

Abstract
Burgeoning evidence supports a role for cyclooxygenase metabolites in regulating membrane excitability in various forms of synaptic plasticity. Two cyclooxygenases, COX-1 and COX-2, catalyze the initial step in the metabolism of arachidonic acid to prostaglandins. COX-2 is generally considered inducible, but in glutamatergic neurons in some brain regions, including the cerebral cortex, it is constitutively expressed. However, the transcriptional mechanisms by which this occurs have not been elucidated. Here, we used quantitative PCR and also analyzed reporter gene expression in a mouse line carrying a construct consisting of a portion of the proximal promoter region of the mouse COX-2 gene upstream of luciferase cDNA to characterize COX-2 basal transcriptional regulation in cortical neurons. Extracts from the whole brain and from the cerebral cortex, hippocampus, and olfactory bulbs exhibited high luciferase activity. Moreover, constitutive COX-2 expression and luciferase activity were detected in cortical neurons, but not in cortical astrocytes, cultured from wild-type and transgenic mice, respectively. Constitutive COX-2 expression depended on spontaneous but not evoked excitatory synaptic activity and was shown to be N-methyl-d-aspartate receptor-dependent. Constitutive promoter activity was reduced in neurons transfected with a dominant-negative cAMP response element binding protein (CREB) and was eliminated by mutating the CRE-binding site on the COX-2 promoter. However, mutation of the stimulatory protein-1 (Sp1)-binding site resulted in an N-methyl-d-aspartate receptor-dependent enhancement of COX-2 promoter activity. Basal binding of the transcription factors CREB and Sp1 to the native neuronal COX-2 promoter was confirmed. In toto, our data suggest that spontaneous glutamatergic synaptic activity regulates constitutive neuronal COX-2 expression via Sp1 and CREB protein-dependent transcriptional mechanisms.
Keywords: cAMP response element-binding protein (CREB); cyclooxygenase (COX); N-methyl-d-aspartate receptor (NMDA receptor, NMDAR); neuron; specificity protein 1 (Sp1); transcription; constitutive expression; transcriptional regulation
Introduction
The first committed reaction in the metabolism of arachidonic acid to prostaglandins and thromboxanes is catalyzed by two related heme-containing bis-oxygenases, cyclooxygenase (COX) 1 and 2. These enzymes share 90% similarity in amino acid sequence and exhibit nearly identical enzyme kinetics (1, 2). Both catalyze two separate reactions, the first metabolizing arachidonic acid to PGG23 (cyclooxygenase reaction), an intermediate that is subsequently reduced in the second reaction to the product, PGH2 (peroxidase reaction). PGH2 is the substrate for various synthases that generate individual biologically active prostaglandins and thromboxanes, often in a cell type-specific manner (3). Although both metabolize arachidonic acid to PGH2, the transcriptional regulation of each isoform differs. The PTGS1 gene encoding COX-1 lacks a TATA box motif in its 5′ promoter region and is generally constitutively active in cells (4, 5). In contrast, the promoter regulatory region of the PTGS2 gene encoding COX-2 is not typically active but can be strongly and rapidly induced under specific conditions by growth factors and proinflammatory mediators (1, 6). An important exception to this occurs in the central nervous system of many species, including rodents and humans. In the brain, for example, COX-2 is constitutively expressed in specific neuronal populations of the cortex and hippocampus (7–9). The molecular and cellular mechanisms that contribute to the constitutive expression of COX-2 in these neurons are not understood. Hence, the goal of this study was to determine the transcriptional mechanisms regulating basal COX-2 expression in cortical neurons and to examine the relationship between transcriptional regulation and neuronal activity.
Results
COX-2 Is Constitutively Expressed in the Brain of PLuc371 Transgenic Mice
Mice harboring the PLuc371 transgenic construct consisting of −371/+70 bp of the PTGS2 gene promoter fused 5′ of a luciferase transgene were used herein to analyze constitutive transcriptional regulation of COX-2 expression. Quantitation of luciferase activity (PTGS2 activity) in different tissues revealed that central nervous system (CNS) possessed the highest level of constitutive activity with little to no luciferase detected in the liver, heart, kidneys, and lungs (Figs. 1A and 2A). Analysis of isolated CNS areas demonstrated high activity in numerous regions including cerebral cortex, hippocampus, olfactory bulb, spinal cord, and cerebellum (Fig. 1A). For the most part, luciferase activity reflected endogenous COX-2 expression assessed by quantitative PCR, with the notable exception of cerebellum and spinal cord (Fig. 1B). Importantly, endogenous COX-2 expression was faithfully represented by the reporter construct in cortex (Fig. 1).
FIGURE 1.
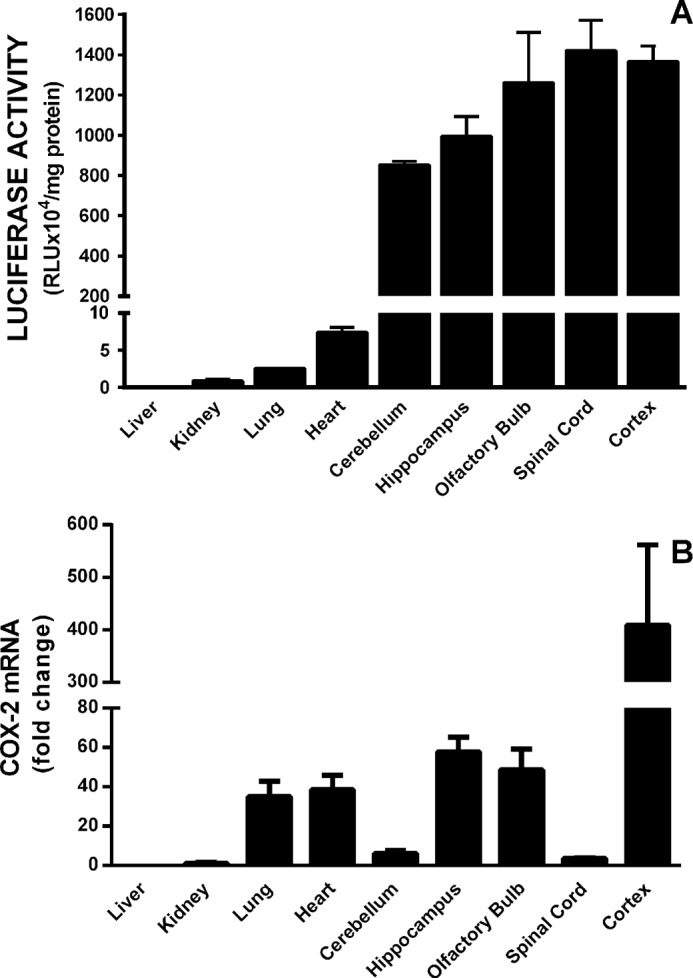
Comparative analysis of constitutive transgenic luciferase levels with native COX-2 mRNA levels. A, three adult male PLuc371 transgenic mice were anesthetized and transcardially perfused with ice-cold saline. Organs were rapidly dissected and frozen, and luciferase activity was measured as described under “Materials and Methods.” Data are expressed as RLU normalized to mg of protein (mean ± S.E.). B, RNA was isolated from tissue homogenates from three adult male mice, and COX-2 mRNA was quantified via quantitative PCR. COX-2 mRNA expression in kidneys was used as the comparator (set to 1).
FIGURE 2.
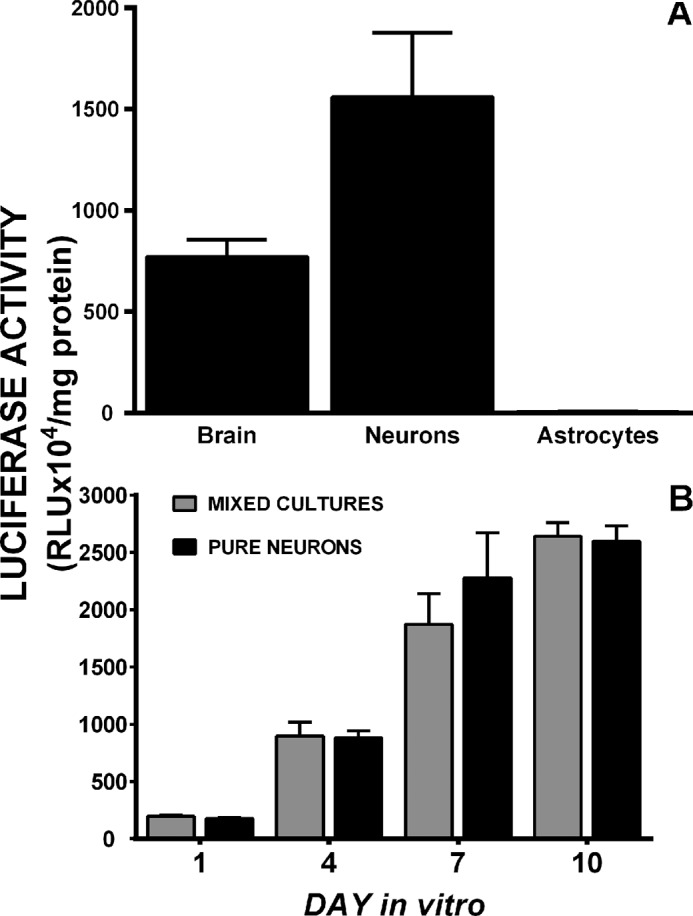
Constitutive COX-2 gene transcription in cultured transgenic cells. A, basal luciferase activity was assessed in brain (cerebral cortex) homogenates from three PLuc371 transgenic mice or whole cell lysates from neurons and astrocytes cultured from PLuc371 brains. Data are expressed as RLU normalized to mg of protein (mean ± S.E.; n = 3–4 cultures per cell type). B, comparison of the temporal pattern of PLuc371-luciferase expression in purified neuronal cultures (pure neurons; black bars) or neuronal cultures plated atop non-transgenic astrocytes (mixed cultures; gray bars) at 1, 4, 7, and 10 days in vitro. Data are expressed as RLU normalized to mg of protein (mean ± S.E.). Between groups (mixed versus pure), there were no significant differences as determined by two-way ANOVA (n = 3 each).
Primary cortical astrocytes cultured from transgenic animals expressed little to no luciferase activity, whereas cortical neuronal cultures, plated atop of non-transgenic astrocytes, showed abundant luciferase activity, suggesting that the majority of the activity in the total brain homogenates was neuronal (Fig. 2A). Astrocyte factors do not appear to be involved in the regulation of constitutive neuronal expression of COX-2 as the temporal expression of luciferase activity in pure neuronal cultures derived from transgenic animals was identical to that found in mixed cultures composed of transgenic neurons and wild-type astrocytes (Fig. 2B). Temporally, activity was low on days in vitro (DIV) 1–4, increased substantially between DIV 4–7, and peaked at DIV 10 (Fig. 2). This was not just a transgenic phenomenon as purified neurons derived from wild-type animals cultured with or without astrocyte conditioned media demonstrated similar COX-2 mRNA levels as do mixed cultures from wild-type animals (data not shown).
PLuc371 Activity Parallels the Physical and Functional Expression of NMDA Receptors
Neuronal luciferase activity near perfectly paralleled the temporal expression of the obligate NMDA receptor (NR) subunit, NR1, as well as its functional activity. Neuronal COX-2 luciferase activity (Fig. 2B) and surface NR1 expression (Fig. 3A) as well as c-Fos mRNA levels (Fig. 3B), the latter used here as marker of neuronal activity (10), increased steadily from DIV 1 through DIV 10. To determine if NMDA receptor activity was required for constitutive neuronal COX-2 expression, COX-2 mRNA levels were measured in purified neuronal cultures (PNCs) that were either wild-type (NR1+/+) or null (NR1−/−) for NR1 (Fig. 4). Strikingly, COX-2 mRNA levels measured in NR1−/− cultures at DIV 7 were dramatically reduced when compared with that found in NR1+/+ cultures (Fig. 4), suggesting that indeed NMDA receptor activity was necessary for constitutive expression of neuronal COX-2 mRNA. In support, the addition of 30 μm NMDA receptor antagonist 5-amino-phosphonovaleric acid (APV), a concentration that reduced NMDA receptor activity in pure neuronal cultures as evidenced by block of c-Fos mRNA production (Fig. 5A), also significantly decreased the levels of COX-2 mRNA (Fig. 5B). Interestingly, neither c-Fos nor COX-2 mRNA expression in cultured neurons was affected by pretreatment with tetanus toxin (300 ng/ml), a Clostridium toxin that prevents evoked but not spontaneous, synaptic vesicle exocytosis by cleaving the SNARE protein, synaptobrevin-2 (11) (Fig. 6).
FIGURE 3.
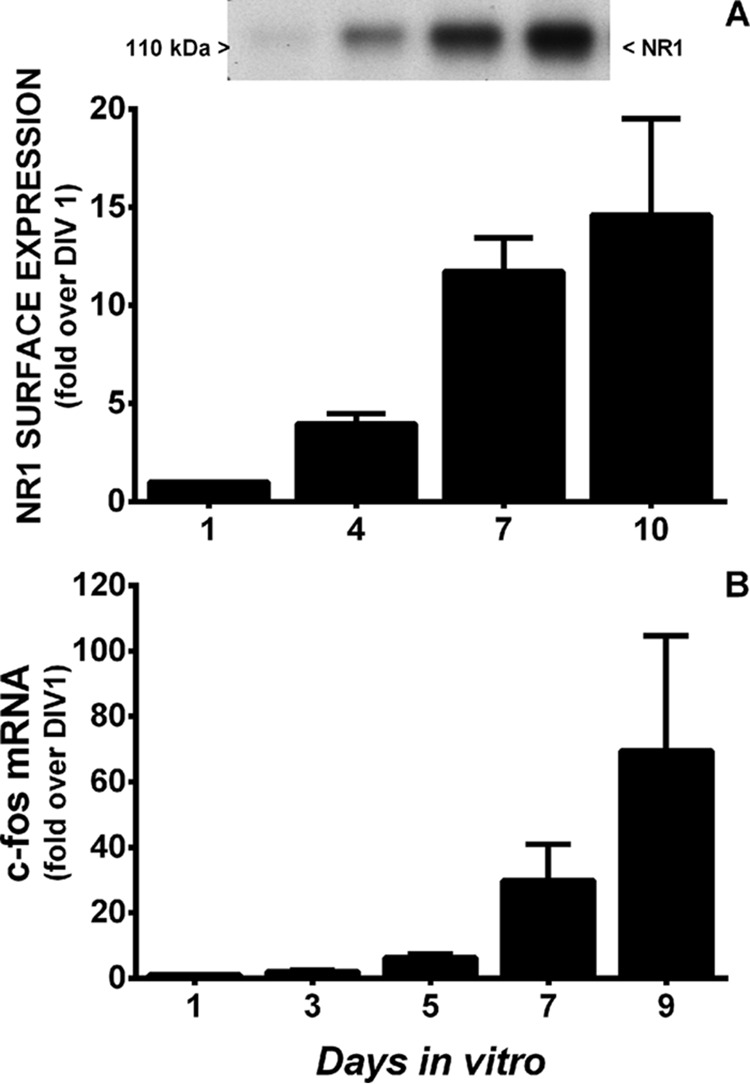
Developmental profile of NMDA receptor surface expression and function. A, NMDAR surface expression. Surface proteins of mixed cortical cell cultures of PLuc371 transgenic neurons and wild-type astrocytes were labeled with biotin at 1, 4, 7, and 10 days in vitro, and the biotinylated proteins were separated from total protein. Western blotting analysis was performed using a mouse monoclonal anti-NR1 antibody. Gel films were scanned, and densitometry was performed on five independent experiments. NR1 levels are expressed as -fold increase over day 1 (set to 1) in vitro. B, NMDA receptor function. Shown is a comparison of the temporal pattern of c-Fos mRNA expression in pure wild-type neurons at 1, 3, 5, 7, and 9 days in vitro as quantified by qRT-PCR. Relative mRNA expression on DIV 1 was set to 1 and used as the comparator (n = 3).
FIGURE 4.
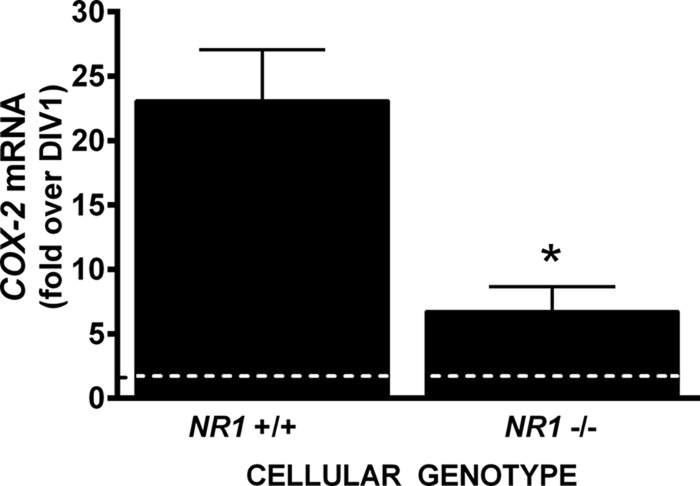
COX-2 mRNA expression in pure neuronal cultures derived from NR1 wild-type and null mutant embryos. Pure cortical neurons wild-type (NR1+/+) or null (NR1−/−) were harvested 1 and 7 days after plating, and COX-2 mRNA was quantified from total RNA isolates by qRT-PCR. The graph represents the mean ± S.E.-fold change in COX-2 mRNA expression at DIV 7 relative to mRNA expression on DIV 1 (represented as a dashed line, set to 1). An asterisk (*) indicates a significant between-group difference as assessed by unpaired Student's t test of the geometric means (p = 0.0204, n = 3).
FIGURE 5.
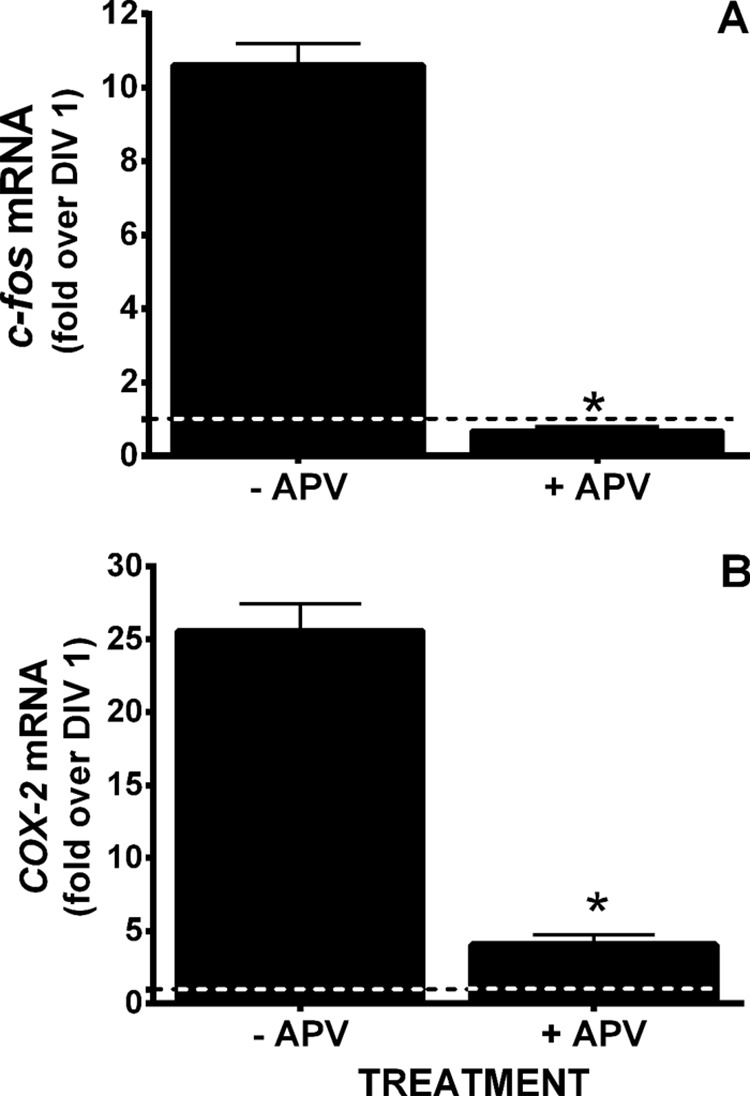
Effect of NMDA receptor antagonism on c-Fos and COX-2 mRNA expression. DIV 5 pure neuronal cultures were treated with 30 μm APV (+ APV) or its vehicle (−APV) and c-Fos (A) and COX-2 (B) mRNA were quantified via qRT-PCR 2 days later. Relative mRNA expression on DIV 1 (dashed lines) was set to 1 and used as comparator. An asterisk (*) indicates a significant between-group (+/−APV) difference as assessed by unpaired Student's t test of the geometric means. p < 0.0001 for both c-fos and COX-2, n = 4.
FIGURE 6.
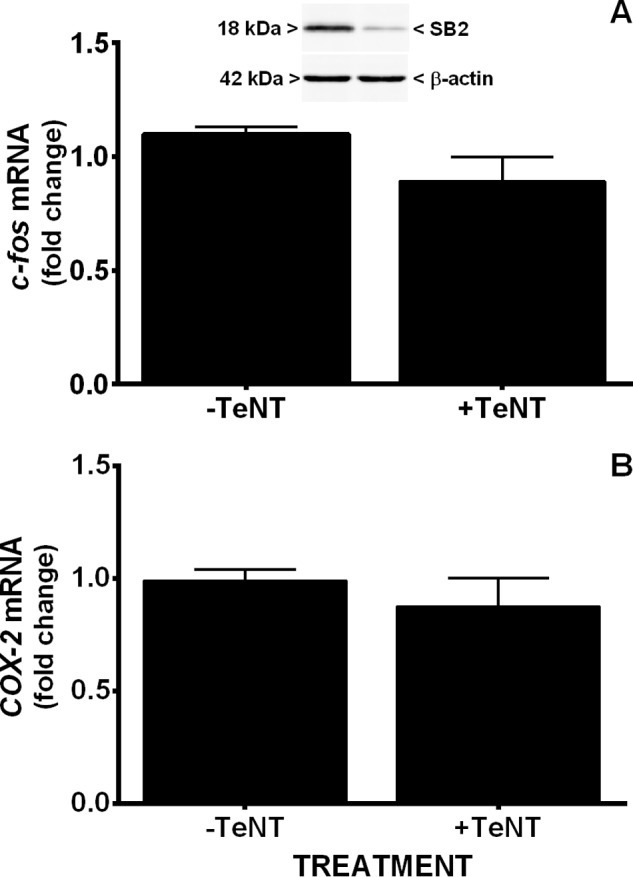
Lack of effect of tetanus toxin on c-Fos and COX-2 mRNA expression. Pure neurons were treated with 300 ng/ml tetanus toxin (+TeNT) or its vehicle (−TeNT) on DIV 5 and c-Fos (A) and COX-2 (B) mRNA quantified via qRT-PCR on DIV 7. Results are expressed as -fold change relative to vehicle (−TeNT, set to 1). There was no significant difference as determined via unpaired Student's t test of the geometric means (n = three from three independent experiments). Top blot: Western blot demonstrating Efficacy of TeNT-induced synaptobrevin-2 (SB2) cleavage (representative of three blots).
Transcriptional Regulation of Constitutive COX-2 Expression in Neuronal Cultures
Several putative cis-acting elements are found in the 5′-flanking region of the PTGS2 gene used to make the transgenic mouse. These include binding sites for nuclear factor-IL-6, glucocorticoid receptor, activator protein-1 and activator protein-2, CAAT box/enhancer-binding protein, cAMP response element-binding protein site (CREB), and stimulatory protein-1 (Sp1). Of these, CREB is known to specifically regulate numerous NMDA receptor-dependent genes (12, 13). To test whether CREB was involved in the constitutive expression of COX-2 in neurons, we co-transfected neurons with the Pluc371 plasmid and a dominant negative CREB (A-CREB) plasmid. The results demonstrate that A-CREB significantly attenuated Pluc371 promoter luciferase activity (Fig. 7). To confirm that the CREB site was functionally involved in the transcriptional regulation of constitutive COX-2 expression in neurons, we mutated the CREB-binding CRE site of the Pluc371 plasmid and measured the effects on luciferase activity. Additionally, as Sp1 has been demonstrated to be essential for the constitutive expression of numerous genes, we chose to mutate its binding site as well. Mutation of the CRE site completely abolished expression of luciferase activity from Pluc371 (Fig. 8), confirming a role for CREB. Surprisingly, mutation of the Sp1-binding site resulted in a significant elevation of luciferase activity, suggesting Sp1 acts as a transcriptional repressor of COX-2 expression in neurons (Fig. 8A). Interestingly, NMDA receptor antagonism reduced the increase mediated by mutation of the Sp1 site back down to control levels (Fig. 8A). To examine a direct association of CREB and Sp1 with the endogenous PTGS2 gene promoter in neurons, ChIP analysis was performed on extracts from primary neurons at DIV 7. As shown in Fig. 8B, PTGS2 gene promoter fragments were successfully amplified from neuron genomic DNA samples using antisera directed against Sp1 or CREB. In each preparation, strong binding was observed for CREB, whereas the SP1 appeared to be considerably weaker (Fig. 8B). When IgG antiserum was used for immunoprecipitation, a faint band was observed in 2/4 gels for Sp1 and 1/4 for CREB (supplemental Figs. 1 and 2). Although, the exact reason for the small amount of template in the IgG control is not known, such background is not unusual (14, 15). Nevertheless, in all cases we see a significant -fold enrichment of the positive binding region for anti-SP1 as well as for anti-CREB antibodies over the IgG control when expressed as a ratio of bound sequence over input (Fig. 8B). Together these data provide compelling evidence for the binding and function of these transcription factors in the control of neuronal constitutive COX-2 gene expression.
FIGURE 7.
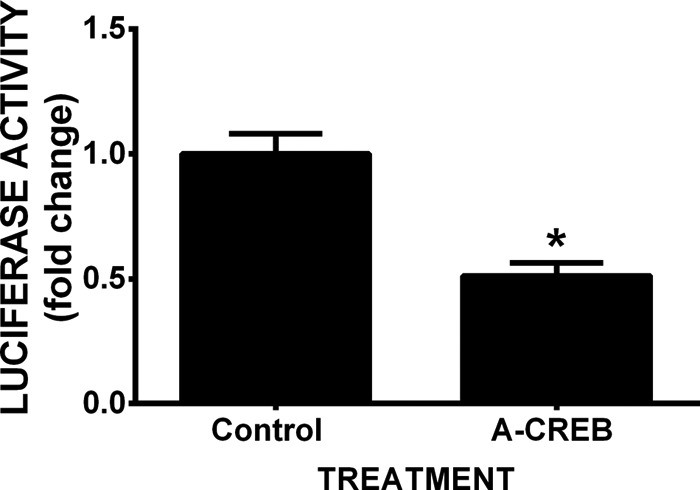
CREB is involved constitutive neuronal COX-2 transcription. Four days after plating, pure neuron cultures from non-transgenic mice were co-transfected with the PLuc371 (luciferase) reporter vector together with A-CREB (dominant negative inhibitor of CREB) and pGIPZ (GFP) expression plasmids as described under “Materials and Methods.” Comparisons were made to cells transfected in parallel with PLuc371 reporter and pGIPZ (GFP) DNA plus empty vector. Luciferase activity in cell lysates was assessed 3 days after transfection (DIV 7) and normalized to GFP intensity, which was quantified via fluorescence microscopy. Luciferase activity in cells transfected with the control plasmid was set to 1, and data are expressed as -fold change from control. The asterisks (*) indicate a value significantly different from control as assessed by Mann-Whitney U test (p = 0.0043, n = 6).
FIGURE 8.
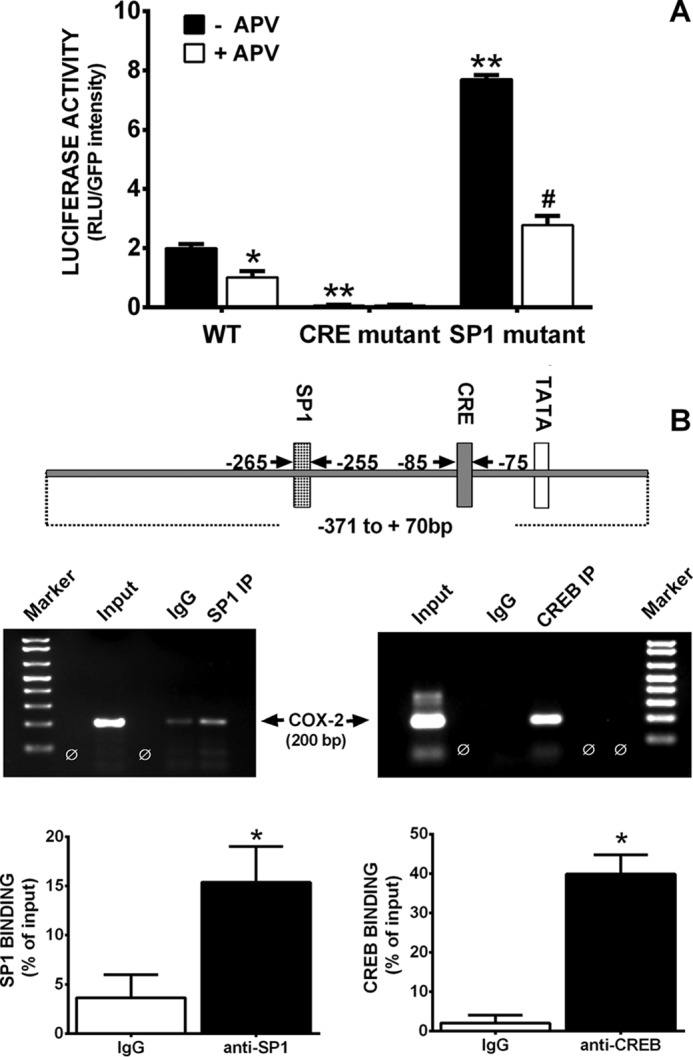
A, role of SP1 or CRE DNA consensus motifs in neuronal COX-2 transcription. Four days after plating, pure neuron cultures were co-transfected with pGIPZ and unmodified PLuc371 or PLuc371 having mutated SP1 or CRE elements. Three days later, GFP intensity was quantified via fluorescence microcopy, after which cells were lysed, and luciferase activity was determined. Data are expressed as RLU + S.E./GFP intensity. An asterisk (*) indicates values that are significantly different from control (−APV) (*, p < 0.02; **, p < 0.0001), whereas a pound signs (#) represents a significant SP1 mutant group difference (p < 0.0001) as determined by two-way ANOVA followed by Bonferroni's t test for multiple comparisons (n = 3). B, ChIP analysis of Sp1 and CREB binding to the native PTGS2 promoter. Top, schematic representation of CREB and Sp1-binding sites in the proximal segment of the PTGS2 promoter. Bottom, cross-linked DNA-protein complexes immunoprecipitated (IP) using antibodies to Sp1 and CREB or non-immune IgG as well as an input chromatin sample were analyzed by PCR using primers specific to the COX-2 promoter. Percent protein binding to DNA was determined by normalizing PCR products derived from non-immune and anti-SP1 or anti-CREB IgG pulldown to Input. Data are expressed as the mean ± S.E. An asterisk (*) represents a significant increase in binding over non-immune IgG as determined via two-tailed t tests (p = 0.035 and 0.0017 for SP1 and CREB, respectively, n = 4 each).
Discussion
In contrast to various other tissues, COX-2 expression occurs constitutively in the central nervous system (7–9, 16, 17). Despite knowing that the PTGS2 gene promoter contains a TATA box motif and a number of cis-acting elements, including CREB, C/EBP, NF-IL6, AP-1, Sp1, and NF-κB consensus sequences (18, 19), the molecular mechanisms underlying constitutive neuronal COX-2 expression remain largely unknown. With respect to this, this study presents with several original findings. First, results indicate that the factor(s) regulating constitutive neuronal COX-2 expression were inherent to neurons, as co-culture with astrocytes or treatment with astrocyte-conditioned media4 fail to regulate either Pluc371 gene reporter activity or COX-2 mRNA levels in neurons. Second, constitutive expression depends largely on the presence of functional NMDA receptors as genetic loss of the protein or functional block of NMDA receptors prevents the constitutive neuronal expression of COX-2. Third, the lack of effect of tetanus toxin reveals the involvement of spontaneous glutamatergic synaptic activity. Fourth, ChIP analysis demonstrates that both Sp1 and CREB bind to native COX-2 promoter, whereas promoter mutation studies indicate that constitutive neuronal COX-2 expression is regulated by both Sp1 and CREB family-dependent mechanisms.
Consistent with the COX-2 mRNA expression data found in the Allen Brain Atlas (20), brain tissue extracts from transgenic mice exhibit high constitutive luciferase activity in hippocampus, olfactory bulbs, and cortex (Fig. 1A). We additionally demonstrate constitutive activity in the cerebellum and spinal cord (Fig. 1A). This expression closely mirrored tissue mRNA levels, with the notable exception of cerebellum and spinal cord, suggesting areas of the promoter that suppress expression may be missing from our construct and/or that native expression in these tissues is influenced by elements in the COX-2 3′-UTR. Indeed, underscoring this point, analysis of basal expression of luciferase activity driven from the endogenous PTGS2 gene in a knock-in mouse also shows high expression in cortex and hippocampus, with little expression in cerebellum; the spinal cord was not assessed (21).
With respect to cortex, where the construct does faithfully reproduce native expression, this activity likely reflects neuronal expression as cultured cortical neurons from transgenic mice exhibit high luciferase activity, whereas activity in purified cultures of transgenic cortical astrocytes is negligible (Fig. 2A). This is congruent with previous immunocytochemical data from our laboratory demonstrating constitutive COX-2 protein expression in neurons but not astrocytes (17) and rat in vivo data demonstrating the same (9). Interestingly, astrocytic factors appear to have little or no influence on neuronal COX-2 expression, as evidenced by the fact that there was no change in the profile of constitutive luciferase expression in neurons when astrocytes were either in direct contact (Fig. 2B) or when astrocyte-conditioned medium was added to purified neuronal cultures.4 These findings suggest that the factors controlling constitutive COX-2 expression are intrinsic to neurons.
The constitutive COX-2 expression found in our cultures occurs in a temporal pattern (Fig. 2B), one that coincides with reported increases in the development of spontaneous post-synaptic currents in cultured cortical neurons (22). Additionally, this increase precisely mirrors the developmental expression of NMDA receptor protein and activity in our cultures as we reported previously (23) and confirm herein (Fig. 3A), raising the intriguing possibility that constitutive COX-2 expression is NMDA receptor-dependent. Indeed, functional block (genetic or pharmacological) of NMDA receptors effectively suppressed neuronal COX-2 mRNA expression (Figs. 4 and 5). The failure of tetanus toxin, a Clostridium toxin that prevents evoked, but not spontaneous, synaptic vesicle exocytosis in our and other systems (23–26), to reduce both constitutive neuronal activity and basal COX-2 mRNA expression in our cultures (Fig. 6) led us to conclude that spontaneous synaptic activity regulates basal neuronal COX-2 expression. This is not just a culture phenomenon, as basal COX-2 mRNA expression increased in rat forebrain and hippocampus over a developmental time frame in vivo and brain COX-2 basal mRNA expression can be reduced by neuronal deafferentation or by NMDA receptor inhibition but only slightly by intrahippocampal administration of tetrodotoxin (9).
Many of the regulatory elements driving constitutive neuronal expression must be found within −371 base pairs of the COX-2 promoter, as we found robust gene expression of constitutive COX-2 using the Pluc371 transgenic construct both in vivo and in vitro. This is an area that lacks the NF-κB site, which is located ∼400 bp upstream of the transcriptional start site (27). Hence, constitutive neuronal PTGS2 transcription appears to be independent of NFκB. This is in line with a recent study demonstrating both negligible constitutive NFκB activity in brain and a lack of effect of NFκB inhibition on PTGS2 promoter activity in knock-in reporter mice (21). Interestingly, stimulus-induced increases in neuronal COX-2 expression do appear to involve NFκB binding. For instance, Kaltschmidt et al. (28) show that exposure of neurons to the phorbol 12-myristate 13-acetate (PMA) increases neuronal COX-2 expression in an NF-κB -dependent manner. Additionally, NFκB is regarded as a strong regulator of inducible COX-2 expression in neurons from diseased brain (29, 30) and spinal cord (31, 32). Thus, synaptic activity-dependent constitutive COX-2 expression is mechanistically different from injury-induced COX-2 expression.
NMDA receptor activation stimulates neuronal gene expression via several signaling pathways, including the Ras-MAP kinase (MAPK) pathway, tyrosine kinases, and via activation of the transcription factors, including Fos, Fra, and CREB (33–40). With respect to the later, sequence analysis revealed that there is one cAMP-response element (CRE) site on the murine COX-2 promoter located between −85 and −75 bp upstream of the transcription start site. CREB and its family members contain a bZIP domain that mediates DNA binding and dimerization to CRE site and enhances gene transcription (41–43). Transfection of our neuronal cultures with a dominant-negative inhibitor of CREB (A-CREB), which interacts with CREB at its bZIP domain binding (44, 45), resulted in a significant decrease in the transgenic PTGS2 promoter-driven luciferase expression (Fig. 7), suggesting an important role for CREB in regulation of constitutive COX-2 expression in neurons. The fact that mutation of the CRE site on the promoter completely abolished luciferase activity (Fig. 8A) further supports this idea.
As a putative negative control, we also mutated the putative Sp1-binding site found between −265 and −255 upstream of the transcriptional start site. We chose Sp1 as it is a ubiquitously expressed transcription factor that is required for the expression of a variety of constitutively expressed genes (46), including endothelial COX-1 expression (47). Sp1 also promoted hypoxia-induced COX-2 expression in the vascular endothelium and oxidative and DNA damage-induced COX-2 expression in neuronal cells (27, 48). To our surprise, this mutation resulted in a significant increase in COX-2 luciferase activity (Fig. 8A), which demonstrates that Sp1 does not promote but rather represses basal COX-2 expression in neuronal cells. Indeed, Sp1 does constitutively bind to the native cortical neuron PTGS2 gene promoter (Fig. 8B). Several other studies describe Sp1 as a negative regulator of transcription as well. For instance, disulfide bond A oxidoreductase-like protein (DsbA-L) in preadipocytes (49) and Myc-associated zinc finger protein (MAZ) expression in hepatocytes (50) have also been demonstrated to be negatively regulated by Sp1. Gene expression can also be repressed through recruitment of histone deacetylase or methyltransferase to Sp1 sites (51, 52). The fact that APV was able to suppress this enhancement supports the involvement of NMDA receptor activity (Fig. 8A).
Finally, ChIP analysis revealed a significant -fold enrichment of the positive binding region for the anti-CREB and anti-SP1 antibodies over the IgG control (Fig. 8B; supplemental Figs. 1 and 2), although SP1 binding was substantially weaker. This latter result could be due to the accessibility of the Sp1-binding site as it is known to be influenced by histone modifications (14, 51–53). Or it could reflect a low affinity of the antibody for the antigen target. Of the two ChIP validated antibodies tested, only the one described herein proved effective.
In sum, we explored the molecular basis for the constitutive COX-2 expression that is found in neurons using a novel transgenic mouse and several cellular and molecular tools. We found that the PTGS2 promoter mouse line as well as our cortical cell culture models near faithfully represent the expression of COX-2 reported in vivo. Specifically, we demonstrate that spontaneous synaptic excitation driven by NMDA receptor activity underlies the enhancement in COX-2 expression seen during development. Additionally, we report that the expression is cell-autonomous in that it does not rely on astrocyte signaling. Finally, we identified two molecular players that govern constitutive PTGS2 transcription in cortical neuron via repression (Sp1) as well as enhancement (CREB). Our ongoing studies are devised to elucidate the signaling mechanism by which CREB positively and Sp1 negatively regulates COX-2 expression in neuronal cells.
Given the burgeoning evidence in support of a role for COX-2 metabolites in regulating membrane excitability in various forms of synaptic plasticity (54–65), the present findings add to our understanding of the dynamic gene expression landscape occurring in brain development and its underlying synaptic connectivity.
Materials and Methods
Animals
This study was conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals and was approved by the Institutional Animal Care and Use Committee of both the University of Connecticut Health Center and Syracuse University.
Transgenic COX-2 Reporter Mice
Mice transgenic for the Pluc371 DNA construct (66, 67), containing the DNA construct −371 to +70 bp of the murine COX-2 promoter fused to a luciferase reporter gene, were developed in a CD-1 background by the Transgenic Animal Facility at the University of Connecticut Health Center, as described previously (66). This region of the murine COX-2 gene contains numerous known cis-acting transcriptional elements whose elements have been demonstrated to contribute to COX-2 transcriptional induction in non-neuronal cells under various conditions (68–70).
Cortical Cell Culture
Purified Primary Astrocytes
Astrocytes were cultured from the cortices of postnatal day 1–3 mouse pups as described in detail (71). Cells were plated 400 μl/well (2 hemispheres/10 ml/plate) (Falcon Primaria 24-well plates). Once confluent, astrocyte monolayers were treated with 8 μm β-d-cytosine arabinofuranoside (AraC) once for 4–7 days to reduce the number of microglia.
Mixed Cortical Cultures
Mixed cortical cultures containing both astrocytes and neurons were prepared from postnatal and fetal mice, respectively, as described in detail previously (17, 72). In brief, confluent astrocyte monolayers (9–11 DIV) were first established. Cortical cells from fetal mice were then plated at a density of 3.0–3.8 hemispheres/plate on an established astrocytic monolayer (12–24 DIV). After 5–7 days in culture, mixed cultures were treated with 8 μm Ara-C once for 2 days. Experiments were performed on mixed cultures containing ∼50% neurons and 50% astrocytes.
PNCs
PNCs were cultured from cortices derived from the embryos of day 15 mouse and plated at 3.0–3.5 hemispheres/plate or 1.0–1.25 million cells/ml in Neurobasal medium supplemented with B27, 2 mm glutamine, and antibiotics as described in detail (73). Pure cortical neurons wild-type (NR1+/+) or null (NR1−/−) for NR1 were cultured from single embryos derived from the breeding of NR1± parents as described (74, 75). Cultures were >98% pure. Media were partially replenished in cultures twice weekly. All cultures were kept at 37 °C in a humidified 6% CO2-containing atmosphere.
Measurement of Luciferase Activity
Transgenic Mice
Luciferase activity from reporter mice tissue was analyzed using the Luciferase Assay System according to the manufacturer's protocol (Promega, Madison, WI). Briefly, adult male animals were anesthetized by i.p. injection of 100 mg/kg nembutal sodium solution (Abbot Laboratories, Chicago, IL) and transcardially perfused with 20 ml of ice-cold saline. Organs were rapidly dissected into ice-cold phosphate-buffered saline (PBS). Tissue was weighed, then homogenized in ice-cold PBS (0.1g of tissue/ml). 400 μl of tissue homogenate was mixed with 100 μl of 5× reporter lysis buffer and immediately frozen at −80 °C. Samples were thawed and spun (10,000 × g; 30 s), and then 10–20 μl was assayed for luciferase activity using an OPTOCOMP II Luminometer (MGM Instruments, Hamden, CT).
Cell Culture
Whole cell lysates were collected in 100 μl if 1X reporter lysis buffer, freeze-thawed, and spun (10,000 × g; 30 s). 10–20 μl of the supernatant was analyzed as described above. Protein concentrations in all samples were quantified with the BCA protein assay (Pierce). Data are expressed as relative light units (RLU) normalized to mg protein.
Drug Exposure
PNCs were exposed to the NMDA receptor antagonist APV (Tocris) or tetanus toxin (TeNT; Calbiochem) for 2 days (DIV 5–7) at 37 °C in a humidified 5.5% CO2-containing normoxic incubator at a final concentration of 30 μm or 300 ng/ml, respectively. Stock solutions of APV (1.2 mm) and tetanus toxin (12 μg/ml) were prepared in H2O.
qRT-PCR
COX-2 and c-Fos mRNA levels were measured using qRT-PCR. Total RNA was extracted from cells using TRIzol reagent (Invitrogen), and first-strand cDNA synthesis was performed as described previously (17, 76). cDNA was subjected to qRT-PCR in a singleplex reaction containing either mouse Fos (Mm00487425_m1), PTGS2 (Mm00478372_m1), or mouse β-actin (Mm01205647_m1) primer pairs with probes (Assay-On-Demand, Applied Biosystems) along with TaqMan Universal PCR Master Mix (Applied Biosystems) according to the manufacturer's protocol. PCR reactions were performed in duplicate or triplicate using an ABI 7500, an ABI 7900HT Fast Real Time PCR System (Applied Biosystems), or an Eppendorf Realplex2 under the following conditions: 50 °C for 2 min and 95 °C for 10 min followed by 40 amplification cycles (95 °C for 15 s and 60 °C for 1 min). Data analysis was performed using the comparative cycle threshold method (ΔΔCT), where CT values for COX-2 and c-Fos mRNAs were normalized to β-actin CT values from the same sample and then compared with a calibrator sample CT value defined in each figure legend.
Plasmid and Mutagenesis of COX-2 Promoter
Dominant negative CREB (A-CREB) was a gift from Charles Vinson (Addgene plasmid #33371) (44). Mutation of the Sp1 or CRE site in the PTGS2 gene promoter of Pluc371 (pTIS10s) was carried out using QuikChange® Site-Directed Mutagenesis kit (Agilent Technologies). The Sp1-binding site was mutated by replacing the GC box sequence GGGCGG with GTTCGG. The CRE binding site was mutated by replacing CTACGTCA with CTGATTCA. Mutations were confirmed by sequencing.
Transfection of Cortical Neuron
Neuron cultures were transfected 4 days after plating. For each transfection, 4 μl of Lipofectamine 2000 (Invitrogen) were added to 0.7 ml Opti-MEM medium (Invitrogen) and incubated at 25 °C for 10 min. PLuc371 (2 μg) reporter plasmid together with 0.5 μg of GFP reporter plasmid (pGIPZ, Open Biosystems) plus 0.5 μg A-CREB or its empty vector were added to the Lipofectamine mixture, briefly vortexed, and incubated for another 20 min (25 °C). In the mutation studies, 2.5 μg of control (wild type), SP1 mutant, or CRE mutant COX-2 luciferase reporter DNA constructs plus 0.5 μg of GFP reporter DNA were added to the Lipofectamine mixture, briefly vortexed, and incubated for another 20 min (25 °C).
A portion of neuronal culture medium (250 μl) was removed and immediately replaced with 175 μl of the transfection medium. Neurons were then incubated at 37 °C for 5–6 h, after which they were washed 4× (250 μl) with neuronal plating medium and placed back into a 37 °C, 5.5% CO2 containing normoxic incubator. Transfection efficiency was assessed in live cultures by measurement of GFP fluorescence intensity. Images from four microscopic fields (10× magnification) were acquired by a DP73 high performance Peltier cooled digital color camera mounted on an Olympus IX50 inverted microscope outfitted with epifluorescence controlled by CellSens Standard software (Olympus, Center Valley, PA). GFP intensity was quantified with NIH ImageJ. Luciferase activity in transfected cells was measured 3 days after transfection (DIV 7) using the Dual-Glo® Luciferase Assay Systems according to the manufacturer's instruction. Plates were freeze-thawed once (−80 °C to 37 °C) after which Dual-Glo® Luciferase Assay Reagent was added to each well. After incubation at 25 °C for 10 min, luciferase activity was determined using a Bio-Tek Synergy2 microplate reader.
Western Blotting
Cell Surface Biotinylation
Cell surface biotinylation was carried out as described (77, 78) with minor modifications (23). Briefly, cells were washed with 3× with ice-cold TBS and lysed by the addition Nonidet P-40 lysis buffer (Thermo Fisher Scientific). Biotinylated proteins were allowed to adsorb for 1 h (4 °C) to streptavidin-agarose beads (1:1 suspension in Nonidet P-40 lysis buffer) after which the beads were pelleted (2000 × g; 5 min; 4 °C). Biotinylated proteins were eluted from the beads by direct addition of 50 μl of reducing loading buffer followed by boiling. Cell supernatant, representing the cytosolic fraction, was removed to a new tube, the eluted beads were washed with lysis buffer (500 μl) and pelleted, and the supernatants were added to the original pool. This was repeated a second time after which the beads received a final wash with Tris-HCl (50 mm, pH 7.5). Proteins in the collected supernatant fraction were precipitated by the addition of ice-cold acetone (5 volumes; −20 °C overnight). The resulting pellet was dissolved in 100 μl of a reducing loading buffer. All samples were boiled (100 °C; 2 min) before separation via SDS-PAGE (8% polyacrylamide gel) under reducing conditions. Proteins were then electrophoretically transferred onto nitrocellulose and immunoblotted for NR1 (mouse monoclonal; 1:4500; Novus Biologicals Littleton, CO) and β-tubulin class III (mouse monoclonal; 1:5000; Sigma) to assess for biotinylation of intracellular proteins should it have occurred. If >15% of the β-tubulin class III was found to be biotinylated, samples were excluded from analysis.
Total Cell Lysates
Purified neuron cultures were washed once with ice-cold PBS and harvested by gentle scraping. Cells were spun (700 × g; 5 min; 4 °C), and the resulting pellet was suspended in lysis buffer containing 20 mm HEPES (pH 7.4), 2 mm EGTA (pH 8.0), 50 mm β-glycerol phosphate (pH 7.2), 1 mm DTT, 1 mm Na3VO4, 5 mm NaF, 1% Triton X-100, 0.2 mm PMSF. and 1× Complete Protease Inhibitor (Roche Applied Science). The resuspended pellet was then incubated on ice (30 min). Cellular debris was removed by centrifugation (12,000 × g; 20 min; 4 °C). 40 μg of protein (BCA assay; Pierce) was separated by 15% SDS-PAGE under reducing conditions and electrophoretically transferred to a PVDF membrane (Bio-Rad). Membranes were blocked (Odyssey® blocking buffer at 25 °C for 1 h) and then probed (4 °C, overnight) with an anti-synaptobrevin-2 rabbit polyclonal antibody (1:1000 dilution; Synaptic System), and a mouse monoclonal antibody was directed against β-actin (1:4000 dilution; Sigma). Species-specific secondary antibodies labeled with spectrally distinct IRDye® fluorescent dyes (LI-COR Biosciences; Lincoln, NE) were used to detect primary antibodies (1 h at 25 °C). Results were recorded on LI-COR ODYSSEY® Fc Imaging system (LI-COR Biosciences).
Chromatin Immunoprecipitation Assay
Cross-linking of protein-DNA complexes for detection of transcription factor binding to the COX2 promoter was accomplished by either ultraviolet irradiation (SP1; 150 mJ/cm2; Stratagene, UV Stratalinker 1800) or 1% formaldehyde fixation (CREB) of purified neuronal cultures at DIV 7 (1.2 × 106 cells/100 mm plate). Subsequently, cells were harvested by gentle scraping and spun at 600 × g for 5 min at 4 °C. The resulting pellet was suspended in ice-cold ChIP lysis buffer (1 ml) containing 50 mm HEPES-KOH (pH 7.5), 140 mm NaCl, 1 mm EDTA (pH 8.0), 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 1× Complete Protease Inhibitor (Roche Applied Science). After 10 min on ice, cell lysates were sonicated (Sonic Dismembrator Model 100, Fisher Scientific) 6 times (3 × 15-s each) with 40-s breaks between each and cellular debris pelleted at 8000 × g (1 min, 4 °C). Supernatants were diluted 1:5 in buffer containing 1 mm EDTA (pH 8.0), 1% Triton X-100, 20 mm Tris-HCL (pH 8.0), and 150 mm NaCl and divided equally into two samples. One sample contained the primary antibodies for CREB or Sp1 (5 μg of mouse monoclonal anti-CREB, (SC-271; Santa Cruz, Dallas, TX) or 5 μg rabbit polyclonal anti-Sp1 (07-645; Millipore, Billerica, MA). and the other contained the respective non-immune antibodies (5 μg, mouse IgG; Santa Cruz, Dallas, TX) and 5 μg of rabbit IgG (Millipore, Billerica, MA). The samples were rotated at 4οC overnight, after which prewashed anti-mouse Ig IP Beads or anti-rabbit Ig IP beads (for CREB and Sp1 pulldown assays, respectively, True Blot®) were added. After rotating for an additional 4 h at 4 °C, beads were pelleted at 10,000 × g (1 min; 4 οC), and the supernatants were discarded. Beads were washed 4× with 1 ml of buffer containing 2 mm EDTA (pH 8.0), 1% Triton X-100, 0.1% SDS, 20 mm Tris-HCL (pH 8.0), and 150 mm NaCl with 1× Complete Protease Inhibitor (Roche Applied Science) then cleared with 1 ml of final wash buffer (2 mm EDTA (pH 8.0), 1% Triton X-100, 0.1% SDS, 20 mm Tris-HCL (pH 8.0) and 500 mm NaCl with 1× Complete Protease Inhibitor (Roche Applied Science)). DNA-protein complexes were eluted from beads by adding 150 μl of warm (30 °C) elution buffer (1% SDS and 100 mm NaHCO3 in H20) followed by slow vortexing for a total of 15 min. DNA was purified with Promega Wizard SV Gel and PCR clean-up kit (product # A9281).
PCR Amplification
cDNA samples (1 μl) were amplified using MJ Mini Personal Thermocycler for 35 cycles (94 °C/45s, 52 °C/30s, 72 °C/60s) using TaqDNA polymerase (Invitrogen) and target-specific primers in a total volume of 25 μl. Amplimers for assessment of COX-2 DNA were 5′-GAGGGGAAGCTGTGACACTC-3′ (Sp1 forward) and 5′-ACGCAAATGAGAGACGAAGG-3′ (Sp1 reverse) and 5′-GGCTTCCTTCGTCTCTCATTT-3′ (CRE forward) and 5′-TGACAACTGGCTGCTAATGG-3′ (CRE reverse). PCR products, separated on a 2% agarose gel containing ethidium bromide (200 μg/ml), were visualized with the LI-COR Odyssey Fc Infrared Imaging System.
Statistical Analysis
Data were analyzed using GraphPad Prism software as described in each figure legend. Non-normal data were either transformed before analysis with parametric statistical tests, or non-parametric tests were employed. Significance was assessed at p < 0.05, individual p values are described in the figure legends.
Author Contributions
C. P. provided the transgenic COX-2 reporter mice, initially noting constitutive COX-2 expression in brain. S. J. H. and J. A. H. conceived and coordinated the study and were responsible for the written content. K. D., Y. G., and J. S. contributed equally to the performance and analysis of experiments except for the study presented in Fig. 4, which was performed by J. A. H. Additionally, these authors contributed to the writing of the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Harvey Herschman (UCLA, Los Angeles, CA) for the DNA construct consisting of −371 to +70 bp of the murine COX-2 promoter fused to a luciferase reporter gene. We thank Drs. Steve Finkbeiner and Sarah Carter (Gladstone Institute of Neurological Disease, University of California, San Francisco) for providing mRNA from NR1+/+ and NR1−/− neuron cultures for analysis.
This work was supported by National Institutes of Health Grants NS036812 (to S. J. H. and J. A. H.), NS082982 (to J. A. H.), and T32 DC005355 (to K. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. 1 and 2.
K. Dhandapani, unpublished observation.
- PG
- prostaglandin
- DIV
- days in vitro
- NR
- NMDA receptor
- PNC
- purified neuronal culture
- APV
- 5-amino-phosphonovaleric acid
- CREB
- cAMP response element (CRE)-binding protein
- Sp1
- stimulatory protein-1
- RLU
- relative light units
- TeNT
- tetanus toxin.
References
- 1. Vane J. R., Bakhle Y. S., and Botting R. M. (1998) Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 38, 97–120 [DOI] [PubMed] [Google Scholar]
- 2. Bhattacharyya D. K., Lecomte M., Dunn J., Morgans D. J., and Smith W. L. (1995) Selective inhibition of prostaglandin endoperoxide synthase-1 (cyclooxygenase-1) by valerylsalicylic acid. Arch. Biochem. Biophys. 317, 19–24 [DOI] [PubMed] [Google Scholar]
- 3. Smith W. L., and Song I. (2002) The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 68, 115–128 [DOI] [PubMed] [Google Scholar]
- 4. Kraemer S. A., Meade E. A., and DeWitt D. L. (1992) Prostaglandin endoperoxide synthase gene structure: identification of the transcriptional start site and 5′-flanking regulatory sequences. Arch. Biochem. Biophys. 293, 391–400 [DOI] [PubMed] [Google Scholar]
- 5. Wang L. H., Hajibeigi A., Xu X. M., Loose-Mitchell D., and Wu K. K. (1993) Characterization of the promoter of human prostaglandin H synthase-1 gene. Biochem. Biophys. Res. Commun. 190, 406–411 [DOI] [PubMed] [Google Scholar]
- 6. Smith W. L., Garavito R. M., and DeWitt D. L. (1996) Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 271, 33157–33160 [DOI] [PubMed] [Google Scholar]
- 7. Adams J., Collaço-Moraes Y., and de Belleroche J. (1996) Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J. Neurochem. 66, 6–13 [DOI] [PubMed] [Google Scholar]
- 8. Kaufmann W. E., Worley P. F., Pegg J., Bremer M., and Isakson P. (1996) COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 93, 2317–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamagata K., Andreasson K. I., Kaufmann W. E., Barnes C. A., and Worley P. F. (1993) Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11, 371–386 [DOI] [PubMed] [Google Scholar]
- 10. Morgan J. I., and Curran T. (1991) Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu. Rev. Neurosci. 14, 421–451 [DOI] [PubMed] [Google Scholar]
- 11. Sweeney S. T., Broadie K., Keane J., Niemann H., and O'Kane C. J. (1995) Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14, 341–351 [DOI] [PubMed] [Google Scholar]
- 12. Zhou X., Moon C., Zheng F., Luo Y., Soellner D., Nuñez J. L., and Wang H. (2009) N-Methyl-d-aspartate-stimulated ERK1/2 signaling and the transcriptional up-regulation of plasticity-related genes are developmentally regulated following in vitro neuronal maturation. J. Neurosci. Res. 87, 2632–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hardingham G. E., Arnold F. J., and Bading H. (2001) Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 4, 261–267 [DOI] [PubMed] [Google Scholar]
- 14. Kuan C. S., See Too W. C., and Few L. L. (2016) Sp1 and Sp3 Are the Transcription Activators of Human ek1 Promoter in TSA-Treated Human Colon Carcinoma Cells. PloS ONE 11, e0147886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chu C., Zavala K., Fahimi A., Lee J., Xue Q., Eilers H., and Schumacher M. A. (2011) Transcription factors Sp1 and Sp4 regulate TRPV1 gene expression in rat sensory neurons. Mol. Pain 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Breder C. D., Dewitt D., and Kraig R. P. (1995) Characterization of inducible cyclooxygenase in rat brain. J. Comp. Neurol. 355, 296–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hewett S. J., Uliasz T. F., Vidwans A. S., and Hewett J. A. (2000) Cyclooxygenase-2 contributes to N-methyl-d-aspartate-mediated neuronal cell death in primary cortical cell culture. J. Pharmacol. Exp. Ther. 293, 417–425 [PubMed] [Google Scholar]
- 18. Fletcher B. S., Kujubu D. A., Perrin D. M., and Herschman H. R. (1992) Structure of the mitogen-inducible TIS10 gene and demonstration that the TIS10-encoded protein is a functional prostaglandin G/H synthase. J. Biol. Chem. 267, 4338–4344 [PubMed] [Google Scholar]
- 19. Appleby S. B., Ristimäki A., Neilson K., Narko K., and Hla T. (1994) Structure of the human cyclo-oxygenase-2 gene. Biochem. J. 302, 723–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T.-M., Chin M. C., Chong J., et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 21. Kirkby N. S., Chan M. V., Zaiss A. K., Garcia-Vaz E., Jiao J., Berglund L. M., Verdu E. F., Ahmetaj-Shala B., Wallace J. L., Herschman H. R., Gomez M. F., and Mitchell J. A. (2016) Systematic study of constitutive cyclooxygenase-2 expression: role of NF-κB and NFAT transcriptional pathways. Proc. Natl. Acad. Sci. U.S.A. 113, 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin Y. C., Huang Z. H., Jan I. S., Yeh C. C., Wu H. J., Chou Y. C., and Chang Y. C. (2002) Development of excitatory synapses in cultured neurons dissociated from the cortices of rat embryos and rat pups at birth. J. Neurosci. Res. 67, 484–493 [DOI] [PubMed] [Google Scholar]
- 23. Fogal B., Trettel J., Uliasz T. F., Levine E. S., and Hewett S. J. (2005) Changes in secondary glutamate release underlie the developmental regulation of excitotoxic neuronal cell death. Neuroscience 132, 929–942 [DOI] [PubMed] [Google Scholar]
- 24. Schiavo G., Poulain B., Rossetto O., Benfenati F., Tauc L., and Montecucco C. (1992) Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc. EMBO J. 11, 3577–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schiavo G., Benfenati F., Poulain B., Rossetto O., Polverino de Laureto P., DasGupta B. R., and Montecucco C. (1992) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359, 832–835 [DOI] [PubMed] [Google Scholar]
- 26. Taylor A. L., and Hewett S. J. (2002) Potassium-evoked glutamate release liberates arachidonic acid from cortical neurons. J. Biol. Chem. 277, 43881–43887 [DOI] [PubMed] [Google Scholar]
- 27. Xu Q., Ji Y. S., and Schmedtje J. F. Jr. (2000) Sp1 increases expression of cyclooxygenase-2 in hypoxic vascular endothelium: implications for the mechanisms of aortic aneurysm and heart failure. J. Biol. Chem. 275, 24583–24589 [DOI] [PubMed] [Google Scholar]
- 28. Kaltschmidt B., Linker R. A., Deng J., and Kaltschmidt C. (2002) Cyclooxygenase-2 is a neuronal target gene of NF-κB. BMC Mol. Biol. 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lukiw W. J., and Bazan N. G. (1998) Strong nuclear factor-κB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer's disease superior temporal lobe neocortex. J. Neurosci. Res. 53, 583–592 [DOI] [PubMed] [Google Scholar]
- 30. Jang J.-H., and Surh Y.-J. (2005) β-Amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen-activated protein kinase-NF-[κ]B signaling pathway. Free Radic. Biol. Med. 38, 1604–1613 [DOI] [PubMed] [Google Scholar]
- 31. Lee K. M., Kang B. S., Lee H. L., Son S. J., Hwang S. H., Kim D. S., Park J. S., and Cho H. J. (2004) Spinal NF-κB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. Eur. J. Neurosci. 19, 3375–3381 [DOI] [PubMed] [Google Scholar]
- 32. Tegeder I., Niederberger E., Schmidt R., Kunz S., Gühring H., Ritzeler O., Michaelis M., and Geisslinger G. (2004) Specific Inhibition of IκB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J. Neurosci. 24, 1637–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. English J. D., and Sweatt J. D. (1996) Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J. Biol. Chem. 271, 24329–24332 [DOI] [PubMed] [Google Scholar]
- 34. Rakhit S., Clark C. J., O'shaughnessy C. T., and Morris B. J. (2005) N-methyl-d-aspartate and brain-derived neurotrophic factor induce distinct profiles of extracellular signal-regulated kinase, mitogen- and stress-activated kinase, and ribosomal s6 kinase phosphorylation in cortical neurons. Mol. Pharmacol. 67, 1158–1165 [DOI] [PubMed] [Google Scholar]
- 35. Monti B., Marri L., and Contestabile A. (2002) NMDA receptor-dependent CREB activation in survival of cerebellar granule cells during in vivo and in vitro development. Eur. J. Neurosci. 16, 1490–1498 [DOI] [PubMed] [Google Scholar]
- 36. Shaywitz A. J., and Greenberg M. E. (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861 [DOI] [PubMed] [Google Scholar]
- 37. Tian X., Gotoh T., Tsuji K., Lo E. H., Huang S., and Feig L. A. (2004) Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk, and CREB. EMBO J. 23, 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xia Z., Dudek H., Miranti C. K., and Greenberg M. E. (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 16, 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sonnenberg J. L., Mitchelmore C., Macgregor-Leon P. F., Hempstead J., Morgan J. I., and Curran T. (1989) Glutamate receptor agonists increase the expression of Fos, Fra, and AP-1 DNA binding activity in the mammalian brain. J. Neurosci. Res. 24, 72–80 [DOI] [PubMed] [Google Scholar]
- 40. Impey S., Mark M., Villacres E. C., Poser S., Chavkin C., and Storm D. R. (1996) Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16, 973–982 [DOI] [PubMed] [Google Scholar]
- 41. Foulkes N. S., Borrelli E., and Sassone-Corsi P. (1991) CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell 64, 739–749 [DOI] [PubMed] [Google Scholar]
- 42. Hai T. W., Liu F., Coukos W. J., and Green M. R. (1989) Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 3, 2083–2090 [DOI] [PubMed] [Google Scholar]
- 43. Lonze B. E., and Ginty D. D. (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 [DOI] [PubMed] [Google Scholar]
- 44. Ahn S., Olive M., Aggarwal S., Krylov D., Ginty D. D., and Vinson C. (1998) A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol. 18, 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lubelski D., Ponzio T. A., and Gainer H. (2012) Effects of A-CREB, a dominant negative inhibitor of CREB, on the expression of c-fos and other immediate early genes in the rat SON during hyperosmotic stimulation in vivo. Brain Res. 1429, 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li L., He S., Sun J. M., and Davie J. R. (2004) Gene regulation by Sp1 and Sp3. Biochem. Cell Biol. 82, 460–471 [DOI] [PubMed] [Google Scholar]
- 47. Xu X. M., Tang J. L., Chen X., Wang L. H., and Wu K. K. (1997) Involvement of two Sp1 elements in basal endothelial prostaglandin H synthase-1 promoter activity. J. Biol. Chem. 272, 6943–6950 [DOI] [PubMed] [Google Scholar]
- 48. Lee J., Kosaras B., Aleyasin H., Han J. A., Park D. S., Ratan R. R., Kowall N. W., Ferrante R. J., Lee S. W., and Ryu H. (2006) Role of cyclooxygenase-2 induction by transcription factor Sp1 and Sp3 in neuronal oxidative and DNA damage response. FASEB J. 20, 2375–2377 [DOI] [PubMed] [Google Scholar]
- 49. Fang Q., Yang W., Li H., Hu W., Chen L., Jiang S., Dong K., Song Q., Wang C., Chen S., Liu F., and Jia W. (2014) Negative regulation of DsbA-L gene expression by the transcription factor Sp1. Diabetes 63, 4165–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Song J., Ugai H., Kanazawa I., Sun K., and Yokoyama K. K. (2001) Independent repression of a GC-rich housekeeping gene by Sp1 and MAZ involves the same cis-elements. J. Biol. Chem. 276, 19897–19904 [DOI] [PubMed] [Google Scholar]
- 51. Doetzlhofer A., Rotheneder H., Lagger G., Koranda M., Kurtev V., Brosch G., Wintersberger E., and Seiser C. (1999) Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol. 19, 5504–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim S., Kang J. K., Kim Y. K., Seo D. W., Ahn S. H., Lee J. C., Lee C. H., You J. S., Cho E. J., Lee H. W., and Han J. W. (2006) Histone deacetylase inhibitor apicidin induces cyclin E expression through Sp1 sites. Biochem. Biophys. Res. Commun. 342, 1168–1173 [DOI] [PubMed] [Google Scholar]
- 53. Zhu W. G., Srinivasan K., Dai Z., Duan W., Druhan L. J., Ding H., Yee L., Villalona-Calero M. A., Plass C., and Otterson G. A. (2003) Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol. Cell. Biol. 23, 4056–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cowley T. R., Fahey B., and O'Mara S. M. (2008) COX-2, but not COX-1, activity is necessary for the induction of perforant path long-term potentiation and spatial learning in vivo. Eur. J. Neurosci. 27, 2999–3008 [DOI] [PubMed] [Google Scholar]
- 55. Yang H., and Chen C. (2008) Cyclooxygenase-2 in synaptic signaling. Curr. Pharm. Des. 14, 1443–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen C., and Bazan N. G. (2005) Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J. Neurophysiol. 93, 929–941 [DOI] [PubMed] [Google Scholar]
- 57. Sang N., Zhang J., Marcheselli V., Bazan N. G., and Chen C. (2005) Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J. Neurosci. 25, 9858–9870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koch H., Huh S. E., Elsen F. P., Carroll M. S., Hodge R. D., Bedogni F., Turner M. S., Hevner R. F., and Ramirez J. M. (2010) Prostaglandin E2-induced synaptic plasticity in neocortical networks of organotypic slice cultures. J. Neurosci. 30, 11678–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Le T. D., Shirai Y., Okamoto T., Tatsukawa T., Nagao S., Shimizu T., and Ito M. (2010) Lipid signaling in cytosolic phospholipase A2α-cyclooxygenase-2 cascade mediates cerebellar long-term depression and motor learning. Proc. Natl. Acad. Sci. U.S.A. 107, 3198–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hewett S. J., Bell S. C., and Hewett J. A. (2006) Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol. Ther. 112, 335–357 [DOI] [PubMed] [Google Scholar]
- 61. Murray H. J., and O'Connor J. J. (2003) A role for COX-2 and p38 mitogen activated protein kinase in long-term depression in the rat dentate gyrus in vitro. Neuropharmacology 44, 374–380 [DOI] [PubMed] [Google Scholar]
- 62. Sang N., and Chen C. (2006) Lipid signaling and synaptic plasticity. Neuroscientist 12, 425–434 [DOI] [PubMed] [Google Scholar]
- 63. Straiker A., Wager-Miller J., Hu S. S., Blankman J. L., Cravatt B. F., and Mackie K. (2011) COX-2 and fatty acid amide hydrolase can regulate the time course of depolarization-induced suppression of excitation. Br. J. Pharmacol. 164, 1672–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Teather L. A., Packard M. G., and Bazan N. G. (2002) Post-training cyclooxygenase-2 (COX-2) inhibition impairs memory consolidation. Learn. Mem. 9, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang H., Zhang J., Andreasson K., and Chen C. (2008) COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol. Cell. Neurosci. 37, 682–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Choudhary S., Kumar A., Kale R. K., Raisz L. G., and Pilbeam C. C. (2004) Extracellular calcium induces COX-2 in osteoblasts via a PKA pathway. Biochem. Biophys. Res. Commun. 322, 395–402 [DOI] [PubMed] [Google Scholar]
- 67. Kujubu D. A., Fletcher B. S., Varnum B. C., Lim R. W., and Herschman H. R. (1991) TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J. Biol. Chem. 266, 12866–12872 [PubMed] [Google Scholar]
- 68. Kim Y., and Fischer S. M. (1998) Transcriptional regulation of cyclooxygenase-2 in mouse skin carcinoma cells: regulatory role of CCAAT/enhancer-binding proteins in the differential expression of cyclooxygenase-2 in normal and neoplastic tissues. J. Biol. Chem. 273, 27686–27694 [DOI] [PubMed] [Google Scholar]
- 69. Wardlaw S. A., Zhang N., and Belinsky S. A. (2002) Transcriptional regulation of basal cyclooxygenase-2 expression in murine lung tumor-derived cell lines by CCAAT/enhancer-binding protein and activating transcription factor/cAMP response element-binding protein. Mol. Pharmacol. 62, 326–333 [DOI] [PubMed] [Google Scholar]
- 70. Kang Y. J., Wingerd B. A., Arakawa T., and Smith W. L. (2006) Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. 177, 8111–8122 [DOI] [PubMed] [Google Scholar]
- 71. Uliasz T. F., Hamby M. E., Jackman N. A., Hewett J. A., and Hewett S. J. (2012) Generation of primary astrocyte cultures devoid of contaminating microglia. Methods Mol. Biol. 814, 61–79 [DOI] [PubMed] [Google Scholar]
- 72. Trackey J. L., Uliasz T. F., and Hewett S. J. (2001) SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J. Neurochem. 79, 445–455 [DOI] [PubMed] [Google Scholar]
- 73. Jackman N. A., Uliasz T. F., Hewett J. A., and Hewett S. J. (2010) Regulation of system x(c)(−)activity and expression in astrocytes by interleukin-1β: implications for hypoxic neuronal injury. Glia 58, 1806–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bradley J., Carter S. R., Rao V. R., Wang J., and Finkbeiner S. (2006) Splice variants of the NR1 subunit differentially induce NMDA receptor-dependent gene expression. J. Neurosci. 26, 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Forrest D., Yuzaki M., Soares H. D., Ng L., Luk D. C., Sheng M., Stewart C. L., Morgan J. I., Connor J. A., and Curran T. (1994) Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron 13, 325–338 [DOI] [PubMed] [Google Scholar]
- 76. Hamby M. E., Uliasz T. F., Hewett S. J., and Hewett J. A. (2006) Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J. Neurosci. Methods 150, 128–137 [DOI] [PubMed] [Google Scholar]
- 77. McIlhinney R. A., Le Bourdellès B., Molnár E., Tricaud N., Streit P., and Whiting P. J. (1998) Assembly intracellular targeting and cell surface expression of the human N-methyl-d-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology 37, 1355–1367 [DOI] [PubMed] [Google Scholar]
- 78. McIlhinney R. A., Molnár E., Atack J. R., and Whiting P. J. (1996) Cell surface expression of the human N-methyl-d-aspartate receptor subunit 1a requires the co-expression of the NR2A subunit in transfected cells. Neuroscience 70, 989–997 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.