

Abstract
Human cytomegalovirus (HCMV) UL84 encodes a 75-kDa protein required for oriLyt-dependent DNA replication and interacts with IE2 in infected and transfected cells. UL84 localizes to the nucleus of transfected and infected cells and is found in viral replication compartments. In transient assays it was shown that UL84 can interfere with the IE2-mediated transactivation of the UL112/113 promoter of HCMV. To determine whether UL84 protein-protein interactions are necessary for lytic DNA synthesis, we purified UL84 and used this protein to generate a monoclonal antibody. Using this antibody, we now show that UL84 forms a stable interaction with itself in vivo. The point of self-interaction maps to a region of the protein between amino acids 151 and 200, a domain that contains a series of highly charged amino acid residues. Coimmunoprecipitation assays determined that UL84 interacts with a protein domain present within the first 215 amino acids of IE2. We also show that an intact leucine zipper domain of UL84 is required for a stable interaction with IE2 and UL84 leucine zipper mutants fail to complement oriLyt-dependent DNA replication. UL84 leucine zipper mutants no longer interfere with IE2-mediated transactivation of the UL112/113 promoter, confirming that the leucine zipper is essential for a functional interaction with IE2. In addition, we demonstrate that both the leucine zipper and oligomerization domains of UL84 can act as transdominant-negative inhibitors of lytic replication in the transient assay, strongly suggesting that both an IE2-UL84 and a UL84-UL84 interaction are required for DNA synthesis.
A common feature of herpesvirus lytic DNA replication is the requirement for six core replication proteins. These proteins—a DNA polymerase, helicase, primase, primase-associated factor, DNA processivity factor, and a single-stranded DNA-binding protein—carry out the necessary enzymatic processes to efficiently synthesize DNA (11, 21, 23). These enzymatic replication events follow the obligatory initiation of DNA synthesis that is mediated by proteins that are unique to each herpesvirus system and have been traditionally referred to as initiator proteins or origin-binding proteins (OBPs). In some cases, these initiator proteins themselves have an inherent catalytic property that may unwind a specific region of DNA and facilitate the assembly of the entire core replication machinery.
Although the exact mechanism by which these OBPs initiate DNA synthesis has not been elucidated, several common features have been defined. (i) It appears that all characterized initiator proteins interact directly or indirectly with DNA within the origin of lytic replication. (ii) All identified initiator proteins have been shown to interact with themselves in transfected cells and contain leucine zipper motifs (5, 7, 8, 14, 15). Stable self-interaction of these proteins is seemingly necessary for their intrinsic enzymatic activity and may serve to regulate the initiation of DNA synthesis.
For HCMV, the transient-cotransfection replication assay identified UL84 and IE2 as two noncore proteins that were required for origin-dependent DNA replication in human fibroblasts (23, 27). UL84 is able to localize to the nucleus in transfected cells, indicating that no other viral protein is needed for this event. In addition, UL84 can be found in replication compartments and is essential for the formation of these structures (27, 32). The IE2 protein has been implicated as the major transactivator of HCMV gene expression (22). Interestingly, these two proteins were shown to interact in infected and transfected cells (30); however, the role of this interaction in either DNA replication or regulation of gene expression is not known. Evidence for a regulatory function for UL84 comes from studies showing that overexpression of UL84 in U373 cells resulted in a transdominant inhibition of viral replication (13). In the same study it was demonstrated that UL84 could interfere with IE2-mediated transactivation of the early gene promoter for UL112/113. Cotransfection of UL84 with IE2 resulted in a marked decrease in the ability of IE2 to stimulate transcription from the UL112/113 promoter. The proposed mechanism for this interference was due to the physical interaction between UL84 and IE2 (13). To date, UL84 has only been evaluated in the context of a regulator of gene expression mediated by IE2 and not with respect to the role of an IE2-UL84 interaction in DNA replication. One question that remains to be answered is if this protein-protein interaction is important for the function of UL84 in lytic DNA replication.
We generated here a monoclonal antibody by using UL84 purified protein and used this reagent to show that UL84 can exist as oligomers. The protein domain involved in this interaction maps to amino acids (aa) 151 to 200 of the protein. Interestingly, the UL84 oligomerization domain is distinct from the identified leucine zipper domain within the protein; however, an intact leucine zipper domain is essential for the interaction of UL84 with IE2. UL84 leucine zipper mutants no longer interact with IE2, as shown in pull-down assays. In addition, leucine zipper mutants of UL84 failed to complement origin-dependent DNA replication and were severely impaired in their ability to interfere with IE2-mediated transactivation of the UL112/113 promoter. Also, using the cotransfection replication assay, we show that expression of either the UL84 leucine zipper region or the UL84 oligomerization domain can act as a transdominant-negative inhibitor of oriLyt-dependent DNA replication, suggesting that a UL84-IE2 and a UL84-UL84 interaction contributes to DNA replication.
MATERIALS AND METHODS
Cells and viruses.
Telomerized human foreskin fibroblasts (T-HFs) (31a) were used for transient-replication assays and were propagated in Dulbecco modified Eagle medium (DMEM) supplemented with 10% (vol/vol) fetal calf serum. The 293 packaging cell line (Clontech) that was used to produce UL84 recombinant adenovirus was propagated in DMEM supplemented with 10% (vol/vol) fetal calf serum. Cos7 cells and Vero cells were propagated in DMEM supplemented with 10% (vol/vol) newborn calf serum. AD169 (ATCC V-538) was propagated as described previously (25).
Construction of a recombinant adenovirus expressing UL84.
Ad84 virus was generated by using AdEasy Adenoviral Vector System (Stratagene). AD169 genomic DNA was used as a template for amplifying the UL84 ORF. PCR was performed with the following primers: 5′-CCGGTACCACACCAAGCATGCCACGCGTCGACCCCAACCT-3′ and 5′-CGCTCGAGCTTAGCCCTACTTATCGTCGTCATCCTTGTAATCGAGATCGCCGCAGACCATGGCTAA-3′.The amplified UL84 open reading frame (ORF) was generated such that the FLAG sequence was fused in the same reading frame context as UL84. The PCR product was cleaved with KpnI and XhoI and ligated into pShuttle-CMV vector (Stratagene). Recombinant adenovirus was generated as recommended by the manufacturer. Virus titers were determined with a 293 packaging cell line.
DNA constructs.
pUL84-TAG(1-586), pUL84-TAG(68-586), pUL84-TAG(83-586), pUL84-TAG(104-586), pUL84-TAG(141-586), pUL84-TAG(201-586), pUL84-TAG(351-586), pUL84-TAG(1-513), and pUL84-EGFP were described previously by Xu et al. (32). The pUL84-HA expression plasmid was made by PCR amplification of AD169 DNA with the primers 5′-CggcgcACACCAAGCATGCCACGCGTCGACCCCAA-3′ and 5′-GGCGTCTTATCAAGCGTAGTCTGGGACGTCGTATGGGTAGAGATCGCCGCAGACCATGGC-3′, which was subsequently ligated into pTarget (Promega).
The pUL84EXPRESS expression plasmid was made by PCR amplification of AD169 DNA with the primers 5′-GcgccgaattcCCAAGCATGCCACGCGTCGACCCC-3′ and 5′-CgcggtcctcgagcTCTGTTTAGAGATCGCCGCAG-3′, which was subsequently digested with EcoRI and XhoI and ligated into pCDNA3.1+ (Invitrogen).
All UL84 peptide subclones were made by PCR amplification of AD169 DNA followed by ligation of the resulting product into pTarget (Promega). PCR products were generated such that the FLAG or hemagglutinin (HA) epitope and the simian virus 40 (SV40) nuclear localization sequences (NLS) were fused in the same reading frame context as the UL84 ORF. The following primers were used for UL84 subclones: p84FR(1-100) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCCAAGCATGCCACGCGTCGACCCCAACCTTCGGAA-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcAGGCTGACGGATGTCGTGTTTATCCGT-3′), p84FR(101-200) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCCAAGCATGCGGGTGCACCGCGGCACCTACCATCT-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcGAAGGCCGGGCGCACGTCAGAATCCTC-3′), p84FR(201-300) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCCAAGCATGTCTCTCTTTCCCGCACGCCCAGGCTG-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcCGTGGGCGTCAGATCCGCGTCCGTTGA-3′), p84FR(301-400) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCTGACGGTGCGCGTACGACACGC-3′ and 5′-TTAGCCCTACTTATCGTCGTCATCCTTGTAATCTCGCACCGTTTCGCGTCGCAGCCGCAGGC-3′), p84FR(401-500) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCGACCCTTCTTTTCGGACGCGCC-3′ and 5′-TTAGCCCTACTTATCGTCGTCATCCTTGTAATCAAAATAGAGGCGCCCCAGGAGCGCGTC-3′), p84FR(501-586) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCCAAGCATGATCTCGTCAAAGCATACGCTGAATCG-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcGAGATCGCCGCAGACCATGGCTAAAGT-3′), and p84OD(151-300) (5′-CGGTGCCACCATGCCTCCAAAAAAGAAGAGAAAGGTCCcgaattcacaccaagcATGGAAGAAGAAACGGCG-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcCGTGGGCGTCAGATCCGCGTCCGTTGA-3′).
The IE2 cDNA expression vector, pON2206, was a gift from Edward Mocarski (Stanford University).
All IE2 subclones were made by PCR amplification of IE2 cDNA with pON2206 as a template and ligating the resulting product into pTarget vector. PCR products were generated such that the HA and SV40 NLS sequences were fused in the same reading frame context as the IE2 subclone. The following primers were used for IE2 subclones: pIE2-HA(1-215) (5′-CggtgccaccatgCCTCCAAAAAAGAAGAGAAAGGTCatgGAGTCCTCTGCCAAGAGAAA-3′and 5′-ggcgtcttatcaagcgtagtctgggacgtcgtatgggtaGGTTTCGACTTCTTCACCCTG-3′), pIE2-HA(216-386) (5′-cggtgccaccatgCCTCCAAAAAAGAAGAGAAAGGTCACCCGCGGTGCTACCGCGTCTTC-3′ and 5′-ggcgtcttatcaagcgtagtctgggacgtcgtatgggtaGGGAATCGTGAAGGGCAGGTT-3′), and pIE2-HA(387-580) (5′-cggtgccaccatgCCTCCAAAAAAGAAGAGAAAGGTCCCCAGTATGCACCAGGTGTTAGA-3′ and 5′-ggcgtcttatcaagcgtagtctgggacgtcgtatgggtaCTGAGACTTGTTCCTCAGGTC-3′).
The construct pUL112/113pr, containing the UL112/113 promoter upstream of the luciferase gene, was generated by PCR amplification with AD169 DNA as a template by using the primers 5′-CCGGTACCAGATCTCCGCGTCACCTTTCATCGAGT-3′ and 5′-CCAAGCTTGGCCGTGGAGCGAGTGCCGCCGCAGCC-3′. The 750-bp PCR product was then cleaved with KpnI and HindIII and subcloned into the luciferase reporter vector pGL2-Basic (Promega).
pEGFP-N1 (Clontech) and pFLAG-control (Stratagene) expression constructs were utilized according to manufacturer's recommendations.
Site-directed mutagenesis.
UL84 leucine zipper mutants were generated by using QuickChange Multi-Site-Directed mutagenesis kit (Stratagene). Construct pUL84 L1+L2, containing valine residues in place of leucine residues at aa 114 and 121, was made by using the mutagenesis primers 5′-ACCTACCATCTGATCCAGTTGCACGTCGACCTCCGACCCGAAGA-3′ and 5′-GACCTCCGACCCGAAGAAGTGCGGGATCCCTTCCAGATTCTG-3′. Construct pUL84 L2+L3, containing valine residues in the place of leucine residues at aa 121 and 128, was generated by using the mutagenesis primers 5′-GACCTCCGACCCGAAGAAGTGCGGGATCCCTTCCAGATTCTG-3′ and 5′-CGGGATCCCTTCCAGATTCTGGTCTCTACGCCGCTGCAA-3′. Construct pUL84 L3+L4, containing valine residues in place of leucine residues at aa 128 and 135, was generated by using the mutagenesis primers 5′-CGGGATCCCTTCCAGATTCTGGTCTCTACGCCGCTGCAA-3′ and 5′-TCTACGCCGCTGCAAGTGGGGGAAGCGAACGACGAGTCT-3′.
UL84 leucine zipper mutant subclone p84-2LZA was made by PCR amplification of a UL84 fragment with pUL84 L1+L2 DNA as a template and ligating the resulting product into pTarget vector. The primers used for the construction of p84-2LZA are 5′-CCAAGCATGCGGGTGCACCGCGGCACCTACCATCT-3′ and 5′-TTAGCCCTActtatcgtcgtcatccttgtaatcGAAGGCCGGGCGCACGTCAGAATCCTC-3′.
All constructs were confirmed by DNA sequence analysis.
Luciferase assay.
U373 cells were cotransfected with a construct containing the UL112-113 promoter, pUL112/113pr, the IE2 expression construct, pON2206, and pUL84-TAG(1-586) or UL84 leucine zipper mutants. Cells were lysed 2 days posttransfection, and the luciferase activity was determined with a luminometer according to the manufacturer's instructions.
UL84 protein purification.
Vero cells were plated to 70 to 90% confluency in 10 100-mm dishes. Cells were infected with Ad84 at a multiplicity of infection of 5. Cells were incubated for 48 h, at which time the cells were washed twice with phosphate-buffered saline (PBS; pH 7.4) and sonicated in 1 ml of lysis buffer A (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, 2 mM MgCl, 0.2 mM EDTA, 10 μl of protease inhibitor cocktail σ[/ml]) with shaking for 30 min at room temperature. Cells were removed from the plate by scraping and passed through a 22-gauge needle three times to shear DNA. Cellular debris was removed by centrifugation at 1,500 × g for 10 min. The resulting lysate was passed through a low-protein-binding 2-μm-pore-size filter (Millipore) before incubation with FLAG affinity beads (Sigma). Protein purification proceeded according to the manufacturer's recommendations for column purification. Protein was eluted with 100 μg of 3X FLAG peptide (Sigma)/ml. Fractions were analyzed by separation on a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and visualized by using Coomassie blue or reacted with antibody in a Western blot. Fractions shown to contain the most protein were concentrated by using an Amicon Ultra-4 30,000 MWCO (Millipore) and analyzed by Western blot before use.
Generation of a UL84 monoclonal antibody.
A UL84 monoclonal antibody (MAb84) was produced by using purified recombinant UL84 protein by the method described in reference 3. All work was performed at the University of Nevada Monoclonal Antibody Core Facility. Antibodies were screened by using purified UL84 recombinant protein as an antigen in an enzyme-linked immunosorbent assay. Several positive hybridoma clones were isolated, the clone used to produce purified antibody was chosen based on relative affinity to antigen and cell line viability. This clone was used to produce large quantities of secreted antibody that was purified by using a protein A column. After the antibody was isolated, the isotype of the antibody was determined to be immunoglobulin G2a.
Antibodies.
The IE2-specific antibody G13-12E2 (Vancouver Biotech), anti-FLAG M2 (Sigma), and anti-HA (Sigma) antibodies were used for coimmunoprecipitation assays.
Coimmunoprecipitation assay.
Cos7 cells were plated to 70 to 90% confluency in 100-mm dishes. Cells were transfected with 10 μg of the appropriate plasmids by using Transfectin reagent (Bio-Rad) per manufacturer's recommendations. At 48 h posttransfection cells were washed twice with PBS (pH 7.4) and lysed by using 1 ml of lysis buffer A by shaking for 30 min at room temperature. Cells were scraped from the plate and passed through a 22-gauge needle three times. Cellular debris was removed by centrifugation at 1,500 × g for 10 min. Lysates containing expressed proteins were mixed together (capture assay) for 1 h at 4°C before incubation with specific antibodies. Coimmunoprecipitations were carried out by using a conjugated or nonconjugated immunoprecipitation system.
(i) Antibody-conjugated coimmunoprecipitation.
Anti-FLAG M2 Affinity Gel Freezer-Safe beads σ were prepared according to the manufacturer's recommendations. Portions (50 μl) of the prepared beads were added for each coimmunoprecipitation and incubated at 4°C overnight. The complexes were washed according to the manufacturer's recommendations, and proteins were eluted by using 100 μg of 3X FLAG peptide/ml (and analyzed by Western blotting).
(ii) Antibody-nonconjugated coimmunoprecipitation.
Lysates were incubated with the appropriate monoclonal antibody for 1 h at 4°C, at which time 40 μl of protein G plus agarose beads were added. The coimmunoprecipitation was again incubated at 4°C overnight. The complexes were washed with ice-cold PBS (pH 7.4) four times. The protein complexes were removed from the beads by the addition of 2X Laemmli sample buffer (Bio-Rad) containing 2-mercaptoethanol and then heated to 95°C for 5 min. Samples were separated by SDS-PAGE and analyzed by Western blot.
For experiments involving full-length UL84 or IE2 expression, constructs were cotransfected into Cos7 cells, followed by incubation of cell lysates by either the conjugated or nonconjugated coimmunoprecipitation method.
Transdominant-negative inhibition of oriLyt-dependent DNA replication.
For transdominant-negative inhibition, telomerized HFs were cotransfected with 1 μg of each plasmid encoding each of the human herpesvirus 8 (HHV-8) core replication proteins under the control of the SV40 promoter (1) and 1 to 2 μg of plasmids expressing various UL84 or IE2 peptides and UL84 and IE2 expression constructs, pUL84-TAG(1-586) and pON2206, and 10 μg of the pGEM-oriLyt, which contained the HCMV cloned oriLyt (23). Total cellular DNA was harvested 5 days posttransfection, and replication products were analyzed as described previously (23, 32). Control transfections contained the same mixture as described above except that instead of including plasmids that expressed UL84 or IE2 peptides, we substituted the parent expression plasmids for these constructs.
For the analysis of UL84 leucine zipper site-directed mutants, cotransfections contained plasmids pUL84 L1+L2, pUL84 L2+L3, or pUL84 L3+L4 substituted for UL84 native expression constructs, and the replication assay was performed as described previously (32).
In vitro cross-linking.
UL84 protein fragments were expressed by using TNT Quick-Coupled Transcription/Translation Kit (Promega). First, 10 μl of translated protein was incubated in a final concentration of 0.008% glutaraldehyde in 0.1 M NaCl for 30 min at room temperature. Then protein was separated through a SDS-12% PAGE gel, fixed, treated with Amplify (Amersham) for 30 min, and exposed to X-ray film for 15 h.
RESULTS
Expression and purification of UL84.
As a first step toward the characterization of UL84 and its interaction with other viral proteins, we developed a system to obtain relatively pure protein from mammalian cells. We chose to isolate UL84 from mammalian cells, as opposed to purification from a bacterial system, because it is not known if the lack of posttranslational modifications would effect protein-protein interactions. We generated a recombinant adenovirus expression construct such that the UL84 ORF was fused in frame with the FLAG epitope. This FLAG tag allowed for affinity purification of UL84 and also provided a mechanism to track the protein during the purification procedure.
High-titer UL84 recombinant adenovirus was produced by using 293 packaging cells. Virus was harvested and Vero cells were infected at a multiplicity of infection of ∼5. Infection was allowed to proceed for 48 h, at which time cells were harvested, and protein lysates were prepared. FLAG-tagged UL84 was purified by incubation of protein lysates with FLAG affinity beads, followed by a wash procedure. UL84 was then eluted by competition with 3X FLAG peptide. Initial protein lysates and column flowthrough samples are shown in Fig. 1 (Fig. 1, lanes 1 and 2, respectively). The column was washed until the flowthrough was cleared of almost all protein (Fig. 1, lane 3). UL84-FLAG protein was then eluted in five fractions (Fig. 1, lanes 4 to 8). The UL84-FLAG protein, which migrates at ca. 75 kDa, was collected and analyzed by Western blotting with the anti-FLAG antibody (data not shown). This procedure indicated that we could isolate UL84 recombinant protein that could be used in further studies.
FIG. 1.

Purification of recombinant UL84 from mammalian cells. Vero cells were infected with a recombinant adenovirus expressing FLAG-tagged UL84. Total protein cell lysates were loaded onto a column containing FLAG affinity beads, and recombinant protein was eluted by using 3X FLAG peptide. Lanes: 1, protein lysate from Ad84-infected Vero cells; 2, column flowthrough; 3, wash; 4, first protein elution fraction with 3X FLAG peptide; 5, second protein elution fraction with 3X FLAG peptide; 6, third protein elution fraction with 3X FLAG peptide; 7, fourth protein elution fraction with 3X FLAG peptide; 8, fifth protein elution fraction with 3X FLAG peptide.
Generation of a monoclonal antibody to UL84 purified protein.
In order to study UL84 and its interaction with itself and other virus-encoded proteins, we required a highly specific antibody. After purifying UL84 from Vero cells infected with an adenovirus expressing FLAG-tagged UL84 protein, we used this purified protein as antigen to produce a monoclonal antibody (see Materials and Methods). As an initial evaluation of this antibody, MAb84, we reacted it with various UL84 expression constructs and HCMV-infected cell protein [Fig. 2A, lanes 1-586(FL), 84-EGFP, and AD169]. MAb84 failed to interact with the FLAG control plasmid (Fig. 2A, lane TAG-Control). In order to determine the approximate location of the point of interaction of MAb84 with the UL84 peptide, we reacted MAb84 with protein lysates prepared from cells transfected with various UL84 truncation mutants (Fig. 2B). Western blot analysis showed that MAb84 was able to interact with four of the six UL84 truncation mutants (Fig. 2B and C). MAb84 failed to recognize mutants 4 (aa 201 to 586) and 5 (aa 351 to 586), indicating that the antibody interacted with an epitope present within the protein between mutant 3 (aa 141 to 586) and mutant 4 (aa 201 to 586) (Fig. 2B and C). All deletion constructs were shown to produce protein, as judged by Western blot analysis with anti-FLAG antibody (data not shown). To further characterize the MAb84 epitope, we expressed 100-aa FLAG-tagged segments of the UL84 protein and incubated cell extracts with MAb84. MAb84 recognized a UL84 fragment encoding aa 101 to 200, indicating that the epitope for this antibody was within this fragment (Fig. 2D). All fragments were shown to express protein when reacted with anti-FLAG antibody (Fig. 2D, anti-FLAG panel). Based on the results from the deletion mutant and the interaction with the expressed small peptides, MAb84 interacts with a protein domain mapping between aa 141 and 200.
FIG. 2.

Generation of a monoclonal antibody to UL84 (MAb84). Purified UL84 recombinant protein isolated from Cos7 cells was used to produce a monoclonal antibody. (A) Western blot of cell protein lysates from either the transfection of various UL84 expression constructs or HCMV infection and reacted with MAb84. Lanes: 1-586(FL), cell lysate from Cos7 cells transfected with pUL84-TAG(1-586); 84-EGFP, cell lysate from Cos7 cells transfected with a plasmid expressing a UL84-EGFP fusion protein; TAG-Control, cell lysate from Cos7 cells transfected with a pFLAG-control expression plasmid; AD169, cell lysate from human foreskin fibroblasts infected with HCMV (AD169) harvested 72 h postinfection. (B) UL84 deletion constructs used to map the epitope for MAb84. (C) Western blot of MAb84 reacted with a series of UL84 deletion mutants. Lanes: 1, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(83-586); 2, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(104-586); 3, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(141-586); 4, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(201-586); 5, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(351-586); 6, cell lysate from Cos7 cells transfected with UL84 deletion mutant pUL84-TAG(1-513). (D) MAb84 interacts with a region of the UL84 protein mapping to aa 101 to 200. Cos7 cells were transfected with expression plasmids expressing ∼100-aa segments of the UL84 protein. Each segment was constructed such that it was fused in frame with the FLAG epitope. Cell lysates were prepared and a Western blot was performed by using MAb84. Lanes correspond to each expression plasmid. Panel anti-UL84 was reacted with MAb84 antibody. Panel anti-FLAG was reacted with anti-FLAG antibody.
UL84 interacts with itself in vivo.
Because UL84 contains a leucine zipper motif and these structures are implicated in protein-protein interactions, we wanted to investigate the possibility that UL84 forms a stable interaction with itself. We cotransfected COS7 cells with plasmid constructs that expressed two differentially tagged (enhanced green fluorescent protein [EGFP] and FLAG) UL84 recombinant proteins. Cells lysates were prepared from cotransfected cells, and proteins were immunoprecipitated with either anti-FLAG or anti-EGFP antibodies. As controls, we cotransfected a plasmid encoding either EGFP (pEGFP-N1) or FLAG control protein (pFLAG-Control), along with pUL84-TAG(1-586) or pUL84-EGFP. Immunoprecipitated proteins were separated by using SDS-PAGE, followed by analysis by Western blots to detect each UL84 protein species. Cell lysates that were immunoprecipitated with the anti-FLAG antibody were reacted with anti-EGFP. Western blot analysis showed that a band was observed in samples reacted with anti-EGFP, indicating that the anti-FLAG antibody pulled down the UL84-EGFP fusion protein (Fig. 3A, lane 1). However, no band was detected when the EGFP control plasmid was used in the cotransfection (Fig. 3A, lane 2). Likewise, when samples were immunoprecipitated with anti-EGFP antibody and reacted with anti-FLAG, a band was detected only in the UL84 cotransfected samples and not when one of the transfected plasmids was a control FLAG expression construct (Fig. 3A, lane 3 and 4, respectively).
FIG. 3.
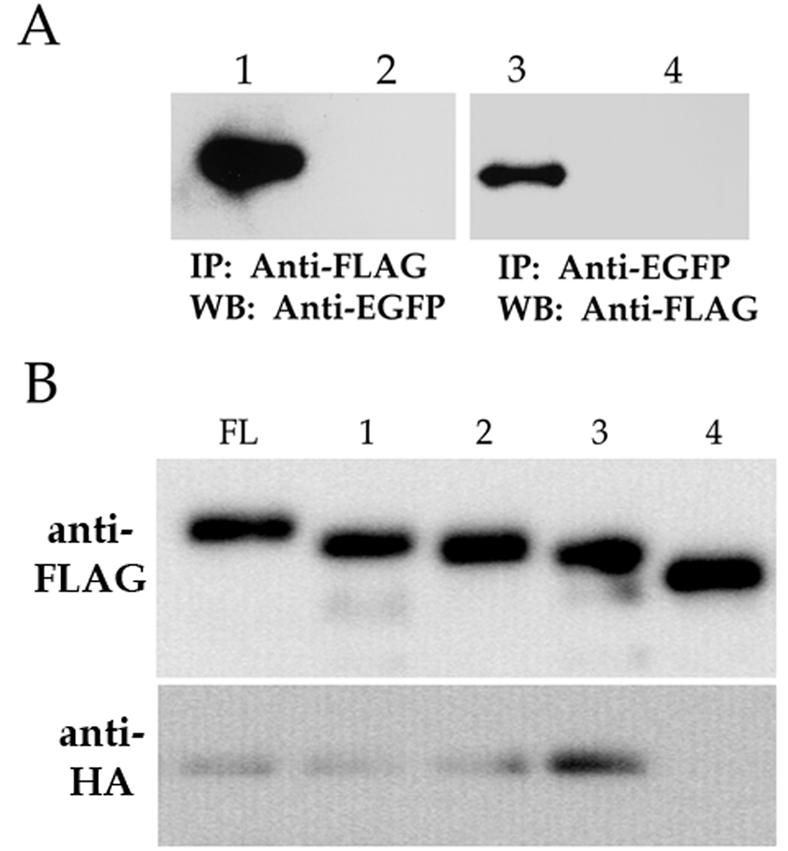
UL84 interacts with itself in vivo. (A) Western blot of coimmunoprecipitated cell lysates transfected with pUL84-EGFP and pUL84-TAG(1-586). Lanes: 1, pUL84-TAG(1-586) and pUL84-EGFP cotransfected cell lysates immunoprecipitated with anti-FLAG and reacted with anti-EGFP; 2, pUL84-FLAG and pEGFP-N1 cotransfected cell lysates immunoprecipitated with anti-FLAG and reacted with anti-EGFP; 3, pUL84-TAG(1-586) and pUL84-EGFP cotransfected cell lysates immunoprecipitated with anti-EGFP and reacted with anti-FLAG; 4, FLAG-control plasmid and pUL84-EGFP cotransfected cell lysates immunoprecipitated with anti-EGFP and reacted with anti-FLAG. (B) Western blot of coimmunoprecipitates from cell lysates containing UL84-FLAG deletion proteins and full-length UL84-HA-tagged protein. Lanes: FL, mixed lysates from cells transfected with pUL84-TAG(1-586) and pUL84-HA; 1, mixed lysates from cells transfected with pUL84-TAG(83-586) and pUL84-HA; 2, mixed lysates from cells transfected with pUL84-TAG(104-586) and pUL84-HA; 3, mixed lysates from cells transfected with pUL84-TAG(141-586) and pUL84-HA; 4, mixed lysates from cells transfected with pUL84-TAG(201-586) and pUL84-HA. The top panel shows a Western blot of coimmunoprecipitated protein reacted with anti-FLAG antibody. The bottom panel shows a Western blot of coimmunoprecipitated protein reacted with anti-HA antibody.
In order to determine which region of the UL84 protein is involved in oligomerization, we transfected Cos7 cells with a series of UL84 deletion constructs (deletions 1 to 4 shown in Fig. 2B). Deletion constructs were transfected into Cos7 cells, and expressed proteins were captured by using FLAG affinity beads. Cell lysates containing full-length UL84-HA protein were then added to the deletion mutants previously captured on the beads. Beads were washed, and proteins were eluted by using 3X FLAG peptide. Full-length UL84 was able to interact with deletion mutants having removed up to aa 141. Mutant 4, which initiates at aa 201, was unable to interact with full-length UL84 (Fig. 3B, panel anti-HA, lane 4). Therefore, the protein domain involved in oligomerization is located in a region between aa 141 and 201. This region is downstream of the putative leucine zipper motif (aa 114 to 135), indicating that the leucine zipper is not involved in a UL84-UL84 interaction.
The UL84 oligomerization domain (OD) lies between aa 151 and 200.
In order to further define the protein domain involved in oligomerization, we initially expressed three FLAG-tagged 100-aa polypeptides [p84FR(1-100), p84FR(101-200), and p84FR(201-300) such that the first 300 aa of the UL84 protein were represented (Fig. 4A) ]. Three transfections were performed; each delivered one of the three plasmids encoding a 100-aa UL84 peptide. Additional transfections delivered full-length UL84 (untagged, pUL84EXPRESS). FLAG-tagged protein was captured on FLAG affinity beads, followed by the addition of cell lysates containing full-length UL84 protein. After incubation, the beads were washed, and samples were eluted with 3X FLAG peptide. Western blots of transfected cell lysates show that full-length UL84 was efficiently expressed in all transfections (Fig. 4B, top panel, UL84 full-length). The middle panel demonstrates that each of the small peptides were efficiently expressed and immunoprecipitated by the FLAG affinity beads in the capture assay (Fig. 4B, middle panel lanes 1, 2, and 3). Only peptides produced from the transfection of p84FR(101-200) were able to bind full-length UL84 in the capture assay (Fig. 4B, bottom panel, lane 2). These data indicated that UL84 interacts with itself by using a protein domain that is located in a region of the protein between aa 101 and 200 (consistent with deletion mapping results), and this expressed polypeptide is sufficient to mediate this interaction.
FIG. 4.

The UL84 oligomerization domain maps to aa 151 to 200 and does not involve the leucine zipper motif. (A) Schematic of UL84 ORF and FLAG-tagged peptides used to define the protein domain involved in UL84 oligomerization and the relative positions of the leucine zipper motif (LZ) and charged region (CR). Fragments labeled 1, 2, 3, and 4 correspond to constructs p84FR(1-100), p84FR(101-200), p84FR(201-300), and p84OD(151-300), respectively. (B) UL84 oligomerization involves a region of the protein mapping between aa 101 and 200. Cell lysates transfected with plasmids expressing small peptides of UL84 were incubated with cell lysates prepared from cells transfected with full-length UL84 (pUL84EXPRESS). The top panel shows a Western blot of cell lysates prepared from cells transfected with pUL84EXPRESS and reacted with MAb84. Lanes in the middle panel show the following: lane 1, mixed cell lysates from cells transfected with pUL84EXPRESS and p84FR(1-100), immunoprecipitated, with anti-FLAG, and reacted with anti-FLAG; lane 2, mixed cell lysates from cells transfected with pUL84EXPRESS and p84FR(101-200), immunoprecipitated with anti-FLAG, and reacted with anti-FLAG; lane 3, mixed cell lysates from cells transfected with pUL84EXPRESS and p84FR(201-300), immunoprecipitated with anti-FLAG, and reacted with anti-FLAG. Lanes in the bottom panel show: lane 1, mixed cell lysates from cell transfected with pUL84EXPRESS and p84FR(1-100), immunoprecipitated with anti-FLAG, and reacted with MAb84; lane 2, mixed cell lysates from cell transfected with pUL84EXPRESS and p84FR(101-200), immunoprecipitated with anti-FLAG, and reacted with MAb84; lane 3, mixed cell lysates from cell transfected with pUL84EXPRESS and p84FR(201-300), immunoprecipitated with anti-FLAG, and reacted with MAb84. (C) The UL84 OD is located between aa 151 and 200. A Western blot of coimmunoprecipitated protein from mixed cell lysates from cells transfected with pUL84-HA and p84OD(151-300) (polypeptide 4) expression plasmids is shown. (Top panel) Western blot reacted with anti-HA antibody. Lanes: 1, lysate from cells transfected with a UL84-HA expression plasmid; 2, immunoprecipitated protein from mixed cell lysates with anti-FLAG antibody and reacted with anti-HA antibody on a Western blot. (Bottom panel) Western blot reacted with anti-FLAG antibody. Lanes: 1, protein lysate from cells transfected with a p84OD(151-300) (polypeptide 4); 2, immunoprecipitated protein from mixed cell lysates with anti-FLAG antibody and reacted with anti-FLAG antibody. (D) UL84 forms a homodimer in vitro. UL84 expression plasmids were used in vitro translation reactions, and the resulting polypeptides were subjected to chemical cross-linking. Lanes: 1, in vitro-translated protein produced from p84OD(151-300); 2, in vitro-translated protein produced from p84OD(151-300) incubated with 0.008% glutaraldehyde; 3, in vitro-translated protein produced from pIE2-HA(387-580); 4, in vitro-translated protein produced from pIE2-HA(387-580) incubated with 0.008% glutaraldehyde.
To further define the OD of UL84, we performed the capture assay using a fragment that encoded aa 151 to 300, fragment 4 in Fig. 4A [construct p84OD(151-300)] plus pUL84-HA. Since it is now known that the OD lies within fragment 2 (aa 101 to 200), if the polypeptide produced from fragment 4 captured full-length UL84, then this would indicate that the OD is located between aa 151 and 200. Figure 4C is a Western blot that shows the result of the capture assay with cell lysates from transfection of pUL84-HA and p84OD(151-300) (Fig. 4A, fragment 4). The polypeptide produced from fragment 4 [p84OD(151-300)] interacted with full-length UL84-HA as shown by reacting coimmunoprecipitated proteins with an anti-HA antibody on a Western blot (Fig. 4C, top panel, lane 2). Polypeptide 4 was produced in cell lysates and was efficiently captured by the FLAG beads, as shown by Western blotting with FLAG antibody (Fig. 4C, bottom panel, lanes 1 and 2, respectively). These results localized the UL84 OD between aa 151 and 200.
To confirm that UL84 forms a stable self-interaction, we performed an in vitro cross-linking experiment with fragment number 4. In vitro-translated protein was treated with glutaraldehyde, and protein species were separated on an SDS-PAGE gel. Chemical cross-linking resulted in the shift from a monomer protein species (∼40 kDa) to a band of ∼80 kDa, suggesting that two subunits of the expressed peptide can exist as a dimer complex (Fig. 4D, lanes 1 and 2, respectively). A control sample with a plasmid expressing amino acid region from aa 387 to 580 of IE2, which contains the oligomerization domain for this protein (6), also was used in a cross-linking experiment. Treatment of this expressed polypeptide gave the expected conversion from monomer to dimer as previously described (Fig. 4D, compare lanes 3 and 4) (6). No other expressed UL84 peptides were shown by these methods to cross-link (data not shown). These results strongly suggest that UL84 full-length protein can exist as a homodimer.
The UL84 putative leucine zipper motif is essential for complementation of oriLyt-dependent DNA replication but does not contribute to oligomerization.
The leucine residues that comprise the putative leucine zipper domain for UL84 are located at aa 114, 121,128, and 135, respectively (Fig. 5A). To confirm that the leucine heptad repeats did not contribute to oligomerization in the context of the full-length UL84 protein, we introduced base pair changes, by site-directed mutagenesis, such that the leucine residues were changed to valine residues. Three constructs were generated, each of which contained mutations in two of the four leucine residues. These constructs were used to determine whether mutations of the leucine residues would affect the ability of UL84 to oligomerize. Coimmunoprecipitation assays were performed with UL84-HA-tagged wild-type protein cotransfected with each of the FLAG-tagged leucine zipper mutants. Figure 5B shows that full-length UL84-HA (Fig. 5B, top panel) was still able to bind to all UL84 leucine zipper mutants L2+L3, L1+L2, and L3+L4 (Fig. 5B, bottom panel). This result showed that the mutation of the putative leucine zipper motif in the context of the full-length UL84 protein did not affect oligomerization. This result also confirms that the leucine zipper is not involved in an UL84-UL84 interaction.
FIG. 5.

The leucine zipper motif of UL84 is not involved in oligomerization but is required for oriLyt-dependent DNA replication. (A) Schematic of the UL84 leucine zipper motif, indicating leucine residues 1 (aa 114), 2 (aa 121), 3 (aa 128), and 4 (aa 135) that comprise the putative leucine zipper. (B) Coimmunoprecipitation of UL84-HA and UL84 leucine zipper mutants. Mutation of the UL84 leucine zipper motif does not interfere with UL84 oligomerization. UL84 FLAG-tagged leucine zipper mutants were cotransfected with full-length UL84-HA-tagged protein, and protein lysates were immunoprecipitated with anti-FLAG antibody. Coimmunoprecipitated protein was analyzed by Western blotting with anti-HA (top panel) and anti-FLAG (bottom panel) antibodies. (Top panel) All lanes contain coimmunoprecipitated UL84-HA protein. (Bottom panel) Lanes: L2+L3, UL84 leucine zipper mutant in which leucine residues 2 and 3 were changed to valine; L1+L2, UL84 leucine zipper mutant in which leucine residues 1 and 2 were changed to valine; L3+L4, UL84 leucine zipper mutant in which leucine residues 3 and 4 were changed to valine. (C) An intact UL84 leucine zipper is required for complementation of oriLyt-dependent DNA replication. A cotransfection replication assay was performed by using UL84 leucine zipper mutants in place of wild-type UL84. T-HFs were cotransfected with the core replication proteins plus IE2 and oriLyt. Total cell DNA was harvested and cleaved with EcoRI and DpnI, and replicated oriLyt DNA was analyzed by Southern blotting. Shown is the replicated oriLyt band. Lanes: Control, DNA from cells cotransfected with the core replication proteins plus IE2, oriLyt, and pUL84-TAG(1-586); L2+L3, same as the control except the UL84 leucine zipper mutant, UL84 L2+L3, was substituted for pUL84-TAG(1-586); L1+L2, same as the control except the UL84 leucine zipper mutant, UL84 L1+L2, was substituted for pUL84-TAG(1-586); L3+L4, same as the control except the UL84 leucine zipper mutant, UL84 L3+L4, was substituted for pUL84-TAG(1-586).
We next chose to test the ability of leucine zipper mutants to complement oriLyt-dependent DNA replication in order to determine whether the leucine zipper was involved in the replication function of UL84. The replication assay was performed with the UL84 leucine zipper mutants in place of wild-type UL84. The core replication proteins for HHV-8 under the control of the SV40 promoter (1), along with HCMV oriLyt and pON2206, were used in the cotransfection replication assay. The HHV-8 core replication proteins, along with IE2 and UL84, efficiently amplified cloned oriLyt in T-HFs (Fig. 5C, lane control). The leucine zipper mutants L2+L3 and L1+L2 were unable to complement DNA replication, whereas the transfection of pUL84 L3+L4 resulted in a weak replication signal (Fig. 5C). Therefore, although UL84 oligomerization was unaffected by the changes in leucine residues, a finding consistent with the mapping of the OD between aa 151 and 200, UL84 leucine zipper mutants were no longer able to complement origin-dependent DNA replication. This suggests that the leucine zipper structure is important for the replication function of UL84. However, the failure of UL84 leucine zipper mutants to complement DNA replication is not due to the interruption of a UL84-UL84 interaction.
UL84 leucine zipper mutants no longer repress IE2-mediated transactivation.
It was reported previously that IE2 can transactivate the UL112/113 promoter and that UL84 expression can abolish this effect in U373 cells (13, 28, 29). The ability of UL84 to dampen the transactivation effects of IE2 was correlated to the first 104 aa and the last 73 aa of the UL84 protein (13). However, since we have now implicated the leucine zipper of UL84 to contribute to DNA replication, we wanted to investigate whether the loss of a replication function for UL84 leucine zipper mutants involved the ability of UL84 to interact with IE2. Consequently, we evaluated the ability of UL84 leucine zipper mutants to downregulate IE2 transactivation of the UL112/113 promoter. U373 cells were cotransfected with a luciferase reporter construct containing the UL112/113 promoter, along with IE2 and either wild-type UL84 or the UL84 leucine zipper mutant expression constructs. UL84 leucine zipper mutants were markedly decreased in their ability to repress the transactivation effect of IE2 on the UL112/113 promoter, whereas wild-type UL84 was fully capable of almost completely abolishing IE2 activation of the promoter (Fig. 6). Interestingly, UL84 leucine zipper mutant L3 +L4, which weakly complemented oriLyt-dependent DNA replication, had an intermediate effect on IE2 transactivation (Fig. 6). These results strongly suggest that the leucine zipper of the UL84 protein is involved in regulating the activity of IE2.
FIG. 6.

UL84 leucine zipper mutants no longer repress IE2-mediated transactivation. Graph of luciferase reporter assay showing the effects various UL84 leucine zipper mutants on the IE2-mediated transactivation of the UL112/113 promoter. Vero cells were cotransfected with the luciferase reporter construct p112/113pr, pON2206, and either pUL84-TAG(91-586) or the mutated leucine zipper constructs pUL84 L1+L2, pUL84 L2+L3, or pUL83 L3+L4. Error bars indicate the standard deviations of three separate experiments.
The leucine zipper motif of UL84 is involved in IE2-UL84 heterodimerization.
Since it was demonstrated that UL84 leucine zipper mutants had a diminished effect on the repression of transactivation of the UL112/113 promoter by IE2, we next investigated whether an intact UL84 leucine zipper was necessary to interact with IE2. We cotransfected Cos7 cells with both an IE2 expression plasmid and the UL84 leucine zipper mutant L1+L2. Protein lysates were prepared and immunoprecipitated with anti-FLAG antibody. Immunoprecipitated protein was separated on an SDS-PAGE gel, and Western blotting was performed to determine whether the UL84 leucine zipper mutants retained their ability to interact with IE2. As demonstrated previously (13), control coimmunoprecipitations from cells cotransfected with wild-type IE2 and pUL84-TAG(1-586) with the FLAG antibody resulted in the detection of both proteins on Western blots reacted with antibodies specific for FLAG and IE2 (Fig. 7, lane 1). However, no IE2 protein was detected when lysates from cells cotransfected with plasmids expressing the UL84 leucine zipper mutant and wild-type IE2 were immunoprecipitated (Fig. 7, lane 2). Both the UL84 leucine zipper mutant and the IE2 protein were shown to be present at approximately the same levels, as determined by Western blot analysis of protein lysates (Fig. 7, lane 3). These data indicated that the leucine zipper motif of UL84 is implicated in the interaction with IE2. This observation, coupled with the data that leucine zipper mutants of UL84 fail to complement oriLyt-dependent DNA replication, suggests that an IE2-UL84 interaction is necessary for DNA replication.
FIG. 7.
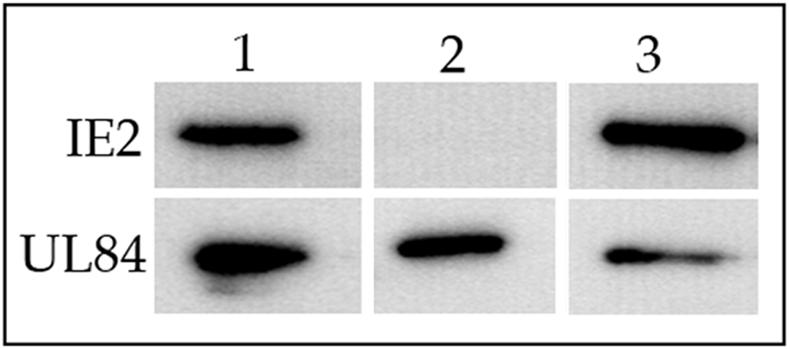
An intact UL84 leucine zipper motif is required for interaction with IE2. Cos7 cells were cotransfected with the IE2 expression construct pON2206 and UL84 leucine zipper mutant L1+L2. Cell lysates were immunoprecipitated with anti-FLAG antibody and coimmunoprecipitates were analyzed by Western blot with C13-12E2 and anti-FLAG antibodies. Lanes: 1, coimmunoprecipitation of wild-type IE2 (top panel) and UL84-TAG(1-586) protein (bottom panel); 2, immunoprecipitation of wild-type IE2 (top panel) and UL84 L1+L2 protein (bottom panel); 3, control cell lysates containing IE2 (top panel) and UL84 L1+L2 protein (bottom panel) reacted with C13-12E2 and anti-FLAG, respectively.
Identification of IE2 protein domain involved in IE2-UL84 heterodimerization.
Previous data showing the interaction between IE2 and UL84 in infected cells, coupled with the known effects of UL84 on IE2 transactivation, led us to investigate which region of the IE2 protein was involved in UL84 heterodimerization. We generated IE2 expression constructs that encoded three regions of the IE2 protein such that the resultant polypeptides had an in-frame HA epitope (Fig. 8A). These three IE2-HA expression constructs were transfected into Cos7 cells, and cell lysates were prepared. The expression of each IE2-HA fragment was confirmed by Western blot analysis performed on the cell lysates (Fig. 8B, top panel). IE2-HA cell lysates were incubated with UL84-FLAG immobilized on FLAG affinity beads. Bound UL84-FLAG protein was eluted with 3X FLAG peptide, and samples were subjected to Western blotting to identify which IE2-HA fragment interacted with UL84. Figure 8B, middle panel, shows that in all cases UL84 was expressed and efficiently eluted from the FLAG affinity beads. Western blots with anti-HA revealed that the IE2 fragment encoding the first 215 aa of the protein, pIE2-HA(1-215), interacted with full-length UL84 (Fig. 8B, bottom panel, lane 1). As a control for our capture system, we also pulled down full-length IE2 by using FLAG affinity bead-immobilized UL84. As expected, we efficiently captured full-length IE2 protein with immobilized UL84 (Fig. 8C). These data indicated that the region of IE2 involved in UL84-IE2 heterodimerization was located in the first 215 aa of the protein.
FIG. 8.

UL84 interacts with a protein domain within the first 215 aa of IE2. DNA fragments of IE2 cDNA were subcloned into an expression vector such that the region of the IE2 protein would be expressed in frame with the HA epitope and the SV40 NLS. These constructs were transfected into Cos7 cells and lysates were prepared and mixed with pUL84-TAG(1-586) protein cell lysates. The mixed cell lysates were immunoprecipitated with anti-FLAG. Coimmunoprecipitates were analyzed by Western blot with anti-HA antibody. (A) Schematic of IE2 expression constructs used for coimmunoprecipitations. (B) Coimmunoprecipitation of UL84 and IE2 peptides. (Top panel) Western blot of cell lysates transfected with IE2 expression plasmids with anti-HA. Lanes: 1, protein lysates from cells transfected with pIE2-HA(1-215); 2, protein lysates from cells transfected with plasmid pIE2-HA(216-386); 3, protein lysates from cells transfected with plasmid pIE2-HA(387-580). (Middle panel) Lanes 1 to 3 are protein lysates from cells transfected with pUL84-TAG(1-586) with anti-FLAG. (Bottom panel) Coimmunoprecipitation of IE2 peptide 1-215 and full-length UL84. Western blot analysis of coimmunoprecipitates from mixed cell lysates containing expressed proteins. Mixed lysates were immunoprecipitated with anti-FLAG antibody and reacted with anti-HA antibody. Lanes: 1, coimmunoprecipitation of mixed lysates from cells expressing pIE2-HA(1-215) and pUL84-TAG(1-586); 2, coimmunoprecipitation of mixed lysates from cells expressing pIE2-HA(216-386) and pUL84-TAG(1-586); 3, coimmunoprecipitation of mixed lysates from cells expressing pIE2-HA(387-580) and pUL84-TAG(1-586). (C) Mixed lysate coimmunoprecipitation of UL84 and IE2. Protein lysates containing pON2206 and pUL84-TAG(1-586) were mixed together and immunoprecipitated with anti-FLAG. Coimmunoprecipitated protein analyzed by Western blot by using either G13-12E2 (right panel) or anti-FLAG (left panel).
Expression of UL84 OD or LZ domain has a transdominant-negative effect on origin-dependent DNA replication.
Three possible protein-protein interactions can occur with respect to UL84 and IE2: (i) the previously described IE2-IE2 interaction (6), (ii) a UL84-UL84 interaction, or (iii) a UL84-IE2 association. Despite these possible interactions, it is not clear which of these possible scenarios contributes to DNA replication. It was previously shown that overexpression of UL84 in U373 cells can inhibit HCMV growth (13). However, the exact nature of this repression was not apparent in that report. We chose to investigate whether the expression of the UL84 oligomerization domain could act as a transdominant inhibitor of origin-dependent DNA replication. This would indicate whether a UL84-UL84 interaction participates in DNA replication. In addition, since we now have shown that a protein domain within the first 215 aa of the IE2 protein interacts with UL84 and that an intact leucine zipper motif of UL84 is required for an interaction with IE2, we also expressed polypeptides containing these domains in the transient replication assay as potential transdominant inhibitors.
We cotransfected T-HFs with the HHV-8 core replication proteins along with oriLyt and pUL84-TAG(1-586) and the IE2 expression plasmid pON2206. We chose to use HHV-8 replication proteins so that we could concentrate only on the UL84-IE2 interaction. Each cotransfection also included one of the expression constructs encoding 100-aa segments of the UL84 ORF fused in frame with the SV40 NLS. Total cellular DNA was harvested and cleaved with EcoRI and DpnI. Replicated plasmid was detected by Southern blot analysis as previously described (24, 32). Addition of the expression vector p84FR(101-200) resulted in the absence of a replication signal (Fig. 9A), lane p84FR(101-200). Replication of oriLyt in the presence of p84FR(1-100), p84FR(201-300), p84FR(301-400), p84FR(401-500), or p84FR(501-586) had no effect on oriLyt-dependent DNA replication (Fig. 9A). This result suggested that a UL84-UL84 and/or a UL84-IE2 interaction was required for DNA synthesis since the inhibitory polypeptide contained both the UL84 OD and an intact leucine zipper motif. In order to determine whether one or both (UL84-UL84 or UL84-IE2) of the protein-protein interactions were required, we repeated the transient replication assay by using an expression construct encoding small peptides of the IE2 protein. No replication signal was detected when pIE2-HA(1-215) was added to the replication mixture (Fig. 9B, lane 2). Transfections that contained plasmids that expressed IE2 fragments spanning aa 216 to 386 [pIE2-HA(216-386)] or aa 387 to 580 [pIE2-HA(387-580)] had no effect on the amplification of oriLyt in the transient assay (Fig. 9B, lanes 3 and 4). This suggests that the oligomerization of IE2 was not a requirement for oriLyt DNA amplification, since the oligomerization domain of IE2 lies between aa 388 and 542 of the IE2 protein (6). This result also corroborates the data that UL84-IE2 interaction is essential for HCMV lytic replication, since the IE2 protein domain expressing the IE2-UL84 heterodimerization domain had a lethal transdominant-negative effect on origin-dependent DNA replication.
FIG. 9.

Transdominant-negative inhibition of oriLyt-dependent DNA replication. T-HFs were cotransfected with the HHV-8 core replication expression plasmids plus pON2206, pUL84-TAG(1-586), HCMV oriLyt, and various UL84 and IE2 plasmids expressing small peptides. DNA was harvested and cleaved with EcoRI and DpnI. Replication products were examined by Southern blot analysis. (A) Expression of UL84 aa 101 to 200 inhibits oriLyt-dependent DNA replication. Lanes: Control, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt; +p84FR(1-100), cotransfection of core replication proteins, pON2206, pUL84 TAG, and HCMV oriLyt plus construct p84FR(1-100); +p84FR(101-200), cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct p84FR(101-200); +p84FR(201-300), cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct p84FR(201-300); +p84FR(301-400), cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586) and HCMV oriLyt plus construct p84FR(301-400); +p84FR(401-500), cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586) and HCMV oriLyt plus construct p84FR(401-500); +p84FR(501-586), cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586) and HCMV oriLyt plus construct p84FR(501-586). (B) Expression of IE2 aa 1 to 215 inhibits oriLyt-dependent DNA replication. Lanes: 1, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt; 2, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct pIE2-HA(1-215); 3, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct pIE2-HA(216-386); 4, cotransfection of core replication proteins, pON2206, pUL84-TAG (1-586), and HCMV oriLyt plus construct pIE2-HA(387-580). (C) Expression of a peptide containing an intact oligomerization domain and a mutated leucine zipper motif (p84-2LZA) inhibits oriLyt-dependent DNA replication. Lanes: 1, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt; 2, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct p84-2LZA; 3, cotransfection of core replication proteins, pON2206, pUL84-TAG(1-586), and HCMV oriLyt plus construct p84OD(151-300).
In order to decipher if both UL84-UL84 and IE2-UL84 interactions contributed to DNA replication, we again repeated the cotransfection replication assay by using constructs that expressed an intact oligomerization domain and a disrupted leucine zipper motif. A construct expressing the same 101 to 200 aa was expressed, except that this new plasmid (p84-2LZA) has the same amino acid changes to the leucine residues within the leucine zipper as used previously(L1+L2), which no longer interacted with IE2. Transfection of this construct resulted in a loss of replication signal (Fig. 9C, lane 2). We next choose to investigate whether the expression of the UL84 oligomerization domain alone, that is, in the absence of the leucine zipper motif, could have a lethal effect on oriLyt-dependent DNA replication. p84OD(151-300) was cotransfected, along with all of the required replication plasmids, including wild-type UL84, pON2206, and the HCMV cloned oriLyt and a replication assay was performed. The addition of p84OD(151-300) to the transfection mixture was also shown to have a lethal effect on oriLyt-dependent DNA replication (Fig. 9C, lane 3). These results gave further evidence that the expression of a peptide containing the UL84 OD or the leucine zipper motif in transfected cells interfered with essential UL84-UL84 and UL84-IE2 interactions and consequently inhibited lytic DNA replication.
DISCUSSION
We have shown that the UL84 protein of HCMV can form a stable interaction with itself and this interaction does not involve the identified leucine zipper motif. The UL84 oligomerization domain is located between aa 151 and 200 of the protein. We believe that UL84 can exist in a dimer conformation based on in vitro studies that demonstrated that chemically cross-linked UL84 peptides migrated at approximately twice the observed molecular weight of uncross-linked protein; however, this conclusion does not eliminate the possibility of the existence of higher order structures such as tetramers.
UL84 was implicated as having a direct role in DNA replication as evidenced by reports demonstrating that it is the only noncore replication protein required for oriLyt-dependent DNA replication in Vero cells (27). UL84 is believed to perform an function analogous to that of the OBPs or initiator proteins found in other herpesvirus systems. Although little is known about the exact role of UL84 in DNA replication, other OBPs have been shown to form homodimers in vivo and in vitro.
In the case of the UL9 protein for herpes simplex virus type 1 (HSV-1) this protein binds to a specific region within HSV-1 oriLyt and can initiate DNA synthesis by unwinding the DNA strands (17-19). It has been shown that UL9 binds to HSV-1 OriS as a homodimer with a stoichiometry of one homodimer per binding site; this conformation is crucial for the initiation of lytic replication (9). In addition, UL9 contains a leucine zipper domain that is similar to the one identified in UL84 but is not implicated in DNA binding and is hypothesized to be involved in protein-protein interactions with other core replication proteins (31). Recently, it was shown that UL9 protein levels might serve to regulate and control DNA replication in HSV-1 (20). In addition, UL9 also tethers other replication proteins and probably facilitates the recruitment of the entire replication machinery to oriLyt (2, 32).
The initiator protein of Epstein-Barr virus lytic DNA replication, Zta, acts as the OBP, as well as a viral transactivator (10). There appear to be two distinct domains in the Zta protein, one responsible for transactivation and the other required for replication, although the exact role of the transactivation function with respect to DNA replication remains controversial (26). The homodimerization domain for Zta was shown, through mutational analysis, to support a coiled-coil structure model (4, 12). For UL84, although there is a leucine zipper motif within the protein, it is clear that this domain is not involved in the formation of a UL84-UL84 protein interaction. The UL84 oligomerization domain appears to encompass a highly charged amino acid region. This highly charged region is also capable of forming a coiled-coil structure, as judged by the COILS program (16). It is important to note, however, that this software program is biased against protein domains containing highly charged amino acids, and we have not confirmed the formation of this structure by biochemical methods. Notwithstanding this bias, our results strongly suggest that this structure is involved in a protein-protein interaction. Interestingly, the monoclonal antibody, MAb84, interacts with the same region as the identified UL84 oligomerization domain. One possible explanation for this is related to the fact that this region is predicted to have the most hydrophilic amino acid composition within the protein and is consequently probably one of the strongest antigenic regions of the protein. The apparent hydrophilic property of the OD is also consistent with this region acting as the point of interaction for protein binding.
With respect to the leucine zipper motif in UL84, this region appears to be involved in binding to the IE2 protein. Indeed, leucine zipper mutants of UL84 fail to complement DNA replication and have a diminished effect on IE2-mediated transactivation of the UL112/113 promoter. Also, transdominant-negative experiments indicated that the interactions of UL84 with itself, as well as the interaction of UL84 with IE2, are both required for efficient amplification of oriLyt in the transient replication assay. The requirement for an intact leucine zipper motif for the interaction with IE2 is in contrast to a previously published report that mapped the domain of interaction of UL84 with IE2 to a region of UL84 between aa 1 and 104, a region upstream of the leucine zipper domain (13). The differences in results could be due to the disruption of native protein folding, since truncated proteins were solely used to map protein-protein interactions (13), whereas our studies of protein interaction involved the introduction of site-directed amino acid changes to leucine residues comprising the leucine zipper, which may have minimal effects on protein folding. However, the results presented here provide clear evidence that the leucine zipper domain is involved in IE2 binding and that this binding is involved in DNA synthesis as demonstrated by expression of this domain having a transdominant-negative effect on oriLyt-dependent DNA replication. Whether or not this binding is facilitated through direct contact with IE2 or through an intermediary protein is unknown at this time; however, mutations in the leucine zipper motif of UL84 abolished any observed IE2 interaction as determined by coimmunoprecipitation assays. Conversely, mutation of the UL84 leucine zipper region had no effect on the ability of these peptides to interact with full-length UL84. This result serves to confirm the data from UL84 deletion mutants that the leucine zipper of UL84 is not involved in the formation of oligomers.
We also identified a 215-aa region of the IE2 protein that interacts with UL84. Although the heterodimerization domain lies within the first 215 aa of IE2, protein binding probably does not involve the first 93 aa of IE2, since this region is also present in IE1, which has not been shown to interact with UL84 (30). The IE2 domain that interacts with UL84 was also shown to act as a transdominant-negative inhibitor of oriLyt-dependent DNA replication. This result provides additional evidence that an interaction between UL84 and IE2 is essential for origin-dependent DNA replication. Interestingly, the addition of a construct expressing the oligomerization domain of IE2 had no effect on amplification of oriLyt, suggesting that the formation of an IE2-IE2 interaction was not required for oriLyt-dependent DNA replication. We, of course, acknowledge that this is only one possible explanation and that the expression of the oligomerization domain of IE2 may not be sufficient to act as a transdominant inhibitor of IE2 oligomerization in the replication assay due to incorrect protein folding or some other undefined reason. However, it could be that the IE2 homodimer conformation is necessary for transactivation of viral early promoters and that this conformation is not important in the context of DNA replication.
The use of transdominant inhibitors of DNA replication in the transient replication assay offers a unique method to understand the significance of protein-protein interactions in DNA synthesis. Since the replication assay involves the transfection of only minimal proteins participating in DNA replication, it is better suited for assaying the effects of transdominant inhibitions of DNA replication than a system that uses infectious virus. The use of HHV-8 core replication proteins was sufficient to amplify the HCMV oriLyt. This was not an entirely surprising result given that both the set of EBV and HSV-1 core proteins also replicated HCMV oriLyt (26, 27). This result confirms that the core replication proteins perform the enzymatic functions of DNA replication in an indiscriminate manner. We observed the same transdominant effects with the entire set of HCMV replication proteins.
We assume that transdominant-negative effects described here are due to the expression of the oligomerization and leucine zipper domains, and a result of the formation of inactive or defective protein complexes. However, inhibition of DNA synthesis could be due to the competition of small peptides for DNA-binding sites. Although this is a possibility, the use of several controls, including the expression of mutated forms of peptides and the expression of analogous oligomerization domains, make this interpretation less likely.
The interaction of IE2 with UL84 closely resembles the scenario observed for HHV-8 lytic DNA replication. The replication proteins K-bZIP and ORF50 (K-Rta) have been shown to be required for origin-dependent DNA replication in addition to the core replication proteins (1). K-bZIP interacts with ORF50 and dampens the transactivation activity of ORF50 on some early promoters (14). K-bZIP is implicated as the OBP, and ORF50 is the major transactivator protein and also interacts with a promoter found within HHV-8 oriLyt (1), once again a situation similar to that found with HCMV (31a).
Based on these similar observations, it appears that initiation of HCMV DNA replication may also involve two proteins. IE2 may serve to transactivate a region within oriLyt and UL84 may modulate this transactivation and therefore regulate the timing of DNA synthesis. Also, the UL84 oligomer, like HHV-8 K-bZIP, may have an as-yet-unidentified enzymatic activity that may contribute to the unwinding of DNA at a specific point within oriLyt. The examination of this possible function will be the goal of future studies.
Acknowledgments
This study was supported by NIH Public Health service grant AI45096.
We thank Bill Welch for help with cross-linking experiments and Thomas Kozel and Suzanne Brandt for assistance with antibody purification and characterization.
REFERENCES
- 1.AuCoin, D. P., K. S. Colletti, S. A. Cei, I. Papouskova, M. Tarrant, and G. S. Pari. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318:542-555. [DOI] [PubMed] [Google Scholar]
- 2.Boehmer, P. E., and I. R. Lehman. 1993. Physical interaction between the herpes simplex virus 1 origin-binding protein and single-stranded DNA-binding protein ICP8. Proc. Natl. Acad. Sci. USA 90:8444-8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt, S., P. Thorkildson, and T. R. Kozel. 2003. Monoclonal antibodies reactive with immunorecessive epitopes of glucuronoxylomannan, the major capsular polysaccharide of Cryptococcus neoformans. Clin. Diagn. Lab. Immunol. 10:903-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, Y. N., D. L. Dong, G. S. Hayward, and S. D. Hayward. 1990. The Epstein-Barr virus Zta transactivator: a member of the bZIP family with unique DNA-binding specificity and a oligomerization domain that lacks the characteristic heptad leucine zipper motif. J. Virol. 64:3358-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, D., and P. D. Olivo. 1994. Expression of the varicella-zoster virus origin-binding protein and analysis of its site-specific DNA-binding properties. J. Virol. 68:3841-3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiou, C. J., J. Zong, I. Waheed, and G. S. Hayward. 1993. Identification and mapping of oligomerization and DNA-binding domains in the C terminus of the IE2 regulatory protein of human cytomegalovirus. J. Virol. 67:6201-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deb, S., and S. P. Deb. 1991. A 269-amino-acid segment with a pseudo-leucine zipper and a helix-turn-helix motif codes for the sequence-specific DNA-binding domain of herpes simplex virus type 1 origin-binding protein. J. Virol. 65:2829-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elias, P., and I. R. Lehman. 1988. Interaction of origin binding protein with an origin of replication of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 85:2959-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fierer, D. S., and M. D. Challberg. 1995. The stoichiometry of binding of the herpes simplex virus type 1 origin binding protein, UL9, to OriS. J. Biol. Chem. 270:7330-7334. [DOI] [PubMed] [Google Scholar]
- 10.Fixman, E. D., G. S. Hayward, and S. D. Hayward. 1995. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 69:2998-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fixman, E. D., G. S. Hayward, and S. D. Hayward. 1992. trans-Acting requirements for replication of Epstein-Barr virus oriLyt. J. Virol. 66:5030-5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flemington, E., and S. H. Speck. 1990. Evidence for coiled-coil dimer formation by an Epstein-Barr virus transactivator that lacks a heptad repeat of leucine residues. Proc. Natl. Acad. Sci. USA 87:9459-9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebert, S., S. Schmolke, G. Sorg, S. Floss, B. Plachter, and T. Stamminger. 1997. The UL84 protein of human cytomegalovirus acts as a transdominant inhibitor of immediate-early-mediated transactivation that is able to prevent viral replication. J. Virol. 71:7048-7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao, W., Y. Tang, S. F. Lin, H. J. Kung, and C. Z. Giam. 2003. K-bZIP of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 77:3809-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin, S. F., D. R. Robinson, G. Miller, and H. J. Kung. 1999. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 73:1909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science 252:1162-1164. [DOI] [PubMed] [Google Scholar]
- 17.Makhov, A. M., P. E. Boehmer, I. R. Lehman, and J. D. Griffith. 1996. The herpes simplex virus type 1 origin-binding protein carries out origin specific DNA unwinding and forms stem-loop structures. EMBO J. 15:1742-1750. [PMC free article] [PubMed] [Google Scholar]
- 18.Makhov, A. M., P. E. Boehmer, I. R. Lehman, and J. D. Griffith. 1996. Visualization of the unwinding of long DNA chains by the herpes simplex virus type 1 UL9 protein and ICP8. J. Mol. Biol. 258:789-799. [DOI] [PubMed] [Google Scholar]
- 19.Makhov, A. M., S. S. Lee, I. R. Lehman, and J. D. Griffith. 2003. Origin-specific unwinding of herpes simplex virus 1 DNA by the viral UL9 and ICP8 proteins: visualization of a specific preunwinding complex. Proc. Natl. Acad. Sci. USA 100:898-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marintcheva, B., and S. K. Weller. 2003. Existence of transdominant and potentiating mutants of UL9, the herpes simplex virus type 1 origin-binding protein, suggests that levels of UL9 protein may be regulated during infection. J. Virol. 77:9639-9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGeoch, D. J., M. A. Dalrymple, A. Dolan, D. McNab, L. J. Perry, P. Taylor, and M. D. Challberg. 1988. Structures of herpes simplex virus type 1 genes required for replication of virus DNA. J. Virol. 62:444-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mocarski, E. S. A. C., T. C. 2001. Cytomegaloviruses and their replication, p. 2629-2674. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott/The Williams & Wilkins Co., Philadelphia, Pa.
- 23.Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pari, G. S., M. A. Kacica, and D. G. Anders. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol. 67:2575-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prichard, M. N., S. Jairath, M. E. Penfold, S. St Jeor, M. C. Bohlman, and G. S. Pari. 1998. Identification of persistent RNA-DNA hybrid structures within the origin of replication of human cytomegalovirus. J. Virol. 72:6997-7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarisky, R. T., Z. Gao, P. M. Lieberman, E. D. Fixman, G. S. Hayward, and S. D. Hayward. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J. Virol. 70:8340-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarisky, R. T., and G. S. Hayward. 1996. Evidence that the UL84 gene product of human cytomegalovirus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz, R., M. Summer, A. Scully, and D. H. Spector. 1994. Site-specific binding of the human cytomegalovirus IE2 86-Kilodalton protein to an early gene promoter. J. Virol. 68:5613-5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommer, M. H., A. L. Scully, and D. H. Spector. 1994. Transactivation by the human cytomegalovirus IE2 86-kilodalton protein requires a domain that binds to both the TATA box-binding protein and the retinoblastoma protein. J. Virol. 68:6223-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spector, D. J., and M. J. Tevethia. 1994. Protein-protein interactions between human cytomegalovirus IE2-580aa and pUL84 in lytically infected cells. J. Virol. 68:7549-7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trego, K. S., and D. S. Parris. 2003. Functional interaction between the herpes simplex virus type 1 polymerase processivity factor and origin-binding proteins: enhancement of UL9 helicase activity. J. Virol. 77:12646-12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31a.Xu, Y., S. A. Cei, K. S. Colletti, A. Rodriguez Huete, and G. S. Pari. Human cytomegalovirus DNA replication requires transcriptional activation via an IE2 and UL84-responsive bidirectional promoter element within oriLyt. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 32.Xu, Y., K. S. Colletti, and G. S. Pari. 2002. Human cytomegalovirus UL84 localizes to the cell nucleus via a nuclear localization signal and is a component of viral replication compartments. J. Virol. 76:8931-8938. [DOI] [PMC free article] [PubMed] [Google Scholar]