 Our data now further underline the importance of mTOR-dependent adaptive podocyte functions. In agreement, rapamycin treatment has been reported to cause proteinuria and FSGS in patients with underlying glomerular disease or after renal transplantation.33–35 Hence, our data may help to better predict situations where mTORC1 sustains an adaptive response and rapamycin-induced glomerulopathy is a more likely scenario to occur. Recently, a role for the mTORC2-Akt2 survival axis was also documented in podocytes. Although rapamycin predominantly inhibits mTORC1, mTORC2 signaling appeared to be as well abolished in patients receiving prolonged rapamycin therapy.36 Similar to the deletion of mTORC1 demonstrated here, the deletion of mTORC212,13 sensitized mice to proteinuria and foot process effacement in models of nephron reduction.36 However, mTORC1-deficient models even developed nephrotic-range proteinuria and renal failure, underlining the critical contribution of impaired mTORC1 signaling to rapamycin-induced glomerulopathy (Figure 6). In any case, mTORC1 and mTORC2 seem to synergistically promote podocyte adaptation and maintenance, as simultaneous deletion of both mTORC complexes resulted in the most severe phenotype.13 Thus, it will be important to test whether novel mTORC1-specific inhibitors or dual inhibitors with simultaneous, but less efficient, inhibitory function on both mTOR complexes will cause less glomerular side effects and proteinuria than currently used mTOR inhibitors.
mTOR-Dependent Metabolic Podocyte Programming
Clarification of downstream events of mTOR signaling should lead to a better understanding of both the etiology and consequences of aberrant podocyte size regulation. In general, cell hypertrophy and an increased protein synthesis is an energy-dependent process that involves ATP hydrolyzation.37 Although the balance of cell size regulation and energy supply is usually tightly controlled,38 pathologic conditions can shift the metabolic profile and mitochondrial efficiency as has been extensively demonstrated in physiologic versus pathologic cardiac hypertrophy.39,40 In podocytes, mTOR seems to be the main driving force linking both cell size and energy supply. mTOR inhibition appears to not only affect mitochondrial energy supply, but also mitochondrial efficiency and functionality, resulting in significantly increased ROS generation in a dose-dependent manner that likely contributes to podocyte injury. This is in line with previous work, demonstrating that TGF-β1 mTOR dependently increased both mitochondrial respiration and ROS production in podocytes.41 Mechanistically, increased ROS production is likely explained by the excessive demands of electron carriers in the presence of persistent mTOR activation,42 whereas the decreased expression of the distal subunits of complex II, SDHB, is a sufficient explanation for the increase in ROS production caused by high-dose mTOR inhibition.43
Surprisingly, the cellular effects of mTOR inhibition are highly dose-dependent and vary from shut-down of oxidative phosphorylation accompanied with high levels of ROS production to a distinct modulation of cellular energy homeostasis, strengthening podocytes against toxic stimuli like ADR or FSGS-inducing factors in human disease. The underlying mechanisms of how low-dose mTOR inhibition preserves mitochondrial function in FSGS are not completely clear yet. In general, we predict a metabolic shift in podocytes to a more resting-like state with increased stress resistance.
Partial mTOR Inhibition as a Novel Therapeutic Concept To Treat FSGS and Progressive Glomerular Disease
Our genetic, as well as metabolic, data clearly implied a rationale for inhibition of mTOR as a therapeutic intervention. Excitingly, rapamycin, even when used at very low doses, could significantly reduce proteinuria and prevent podocyte loss and glomerulosclerosis. In contrast to previously published work, high-dose rapamycin treatment also exerted positive effects on halting the development of FSGS-like lesions in our ADR model, suggesting that the potential podocyte-toxic effects of higher doses of rapamycin vary depending on the mouse background, the pharmacodynamics, and the dose of ADR.44 Nonetheless, there is need for further studies to define the subpopulation of human patients with FSGS who may benefit from pharmacologic inhibition of mTORC1, as well as the dose dependency of renal adverse events.
These data are now shedding new light on an interesting contradiction concerning the effects of mTOR inhibition in glomerular diseases. Although some human as well as animal studies evidenced beneficial effects, other studies targeting the same disease entities documented a worse outcome in response to mTOR inhibition. Supplemental Table 1 summarizes some of the recent human mTOR-inhibitor–based studies targeting steroid-resistant FSGS, ranging from massive worsening of proteinuria and renal function to complete remission of disease. Although this cannot be tested, our results may suggest that different treatment responses might correlate with the underlying severity of podocyte injury, levels of adaptive mTOR activation, and interindividual efficiency of mTOR inhibition. In any case, mTOR inhibitors used at high, fully immunosuppressive dosages appear to always bear the risk of causing a worsening of an underlying podocyte disease.
In summary, our data indicate clear experimental evidence for a novel treatment regimen on the basis of partial mTOR inhibition to prevent progressive glomerulosclerosis and FSGS (Figure 6B). Clinical studies will be required to confirm as well as to titrate the exact levels of an mTOR inhibitor therapy. Ideally, treatment protocols should also be directly adapted to the individual mTOR activity in biopsy specimens. The required serum levels of an mTOR-inhibitor–based therapy are likely very well below the current immunosuppression dosing regimens, which would also significantly reduce side effects of mTOR pharmacotherapy. Thus, these data suggest a novel targeted, better tolerated, and individualized mTOR-based therapy to prevent FSGS and progressive renal disease.
Concise Methods
A detailed description including information about materials and reagents, histology, immunofluorescence, immunohistochemistry electron microscopy, western blotting, and isolation of glomeruli is included in the Complete Methods supplemental material of this paper.
Human Kidney Samples, Study Approval, and Microarray Analysis
Microarray analysis was performed with human renal biopsy specimens collected within the framework of the European Renal cDNA Bank–Kröner-Fresenius Biopsy Bank. Microdissection of glomeruli, RNA hybridization, and analysis were performed as reported previously.45 Please see our Supplemental Material for details.
Patients with recurrent FSGS were recruited at Charité-Universitätsmedizin Berlin. The Ethics Committee of the Charité-Universitätsmedizin Berlin approved the study. Please see our Supplemental Material for details.
Formalin-fixed, paraffin-embedded renal tissue specimens were obtained from the Hôpital Européen Georges Pompidou, Assistance Publique-Hôpitaux de Paris, Paris, France. Human tissue was used after approval from, and following the guidelines of, the local Ethics Committee (IRB00003888, FWA00005831). We chose representative images at high magnification. Samples from three patients each with FSGS and MCD were analyzed.
Mouse Experiments
For all models of acquired FSGS (ADR nephropathy and subtotal nephrectomy), mice were on or backcrossed to an ICR/CD1 background because of its known sensitivity toward the development of FSGS-like lesions.
We initially purchased CD1 (Crl: CD1 [ICR]) from Charles River (Europe) for ADR nephropathy experiments on a wild-type background. Later on, we used mice on a cognate ICR background (IcrTac: ICR; Taconic) for ADR injection and backcrossing from ADR-resistant genetic backgrounds (C57BL/6J). Doxorubicin (ADR) was aseptically prepared by our hospital pharmacy. A single dose of 12 µg/g ADR was intravenously injected once in the retro-orbital plexus under isoflurane anesthesia.
Sirolimus was injected at a dose of 0.5 µg (low dose) or 4 µg (high dose) (Pfizer, Germany) per gram of mouse body weight intraperitoneally, in a TWEEN 20 and PEG-400 solution (Sigma-Aldrich, Germany), three times a week for a follow-up course of 6 (delayed treatment) or 8 weeks (simultaneous treatment). Trough levels were obtained 24 hours after the last injection using liquid chromatography–mass spectrometry.
RaptorHet Podocyte mice were generated by crossing Raptorflox/flox with NPHS2.Cre mice after backcrossing for six generations on an ICR background (IcrTac: ICR; Taconic) as previously described.13 Raptorflox/flox; NPHS2.rtTA; tetO.CreNPHS2.rtTA mice have been backcrossed on an ICR background as previously described.13 Induction of Raptor deletion in NPHS2.rtTA; tetO.Cre mice was performed as previously described.13,46–48 In general, we used littermates as controls in experimental groups. Tsc1Δpodocyte (Tsc1flox/flox; NPHS2.Cre) mice were generated by backcrossing of Tsc1fl/+ (129S4/SvJae; C57BL/6J) mice for 6 generations on a C57BL/6J background as previously described.12 These animal studies were approved by the Committee on Research Animal Care, Regierungspräsidium Freiburg.
Subtotal nephrectomy (Nx) was performed as previously described.36 After surgery, mice were fed a defined diet containing 30% casein and 0.5% sodium. Twenty-four hours before euthanasia, blood was collected from the tail veins of overnight-fasted mice for determination of urea concentrations. Urine was collected at week 5 of the experiment for determination of proteinuria and albuminuria. Animal procedures for subtotal nephrectomy were approved by the Departmental Director of Services Vétérinaires de la Préfecture de Police de Paris and the ethical committee of Paris Descartes University. Plasma urea and urinary protein, albumin, and creatinine concentrations were measured using an Olympus multiparametric analyzer (Instrumentation Laboratory).
Quantification of Podocytes and Glomerular Tuft Volume
To determine the number of podocytes per glomerulus as well as the glomerular tuft volume (6 weeks after ADR injection in wild-type animals [Figure 1], and 8 weeks after ADR injection or nephrectomy for other experiments; Tsc1Δpodocyte and control animals at 6 weeks of age), we modified the approach of Hodgin et al.49 using frozen kidney sections, which were stained for WT1, Nidogen, and Hoechst 33342. Please see our Supplemental Material for a detailed description of data analysis.
Quantification of Immunofluorescence Results
Glomeruli were selected as region of interest and a macro (consisting of a color threshold procedure, followed by filtering and Danielsson algorithm; AxioVision software; Carl Zeiss SpA) was applied to select stained areas and calculate them as percentage of the region of interest area. Please see our Supplemental Material for details.
Histologic Analysis
Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin and further processed for PAS staining. Sclerosis index was done as described previously by el Nahas et al.50 Please see our Supplemental Material for a detailed description of data analysis.
Quantitative Stereologic Analysis
Quantitative stereologic analysis of kidney sections was performed as described previously.51 Briefly, the mean glomerular volume (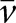 [Glom]) was determined from the mean glomerular profile area (
[Glom]) was determined from the mean glomerular profile area (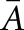 Glom),52 which was obtained by measuring 100 systematically sampled glomerular profiles per animal. The physical dissector principle was applied for counting podocytes (Q−) as described, using semithin sections.53,54 The numerical density of podocytes in glomeruli (NV[P/Glom]) was calculated as the quotient of the sum of Q− divided by the dissector volume. The mean podocyte volume,
Glom),52 which was obtained by measuring 100 systematically sampled glomerular profiles per animal. The physical dissector principle was applied for counting podocytes (Q−) as described, using semithin sections.53,54 The numerical density of podocytes in glomeruli (NV[P/Glom]) was calculated as the quotient of the sum of Q− divided by the dissector volume. The mean podocyte volume, 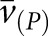 , was calculated by dividing Vv(P/Glom) by NV(P/Glom). All results were corrected for embedding shrinkage.
, was calculated by dividing Vv(P/Glom) by NV(P/Glom). All results were corrected for embedding shrinkage.
Cell Culture
For in vitro experiments, immortalized human podocytes were kindly provided by Moin Saleem (Bristol, UK), and directly isolated primary murine podocytes were used. Primary cells were isolated as previously described.55 Please see our Supplemental Material for a detailed description.
Assessment of Oxidative Stress
Oxidative stress was assessed using CellROX Deep Red Flow Cytometry Assay Kit (#C10491, Molecular Probes; Life technologies, Eugene) following the manufacturer’s instructions. Please see our Supplemental Material for a detailed description.
ATP Measurement
ATP was measured using ADP/ATP Ratio Assay Kit (bioluminescent) (ab 65313; Abcam, Cambridge, UK) according to the manufacturer’s instructions. Please see our Supplemental Material for a detailed description.
Mitochondrial Analysis
Mitochondrial analysis was performed using Seahorse XFp (Seahorse Bioscience Billerica, MA) according to the user’s manual provided by the manufacturer. Presented data were normalized for cellular counts. Please see our Supplemental Material for a detailed description.
Urine and Serum Analyses
Urinary albumin and urinary or serum creatinine, respectively, were measured as previously described.13 Proteinuria was expressed as milligram albumin per milligram creatinine.
Statistical Analyses
All data are expressed as the mean±SEM if not stated otherwise. All shown data reflect a minimum of three subjects per test. Statistical comparisons were performed using two-tailed paired t test if not stated otherwise. P=0.05 was set as statistical significance level for rejection of the null hypothesis if not stated otherwise.
Disclosures
T.B.H. and S.Z. declare that they received project-specific grant funding from Pfizer Pharma GmbH (2010–2012).
Supplementary Material
Acknowledgments
We would like to thank Betina Kiefer, Charlotte Meyer, and Temel Kilic for expert technical assistance. In addition, we would like to express our gratitude to all members of the Huber laboratory for helpful discussions and support. We are grateful to Marie-Claire Gubler for critical advice. We thank all participating centers of the European Renal cDNA Bank–Kröner-Fresenius biopsy bank and their patients for their cooperation. We thank Dr. Fabian Halleck and Dr. Klemens Budde from the Department of Nephrology, Charité-Universitätsmedizin, Berlin, Germany for providing recurrent FSGS samples.
This study was supported by the German Research Foundation (DFG): CRC 1140 (to T.B.H.) and CRC 992 (to T.B.H.), the Heisenberg program (to T.B.H.) and HU 1016/8-1, by the European Research Council–ERC grant 616891 (to T.B.H.), by the Bundesministerium für Bildung und Forschung (BMBF) grant STOP-FSGS 01GM1518C (to T.B.H.), by the Else-Kröner Fresenius Stiftung Forschungskolleg Nierenfunktionsstörungen als Komplikation von Systemerkrankungen (NAKSYS; to T.B. and T.B.H.), by the Excellence Initiative of the German Federal and State Governments (GSC-4, Spemann Graduate School and EXC294, Biological Signalling Studies (BIOSS) II to T.B.H.), and by the H2020-IMI2 consortium Biomarker Enterprise to Attack Diabetic Kidney Disease (BEAt-DKD; 115974).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2016050519/-/DCSupplemental.
References
- 1.United States Renal Data System : 2016 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States, Bethesda, MD, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2016 [Google Scholar]
- 2.D’Agati VD, Kaskel FJ, Falk RJ: Focal segmental glomerulosclerosis. N Engl J Med 365: 2398–2411, 2011 [DOI] [PubMed] [Google Scholar]
- 3.Dantal J, Bigot E, Bogers W, Testa A, Kriaa F, Jacques Y, Hurault de Ligny B, Niaudet P, Charpentier B, Soulillou JP: Effect of plasma protein adsorption on protein excretion in kidney-transplant recipients with recurrent nephrotic syndrome. N Engl J Med 330: 7–14, 1994 [DOI] [PubMed] [Google Scholar]
- 4.Ponticelli C, Moroni G, Glassock RJ: De novo glomerular diseases after renal transplantation. Clin J Am Soc Nephrol 9: 1479–1487, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schachter ME, Monahan M, Radhakrishnan J, Crew J, Pollak M, Ratner L, Valeri AM, Stokes MB, Appel GB: Recurrent focal segmental glomerulosclerosis in the renal allograft: Single center experience in the era of modern immunosuppression. Clin Nephrol 74: 173–181, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Grahammer F, Schell C, Huber TB: The podocyte slit diaphragm--from a thin grey line to a complex signalling hub. Nat Rev Nephrol 9: 587–598, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, Kuick RD, Wiggins RC: Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J Am Soc Nephrol 16: 2953–2966, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Deegens JK, Steenbergen EJ, Wetzels JF: Review on diagnosis and treatment of focal segmental glomerulosclerosis. Neth J Med 66: 3–12, 2008 [PubMed] [Google Scholar]
- 9.Jafar TH, Schmid CH, Landa M, Giatras I, Toto R, Remuzzi G, Maschio G, Brenner BM, Kamper A, Zucchelli P, Becker G, Himmelmann A, Bannister K, Landais P, Shahinfar S, de Jong PE, de Zeeuw D, Lau J, Levey AS: Angiotensin-converting enzyme inhibitors and progression of nondiabetic renal disease. A meta-analysis of patient-level data. Ann Intern Med 135: 73–87, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Fukuda A, Chowdhury MA, Venkatareddy MP, Wang SQ, Nishizono R, Suzuki T, Wickman LT, Wiggins JE, Muchayi T, Fingar D, Shedden KA, Inoki K, Wiggins RC: Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol 23: 1351–1363, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoki K, Huber TB: Mammalian target of rapamycin signaling in the podocyte. Curr Opin Nephrol Hypertens 21: 251–257, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL: mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121: 2181–2196, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstädt H, Kerjaschki D, Cohen CD, Hall MN, Rüegg MA, Inoki K, Walz G, Huber TB: Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao J, Zeng Z, Xu Z, Li J, Jiang L, Fang Y, Xu X, Hu Z, He W, Yang J, Dai C: Mammalian target of rapamycin complex 1 activation in podocytes promotes cellular crescent formation. Am J Physiol Renal Physiol 307: F1023–F1032, 2014 [DOI] [PubMed] [Google Scholar]
- 15.Laplante M, Sabatini DM: mTOR signaling in growth control and disease. Cell 149: 274–293, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K: Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Holz MK, Ballif BA, Gygi SP, Blenis J: mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Ma XM, Blenis J: Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 10: 307–318, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Tumlin JA, Miller D, Near M, Selvaraj S, Hennigar R, Guasch A: A prospective, open-label trial of sirolimus in the treatment of focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 1: 109–116, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Tsagalis G, Psimenou E, Iliadis A, Nakopoulou L, Laggouranis A: Rapamycin for focal segmental glomerulosclerosis: A report of 3 cases. Am J Kidney Dis 54: 340–344, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Cho ME, Hurley JK, Kopp JB: Sirolimus therapy of focal segmental glomerulosclerosis is associated with nephrotoxicity. Am J Kidney Dis 49: 310–317, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Letavernier E, Pe’raldi MN, Pariente A, Morelon E, Legendre C: Proteinuria following a switch from calcineurin inhibitors to sirolimus. Transplantation 80: 1198–1203, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Letavernier E, Bruneval P, Mandet C, Duong Van Huyen JP, Péraldi MN, Helal I, Noël LH, Legendre C: High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol 2: 326–333, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Liern M, De Reyes V, Fayad A, Vallejo G: Use of sirolimus in patients with primary steroid-resistant nephrotic syndrome. Nefrologia 32: 321–328, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Fervenza FC, Fitzpatrick PM, Mertz J, Erickson SB, Liggett S, Popham S, Wochos DN, Synhavsky A, Hippler S, Larson TS, Bagniewski SM, Velosa JA; Mayo Nephrology Collaborative Committee : Acute rapamycin nephrotoxicity in native kidneys of patients with chronic glomerulopathies. Nephrol Dial Transplant 19: 1288–1292, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Wiggins RC: The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int 71: 1205–1214, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC: Podocyte depletion causes glomerulosclerosis: Diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Sato Y, Wharram BL, Lee SK, Wickman L, Goyal M, Venkatareddy M, Chang JW, Wiggins JE, Lienczewski C, Kretzler M, Wiggins RC: Urine podocyte mRNAs mark progression of renal disease. J Am Soc Nephrol 20: 1041–1052, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grahammer F, Wanner N, Huber TB: mTOR controls kidney epithelia in health and disease. Nephrol Dial Transplant 29[Suppl 1]: i9–i18, 2014 [DOI] [PubMed] [Google Scholar]
- 30.Eid AA, Ford BM, Bhandary B, de Cassia Cavaglieri R, Block K, Barnes JL, Gorin Y, Choudhury GG, Abboud HE: Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes 62: 2935–2947, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstädt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB: Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogelbacher R, Wittmann S, Braun A, Daniel C, Hugo C: The mTOR inhibitor everolimus induces proteinuria and renal deterioration in the remnant kidney model in the rat. Transplantation 84: 1492–1499, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Amer H, Cosio FG: Significance and management of proteinuria in kidney transplant recipients. J Am Soc Nephrol 20: 2490–2492, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Torras J, Herrero-Fresneda I, Gulias O, Flaquer M, Vidal A, Cruzado JM, Lloberas N, Franquesa M, Grinyó JM: Rapamycin has dual opposing effects on proteinuric experimental nephropathies: Is it a matter of podocyte damage? Nephrol Dial Transplant 24: 3632–3640, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Munivenkatappa R, Haririan A, Papadimitriou JC, Drachenberg CB, Dinits-Pensy M, Klassen DK: Tubular epithelial cell and podocyte apoptosis with de novo sirolimus based immunosuppression in renal allograft recipients with DGF. Histol Histopathol 25: 189–196, 2010 [DOI] [PubMed] [Google Scholar]
- 36.Canaud G, Bienaimé F, Viau A, Treins C, Baron W, Nguyen C, Burtin M, Berissi S, Giannakakis K, Muda AO, Zschiedrich S, Huber TB, Friedlander G, Legendre C, Pontoglio M, Pende M, Terzi F: AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med 19: 1288–1296, 2013 [DOI] [PubMed] [Google Scholar]
- 37.Dayel MJ, Holleran EA, Mullins RD: Arp2/3 complex requires hydrolyzable ATP for nucleation of new actin filaments. Proc Natl Acad Sci USA 98: 14871–14876, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosca MG, Tandler B, Hoppel CL: Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol 55: 31–41, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN: Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 32: 2361–2367, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL: Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res 80: 30–39, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abe Y, Sakairi T, Beeson C, Kopp JB: TGF-β1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am J Physiol Renal Physiol 305: F1477–F1490, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.James AM, Collins Y, Logan A, Murphy MP: Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol Metab 23: 429–434, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT: Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol 28: 718–731, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stylianou K, Petrakis I, Mavroeidi V, Stratakis S, Kokologiannakis G, Lioudaki E, Liotsi C, Kroustalakis N, Vardaki E, Stratigis S, Perakis K, Kyriazis J, Nakopoulou L, Daphnis E: Rapamycin induced ultrastructural and molecular alterations in glomerular podocytes in healthy mice. Nephrol Dial Transplant 27: 3141–3148, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, Schmid H, Merkle M, Saleem MA, Koller KP, Werner T, Gröne HJ, Nelson PJ, Kretzler M: Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc Natl Acad Sci USA 103: 5682–5687, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN: Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8: 399–410, 2008 [DOI] [PubMed] [Google Scholar]
- 47.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB: Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39–42, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE: VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodgin JB, Bitzer M, Wickman L, Afshinnia F, Wang SQ, O’Connor C, Yang Y, Meadowbrooke C, Chowdhury M, Kikuchi M, Wiggins JE, Wiggins RC: Glomerular aging and focal global glomerulosclerosis: A podometric perspective. J Am Soc Nephrol 26: 3162–3178, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.el Nahas AM, Bassett AH, Cope GH, Le Carpentier JE: Role of growth hormone in the development of experimental renal scarring. Kidney Int 40: 29–34, 1991 [DOI] [PubMed] [Google Scholar]
- 51.Herbach N, Schairer I, Blutke A, Kautz S, Siebert A, Göke B, Wolf E, Wanke R: Diabetic kidney lesions of GIPRdn transgenic mice: Podocyte hypertrophy and thickening of the GBM precede glomerular hypertrophy and glomerulosclerosis. Am J Physiol Renal Physiol 296: F819–F829, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Weibel ER, Gomez DM: A principle for counting tissue structures on random sections. J Appl Physiol 17: 343–348, 1962 [DOI] [PubMed] [Google Scholar]
- 53.Hoeflich A, Weber MM, Fisch T, Nedbal S, Fottner C, Elmlinger MW, Wanke R, Wolf E: Insulin-like growth factor binding protein 2 (IGFBP-2) separates hypertrophic and hyperplastic effects of growth hormone (GH)/IGF-I excess on adrenocortical cells in vivo. FASEB J 16: 1721–1731, 2002 [DOI] [PubMed] [Google Scholar]
- 54.Sterio DC: The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc 134: 127–136, 1984 [DOI] [PubMed] [Google Scholar]
- 55.Schell C, Baumhakl L, Salou S, Conzelmann AC, Meyer C, Helmstädter M, Wrede C, Grahammer F, Eimer S, Kerjaschki D, Walz G, Snapper S, Huber TB: N-wasp is required for stabilization of podocyte foot processes. J Am Soc Nephrol 24: 713–721, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.