

Abstract
Although resistance towards small molecule chemotherapeutics has been well studied, the potential of tumor cells to avoid destruction by membrane-lytic compounds remains unexplored. Anticancer peptides (ACPs) are a class of such agents that disrupt tumor cell membranes through rapid and non-stereospecific mechanisms, encouraging the perception that cellular resistance towards ACPs is unlikely to occur. We demonstrate that eukaryotic cells can, indeed, develop resistance to the model oncolytic peptide SVS-1, which preferentially disrupts the membranes of cancer cells. Utilizing fission yeast as a model organism, we show that ACP resistance is largely controlled through the loss of cell-surface anionic saccharides. A similar mechanism was discovered in mammalian cancer cells where removal of negatively-charged sialic acid residues directly transformed SVS-1 sensitive cell lines into resistant phenotypes. These results demonstrate that changes in cell-surface glycosylation play a major role in tumor cell resistance towards oncolytic peptides.
eTOC Blurb
Although resistance towards small molecule chemotherapeutics has been well studied, the potential of tumor cells to avoid destruction by membrane-lytic compounds remains unexplored. Ishikawa and Medina et al. show that alteration of cell-surface glycan imparts resistancetowards a model oncolytic peptide.
INTRODUCTION
Resistance to anticancer agents remains a significant challenge to the long-term use of chemotherapeutics in oncology. Understanding mechanisms that influence resistance has led to the improvement of chemotherapeutic potency through medicinal chemistry, and provided insight into cellular processes that contribute to drug resistance and sensitivity (Gottesman, 2002). These studies have also informed the selection of drug cocktails for combination chemotherapy, aimed at reducing the potential for development of resistant cell sub-populations (Pluchino et al., 2012). Importantly, this field of research has predominantly focused on mechanisms that govern resistance towards small molecule chemotherapeutics that act on intracellular targets. For example, it is now well established that cancer cells can gain resistance to chemotherapy by upregulating energy-dependent drug transporters, reducing drug uptake, loss of apoptotic signaling, activation of drug metabolism pathways, and mutation of the target molecule structure (Gottesman, 2002). In contrast, the ability of cancer cells to gain resistance towards membrane-lytic anticancer agents has not been widely explored (Figure 1A).
Figure 1. Identification of SVS-1 resistance in a model organism.
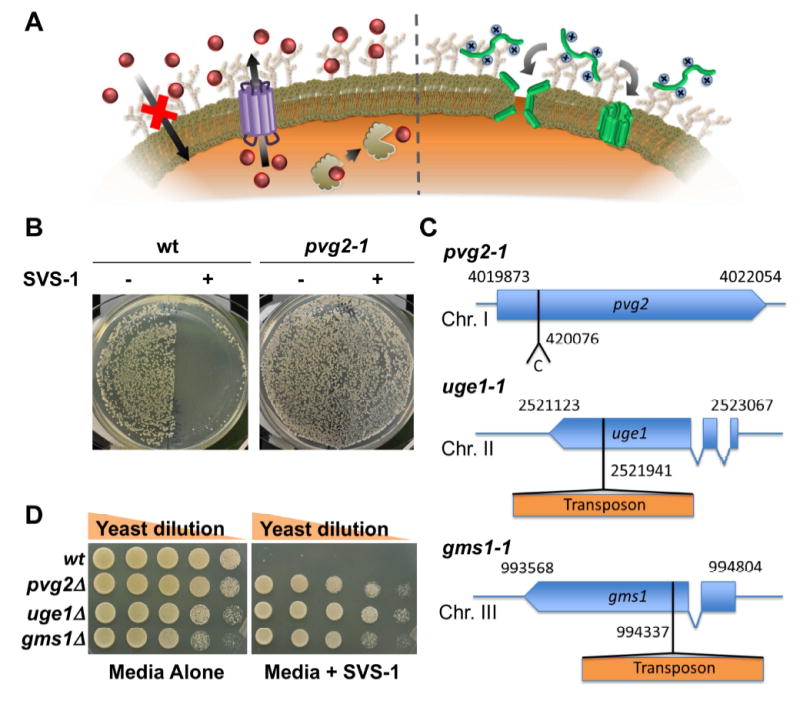
(A) Left: Resistance mechanisms toward small molecule chemotherapeutics (red) have been widely explored, and include increased drug efflux, decreased cellular uptake and mutation of the target molecule. Right: Conversely, little is known about the potential of cells to gain resistance towards membrane-lytic anticancer peptides (green). (B) Resistance towards the oncolytic peptide SVS-1 was explored in yeast as a model organism. Colonies of wild-type yeast (wt; left) transferred to one-half of the plate without SVS-1 (-) grow as expected, while those replica plated onto a surface coated with SVS-1 (+) are killed. Displayed on the right, a spontaneous yeast mutant with loss of function of the pvg2 gene (labeled pvg2-1) exhibited strong resistance to the peptide. Colonies were grown from random spores of indicated strains. (C) Identified yeast mutations which lead to SVS-1 resistance. (D) Deletion mutants, constructed for pvg2, uge1 and gms1 genes, grew as well as the wt strain on regular yeast extract agar growth media, while only the deletion mutants grew on plates coated with the SVS-1 peptide.
Anticancer peptides (ACPs) are a class of charged amphiphiles that exert their action by associating with the outer-leaflet of cancer cell membranes and subsequently disrupting the integrity of the lipid bilayer. The ability of ACPs to perturb cancer cell membranes is rapid and non-stereospecific, encouraging the perception that cellular resistance to these biopolymers is difficult, or unlikely to occur altogether. However, to date, this assumption has not been thoroughly tested in relevant cellular models of cancer. This is of particular significance as there continues to be vested interest in the development of ACPs as potential therapies, where resistance mechanisms will be important for translational studies.
In this work, we explore the ability of eukaryotic cells to develop resistance towards the action of a model cationic oncolytic peptide, and explore causative mutations responsible for the emergence of resistant populations. We selected the de-novo designed SVS-1 peptide (KVKVKVKVDPPTKVKVKVK-NH2), which has been shown to kill cancer cells via membrane disruption. To rapidly identify possible pathways that could lead to resistance, we first utilized the model fission yeast system Schizosaccharomyces pombe. These experiments identified three different gene mutations, each resulting in resistance. Examination of the genetic and molecular mechanisms of resistance in yeast facilitated discovery of similar mechanisms operating in mammalian cancer cells. Interestingly, both organisms developed resistance by altering cell-surface glycans rather than directly modulating the composition of their lipid membranes, the target of ACP action.
RESULTS
Isolation of SVS-1 Resistant Yeast Mutants
The potential of fission yeast to serve as a suitable model organism to investigate SVS-1 resistance was evaluated by testing their ability to form colonies on peptide-coated growth medium. Wild-type yeast (wt) cultured on normal growth media readily form colonies, while cells transferred to the medium surface treated with the SVS-1 peptide are killed (Figure 1B, left). This experiment demonstrates that fission yeast can be used as a model system to explore the cellular behavior of ACPs. In fact, by such experiments, spontaneously arising peptide-resistant colonies were found, indicating that yeast could acquire resistance to the SVS-1 peptide. One particular strain, named pvg2-1 mutant, showed strong resistance as indicated by its ability to grow on SVS-1 coated media (Figure 1B, right).
In general, resistance towards toxic agents is acquired through genetic mutations, amplification of a particular gene, or change of gene expression caused by epigenetic alterations (Calo et al., 2014). To test for these possibilities, we analyzed the survival of colonies originating from random spores of the putative pvg2-1 mutant. Should resistance result from DNA rearrangements and/or epigenetic changes, it is likely that some segregants would become sensitive to SVS-1 following meiosis required to produce spores. However this was not the case, as all of the several hundred meiotic mutant segregants tested exhibited peptide resistance, suggesting that resistance is likely conferred by a genetically stable mutation in a single gene. This single gene supposition was later confirmed through a backcross experiment (Figure S1). Whole genome sequencing of the mutant identified a single nucleotide insertion in the coding region of the pvg2 gene as the causative mutation (Figure 1C). Ultimately this genetic alteration is predicted to encode for a truncated non-functioning protein. To systematically identify other genes that cause SVS-1 sensitivity, we prepared a mutant yeast library through the random transposon mutagenesis procedure of Park et al (Park et al., 2009). This yeast library was then challenged against SVS-1 coated media, resulting in the identification of 68 different SVS-1 resistant strains. These mutants were then subjected to a secondary enzymatic screen to identify mutations in genes different from pvg2. Design of this screen was based on our initial experiments with the pvg2-1 mutant cells, which were found to be resistant to treatment with glusulase, a commonly used enzyme cocktail that digests the yeast cell wall (Figure S2). Six out of the 68 mutant lines were found to be sensitive to the action of glusulase, suggesting involvement of resistance pathways distinct from pvg2 mutation. Genetic analysis of these mutants indicated that SVS-1 resistance was linked to a transposon insertion in three among six of the identified strains. Here, the known sequence of the transposon insert acts as a marker to directly identify genes that cause peptide resistance following transposon-mediated disruption. These three transposon mutants were selected for further investigation. Insertion sites for two of these mutants were determined by thermal asymmetric interlaced PCR and Sanger-sequencing procedures (see experimental procedures section), revealing that transposons were inserted in protein coding regions of the uge1 and gms1 genes (Figure 1C). The third mutant had multiple transposon insertions in the genome, and was therefore not investigated further. Collectively, these results suggest that loss of pvg2, uge1, and gms1 gene functions independently confer resistance of yeast cells to the SVS-1 peptide. Preparation of deletion mutants of these genes, and subsequent treatment with the peptide, confirmed this assertion (Figure 1D).
Interestingly, all three identified genes have functional roles in the protein glycosylation pathway of the yeast cell wall. The pvg2 gene encodes for a glycan biosynthesis protein, Pvg2, responsible for ligating the anionic saccharide pyruvylated galactose (PvGal) to the termini of protein-linked cell-surface glycans (Andreishcheva et al., 2004). Similarly, the uge1 and gms1 gene functions are required for protein galactosylation (Suzuki et al., 2010; Tabuchi et al., 1997). The uge1 gene codes UDP-glucose 4-epimerase which is involved in the synthesis of UDP-galactose, a sugar donor for protein galactosylation. The gene product of gms1 is a transporter of UDP-galactose from the cytoplasm to the golgi, where the galactose residue is transferred to glycans. Thus, defects of the uge1 and gms1 genes lead to an overall loss of galactose at the cell surface. Because PvGal is displayed at the termini of galactose-containing glycan branches (Gemmill and Trimble, 1998), the amount of this anionic saccharide present on the surface of yeast cells will be reduced when protein galactosylation is compromised by gene loss of function.
Importantly, PvGal is the only negatively charged moiety displayed from N-linked oligosaccharides in S. pombe (Gemmill and Trimble, 1996, 1998). Thus, it is possible that loss of PvGal will significantly decrease the negative electrostatic potential of the mutant yeast’s cell surface. Relevant to the mechanism of SVS-1 resistance, this suggests that electrostatic engagement of the peptide to cell-surface glycans is an important step en route to its membrane lytic action. Therefore, we next tested whether reduction of cell-surface negative charge via glycan augmentation would directly inhibit engagement of the cationic SVS-1 peptide with the yeast cell wall, and thereby confer resistance.
Reduction in Cell-Surface Electronegativity Imparts Resistance to the SVS-1 Peptide
First, we assessed whether cell-surface charge is influenced by disruption of the pvg2, uge1 or gms1 gene. Individual yeast deletion mutants were subjected to a simple cell-binding assay using Q-sepharose beads. Here, fission yeast cells are mixed with the cationic beads, and the number of bound cells is quantified by counting under a light microscope. This assay provides a quick visual method by which to assess changes in cell-surface charge as a result of gene deletion. Wild-type fission yeast readily bound to the beads, while the pvg2 deletion mutant weakly interacted (Figure 2A). This suggests that pvg2 gene deletion leads to a reduction in cell-surface negative charge due to changes in PvGal expression; a finding that is consistent with similar experiments previously reported (Andreishcheva et al., 2004). Reduced Q-sepharose binding was likewise observed for the other SVS-1 resistant mutants (Figure 2B). These results indicate that the negative charge displayed at the surface of the deletion mutants is significantly reduced in comparison to wild-type cells. To confirm this, we quantified the yeast’s cell-surface charge using zeta potential measurements (Figure 2C). All of the deletion mutants showed a significant reduction in their cell-surface negative potential compared to wt cells, each to a similar degree as observed in the bead-binding assay (Figure 2B).
Figure 2. Reduction in yeast cell-surface negative charge due to gene mutation.
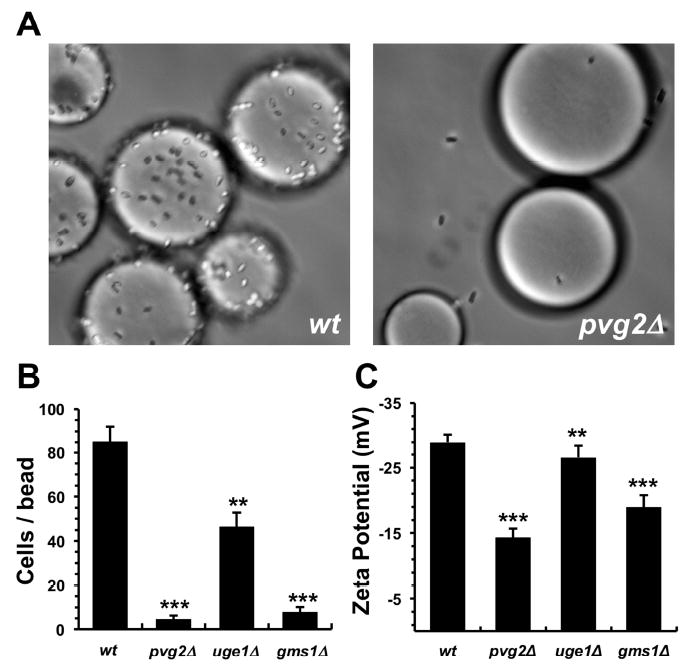
(A) Wild-type cells (wt) avidly bind to cationic Q-sepharose beads, while the pvg2 deletion mutant (pvg2∆) displayed weak binding interactions. Beads appear as large spheres, while cells look like short rods or ellipses. (B) Number of cells bound per bead for the wt yeast and the deletion mutants. (C) Cell-surface charge of each yeast strain as measured by zeta potential analysis. For panels B and C, results are shown as mean ± standard deviation for three replicates. Statistical significance compared to wt cells is shown as ** indicating p ≤ 0.01, and *** indicating p < 0.001.
Next, to evaluate whether changes in cell-surface negative charge are responsible for SVS-1 resistance in yeast, we measured the toxicity of the peptide towards cell sub-populations with reduced surface anionic charge. This was achieved through an iterative selection protocol where yeast cells from the transposon mutant library were incubated with Q-sepharose beads, followed by collection of the unbound cells and growth amplification (Figure 3A). Enrichment was repeated five times, with each recovered mutant mixture plated onto SVS-1 coated surfaces to measure the proportion of peptide resistant colonies. This simple protocol gave very interesting and conclusive results. Nearly all of the mutant cells from the initial transposon library (cycle number 0) were killed by SVS-1 (Figure 3B). However, an enhancement in the fraction of cells exhibiting resistance to the peptide was observed with increasing rounds of enrichment. In fact, all of the cells isolated after four enrichment cycles exhibited robust SVS-1 resistance. Quantification of these results identified an inverse correlation between the percentage of colonies which survive seeding onto SVS-1 coated plates and their propensity to bind Q-sepharose beads, as a function of increasing enrichment cycles (Figure 3C). Collectively, these results strongly support the assertion that yeast cells with reduced cell-surface negative charge display resistance to SVS-1 mediated lysis.
Figure 3. Influence of yeast cell-surface charge on SVS-1 resistance.
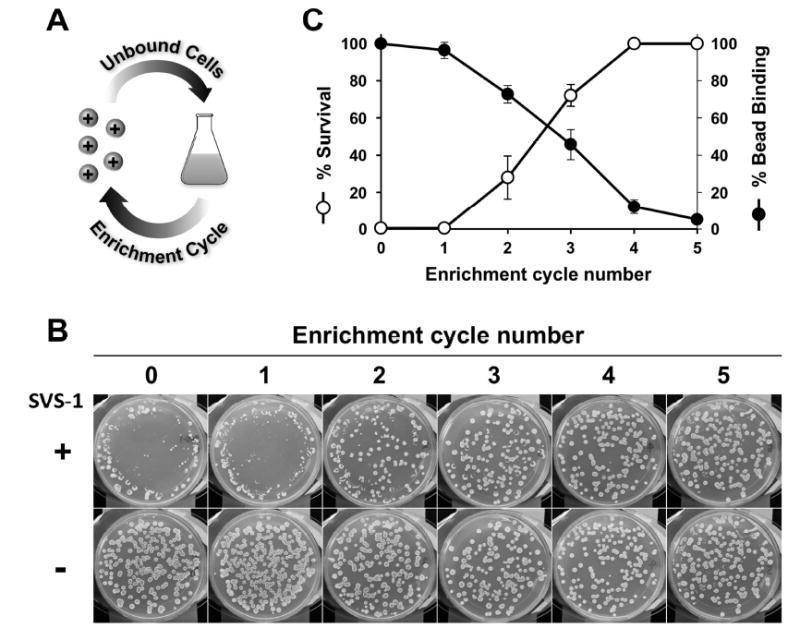
(A) Using an iterative enrichment protocol, yeast cell sub-populations with reduced negative charge were selected. (B) Representative images that show cells selected from each successive enrichment cycle displayed increased resistance to SVS-1 mediated lysis. (C) Correlation of the percentage of cells bound to Q-sepharose beads and their survival on SVS-1 coated plates, as a function of enrichment cycle number. Mean ± standard deviation from three independent experiments are shown.
Increased Cell-Cell Interactions Do Not Impart Peptide Resistance
During the course of our studies we observed an increased propensity of the SVS-1 resistant mutants to form large flocculates when growing in liquid medium. Quantification of cell-clump frequency of liquid log-phase cell cultures show that all three of the SVS-1 resistant mutants have a significantly higher frequency of flocculation in comparison to the wt control (Figure S3A). It is known that cell-surface PvGal plays a role in nonsexual cellular flocculation in fission yeast (Matsuzawa et al., 2011), where electrostatic repulsion between negatively charged cells inhibits their aggregation. A loss of cell-surface PvGal content due to genetic mutation would reduce cell-cell electrostatic repulsion and thereby facilitate flocculation. Important to our study, yeast flocculation may protect cells in the interior of the colony from exposure to the SVS-1 peptide. Thus, resistance may be a consequence of mutant cell aggregation, rather than inhibition of the peptide’s ability to electrostatically engage the surface glycans of individual yeast cells. We tested this possibility by evaluating the SVS-1 sensitivity of different deletion mutants selected for their known potential to readily flocculate (Dekker et al., 2004; Drewes and Nurse, 2003; Kim et al., 2001; Linder et al., 2008; Pan et al., 2012; Prevorovsky et al., 2009; Watson and Davey, 1998). All of these mutants were sensitive to the SVS-1 peptide (Figure S3B), indicating that flocculation propensity alone does not confer resistance to yeast. Taken together, the data thus far suggests that alteration of cell-surface electrostatic potential, via changes in glycan composition, is responsible for resistance, a mechanism we next investigated in mammalian cells.
Isolation and Characterization of SVS-1 Resistant Tumor Cells
The potential for peptide resistance in A549 human lung carcinoma cells was studied by serially culturing cells in media supplemented with increasing concentrations of the peptide over a six month period. This procedure generated an SVS-1 resistant cell line, which grew normally in media containing 50 μM of the peptide. Further increase in the concentration of SVS-1 led to rapid and complete lysis of the isolated cells. Challenge of the resistant cell line, named A549RES, with SVS-1 confirmed that these cells were significantly less susceptible to lysis by the peptide compared to control cells (Figure 4A). In these experiments, A549 cells used to initiate the resistance culture (A549INT) at day 0, as well as the parent cell line cultured for over six months in blank media (A549CUL), were included as controls to confirm that random genetic drift did not contribute to resistance. The results show that SVS-1 has an IC50 ~ 7 μM towards both the A549INT and A549CUL cell lines following peptide treatment in serum-free media (Figure 4B). A549RES cells, however, showed a 5-fold reduction in SVS-1 potency, with an IC50 of 32 ± 4 μM under similar conditions. This is significant as many ACPs have low therapeutic indexes (Gaspar et al., 2013), suggesting that dose escalation in response to this magnitude of peptide resistance could lead to considerable off-target toxicity in a clinical setting. However, this issue may be circumvented as the membrane-specific mechanisms of many ACPs should allow resistant cells to retain sensitivity towards small molecule chemotherapeutics. To test this possibility, we treated SVS-1 resistant A549RES cells with doxorubicin (DOX) and paclitaxel (PTX), two commonly utilized drugs in oncology (Figure S4). Results show both small molecular drugs led to potent A549RES cell killing, with nanomolar IC50 values. Previous results from our lab show the reverse is also true, that cells (OVCAR-3 and NCI/ADR-RES) which have gained resistance to chemotherapy are sensitive to the lytic action of SVS-1 with equal potency as drug-susceptible lines (Medina and Schneider, 2015).
Figure 4. Characterization of SVS-1 resistant A549 tumor cells.
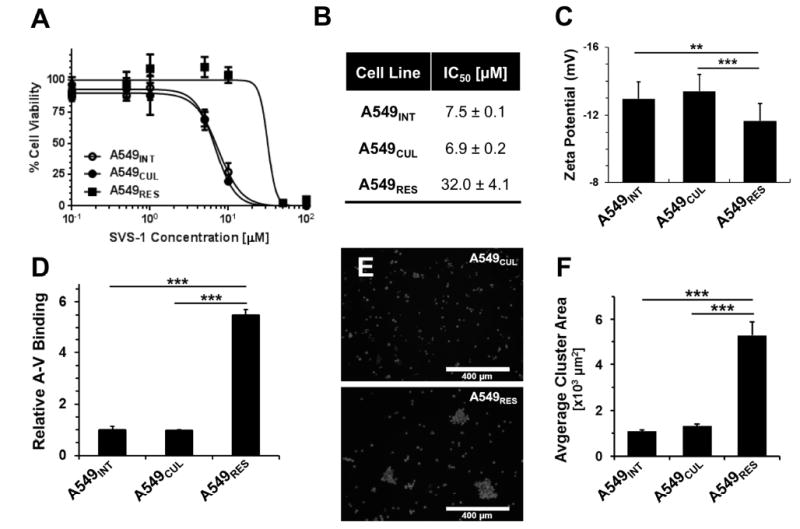
(A) Peptide cytotoxicity towards the resistant A549RES cell line derived through SVS-1 selection in culture. Parent cells used to initiate the resistant line (A549INT) or the same cells cultured over six months in blank media (A549CUL) were included as controls. (B) IC50 values from the viability data were calculated using non-linear regression. (C) Surface zeta potential values of the three cell lines in PBS buffer at pH7.4, 37°C. (D) Relative binding of fluorescently-labeled Annexin-V (A-V) to cell-surface phosphatidylserine was measured by flow cytometry. (E) Fluorescent microscopy images showing increased aggregation of A549RES cells (bottom) compared to the parent A549CUL cell line (top), after staining with a nuclear dye (10x magnification; scale bar = 400 μm). (F) Average cluster area of cell aggregates measured from the microscopy images for each cell line. Quantifications are shown as mean ± standard deviation. Statistical analysis was performed using the Student’s t-test, assuming unequal variance, with ** indicating p ≤ 0.01, and *** indicating p < 0.001.
Importantly, like yeast, our results show that mammalian cells can also gain resistance to the lytic action of the peptide. Based on the strong influence of cell-surface charge on SVS-1 resistance in fission yeast, we next quantified the electrostatic charge of A549RES cells using zeta potential analysis. A549INT and A549CUL parent cell lines had a similar surface zeta potential, while the negative charge of A549RES cells was significantly reduced in comparison (Figure 4C). This suggests that A549RES cells may share an analogous mechanism of resistance to that discovered in mutant yeast, namely a change in cell-surface glycosylation. Similar to yeast, the surfaces of mammalian cells are decorated with negatively charged saccharides (e.g. sialic acid) displayed from membrane-anchored branched glycans (Fuster and Esko, 2005). However, unlike yeast cells, the outer leaflet of mammalian tumor cells also contains the negatively charged lipid phosphatidylserine. Contribution of this lipid to the change in electrostatic potential of A549RES cells was evaluated following incubation of each cell line with fluorescently-labeled Annexin-V, a protein that selectively binds to surface exposed phosphatidylserine. Unexpectedly, expression of this anionic lipid at the cell membrane was increased by nearly 5-fold for the A549RES cells compared to the parent cell lines (Figure 4D). These results suggest that the overall decrease in A549RES anionic charge (Figure 4C) is a result of changes in glycosylation, rather than a reduction of cell-surface phosphatidylserine content. In addition, it was observed that A549RES cells had a tendency to form larger and more frequent aggregates in suspension relative to the parent cell lines (Figure 4, E and F). Interestingly, this behavior is similar to the pronounced flocculation phenotype observed for the resistant yeast mutants (Figure S3).
Changes in Cell Surface Sialic Acid Content Modulates SVS-1 Resistance
The distinct structural features of sialic acid (SA), including an anionic carboxyl group, enable it to play a variety of roles in cellular functions, such as transport of positively charged compounds, cell-cell repulsion and masking of antigenic fragments on receptor molecules (Narayanan, 1994). It is also an established tumor marker, as cancer cells are often hypersialylated compared to their normal counterparts (Narayanan, 1994), and mediate the activation of glycosyl transferases characteristic of tumor cells (Narayanan, 1994).
Through a series of cell-based assays, we tested the possibility that changes in cell-surface SA content can also modulate the lytic activity of SVS-1. Initially, basal levels of SA displayed at the surface of each cell line were measured (Figure 5A). Results show that A549RES cells had approximately 65% less SA on their surfaces compared to that of both the A549INT and A549CUL cell lines. To determine whether this change in glycosylation is responsible for SVS-1 resistance, we prepared SA-deficient cells through a combination of sialyltransferase chemical inhibition and enzymatic hydrolysis using the parent A549CUL cell line. This procedure led to ~80% reduction in cell-surface SA expression compared to that of untreated controls (Figure 5A). The susceptibility of these altered cells to SVS-1 was determined and compared to the A549CUL and A549RES lines. Figure 5B shows that, following SA inhibition, A549CUL cells had an IC50 value that was significantly greater (IC50 = 38 ± 6 μM) than SA-uninhibited cells (IC50 = 17 ± 3 μM), and nearly recapitulated the resistance of the A549RES cell line. Further, two additional human tumor cell lines that vary in their sialic acid content were next examined. Jurkat cells, which express a high level of cell-surface SA, gain resistance towards the lytic action of SVS-1 after treatment via sialyltransferase chemical inhibition and enzymatic hydrolysis (Figure S5). Next, MCF-7 cells, which contain a low amount of SA, show no resistance after SA-removal treatment. When taken together, the results of our SA inhibition studies suggest that the ability of cells to gain resistance towards SVS-1 may be directly related to their basal expression levels of SA. That is, cells which express high levels of SA (e.g. A549 and Jurkat) appear to be more capable of gaining resistance to the lytic action of the peptide when this carbohydrate is removed, while cells characterized by low SA expression (e.g. MCF-7) are not.
Figure 5. Reduction of cell-surface sialic acid (SA) modulates resistance to peptide- sensitive tumor cells.
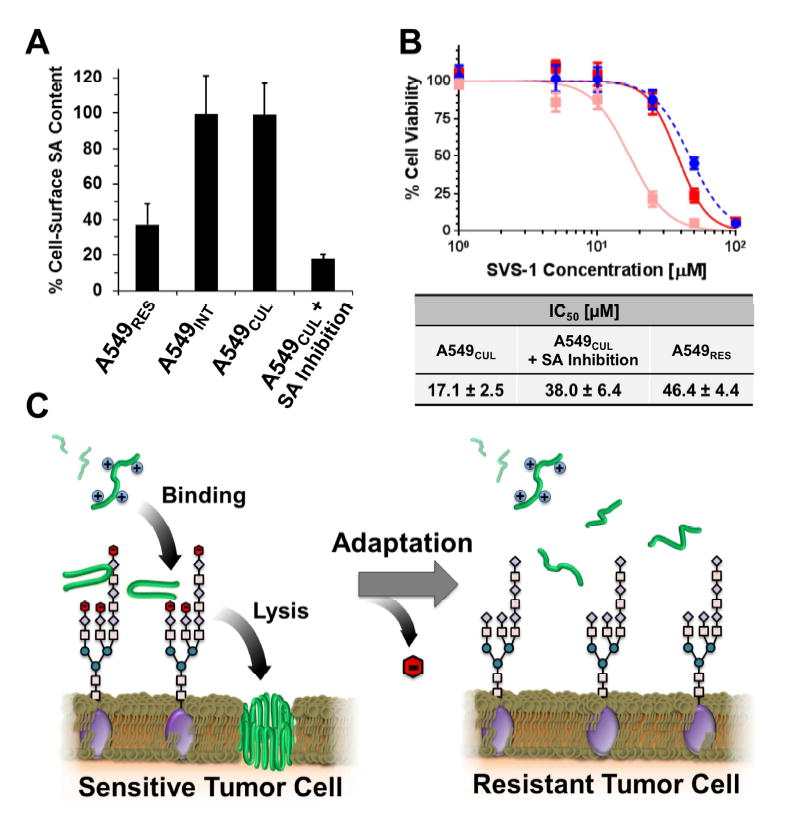
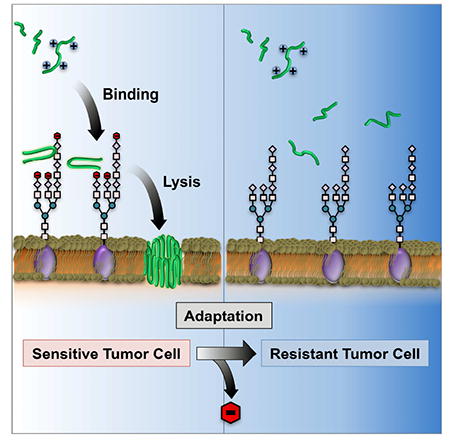
(A) Percentage of SA measured at the surface of A549RES, A549INT and A549CUL cell lines. Treatment of A549CUL cells with the sialyltransferase inhibitior 3Fax-Peracetyl Neu5c, followed by enzymatic hydrolysis of residual carbohydrate, reduced surface SA content by ~80% compared to controls. All data is shown normalized to basal SA expression of untreated A549CUL cells. (B) Cytotoxicity of the SVS-1 peptide towards A549CUL cells before (pink) and after (red) removal of cell-surface SA. Peptide toxicity towards A549RES cells is included as a positive control (blue). Mean ± standard deviation from three independent experiments are shown. IC50 values were calculated using non-linear regression. (C) Model of cellular resistance towards the cationic ACP, SVS-1 (green). Electrostatic binding of SVS-1 with negatively charged saccharides at the cell surface (e.g. SA; red) promotes its partitioning to the membrane where it elicits its lytic function. Adaptation of cells through the loss of SA leads to reduced electrostatic engagement of the peptide with cell-surface glycans, thereby diminishing its potential to access the membrane and conferring resistance.
DISCUSSION
Herein we explore cellular resistance mechanisms towards a model membrane lytic ACP, SVS-1, and demonstrate that changes in cell-surface glycosylation, leading to a concomitant reduction in electrostatic charge, confers peptide resistance to both fission yeast and human cancer cell lines. Anionic saccharides displayed from the terminal ends of cell-surface glycans, in particular PvGal for yeast and its structural analogue SA for human cancer cells, play a critical role in the peptide’s lytic activity. Presumably, this activity is mediated by electrostatic binding of the cationic peptide to glycans on the cell surface (Figure 5C), which may serve to accumulate peptide to the cell’s outer corona, where they then can partition into, and disrupt, the plasma membrane. This mechanism of resistance highlights the importance of cell-surface glycans on the molecular action of ACPs. Given the plethora of studies in the field using model membranes to investigate the mechanisms of ACP action, our work demonstrates that the role of glycans may not have been fully appreciated.
The five-fold decrease in cellular response to the peptide observed for resistant cells (Figure 4) may seem modest, which raises the question whether this magnitude of change would correlate to clinical resistance. Unfortunately, there are currently no ACPs in clinical use, and therefore relationships between cellular susceptibility and clinical resistance have not been established. However, these relationships have been well-studied in HIV-AIDS, where as small as a 3-fold change in viral susceptibility to drug in phenotypical assays has been correlated with patient resistance (Bacheler et al., 2001; Parkin et al., 1999; Rhee et al., 2003). Thus, in the case of viral infectivity, seemingly small changes in drug susceptibility can lead to meaningful resistance in the clinic. Although these examples involve viruses, it is reasonable to expect that similar changes in the responses of cells to membrane-lytic compounds could also lead to clinical resistance, which would be especially problematic in cases where a drug’s therapeutic index is narrow and high doses are necessary to achieve the desired clinical outcome. Aside from the clinical relevance, uncovering this mechanism is not only important to understanding and overcoming the resistance phenomena, but may play a role in altering the selectivity and pharmacokinetics of oncolytic ACPs.
Further, it has been proposed that high levels of negatively charged SA on the surface of tumor cells increases charge repulsion between cells, and therefore may promote cell detachment, tissue invasion and subsequent metastasis (Fuster and Esko, 2005). In fact, increasing cell-surface sialylation has been shown to decrease cell-cell adhesion in cancer, leading to enhanced tumorigenicity and cellular migration (Julien et al., 2006; Lin et al., 2002). In the present study, we found that long term SVS-1 treatment selects cancer cells with dramatically reduced SA levels (Figure 5A), ultimately leading to enhanced cell-cell adhesion (Figure 4, E and F). Therefore, it is possible that cancer cells which gain resistance to ACPs, fortunately, may have a decreased potential for metastasis. In addition, we show that cancer cells which have gained resistance to the SVS-1 peptide retain their sensitivity towards standard chemotherapeutic agents (Figure S4). Conversely, we have also previously shown that cells resistant to small molecule chemotherapeutics can be effectively killed with oncolytic peptides (Medina and Schneider, 2015). This suggests combinatorial treatments using ACPs and anti-cancer drugs may be a potentially successful strategy in the clinic to limit resistance. Collectively, these findings may aid in the design of ACPs in the future, and provide insights into the potential clinical translation of these agents.
SIGNIFICANCE
Anticancer peptides (ACPs) represent an emerging class of potential therapeutics that impart their activity via membrane-lytic mechanisms. Despite broad interest in the design and application of these agents, tumor cell resistance towards ACPs has not been widely explored. Here, for the first time the potential for eukaryotic cells to develop resistance towards a model ACP is systematically studied. We show that changes in cell-surface glycosylation patterns play a dominant role in cellular resistance towards oncolytic peptides, and thus represent an important new criterion in the design of future ACPs.
EXPERIMENTAL PROCEDURES
Materials
Fmoc-protected amino acids were purchased from Novabiochem. PL-Rink resin was purchased from Polymer Laboratories. 1H-Benzotriazolium 1-[bis(dimethylamino) methylene]-5chloro-hexafluorophosphate (1-),3-oxide (HCTU) was obtained from Peptides International. Trifluoroacetic acid was obtained from Acros organics. 1,2-ethanedithiol was purchased from Fluka. Lab-Tek™ 4-well chambered #1 borosilicate glass slides, diethyl ether, N-Methylpyrrolidone (NMP), tissue culture treated flasks and serological pipettes were purchased from Fisher Scientific. Thioanisole, anisole, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 200 mM glutamine solution were obtained from Sigma-Aldrich. RPMI-1640 media and Hoechst 33342 trihydrochloride dye was purchased from Invitrogen. Heat inactivated fetal bovine serum (FBS) and trypsin EDTA were obtained from Hyclone Laboratory Inc. The parent A549 and MCF-7 cancer cell lines were obtained from the NCI-60 repository. Jurkat cells were kindly provided by Dr. Eric Freed (National Cancer Institute).
Peptide Synthesis and Purification
SVS-1 was synthesized on PL-Rink resin using an automated ABI 433A peptide synthesizer. Synthesis was carried out via Fmoc-based solid-phase peptide chemistry with HCTU activation. Dried resin-bound peptide was cleaved from the resin and simultaneously side-chain deprotected using a trifluoroacetic acid/thioanisole/1,2-ethanedithiol/anisole (90:5:3:2) cocktail for 2 hours under argon atmosphere. Crude SVS-1 was precipitated with cold diethyl ether and then lyophilized. Reverse-phase HPLC equipped with a semi-preparative Vydac C18 column was employed to purify the peptides. HPLC solvents consisted of solvent A (0.1% TFA in water) and solvent B (0.1% TFA in 9:1 acetonitrile/water). A linear gradient of 0-100% solvent B over 100 min. was utilized for purification, with the peptide solution collected and then lyophilized. The purity of SVS-1 was verified by analytical HPLC and electrospray ionization (ESI-positive mode) mass spectrometry. Analytical HPLC chromatograms and ESI (+) mass spectra for the pure peptides are shown in Figure S6.
Isolation of SVS-1 Resistant Yeast Mutants
Fission yeast, Schizosaccharomyces pombe, strains were constructed by standard genetic crosses and by PCR-based Gene targeting (Bahler et al., 1998) (Supplementary Table 1), with media and culture conditions followed as previously described (Moreno et al., 1991). In order to isolate spontaneous mutants, approximately 108 cells of a wild type strain were transferred to a YEA plate adsorbed with 850 μL of a 4 mM SVS-1 solution, followed by incubation at 30°C for two days. Survivors were rep licated to a freshly prepared SVS-1 plate, cultured overnight and surviving colonies were isolated.
In a parallel experiment, a transposon mutant library was utilized to screen for additional SVS-1 resistant mutants. The transposon yeast library was prepared as previously reported (Park et al., 2009). In brief, a plasmid carrying a transposable cassette with kanMX6 marker, and another plasmid containing a transposase coding gene under control of the nmt1 promoter, was introduced into cells of a wild type fission yeast strain. Thiamine-deficient medium was used to induce transposase expression in cells. Transposon inserted mutants were selected by their G418 resistance phenotype. Three independently prepared mutant libraries were pooled and stocked for screening. To isolate SVS-1 resistant mutants, approximately 3 × 104 colonies resulting from growth of transposon-mutagenized cells were analyzed.
Identification of Causative Mutations that Impart SVS-1 Resistance in Yeast
The pvg2-1 mutation was identified by the whole genome sequencing procedure as described previously (Iida et al., 2014). For these experiments, the pvg2-1 mutant was subjected to a backcross with a wild-type strain (Figure S1). Genomic DNA of thirty SVS-1 sensitive segregants and thirty resistant segregants were separately pooled. Samples for sequencing were prepared by the Nextera XT DNA sample prep kit (illumina). Mixture of four libraries containing a 1% spike of 12 pM phi-X control library v3 (illumina) was sequenced using the MiSeq reagent kit v2 300 cycles (illumina) in 2 × 150 pair-end. The resulting FASTQ files were uploaded to an online mutation identification tool (Mudi; http://naoii.nig.ac.jp/mudi_top.html), which identified mutation candidates.
For the analysis of transposon inserted mutations, transposon insertion sites were determined by the thermal asymmetric interlaced PCR and Sanger sequencing (Liu and Whittier, 1995). In brief, DNA fragments with transposon insertion site were amplified with the transposon specific primers and the arbitrary degenerate primer. The amplified fragments that contained transposon-chromosome junction sites were sequenced. Linkage of SVS-1 resistance trait with the transposon insertion sites was subjected to tetrad analysis. In cases where transposons were inserted at multiple sites, individual segregant that contained a single peptide resistance causative insertion was used for the analysis.
Evaluation of Cell-Surface Negative Charge and Its Effect on Yeast Mutant Resistance
Binding of yeast cells to positively charged Q-sepharose beads (GE healthcare) was determined by following previously reported methods, with minor modifications (Andreishcheva et al., 2004). Briefly, yeast cells were washed and suspended in water to approximately 1.0 OD600. 1.0 mL of the cell suspension was mixed with 30 μL of equilibrated 50% Q-sepharose slurry, followed by 2 washes in water. The number of yeast cells bound to each bead was counted by observing cells with a phase-contrast microscope.
Selection of SVS-1 resistant yeast mutants with reduced negative charge was performed as follows. The transposon mutant library was grown overnight in YES broth, supplemented with 500 μg/mL of G418. The cells were harvested, washed twice and suspended in water containing Q-sepharose beads. Cells that bound to the beads, and thus showed high negative cell-surface charge, were removed from the cell suspension. Remaining cells were multiplied through overnight growth in fresh medium. This enrichment cycle was repeated 5 times and cultures from each cycle were employed for SVS-1 sensitivity and Q-sepharose binding assays. For the SVS-1 sensitivity assay, cells were spread on YEA plates to form ~500 colonies/plate. These colonies were replicated to a SVS-1 coated plate and a YEA plate to calculate percentage of SVS-1 resistant colonies of each sample. Q-sepharose binding was measured as described above.
Isolation of SVS-1 Resistant A549 Tumor Cells
To establish an SVS-1 resistant human tumor cell line, A549 lung carcinoma cells were initially cultured in media containing 11 μM of SVS-1, which represents the IC10 towards the parent cell line in serum-containing media. The concentration of SVS-1 was then gradually increased from 11 - 50 μM in approximately 5 μM increments. Cells were passaged twice at each dose before moving to a higher concentration. Over a period of 6 months, SVS-1 resistant A549 cells (A549RES) were isolated and found to grow uninhibited in media containing up to ~50 μM of SVS-1. Attempts to continue growing these cells at higher SVS-1 concentrations failed due to immediate lysis of the culture.
Resistance of A549RES cells was confirmed through an SVS-1 cytotoxicity assay. The parent A549 cell lines from the initial passage (A549INT), as well as cells grown in SVS-1 deficient media over the same 6 months (A549CUL), were included as controls. Briefly, 5 × 103 cells/well were plated in 96 well plates and allowed to adhere overnight at 5% CO2, 37 °C. The culture media was then removed and replaced with fresh serum-free media containing with 0.1 – 100 μM concentrations of SVS-1. Blank media and 20% DMSO containing media were used as negative and positive controls, respectively. After 24 hours of incubation, cells were washed and 100 μL of fresh media added to each well. 10 μL of MTT solution (5 mg/mL in PBS) was added to each well and culture incubated for 2 hours. The supernatant was then removed and replaced with 150 μL of DMSO to dissolve the formazan product. Absorbance was read at 540 nm using a UV plate reader (Biotek, Winooski, VT). The absorbance of the positive controls was subtracted from each sample as a blank, and percent viability calculated using the equation: (Absorbancepeptide-treated cells/Absorbanceuntreated cells) × 100. IC50 values were computed using the Graphpad 5.0 software package and represented as the average of three independent experiments ± standard deviation.
Chemotherapeutic Activity Assay
A549RES cells were plated at 5 × 103 cells/well in a 96 well plate, and allowed to adhere overnight at 5% CO2, 37 °C. The culture media was then replaced with 1 00 μL of media containing 5% FBS and 0.001 – 100 μM of doxorubicin or 0.0001 – 1 μM of paclitaxel. After 72 hours of incubation, cells were washed with 100 μL of fresh media and viability measured as described above.
Zeta Potential Measurements
Yeast or A549 tumor cell-surface charge was characterized using a Malvern Zetasizer Nano-ZS particle analyzer (Worcestershire, United Kingdom). 1 × 106 cells were dispersed in 1 mL of sterile milliQ water or PBS for yeast and A549 cells, respectively. 750 μL of each cell suspension was loaded into a disposable folded capillary cell (Malvern DTS1070), warmed to 37°C over 2 minutes, and zeta potential measured. C ells were treated as spheres, with material refractive index and absorbance set to 1.450 and 0.001 respectively. Dispersant viscosity at 25°C was set to 0.89 and 0.91 for pure water and PB S, respectively, with a refractive index of 1.332 for both solutions. Experiments were performed in triplicate for each cell type, with a maximum of 30 runs per sample.
Phosphatidylserine Characterization of A549RES Cells
The phosphatidylserine expression levels of A549 cells was measured by the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Life Technologies, NY). Briefly, 2 × 105 cells were suspended in 1 mL cold PBS and centrifuged at 2,000 rpm for 5 minutes. Supernatant was aspirated and 200 μL of 1X binding buffer was added. Cells were gently vortexed and 10 μL of fluorescently-labeled Annexin-V solution was added to each tube. Samples were incubated for 15 minutes at room temperature to allow binding, followed by addition of 300 μL 1X binding buffer. Cells were kept on ice before analysis by using a Beckman Coulter FACsCalibur flow cytometer (488 nm excitation laser). Gating was based on the fluorescence of untreated cells, and average mean fluorescence for each sample normalized to the controls to calculate relative Annexin-V binding. Three replicates for each experimental condition were performed over three independent experiments.
Cell Clumping Assay
Yeast clumping assay was performed on wild type and deletion mutants by growing yeast cells to log phase in YEA broth. The cells were observed under microscope to count cell aggregates. Flocculates containing three or more cells were considered a “cell clump”. To measure the flocculation of A549CUL and A549RES cells, cultures were trypsinized and centrifuged at 2,000 rpm for 5 minutes. After aspiration of the supernatant, 5 mL of fresh media was added and the cell pellet triturated 20 times by pipette to re-suspend the cells, followed by gentle vortexing for 5 seconds. The cells were then counted and diluted to 1 × 105 cells/mL in serum-containing media containing 2 μg/mL Hoechst 33342 dye, and incubated for 20 minutes at 37°C. 1.0 mL of the cell suspension was then add ed to a 4-well chambered glass slide, mounted onto a EVOS FL Auto fluorescent microscope, and imaged using an LED DAPI light cube (357/44 nm excitation, 447/60 nm emission). To determine average cluster area, the sizes of cell aggregates (composed of ≥5 nuclei) were measured using ImageJ software. For each cell line, three independent samples were imaged, with three fields of view were captured per sample.
Inhibition of Cell-Surface Sialic Acid (SA) and Influence on SVS-1 Resistance
Before inhibition experiments, basal SA expression levels for the A549RES, A549INT and A549CUL cell lines were determined by plating 20 × 103 cells/well in a 96 well plate, and cultured for 5 days at 5% CO2 and 37°C. The media was then replaced with 100 µL of PBS containing 1.5 mU of α2-3,6,8,9-Neuraminidase (Millipore, Darmstadt, Germany) and the plate incubated for 1 hour at 37°C to remove cell-surface bound SA. The concentration of sialic acid in the supernatant was quantified using the Sialic Acid (NANA) Assay Kit (Abcam, Cambridge, MA) by following the manufacturer’s protocol. Briefly, 50 μL from each sample well was transferred to a 96 well plate and mixed with an equal volume of assay buffer containing the converting enzyme, development mixture and probe. The plate was incubated in the dark at room temperature for 30 minutes. Absorbance was read at 570 nm using a UV plate reader (Biotek, Winooski, VT), and results compared to a standard curve to calculate the total amount of SA.
In separate experiments, cell-surface SA expression was measured after treatment of A549CUL cells with the sialyltransferase inhibitor 3Fax-Peracetyl Neu5Ac (Millipore, Darmstadt, Germany). Here, 2 × 104 cells/well were cultured for 5 days in media containing 800 μM of the sialyltransferase inhibitor. Preliminary experiments found that inhibition alone did not significantly reduce the cell-surface SA content when compared to controls (~10%-15%). Therefore, following this inhibition procedure the cells were treated with 1.5 mU of α2-3,6,8,9-Neuraminidase for 15 minutes to remove the remaining cell-surface saccharide, and SA content quantified as previously described. All results were normalized to basal saccharide expression in the control A549CUL cells to calculate the percentage of relative surface sialic acid content.
To determine the influence of SA content on SVS-1 resistance, A549CUL cells were treated with the peptide following sialyltransferase inhibition. Cytotoxicity was performed as previously described, with the exception that SVS-1 treatment solutions contained 800 μM of the sialyltransferase inhibitor. This treatment was done to ensure cell-surface SA was not replenished during the assay. A549RES and A549CUL cells not subjected to the sialic acid inhibition protocol were included as positive and negative controls, respectively.
For MCF-7 cells, the same procedure was used with the exception that the sialyltransferase inhibitor was not included at the time SVS-1 treatment was performed. This protocol was slightly modified for Jurkat cells. Briefly, 1×05 cells were treated with 5 mL of serum containing media with or without 800 μM sialyltransferase inhibitor for 6 days. Inhibited cells were then treated with 80 μL of Vibrio cholerae neuraminidase (Sigma-Aldrich, MO) in 400 μL of serum containing media supplemented with 3 mM of CaCl2 for 30 min at 37°C, followed by washing with 4 mL of serum containing media. Cells were then seeded in 96 well plates at 1×105 cells/well and treated with SVS-1 in the absence of the inhibitor. Viability of Jurkat cells was measured using the cell counting kit-8 (Dojindo Molecular Technologies, MD) according to manufacturer’s instructions with a slight modification. Here, SVS-1 treated cell suspensions were mixed with 100 μL of serum free media containing 20 μL of the kit solution, followed by incubation for 2 hours at 5% CO2 and 37°C. Absorbance of the water soluble formazan product was measured at a wavelength of 450 nm, and relative viability calculated as described above. The reported IC50 value represents the average ± standard deviation of two independent experiments with n=3 in each.
Statistical Analysis
Quantitative data with errors are shown as mean ± standard deviation. Statistical significance was tested using the Student’s t-test.
Supplementary Material
Highlights.
Discovery of resistance mechanism towards lytic peptide
Altered glycosylation reduces cell-surface negative charge and yields resistance
Conserved mechanism of resistance in yeast and cancer cells
Removing cell-surface sialic acid transforms cells into a resistant phenotype
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. We thank Mike Bonaduce, Shuo Gu, Lisheng Dai, Kimberley Peifley, and Stephen Lockett for technical assistance and/or for the use of their facilities, as well as Henry Levin for providing reagents for the yeast transposon mutagenesis procedure. Bulk quantities of the sialyltransferase inhibitor 3Fax-Peracetyl Neu5Ac was synthesized and provided by Gary Pauly. We thank Sherimay Ablan and Eric Freed for providing Jurkat cell line. We thank our colleagues Yuji Yamada, Stephen Hughes and Jeffrey Strathern for valuable discussions.
Footnotes
Author contributions
K. I. and S.H.M. designed and performed experiments, as well as analyzed data. S.H.M., K.I., J.P.S. and A.J.S.K. wrote and edited the paper.
Competing information
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreishcheva EN, Kunkel JP, Gemmill TR, Trimble RB. Five genes involved in biosynthesis of the pyruvylated Galbeta1,3-epitope in Schizosaccharomyces pombe N-linked glycans. J Biol Chem. 2004;279:35644–35655. doi: 10.1074/jbc.M403574200. [DOI] [PubMed] [Google Scholar]
- Bacheler L, Jeffrey S, Hanna G, D’Aquila R, Wallace L, Logue K, Cordova B, Hertogs K, Larder B, Buckery R. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J Virol. 2001;75:4999–5008. doi: 10.1128/JVI.75.11.4999-5008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Calo S, Shertz-Wall C, Lee SC, Bastidas RJ, Nicolas FE, Granek JA, Mieczkowski P, Torres-Martinez S, Ruiz-Vazquez RM, Cardenas ME, et al. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature. 2014;513:555–558. doi: 10.1038/nature13575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker N, Speijer D, Grun CH, van den Berg M, de Haan A, Hochstenbach F. Role of the alpha-glucanase Agn1p in fission-yeast cell separation. Mol Biol Cell. 2004;15:3903–3914. doi: 10.1091/mbc.E04-04-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drewes G, Nurse P. The protein kinase kin1, the fission yeast orthologue of mammalian MARK/PAR-1, localises to new cell ends after mitosis and is important for bipolar growth. FEBS Lett. 2003;554:45–49. doi: 10.1016/s0014-5793(03)01080-9. [DOI] [PubMed] [Google Scholar]
- Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nature Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- Gaspar D, Veiga AS, Castanho MARB. From antimicrobial to anticancer peptides. A review Front Microbiol. 2013;4:294. doi: 10.3389/fmicb.2013.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmill TR, Trimble RB. Schizosaccharomyces pombe produces novel pyruvate-containing N-linked oligosaccharides. J Biol Chem. 1996;271:25945–25949. doi: 10.1074/jbc.271.42.25945. [DOI] [PubMed] [Google Scholar]
- Gemmill TR, Trimble RB. All pyruvylated galactose in Schizosaccharomyces pombe N-glycans is present in the terminal disaccharide 4, 6-O-[(R)-(1-carboxyethylidine)]-Galbeta1,3Galalpha1. Glycobiology. 1998;8:1087–1095. doi: 10.1093/glycob/8.11.1087. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Iida N, Yamao F, Nakamura Y, Iida T. Mudi, a web tool for identifying mutations by bioinformatics analysis of whole-genome sequence. Genes Cells. 2014;19:517–527. doi: 10.1111/gtc.12151. [DOI] [PubMed] [Google Scholar]
- Julien S, Adriaenssens E, Ottenberg K, Furlan A, Courtand G, Vercoutter-Edouart AS, Hanisch FG, Delannoy P, Le Bourhis X. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology. 2006;16:54–64. doi: 10.1093/glycob/cwj033. [DOI] [PubMed] [Google Scholar]
- Kim KH, Cho YM, Kang WH, Kim JH, Byun KH, Park YD, Bae KS, Park HM. Negative regulation of filamentous growth and flocculation by Lkh1, a fission yeast LAMMER kinase homolog. Biochem Biophys Res Comm. 2001;289:1237–1242. doi: 10.1006/bbrc.2001.6128. [DOI] [PubMed] [Google Scholar]
- Lin S, Kemmner W, Grigull S, Schlag PM. Cell surface alpha 2,6 sialylation affects adhesion of breast carcinoma cells. Exp Cell Res. 2002;276:101–110. doi: 10.1006/excr.2002.5521. [DOI] [PubMed] [Google Scholar]
- Linder T, Rasmussen NN, Samuelsen CO, Chatzidaki E, Baraznenok V, Beve J, Henriksen P, Gustafsson CM, Holmberg S. Two conserved modules of Schizosaccharomyces pombe Mediator regulate distinct cellular pathways. Nucleic Acids Res. 2008;36:2489–2504. doi: 10.1093/nar/gkn070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YG, Whittier RF. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics. 1995;25:674–681. doi: 10.1016/0888-7543(95)80010-j. [DOI] [PubMed] [Google Scholar]
- Matsuzawa T, Morita T, Tanaka N, Tohda H, Takegawa K. Identification of a galactose-specific flocculin essential for non-sexual flocculation and filamentous growth in Schizosaccharomyces pombe. Mol Microbiol. 2011;82:1531–1544. doi: 10.1111/j.1365-2958.2011.07908.x. [DOI] [PubMed] [Google Scholar]
- Medina SH, Schneider JP. Cancer cell surface induced peptide folding allows intracellular translocation of drug. J Controlled Release. 2015;209:317–326. doi: 10.1016/j.jconrel.2015.05.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Narayanan S. Sialic acid as a tumor marker. Ann Clin Lab Sci. 1994;24:376–384. [PubMed] [Google Scholar]
- Pan X, Lei B, Zhou N, Feng B, Yao W, Zhao X, Yu Y, Lu H. Identification of novel genes involved in DNA damage response by screening a genome-wide Schizosaccharomyces pombe deletion library. BMC Genomics. 2012;13:662. doi: 10.1186/1471-2164-13-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Evertts AG, Levin HL. The Hermes transposon of Musca domestica and its use as a mutagen of Schizosaccharomyces pombe. Methods. 2009;49:243–247. doi: 10.1016/j.ymeth.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin NT, Lie YS, Hellmann N, Markowitz M, Bonhoeffer S, Ho DD, Petropoulos CJ. Phenotypic changes in drug susceptibility associated with failure of human immunodeficiency virus type 1 (HIV-1) triple combination therapy. J Infect Dis. 1999;180:865–870. doi: 10.1086/314928. [DOI] [PubMed] [Google Scholar]
- Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updates. 2012;15:98–105. doi: 10.1016/j.drup.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevorovsky M, Grousl T, Stanurova J, Rynes J, Nellen W, Puta F, Folk P. Cbf11 and Cbf12, the fission yeast CSL proteins, play opposing roles in cell adhesion and coordination of cell and nuclear division. Exp Cell Res. 2009;315:1533–1547. doi: 10.1016/j.yexcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Rhee S-Y, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31:298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Matsuzawa T, Nukigi Y, Takegawa K, Tanaka N. Characterization of two different types of UDP-glucose/-galactose 4-epimerase involved in galactosylation in fission yeast. Microbiology. 2010;156:708–718. doi: 10.1099/mic.0.035279-0. [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Tanaka N, Iwahara S, Takegawa K. The Schizosaccharomyces pombe gms1+ gene encodes an UDP-galactose transporter homologue required for protein galactosylation. Biochem Biophys Res Comm. 1997;232:121–125. doi: 10.1006/bbrc.1997.6239. [DOI] [PubMed] [Google Scholar]
- Watson P, Davey J. Characterization of the Prk1 protein kinase from Schizosaccharomyces pombe. Yeast. 1998;14:485–492. doi: 10.1002/(SICI)1097-0061(19980330)14:5<485::AID-YEA239>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.