

Abstract
Following traumatic brain injury, complex cerebral energy perturbations occur. Correlating with unfavourable outcome, high brain extracellular lactate/pyruvate ratio suggests hypoxic metabolism and/or mitochondrial dysfunction. We investigated whether focal administration of succinate, a tricarboxylic acid cycle intermediate interacting directly with the mitochondrial electron transport chain, could improve cerebral metabolism. Microdialysis perfused disodium 2,3-13C2 succinate (12 mmol/L) for 24 h into nine sedated traumatic brain injury patients' brains, with simultaneous microdialysate collection for ISCUS analysis of energy metabolism biomarkers (nine patients) and nuclear magnetic resonance of 13C-labelled metabolites (six patients). Metabolites 2,3-13C2 malate and 2,3-13C2 glutamine indicated tricarboxylic acid cycle metabolism, and 2,3-13C2 lactate suggested tricarboxylic acid cycle spinout of pyruvate (by malic enzyme or phosphoenolpyruvate carboxykinase and pyruvate kinase), then lactate dehydrogenase-mediated conversion to lactate. Versus baseline, succinate perfusion significantly decreased lactate/pyruvate ratio (p = 0.015), mean difference −12%, due to increased pyruvate concentration (+17%); lactate changed little (−3%); concentrations decreased for glutamate (−43%) (p = 0.018) and glucose (−15%) (p = 0.038). Lower lactate/pyruvate ratio suggests better redox status: cytosolic NADH recycled to NAD+ by mitochondrial shuttles (malate-aspartate and/or glycerol 3-phosphate), diminishing lactate dehydrogenase-mediated pyruvate-to-lactate conversion, and lowering glutamate. Glucose decrease suggests improved utilisation. Direct tricarboxylic acid cycle supplementation with 2,3-13C2 succinate improved human traumatic brain injury brain chemistry, indicated by biomarkers and 13C-labelling patterns in metabolites.
Keywords: Traumatic brain injury (human), microdialysis, nuclear magnetic resonance spectroscopy, cerebral metabolism, succinate
Introduction
After traumatic brain injury (TBI), complex pathology follows, intertwining intracranial dynamics, electrophysiological responses and cerebral metabolism.1–4 Despite modern advances, many TBI survivors experience long-term disability. Better neurocritical care requires better understanding of post-injury brain pathophysiology. Early studies focussed on ischaemia, nowadays minimised by protocol-driven therapy maintaining adequate cerebral perfusion with intracranial pressure below a critical threshold.5
Despite seemingly adequate provision of metabolic fuels and oxygen, the injured brain sometimes cannot utilise them efficiently: termed ‘mitochondrial dysfunction’, the exact basis is not understood. One potentially relevant process is the reduced Ca2+ uptake state after TBI,6 which will depress the activities of several mitochondrial dehydrogenases,7 leading to suboptimal mitochondrial metabolism. Mitochondria are the cells’ ‘powerhouses’ performing high-yielding energy metabolism. Glycolysis in the cytoplasm produces pyruvate, taken up by mitochondria and processed via the tricarboxylic acid (TCA) cycle, which interacts with the mitochondrial electron transport chain (ETC; also termed respiratory chain) driving ATP synthase. The ETC requires molecular oxygen as terminal electron acceptor. Correlating with unfavourable clinical outcome, high brain extracellular lactate/pyruvate ratio (LPR) suggests high glycolytic activity3: glucose converted to pyruvate generating a low ATP yield, then lactate dehydrogenase (LDH) converts pyruvate plus NADH to lactate plus NAD+, allowing further glycolysis.
Succinate is energetically important – a TCA cycle intermediate and a direct contact between the TCA cycle and the mitochondrial ETC. Succinate conversion to fumarate in the TCA cycle is by ETC complex II (succinate dehydrogenase (SDH)), on the mitochondrial inner membrane. Soluble mitochondrial enzymes perform all the other TCA cycle steps; the NADH produced interacts with ETC complex I, then electron transport runs sequentially (missing out complex II) to Coenzyme Q (CoQ; also termed ubiquinone), complex III, Cytochrome C and complex IV.8 Succinate, contrastingly, misses out complex I, and electron transport runs sequentially from complex II to CoQ, complex III, Cytochrome C and complex IV with molecular oxygen as terminal electron acceptor, culminating in conversion of oxygen to water.8 Complexes I, III and IV export protons across the mitochondrial inner membrane creating a proton electrochemical potential gradient, driving ATP synthesis at complex V (ATP synthase), converting ADP to ATP.8
Succinate can support compromised mitochondria, as it bypasses complex I that can be more vulnerable to inhibition/damage than complexes II, III and IV.9 Optimization of residual energy-producing ability may prevent the ATP level from dropping critically low initiating cell death pathways. ‘Bypassing’ defective ETC components sustained muscle ATP synthesis in a patient with genetically defective complex III, using vitamins C and K.10 In experimental sepsis (which involves energy dysfunction), succinate infusion prevented fall in liver ATP content,11 and prolonged survival.12
Succinate uptake is via the SLC13 family of Na+-coupled di-carboxylate and tri-carboxylate transporters.13–15 SLC13 occur widely, including in brain, astrocytes and neurons where succinate uptake and metabolism were also shown using radio-labelling.16–18 Additionally, nonspecific uptake might occur in any cells with increased plasma membrane permeability. Succinate is metabolised to fumarate by SDH, ETC complex II on the inner mitochondrial membrane. SDH occurs at high concentrations only within mitochondria.19 Hence, appearance of metabolites with characteristic 13C nuclear magnetic resonance (NMR) doublet signals will clearly indicate uptake and mitochondrial metabolism of 2,3-13C2 succinate. For a schematic of labelling via the TCA cycle, see Figure 1. In the TCA cycle, succinate’s metabolite fumarate is converted to malate and then to oxaloacetate (OAA) that can proceed in the TCA cycle to alpha-ketoglutarate, and then spin out (via glutamate) to glutamine. Also, OAA can spin out aspartate, which can exit mitochondria (via the glutamate/aspartate carrier) via the malate-aspartate shuttle mechanism.20 In the cytosol, aspartate is converted to OAA and thence to malate, which can re-enter mitochondria (via the malate/alpha-ketoglutarate carrier) and/or may emerge from the TCA cycle via this pathway.
Figure 1.

Schematic of metabolism of 2,3-13C2 succinate via the TCA cycle and spin-out pathways. Blue-filled circles indicate 13C atoms. Red rectangular outlines indicate metabolites detected by 13C NMR in microdialysates. For further details, see Results and Discussion sections. ME: malic enzyme; PC: pyruvate carboxylase; PDH: pyruvate dehydrogenase; PEPCK: phosphoenolpyruvate carboxykinase; PK: pyruvate kinase.
We tested the hypothesis that 2,3-13C2 succinate (disodium salt) administered by cerebral microdialysis would fuel and enhance the TCA cycle and improve cerebral metabolism focally in TBI patients. We analysed metabolites using NMR of the emerging microdialysates. We successfully used 13C-labelled microdialysis (Figure 2) previously with different substrates.21–23 Microdialysates were also analysed for conventional clinical biomarkers of brain chemistry,3 at baseline and during succinate perfusion. Double 13C-labelling proves unambiguously that substrate molecules crossed from the perfusate into the brain extracellular space, entered cells and were metabolised, exported into the extracellular fluid, and recovered by the microdialysis catheter.
Figure 2.
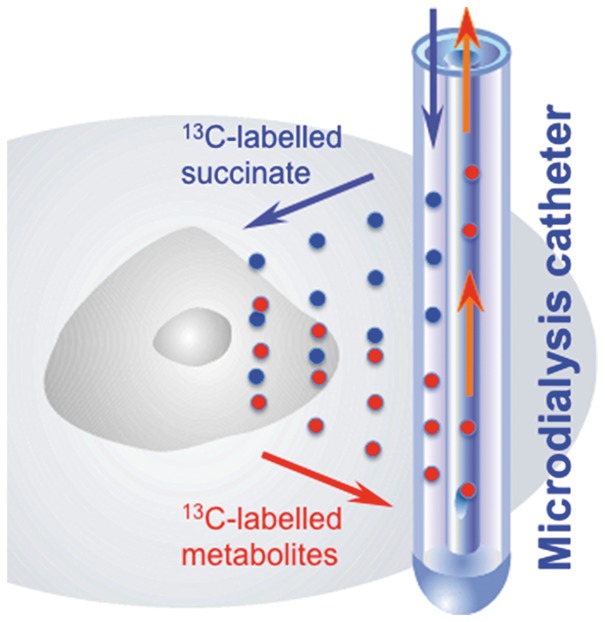
Schematic of 13C-labelled microdialysis. Microdialysis is used both to deliver 13C-labelled succinate focally into the brain extracellular space and simultaneously collect those metabolite molecules that exit from the cells.
Source: Adapted from Carpenter et al.23 ©2014 The Authors. Published by Elsevier B.V. Open access under a CC–BY licence.
Patients and methods
Patients
The protocol was approved by the National Research Ethics Service (NRES) Committee East of England – Cambridge Central (REC Reference No. 11/EE/0463). Written, informed consent was obtained from the next of kin for all subjects. The study was carried out in conformation with the spirit and the letter of the Declaration of Helsinki 1975 (and revised in 1983). Patients (age over 16 years) with severe TBI, defined as cranial trauma with consistent CT scan findings and a post-resuscitation Glasgow Coma Scale ≤ 8 were recruited. Patients were treated according to local TBI management protocols, including endotracheal intubation, ventilation, sedation, muscular paralysis and maintenance of blood sugar (serum glucose) concentration within the target range 4–7 mmol/L.5
Microdialysis technique
CMA 71 microdialysis catheters (membrane 10 mm, cut-off 100 kDa) (M Dialysis AB, Stockholm, Sweden) were placed via a triple lumen cranial access device (Technicam, Newton Abbot, UK) into frontal lobe. For patients with diffuse injury, the right frontal region was chosen. If one hemisphere was more injured than the other, the more injured hemisphere was monitored. Catheters were placed neither into, nor adjacent to, lesions identified on neuroimaging, e.g. contusions. Licox brain tissue oxygen concentration (PbtO2) monitoring probes (Integra LifeSciences Corporation, Plainsboro, NJ) were inserted alongside the microdialysis catheters via the same cranial access device.
Catheters were perfused with CNS Perfusion Fluid (M Dialysis AB), composed of NaCl (147 mmol/L), KCl (2.7 mmol/L), CaCl2 (1.2 mmol/L) and MgCl2 (0.85 mmol/L) in water supplemented with 12 mmol/L disodium 2,3-13C2 succinate (isotopic enrichment 99%, chemical purity 99%) from Cambridge Isotope Laboratories, Inc (Tewksbury, MA) and formulated in CNS perfusion fluid by the Manufacturing Unit, Department of Pharmacy, Ipswich Hospital NHS Trust (Ipswich, UK), who then tested the formulations for purity, sterility, freedom from endotoxins and absence of pyrogenicity, before release for use in patients. The 12 mmol/L concentration was chosen for 2,3-13C2 succinate, because it represents the same number of carbon atoms (12 mmol/L × 4 carbon atoms per succinate molecule = 48 mmol of carbon atoms/L) equivalent to glucose at the upper end24 of its microdialysate concentration range (8 mmol/L × 6 carbon atoms per glucose molecule = 48 mmol of carbon atoms/L). Glucose concentrations of 8 mmol/L in microdialysates are rare; we deliberately chose this high level as being above the more frequently seen levels (which are usually under 5 mmol/L) to ensure that there was no substrate limitation and we could augment the TCA cycle effectively. Moreover, with microdialysis delivery of a substrate, a diffusion gradient will exist such that the tissue levels achieved will not exceed the perfusate concentration.
Microdialysate collection vials were changed and analysed hourly on a bedside ISCUS analyser (M Dialysis AB) employing enzymatic colorimetric assays for glucose, lactate, pyruvate, glutamate and glycerol, during perfusion with 2,3-13C2-succinate, and for a baseline perfusion period with unsupplemented CNS perfusion fluid. The latter was either before or after the 2,3-13C2 succinate perfusion period, with a 2 h exclusion margin applied to the ISCUS data analysis to allow for run-in or wash-out. By testing standard solutions of the five ISCUS analytes at concentrations representative of those in vivo, in CNS-perfusion fluid, with and without disodium succinate standard (12 mmol/L), we established that succinate did not interfere with the ISCUS enzymatic colorimetric chemistry.
NMR analysis
Brain microdialysate samples analysed on the ISCUS at the bedside were then stored at 4℃ (or –80℃ if storage for longer than a few days was necessary) prior to NMR analysis. Microdialysate samples from a 24-h period during 2,3-13C2 succinate perfusion were pooled for each individual patient, due to sensitivity limitations of NMR equipment (see ‘Limitations’ subsection of the Discussion for further explanation). For NMR analysis, 20 µL of deuterium oxide (D2O) and 50 µL internal reference standard from a stock solution of 24.0 mmol/L 3 -(trimethylsilyl)-1-propanesulfonic acid sodium salt (also termed 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt or 4,4-dimethyl-4-silapentane-1-sulfonate sodium salt; DSS) (Sigma-Aldrich, Gillingham, UK) in CNS perfusion fluid was added to 180 µL of the pooled microdialysate sample, then transferred into 3 mm NMR tubes (Hilgenberg GmbH, Malsfeld, Germany).
13C and 1H NMR spectra were acquired on a Bruker Avance III HD 500 MHz spectrometer (Bruker BioSpin GmbH, Karlsruhe, Germany) with a dual 1H/13C cryoprobe (CP DUL500C/H, Bruker BioSpin GmbH), and TopSpin software (Bruker GmbH). For further details of NMR measurements, see Jalloh et al.22 Direct-observe 13C NMR is a wide-ranging screening technology not requiring operators to make prior decisions of what to seek. NMR does not involve physical contact with the sample, which is enclosed in an individual glass tube throughout measurement, eliminating cross-contamination or sample carry-over.
Fractional enrichment (%) is defined as 100 × [13C]/([13C] + [12C]) where square brackets indicate concentrations. [13C] was determined from the 13C NMR spectra and [12C] for lactate from the 1H spectra, using the calibration principles described previously.22 Total glutamine concentration (12C and 13C) was quantified by reverse-phase high-performance liquid chromatography.25 The natural abundance of 13C is 1.1% of all carbon atoms, and 13C results presented for lactate and glutamine were expressed after subtracting this natural background from the 13C singlet NMR signals. 13C doublet NMR signals for lactate and glutamine were not background-subtracted, because the probability of two natural endogenous 13C atoms being next to each other is 1.1% × 1.1% = 0.01%.
Statistical analysis
Changes between baseline and succinate perfusion periods were assessed using Wilcoxon signed rank test (StatView version 5, SAS Institute, Cary, NC), with p < 0.05 for significance. The corresponding effect sizes (matched-pairs rank-biserial correlation r) were calculated by the method described by Kerby.26
Results
Demography
Nine severe TBI patients (seven male, two female) aged 18–60 years (median 47 years) were studied using 2,3-13C2 succinate (disodium salt; 12 mmol/L) perfused via the microdialysis catheter, and for a baseline period with plain unsupplemented perfusion fluid (without 2,3-13C2 succinate). Table 1 describes demography, catheter positions and PbtO2 measured alongside microdialysis in 7/9 of the patients. Median PbtO2 measurements were above 20 mmHg in five patients and above 15 mmHg in the other two patients.
Table 1.
Patient demography.
| TBI Patient i.d. number | Age (years) | Sex | Injury mechanism | GCS at scene/15 | 2,3-13C2 succinate perfusion period start time (hours from injury) | Marshall grade (admission) | Catheter locationa | CT description | PbtO2 at baseline and during 2,3-13C2 succinate perfusion (mmHg) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 56 | F | Fall | 10 | 26.0 | 6a | L frontal | EDH, contusions | 25.3, 31.1 |
| 2 | 52 | M | RTC | 4 | 121.3 | 3 | R frontal | DAI | – |
| 3 | 18 | M | RTC | 8 | 41.9 | 3 | R frontal | DAI, contusions | 21.8, 20.4 |
| 4 | 47 | M | RTC | 8 | 36.9 | 2b | R frontal | DAI, contusions | 19.9, 27.4 |
| 5 | 41 | F | Fall | 3 | 67.5 | 6b | R frontal | R ASDH | –, 35.7 |
| 6 | 20 | M | Assault | 14 | 83.0 | 3 | R frontal | DAI, contusions | –, 15.8 |
| 7 | 60 | M | Fall | 11 | 103.9 | 2d | R frontal | DAI, contusions | 18.6, 17.9 |
| 8 | 47 | M | RTC | 9 | 160.4 | 2b | R frontal | DAI | – |
| 9 | 46 | M | RTC | 6 | 79.5 | 3 | R frontal | L ASDH | 34.9, 34.9 |
M: male; F: female; RTC: road traffic collision; GCS: Glasgow Coma Scale score; L: left; R: right; EDH: extradural haematoma; DAI: diffuse axonal injury; ASDH: acute subdural haematoma; PbtO2: brain tissue oxygen tension (median values; – not measured).
Catheter (microdialysis) location: for patients with a diffuse injury, the right frontal region was chosen; if there was greater injury to one hemisphere rather than the other, the side with the greater burden of injury was monitored. Catheters were inserted via a cranial access device and were placed neither into nor adjacent to lesions (such as contusions) identified on neuroimaging. PbtO2 monitoring probes were inserted alongside the microdialysis catheters via the same cranial access device.
Bedside analysis results
Microdialysate measurements (ISCUS analyser) of glucose, lactate, pyruvate, glutamate, glycerol and LPR are shown in Figure 3 and Supplementary Table 1. The ISCUS cannot measure other species. ISCUS measurements were acquired for the nine TBI patients for a 24-h period while the microdialysis catheter was perfused with plain unsupplemented CNS perfusion fluid directly before or after a 24-h perfusion period with 2,3-13C2 succinate (12 mmol/L). Each data-point shown is the median concentration for that patient during the relevant 24 h period (24 × 1 h vials). Paired (baseline vs. succinate) data were available in all nine patients for glucose, lactate, pyruvate and LPR, and in 7/9 patients for glutamate and 6/9 patients for glycerol. We chose pair-wise statistics (Wilcoxon signed rank test) for comparing baseline vs. succinate perfusion, so that each patient was represented by a pair of data-points preserving data connectivity within each patient. This avoided risk of bias, rather than pooling data and using unpaired statistics that would have incorrectly assumed too many degrees of freedom and over-estimated significance. In addition to Wilcoxon signed rank test significance p values, we also report effect size (matched-pairs rank-biserial correlation r).26
Figure 3.

ISCUS clinical microdialysis analyser measurements. Results are during 24 h baseline perfusion (with plain unsupplemented CNS perfusion fluid) and during 24 h perfusion with 2,3-13C2 succinate (disodium salt; 12 mmol/L). Symbols joined by lines represent individual patients (TBI Patients 1–9). Each pair of data-points indicates median levels at baseline and during succinate perfusion, respectively, for that patient. Note that, for Patients 6 and 7, the lactate concentrations were similar (but not identical), so their symbols and lines are very close to each other. For Patients 1 and 2, the baseline period was post-succinate, while for the other seven patients, the baseline period was pre-succinate. Changes between baseline and succinate perfusion were significant for lactate/pyruvate ratio (p = 0.0152), glucose (p = 0.038) and glutamate (p = 0.018) by Wilcoxon’s signed rank test.
Versus baseline, succinate perfusion significantly decreased the LPR (p = 0.015; effect size r = 0.91), decreasing in 8/9 patients and increasing in 1/9, mean difference −12%. This was due to increased pyruvate concentration, increasing in 7/9 and decreasing in 2/9, mean difference + 17%, missing significance (p = 0.11; effect size r = 0.60). Lactate concentration changed insignificantly (p = 0.86; effect size r = 0.07), increasing in 4/9 and decreasing in 5/9, mean difference −3%. Glycerol concentration change did not reach significance (p = 0.075; effect size r = 0.81), increasing in 5/6 and decreasing in 1/6. Glutamate concentration decreased significantly (p = 0.018; effect size r = 1.00) in 7/7, mean difference−43%. Glucose also decreased significantly (p = 0.038; effect size r = 0.78), decreasing in 7/9 and increasing in 2/9, mean difference −15%. The greatest decreases in microdialysate glucose were in the four individuals (ranging from −22.9% to −31.7%) whose microdialysate glucose baseline concentration was above 3 mmol/L. In the two patients with the highest baseline LPRs of 31.3 and 26.8, these decreased to 21.5 and 23.3, respectively, during succinate perfusion, thus bringing them below 25, regarded as a crucial threshold.3,27 These results are graphed in Figure 3 and tabulated in Supplementary Table 1.
Changes in ISCUS data are modest rather than extreme, as we micro-dosed a focal region of brain. While we cannot rule out some spontaneous changes in brain chemistry, a much larger unsupplemented microdialysis TBI study showed no clear longitudinal trends.24 Comparing changes within patient (baseline vs. succinate supplementation) was preferable to comparing pooled data for the supplemented patients with a separate control unsupplemented group, as is well known that there is much variation between patients for ISCUS data. Using each patient as his/her own control was logical in this small group.
NMR results
NMR analysis of brain microdialysates was performed for six TBI patients who received 2,3-13C2 succinate microdialysis perfusion (Figure 4 illustrates examples). To achieve this, 24 × 1 h microdialysate vials were pooled for each patient, due to sensitivity limitations of our NMR equipment (see ‘Limitations’ subsection of the Discussion for further explanation). It was therefore not possible for us to obtain time-course data by NMR. All six patients showed a strong signal at 36.9 ppm for succinate C2 and C3 (2,3-13C2 succinate). This is a singlet because succinate is a symmetrical molecule so the C2 and C3 positions are equivalent. The strongest metabolite signals were doublets (J = 37.3 Hz) for malate C3 at 45.2 ppm and C2 at 73.1 ppm, in all six patients, indicating 2,3-13C2 malate. There was no discernable singlet for malate. These results – malate doublets, but no singlets – indicate that the malate was entirely derived, without breaking the covalent bond between the two 13C atoms, from the 2,3-13C2 succinate administered. A singlet at 138.0 ppm for C2 and C3 of fumarate (indicating 2,3-13C2 fumarate) was seen in all six patients. This is a singlet because fumarate is a symmetrical molecule. Doublets (J = 36.8 Hz) for lactate C3 (methyl group) and for lactate C2 were observed in all six patients, at 22.8 and 71.3 ppm, respectively, indicating the presence of 2,3-13C2 lactate, together with singlets for lactate C3 and C2.
Figure 4.

Illustrative examples of 13C NMR spectra. Upper two spectra are for microdialysates from TBI Patients 3 and 5 who received perfusion with 2,3-13C2 succinate disodium salt (12 mmol/L). For comparison, the 2,3-13C2 succinate solution (before perfusion) (third spectrum) and microdialysate from an unlabelled TBI patient (using plain unsupplemented CNS perfusion fluid) (fourth spectrum) are also shown. Glc: glucose; Lac: lactate; Gln: glutamine; Mal: malate; DSS: 4,4-dimethyl-4-silapentane-1-sulfonate sodium salt (the internal reference standard). Spectra were run from −20 ppm to + 250 ppm. The main reference DSS signal at 0 ppm, and fumarate (138 ppm singlet for equivalent C2 and C3, in the spectra of patients with 2,3-13C2 succinate perfusion) are not shown in the ranges illustrated.
Doublets (J = 34.8 Hz) were observed at 57.4 and 30.4 ppm, representing glutamine C2 and C3, respectively, indicating the presence of 2,3-13C2 glutamine in five of six patients, together with singlets for glutamine C2 and C3. The remaining patient (TBI Patient 1) showed no clear doublet or singlet signals discernable above noise for Gln C2 or Gln C3. Peak identities were corroborated on NMR by two-dimensional 1H-13C HSQC spectra. Glutamine is synthesised from glutamate that is a spin-off from the TCA cycle intermediate alpha-ketoglutarate. The presence of doublets for glutamine C2 and C3 indicates metabolism of 2,3-13C2 succinate by the TCA cycle with the covalent bond between the two 13C atoms intact. No other doublet signals were observed for glutamine (or glutamate).
None of the six patients showed a clear glutamine C4 singlet above baseline noise. Quantification of the C2 and C3 glutamine singlets (after subtracting background natural abundance 13C) revealed small degrees of fractional enrichment with medians of 1.1% (0%–3.6%) and 3.5% (1.1%–6.9%), respectively.
Pyruvate was undetectable in our 13C NMR spectra. Pyruvate’s chemical shifts (C3 at 29.15 ppm, C2 at 207.7 ppm and C1 at 172.8 ppm, in spectra of standards) do not coincide with lactate or any other of our analytes. Pyruvate is at the crossroads of several biochemical pathways, being synthesised and consumed, so its concentration is never high, and microdialysis only samples the extracellular compartment. In the literature, 13C-labelled pyruvate is usually not itself detectable as a metabolite in NMR spectra of brain tissue extracts, and pyruvate’s involvement as an intermediate is inferred from detection of labelling in its products.28,29 Moreover, exogenous 13C-labelled pyruvate is very rapidly metabolised by brain.30
Results for fractional enrichment (%) in 2,3-13C2 glutamine are shown in Figure 5(a). The median was 16.2% (5.3%–22.6%) measured using the C3 doublet. Similar fractional enrichment was seen for the glutamine C2 doublet, as expected. The 13C doublet signals at glutamine C2 and C3 were not background-subtracted because the probability of two natural endogenous 13C atoms occurring next to each other is 0.01%.
Figure 5.

13C enrichment in glutamine and lactate. (a) Fractional enrichment values (%) for microdialysate 2,3-13C2 glutamine (blue bars; based on the glutamine C3 doublet signal) and 2,3-13C2 lactate (red bars; based on the lactate C3 doublet signal). (b) Corresponding concentrations (µmol/L) of 2,3-13C2 glutamine and 2,3-13C2 lactate.
Fractional enrichment (%) results in 2,3-13C2 lactate are shown in Figure 5(a). Median (IQR) fractional enrichments of the lactate C3 and C2 doublets were 2.3% (1.1%–4.5%) and 2.0% (0.8%–4.4%).
The corresponding concentrations (in µmol/L) of 2,3-13C2 glutamine and 2,3-13C2 lactate are shown in Figure 5(b). Singlets for lactate C3 and C2 revealed no significant 13C enrichment above background natural abundance 13C. In five of six patients, enrichment was greater in 2,3-13C2 glutamine than in 2,3-13C2 lactate, while in the remaining patient (TBI Patient 1), 2,3-13C2 glutamine was undetectable although 2,3-13C2 lactate was present. The median (IQR) 2,3-13C2 lactate/2,3-13C2 glutamine enrichment ratio was 19.9% (12.9%–21.7%).
The 13C-labelling patterns identified are thus clear evidence for the metabolic pathways shown in Figure 1.
Discussion
We have shown that 2,3-13C2 succinate (disodium salt) perfused via microdialysis catheters in TBI patients entered the TCA cycle evidenced by 13C-labelling patterns in metabolites (Figures 1 and 4), and also potentiated several aspects of brain chemistry: importantly decreases in extracellular LPR and glutamate concentration.
Significance of double labelling patterns in metabolites
The characteristic NMR doublets arising from two adjacent 13C atoms prove cellular uptake and mitochondrial metabolism of 2,3-13C2 succinate revealing downstream metabolites. Doublets are clearly distinct from native endogenous molecules, because the probability of two endogenous 13C atoms being adjacent to each other naturally is 0.01% (a 1 in 10,000 chance) and indistinguishable from baseline ‘noise’ in these spectra. As SDH is exclusively mitochondrial, downstream metabolites of 2,3-13C2 succinate must be derived from pathways stemming from mitochondrial metabolism. In microdialysates, the extracellular concentrations and 13C labelling patterns allow deduction of cellular metabolic pathways. The doubly 13C-labelled metabolites are unambiguous evidence the 2,3-13C2 succinate molecules crossed from the perfusate into the brain extracellular space, entered the cells and were metabolised, exported into the extracellular fluid and recovered by the microdialysis catheter.
13C NMR clearly demonstrated that 2,3-13C2 malate, 2,3-13C2 glutamine and 2,3-13C2 lactate were among the metabolites, indicating TCA cycle metabolism with the C2-C3 carbon–carbon bond intact (Figure 5). 2,3-13C2 glutamine is a TCA spinout product originating from alpha-ketoglutarate, a TCA cycle intermediate exiting as glutamate, enzymatically inter-convertible with glutamine that predominates extracellularly.
The TCA cycle intermediate OAA can spin out aspartate, which can exit mitochondria (via the glutamate/aspartate carrier) via the malate-aspartate shuttle mechanism.20 In the cytosol, aspartate is converted to OAA and thence to malate, which can re-enter mitochondria (via the malate/alpha-ketoglutarate carrier). The 2,3-13C2 malate detected in microdialysates may have emerged from the TCA cycle via this pathway.
Our identification of 2,3-13C2 lactate among the metabolites of 2,3-13C2 succinate is the first demonstration of the TCA cycle spin-out route of lactate production in human TBI brain, and merits investigation. Previous indications were in animal brains.28,29,31 The TCA cycle intermediates malate and OAA can be converted to pyruvate by malic enzyme (ME); also OAA can be converted by phosphoenolpyruvate carboxykinase (PEPCK) plus pyruvate kinase (PK) to pyruvate, which LDH can convert to lactate. Our finding of TCA cycle-derived lactate may have implications for interpreting high extracellular levels of lactate and LPR, hitherto simply attributed to glycolysis. An LPR > 25 is interpreted as either hypoxia or ‘mitochondrial dysfunction’. The latter remains poorly understood, and this study raises questions of whether the lactate (and implicitly pyruvate) derived from TCA-cycle spin-out, termed cataplerosis31 forms a significant proportion of total extracellular lactate (and pyruvate), whether cataplerotic levels change in injured vs. normal brain, and biological implications.
Pyruvate recycling – Partial versus full
Lactate generation by TCA cycle cataplerosis is also termed ‘partial pyruvate recycling’, as the pyruvate spun out from the TCA cycle does not re-enter it but proceeds to lactate that exits from cells.31 This appears distinct from full pyruvate recycling28,32,33 whereby spun-out pyruvate is converted by pyruvate dehydrogenase (PDH) to acetate that re-enters the TCA cycle. We found no evidence for full recycling, as spun-out 2,3-13C2 pyruvate conversion by PDH to 1,2-13C2 acetate entering the TCA cycle would have produced 4,5-13C2 glutamine,32 which we did not detect. Neither did we detect 1,2-13C2 lactate, which would have resulted from subsequent spin-out from 1,2-13C2 oxaloacetate, and there was no significant enrichment for 3-13C lactate, which would have arisen from spin-out from 3,4-13C2 oxaloacetate.
Re-entry of 2,3-13C2 pyruvate via anaplerotic pathways pyruvate carboxylase (PC) or ME into the TCA cycle would produce 2,3-13C2 glutamine, the same 13C-pattern as if 2,3-13C2 succinate metabolism proceeds directly (without exit of intermediates and re-entry) round the TCA cycle to 2,3-13C2 alpha-ketoglutarate and spins out 2,3-13C2 glutamine. Re-entry via PC or ME seems unlikely in the absence of evidence for re-entry via PDH. Therefore, the 2,3-13C2 glutamine detected is probably from 2,3-13C2 succinate metabolism proceeding directly round the TCA cycle, further supported by fractional enrichment (%) for 13C being greater (in five of six patients) in 2,3-13C2 glutamine than in 2,3-13C2 lactate (Figure 5(a)).
Succinate perfusion potentiates local brain chemistry
Conventional measures (regardless of labelling), on an ISCUS microdialysis analyser, at baseline and during 2,3-13C2 succinate perfusion, showed glucose concentration decrease suggesting greater utilisation, a lower LPR due to relatively raised pyruvate, and glutamate decrease. LPR ‘Type 2’ elevation characterised by low pyruvate without hypoxia or ischaemia is attributed to metabolic disturbance.34 Increasing pyruvate and thereby lowering LPR thus concurs with better brain metabolism, consistent with TCA cycle operation indicated by the 13C-labelling patterns.
To generate 1 mole of pyruvate by glycolysis, 1 mole of NAD+ is converted into NADH, which must be recycled (oxidised) back to NAD+ to sustain glycolysis. Recycling can occur by the mitochondrial ETCs (if operational). NADH cannot cross the mitochondrial membrane, so hydrogens and electrons are transferred indirectly by shuttles (malate-aspartate and/or glycerol 3-phosphate).20 Malate-aspartate shuttle operation concurs with 2,3-13C2 malate in microdialysates. In mitochondrial dysfunction or hypoxia, NADH can be recycled to NAD+ by LDH-mediated conversion of pyruvate to lactate in the cytosol, producing a high extracellular LPR.
Lowering of brain extracellular glucose during succinate perfusion, suggesting improved glucose utilisation, together with a lower extracellular LPR, may stem from better redox behaviour by aforementioned NADH to NAD+ recycling by mitochondrial shuttles,20 lessening LDH-mediated conversion of pyruvate to lactate. The lower extracellular glutamate during succinate perfusion may result from the malate-aspartate shuttle, drawing glutamate into mitochondria for consumption via the TCA cycle, preventing excitotoxic glutamate build-up. High extracellular glutamate and glucose levels are associated with adverse clinical outcomes.3
Conventionally, the microdialysis literature ascribes glycerol increase to phospholipid degradation suggesting cell death.35 However, debate exists34–36 because glycerol is a glucose metabolite37–39 and glycerol increase during succinate perfusion may arise from increased glycolysis. Glycerol decreased after succinate perfusion ceased, in the two patients whose baseline period was post-succinate (Figure 3), concurring with metabolically derived glycerol, rather than cell death. Conceivably, glycerol may increase by succinate fuelling the mitochondrial ETC enhancing the glycerol 3-phosphate (G3P) shuttle, recycling NADH to NAD+ in the cytosol when dihydroxyacetone phosphate (DHAP; a glycolysis intermediate) is reduced by cytosolic glycerol 3-phosphate dehydrogenase (GPDH) to G3P.20 This is re-oxidized to DHAP by mitochondrial GPDH and electrons transferred to CoQ and enter the mitochondrial ETC.20 Like the malate-aspartate shuttle (above), enhancing the G3P shuttle would improve recycling of NADH to NAD+ by mitochondrial ETC, giving lower LPR and more efficient glucose utilisation.
Importance of oxygen
PbtO2 measurements indicated oxygen supply to the region that received disodium succinate. Without oxygen, the mitochondrial ETC cannot function properly. In experimental ischaemia, SDH (ETC complex II and part of the TCA cycle) ran ‘backwards’, building up succinate and reverse electron transport to complex I.40 Reperfusion produced a surge in reactive oxygen species (via complex I) and cell death.40 Dimethyl succinate (an artificial, lipophilic derivative much more taken up by cells than disodium succinate) exacerbated ischaemia–reperfusion damage, while malonate, a TCA cycle inhibitor that blocks SDH, protected against ischaemia–reperfusion injury.40 Our present study did not involve ischaemia–reperfusion. We suggest that using succinate to support mitochondrial metabolism should be performed in the presence of adequate oxygenation, not ischaemia–reperfusion.
In circumstances like our study, where oxygen appears not limiting, oxidative phosphorylation is limited by mitochondrial efficiency, which appears improved (lower microdialysate LPR) by succinate administration. The doubly 13C-labelled metabolites unambiguously prove that the disodium 2,3-13C2 succinate was taken up and metabolised by mitochondria.
Limitations
The reason we combined brain microdialysates for 24 h per patient was to provide enough material for 13C NMR within a workable spectrum acquisition time on the NMR spectrometer. Our 500 MHz NMR spectrometer has a cryoprobe21,22 that is more sensitive than non-cryo probe technology, but, nevertheless, limits how short a microdialysis timeframe we can analyse. NMR micro-cryoprobes are commercially available, for smaller samples, and future adoption, with higher-field NMR spectrometers for even greater sensitivity, if accessible, will improve time-resolution for tracking biochemical pathways. We did not analyse blood. Focal brain micro-dosing is unlikely to yield measurable labelling in blood, due to dilution of 13C-labelled species with unlabelled endogenous species and physical volume of blood. Neurocritical care protocol-driven therapy includes maintaining a serum glucose 4–7 mmol/L target range. While serum glucose and glycaemic control can influence brain glucose, this relationship is not necessarily straightforward and may be lost in injured brain,27,41 meriting dedicated study with continuous monitoring of both blood and brain.
Cerebral microdialysis is an invasive technique and is thus effectively limited to patients being monitored following severe brain injury. These patients are sedated (see ‘Patients and methods’ section), so there is no placebo effect – they are unconscious. On account of its invasive nature, there are no truly normal controls for cerebral microdialysis in humans. It has sometimes been possible to perform microdialysis in radiologically normal brain areas in patients undergoing surgery for removal of benign tumours in a different part of the brain,22,42 but patient considerations and practical matters make such sampling rare. In the present study, we used each TBI patient as his or her own control (see ‘Patients and methods’ and ‘Results’ sections). In further support of this approach, it has been noted in the literature that microdialysis data are highly auto-correlated even at up to a future 30 h, and subject identity alone explains 52% to 75% of microdialysis marker variance, determined in a large study (comprising more than 7350 hourly samples of complete microdialysis sets from 90 TBI patients) using statistical and pattern-recognition computer models.24 Thus, microdialysate data are highly individualised and it was therefore logical to use each patient as his/her own control rather than comparing with a separate control group of different TBI patients.
Mitochondrial dysfunction is defined as an inability to generate energy despite ‘adequate’ provision of metabolic fuels and oxygen. This suggests a fundamental biochemical failure at the mitochondrial level following TBI. Mitochondrial dysfunction is undoubtedly a complex phenotype and exploring its full ramifications is outside the scope of the present study. We have established that TBI brain retains sufficient mitochondrial capability to metabolise exogenous succinate by the TCA cycle, proved using double 13C-labelling. This exogenous fuelling of the TCA cycle resulted in apparent improvements in brain chemistry judged by surrogate endpoints including LPR decrease. Whether such changes would translate to better clinical outcome would need a more extensive, larger study in TBI patients. Further support for benefits of augmenting brain mitochondrial oxidative metabolism comes from studies in rats with methylene blue,43 or with kaempferol,44 which enhances mitochondrial matrix uptake of Ca2+ and boosts oxidative metabolism, which led to improved brain function and behavioural outcomes after experimental brain injury in a rat TBI model.45
Conclusion
Here we have shown that 2,3-13C2 succinate delivered by microdialysis can interrogate the TCA cycle’s role in producing 13C-labelled metabolites exported into the interstitium, proof-of-concept that the TCA cycle can be directly supplemented and TBI brain chemistry potentiated. Our novel discovery of the TCA cycle as a source of lactate in human TBI brain calls for revision of lactate’s significance, hitherto regarded as simply glycolytic, with possible implications for other diseases (e.g. cancer). Lower LPR suggests that succinate improves redox balance, conceivably by boosting shuttles utilising mitochondrial ETCs to recycle NAPH to NAD+, possibly promoting glucose utilisation and glutamate clearance from the interstitium. Whether such metabolic shifts would ameliorate ATP generation merits investigation. Specific intervention that modifies the LPR provides a promising future avenue.27 This study opens the prospect of enhancing mitochondrial metabolism by using TCA cycle products/substrates such as succinate or other species as a potential therapeutic strategy, for brain injury and other neurological disorders involving mitochondrial dysfunction.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Medical Research Council (Grant Nos. G0600986 ID79068 and G1002277 ID98489) and National Institute for Health Research Biomedical Research Centre, Cambridge (Neuroscience Theme; Brain Injury and Repair Theme). Authors’ support: IJ – Medical Research Council (Grant no. G1002277 ID 98489) and National Institute for Health Research Biomedical Research Centre, Cambridge; KLHC – National Institute for Health Research Biomedical Research Centre, Cambridge (Neuroscience Theme; Brain Injury and Repair Theme); CG – the Canadian Institute of Health Research; AH – Medical Research Council/Royal College of Surgeons of England Clinical Research Training Fellowship (Grant no. G0802251) and Raymond and Beverly Sackler Fellowship; DKM and JDP – National Institute for Health Research Senior Investigator Awards; PJH – National Institute for Health Research Professorship, Academy of Medical Sciences/Health Foundation Senior Surgical Scientist Fellowship and the National Institute for Health Research Biomedical Research Centre, Cambridge.
Supplementary Material
Acknowledgements
The authors thank Mr John Harwood (Manufacturing Unit, Department of Pharmacy, Ipswich Hospital NHS Trust) for supervising the formulation of the 13C substrate.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JDP and PJH are Directors of Technicam, the company that manufactures the triple lumen cranial access device used in this study.
Authors’ contributions
Concept and study design: PJH, IJ, KLHC, AH, TAC, MPM, CNG, PG, DKM, JDP. Data acquisition and analysis: IJ, DJH, KLHC, TAC, AM, PG, SV, MGS, RJS. Drafting of the manuscript and figures: IJ, KLHC, AH, PJAH, SV. Review and editing of the manuscript: All authors.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Timofeev I, Czosnyka M, Carpenter KL, et al. Interaction between brain chemistry and physiology after traumatic brain injury: impact of autoregulation and microdialysis catheter location. J Neurotrauma 2011; 28: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkin M, Hopwood S, Jones DA, et al. Dynamic changes in brain glucose and lactate in pericontusional areas of the human cerebral cortex, monitored with rapid sampling on-line microdialysis: relationship with depolarisation-like events. J Cereb Blood Flow Metab 2005; 25: 402–413. [DOI] [PubMed] [Google Scholar]
- 3.Timofeev I, Carpenter KL, Nortje J, et al. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain 2011; 134: 484–494. [DOI] [PubMed] [Google Scholar]
- 4.Vespa P, Tubi M, Claassen J, et al. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann Neurol 2016; 79: 279–590. [DOI] [PubMed] [Google Scholar]
- 5.Helmy A, Vizcaychipi M, Gupta AK. Traumatic brain injury: intensive care management. Br J Anaesth 2007; 99: 32–42. [DOI] [PubMed] [Google Scholar]
- 6.Verweij BH, Muizelaar JP, Vinas FC, et al. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J Neurosurg 2000; 93: 815–820. [DOI] [PubMed] [Google Scholar]
- 7.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 1990; 70: 391–425. [DOI] [PubMed] [Google Scholar]
- 8.Rich PR, Marechal A. The mitochondrial respiratory chain. Essays Biochem 2010; 47: 1–23. [DOI] [PubMed] [Google Scholar]
- 9.Protti A, Singer M. Bench-to-bedside review: potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit Care 2006; 10: 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eleff S, Kennaway NG, Buist NR, et al. 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc Natl Acad Sci USA 1984; 81: 3529–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malaisse WJ, Nadi AB, Ladriere L, et al. Protective effects of succinic acid dimethyl ester infusion in experimental endotoxemia. Nutrition 1997; 13: 330–341. [PubMed] [Google Scholar]
- 12.Ferreira FL, Ladriere L, Vincent JL, et al. Prolongation of survival time by infusion of succinic acid dimethyl ester in a caecal ligation and perforation model of sepsis. Horm Metab Res 2000; 32: 335–336. [DOI] [PubMed] [Google Scholar]
- 13.Schlessinger A, Sun NN, Colas C, et al. Determinants of substrate and cation transport in the human Na+/dicarboxylate cotransporter NaDC3. J Biol Chem 2014; 289: 16998–17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kekuda R, Wang H, Huang W, et al. Primary structure and functional characteristics of a mammalian sodium-coupled high affinity dicarboxylate transporter. J Biol Chem 1999; 274: 3422–3429. [DOI] [PubMed] [Google Scholar]
- 15.Pajor AM. Sodium-coupled transporters for Krebs cycle intermediates. Annu Rev Physiol 1999; 61: 663–682. [DOI] [PubMed] [Google Scholar]
- 16.Lamp J, Keyser B, Koeller DM, et al. Glutaric aciduria type 1 metabolites impair the succinate transport from astrocytic to neuronal cells. J Biol Chem 2011; 286: 17777–17784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergeron MJ, Clemencon B, Hediger MA, et al. SLC13 family of Na(+)-coupled di- and tri-carboxylate/sulfate transporters. Mol Aspects Med 2013; 34: 299–312. [DOI] [PubMed] [Google Scholar]
- 18.Pajor AM. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch 2014; 466: 119–130. [DOI] [PubMed] [Google Scholar]
- 19.Aldridge WN, Johnson MK. Cholinesterase, succinic dehydrogenase, nucleic acids, esterase and glutathione reductase in sub-cellular fractions from rat brain. Biochem J 1959; 73: 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna MC, Waagepetersen HS, Schousboe A, et al. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol 2006; 71: 399–407. [DOI] [PubMed] [Google Scholar]
- 21.Gallagher CN, Carpenter KL, Grice P, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 2009; 132: 2839–2849. [DOI] [PubMed] [Google Scholar]
- 22.Jalloh I, Carpenter KL, Grice P, et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-(13)C2 glucose. J Cereb Blood Flow Metab 2015; 35: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpenter KL, Jalloh I, Gallagher CN, et al. (13)C-labelled microdialysis studies of cerebral metabolism in TBI patients. Eur J Pharm Sci 2014; 57: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nelson DW, Thornquist B, MacCallum RM, et al. Analyses of cerebral microdialysis in patients with traumatic brain injury: relations to intracranial pressure, cerebral perfusion pressure and catheter placement. BMC Med 2011; 9: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shannon RJ, Timofeev I, Nortje J, et al. Monitoring vigabatrin in head injury patients by cerebral microdialysis: obtaining pharmacokinetic measurements in a neurocritical care setting. Br J Clin Pharmacol 2014; 78: 981–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerby DS. The simple difference formula: an approach to teaching nonparametric correlation. Innov Teach 2014; 3: Article 1. [Google Scholar]
- 27.Hutchinson PJ, Jalloh I, Helmy A, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med 2015; 41: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruz F, Cerdan S. Quantitative 13C NMR studies of metabolic compartmentation in the adult mammalian brain. NMR Biomed 1999; 12: 451–462. [DOI] [PubMed] [Google Scholar]
- 29.Tyson RL, Gallagher C, Sutherland GR. 13C-Labeled substrates and the cerebral metabolic compartmentalization of acetate and lactate. Brain Res 2003; 992: 43–52. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez SV, Nguyen NH, Rise F, et al. Brain metabolism of exogenous pyruvate. J Neurochem 2005; 95: 284–293. [DOI] [PubMed] [Google Scholar]
- 31.Sonnewald U. Glutamate synthesis has to be matched by its degradation - where do all the carbons go? J Neurochem 2014; 131: 399–406. [DOI] [PubMed] [Google Scholar]
- 32.Cerdan S, Kunnecke B, Seelig J. Cerebral metabolism of [1,2-13C2]acetate as detected by in vivo and in vitro 13C NMR. J Biol Chem 1990; 265: 12916–12926. [PubMed] [Google Scholar]
- 33.Cruz F, Scott SR, Barroso I, et al. Ontogeny and cellular localization of the pyruvate recycling system in rat brain. J Neurochem 1998; 70: 2613–2619. [DOI] [PubMed] [Google Scholar]
- 34.Hillered L, Persson L, Nilsson P, et al. Continuous monitoring of cerebral metabolism in traumatic brain injury: a focus on cerebral microdialysis. Curr Opin Crit Care 2006; 12: 112–118. [DOI] [PubMed] [Google Scholar]
- 35.Hillered L, Valtysson J, Enblad P, et al. Interstitial glycerol as a marker for membrane phospholipid degradation in the acutely injured human brain. J Neurol Neurosurg Psychiatry 1998; 64: 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hillered L, Vespa PM, Hovda DA. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma 2005; 22: 3–41. [DOI] [PubMed] [Google Scholar]
- 37.Lin EC. Glycerol utilization and its regulation in mammals. Annu Rev Biochem 1977; 46: 765–795. [DOI] [PubMed] [Google Scholar]
- 38.Clausen F, Hillered L, Gustafsson J. Cerebral glucose metabolism after traumatic brain injury in the rat studied by 13C-glucose and microdialysis. Acta Neurochir (Wien) 2011; 153: 653–658. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen NH, Gonzalez SV, Hassel B. Formation of glycerol from glucose in rat brain and cultured brain cells. Augmentation with kainate or ischemia. J Neurochem 2007; 101: 1694–1700. [DOI] [PubMed] [Google Scholar]
- 40.Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014; 515: 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patet C, Quintard H, Suys T, et al. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma 2015; 32: 1560–1566. [DOI] [PubMed] [Google Scholar]
- 42.Reinstrup P, Stahl N, Mellergard P, et al. Intracerebral microdialysis in clinical practice: baseline values for chemical markers during wakefulness, anesthesia, and neurosurgery. Neurosurgery 2000; 47: 701–709. discussion 709–710. [DOI] [PubMed] [Google Scholar]
- 43.Lin AL, Poteet E, Du F, et al. Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS One 2012; 7: e46585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanganahalli BG, Herman P, Hyder F, et al. Mitochondrial calcium uptake capacity modulates neocortical excitability. J Cereb Blood Flow Metab 2013; 33: 1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murugan M, Santhakumar V, Kannurpatti SS. Facilitating mitochondrial calcium uptake improves activation-induced cerebral blood flow and behavior after mTBI. Front Syst Neurosci 2016; 10: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.