

Significance
O-linked GlcNAcylation is a reversible posttranslational protein modification on serine or threonine residues and regulates multiple cellular signaling pathways. We discovered elevated O-GlcNAc and O-GlcNAc transferase (OGT) in HPV-associated cervical neoplasms relative to the normal cervix. We show that HPV E6 upregulates OGT, increases O-GlcNAc, stabilizes c-MYC via O-GlcNAc, and enhances HPV oncogene activities. Conversely, suppression of O-GlcNAc in HPV-transformed cells by knocking down or inhibiting OGT impairs HPV oncogene-induced activities and impedes tumor growth in animal models. Thus, O-GlcNAc plays a critical role in HPV-induced carcinogenesis, and targeting O-GlcNAc might prove to be a potential therapeutic approach.
Keywords: O-linked GlcNAcylation, HPV E6, c-MYC, cervical cancer, HPV oncogenicity
Abstract
High-risk human papillomaviruses (HPVs) are causative agents of anogenital cancers and a fraction of head and neck cancers. The mechanisms involved in the progression of HPV neoplasias to cancers remain largely unknown. Here, we report that O-linked GlcNAcylation (O-GlcNAc) and O-GlcNAc transferase (OGT) were markedly increased in HPV-caused cervical neoplasms relative to normal cervix, whereas O-GlcNAcase (OGA) levels were not altered. Transduction of HPV16 oncogene E6 or E6/E7 into mouse embryonic fibroblasts (MEFs) up-regulated OGT mRNA and protein, elevated the level of O-GlcNAc, and promoted cell proliferation while reducing cellular senescence. Conversely, in HPV-18–transformed HeLa cervical carcinoma cells, inhibition of O-GlcNAc with a low concentration of a chemical inhibitor impaired the transformed phenotypes in vitro. We showed that E6 elevated c-MYC via increased protein stability attributable to O-GlcNAcylation on Thr58. Reduction of HPV-mediated cell viability by a high concentration of O-GlcNAc inhibitor was partially rescued by elevated c-MYC. Finally, knockdown of OGT or O-GlcNAc inhibition in HeLa cells or in TC-1 cells, a mouse cell line transformed by HPV16 E6/E7 and activated K-RAS, reduced c-MYC and suppressed tumorigenesis and metastasis. Thus, we have uncovered a mechanism for HPV oncoprotein-mediated transformation. These findings may eventually aid in the development of effective therapeutics for HPV-associated malignancies by targeting aberrant O-GlcNAc.
Persistent infections of high-risk (HR) human papillomavirus (HPV)-16, HPV-18, and closely related genotypes are etiologically associated with the development of several human cancers, including anogenital and head and neck cancers (1–3). Two HR HPV genes, E6 and E7, are potent oncogenes based on their immortalizing and transforming activities in cell culture systems and their capacities to induce tumors in animal models. The HR HPV E7 oncoprotein binds to more than 20 cellular targets and interferes with multiple cellular processes, leading to deregulated cell cycle, centrosome amplification, DNA damage, anoikis resistance, anchorage-independent cell growth and malignant transformation as well as immune surveillance evasion. E6 is also a multifunctional protein. Constitutive expression of the HR HPV E6 abrogates cell growth arrest and apoptosis, induces genomic instability and somatic mutations, activates telomerase to promote immortalization, disrupts cell polarity, and prevents anoikis. These properties suggest that HPV-associated carcinogenesis involves a coordinated targeting of multiple pathways with each pathway having a distinct but complementing role in malignant transformation (4).
O-linked GlcNAcylation (O-GlcNAc) is a reversible posttranslational modification, transferring an amino sugar moiety to serine/threonine residues of cytosolic or nuclear proteins (5). The hexosamine biosynthetic pathway (HBP) converts intracellular glucose to UDP-N-acetylglucosamine (UDP-GlcNAc), the donor for the O-GlcNAc modification (6). The enzyme O-GlcNAc transferase (OGT) catalyzes the addition of the amino sugar to target proteins, whereas the enzyme O-GlcNAcase (OGA) catalyzes the removal of the sugar (5, 6). Some substrates of O-GlcNAc are alternatively targeted by kinases (7–9). Thus, there is an extensive crosstalk between O-GlcNAc and pathways or mechanisms that are regulated by protein phosphorylation-signaling cascades (7–9).
Because the biosynthesis of UDP-GlcNAc involves products from glucose, amino acid, fatty acid, and nucleotide metabolism, it has been proposed that O-GlcNAc serves primarily to modulate cellular signaling and transcription regulatory pathways in response to metabolic regulation (10, 11). As a nutrient sensor, O-GlcNAc relays the effects of excessive nutritional intake, an important cancer risk factor, onto protein activities and cellular functions (8). Indeed, major tumor suppressors and oncoproteins, such as p53, MYC, NF-κB, and β-catenin are direct targets of O-GlcNAc (12–17). Chromatin dynamics is also modulated by O-GlcNAc. For example, DNA methylation enzymes of the Tet family, which is involved in epigenetic alterations and cancer, interact with and target OGT to multiple chromatin-remodeling complexes (9). Moreover, histones are subject to O-GlcNAc modification, leading to the alteration of their functions (18). O-GlcNAc and OGT levels are elevated in breast, prostate, colon, bladder, and several other cancers (16, 19, 20), but neither has been examined previously in the context of HPV oncogene expression or cervical neoplasms. In this work, we asked whether O-GlcNAc might play a role in HPV-induced transformation and carcinogenesis.
Results
O-GlcNAc and OGT Are Elevated in Cervical Neoplasms.
To detect O-GlcNAc in the spectrum of cervical lesions attributed to HPV infections, we used immunohistochemistry (IHC) to probe the presence of O-GlcNAc in a large collection of normal, cervical intraepithelial neoplasias (CINs) and carcinomas of the cervix. As shown in Fig. 1 A–C, normal cervical tissues bore a low level of O-GlcNAc, whereas tissues from CINs and carcinomas exhibited significantly higher signals. High-grade CINs exhibited stronger signals and higher H scores compared with low-grade lesions (Fig. S1). Elevated OGT, reduced OGA, or both may have contributed to the elevated O-GlcNAc. To distinguish these possibilities, we probed for OGT and OGA in serial sections of cervical cancer tissues using IHC. Consistently, both O-GlcNAc and OGT were strongly positive in malignant foci (Fig. 1D) compared with the adjacent normal epithelia. In contrast, there was no appreciable difference in OGA signals between malignant and normal tissues (Fig. 1D). Characterization of additional tissue specimens confirmed these initial findings (Fig. 1 E and F). Thus, elevated OGT is primarily responsible for increased O-GlcNAc in CINs and cervical cancers. Additionally, the staining patterns of OGT and O-GlcNAc mostly overlapped with those from p16INK4A (Fig. S2), a surrogate marker for HR HPV-associated high-grade lesions and cancers (21), suggesting that O-GlcNAc may contribute to HPV oncogenic activity.
Fig. 1.

O-GlcNAc and OGT are increased in cervical lesions. Human cervical tissues from healthy controls (n = 22), cervical intraepithelial neoplasias (n = 43), and cervical carcinomas (n = 229) were examined. (A) Representative IHC staining of O-GlcNAc with antibody RL2. (Scale bar, 50 µm.) (B) Quantification of O-GlcNAc antigen positivity in the above tissue specimens. Masked reading was performed by two investigators using the same criteria to evaluate the staining (low: overall negative or weak staining; high: overall moderate or strong staining). The Pearson’s χ2-test was used to analyze the distribution difference of O-GlcNAc among human cervical tissues (P < 0.01). (C) H-scores of O-GlcNAc staining in tissues from normal cervix, CINs, and carcinomas (Pearson’s χ2-test, *P < 0.01). (D–F) OGT is elevated in cervical lesions. (D) Representative IHC staining of O-GlcNAc, OGT, and OGA in serial sections of a cervical cancer tissue. (Scale bar, 50 µm.) Black arrows point to the tumor tissue. Red arrows point to the normal cervix. (E) Quantification of OGT and OGA antigen positivity in cervical tissues following the approaches in B (P < 0.01). (F) H-scores of OGT and OGA in normal tissues and cervical diseases were calculated (*P < 0.01).
Fig. S1.

Detection of O-GlcNAc in CIN lesions. CIN1 (n = 4), CIN2 (n = 16), and CIN3 (n = 23) were examined. (A) Representative IHC staining of O-GlcNAc in CIN1, -2, and -3 were shown. (Scale bar, 50 µm.) (B) H scores of O-GlcNAc staining in tissues from the CIN lesions were calculated (Pearson’s χ2-test, *P < 0.01).
Fig. S2.

Staining patterns of O-GlcNAc, OGT, and p16INK4A in a cervical carcinoma. Three serial tissues from a cervical squamous cell carcinoma were stained with anti–O-GlcNAc (RL2), anti-OGT, and anti-p16INK4A antibodies. Representative IHC staining fields were shown. (Scale bar, 50 µm.)
O-GlcNAc Is Induced by and Mediates the Oncogenic Activity of E6 and E6/E7.
To determine whether increased O-GlcNAc in CINs and cervical cancers is directly linked to HPV infection, we transduced primary mouse embryonic fibroblasts (MEFs) (at passage 3) with recombinant lentiviruses or retroviruses expressing HPV16 or HPV18 E6, E7, E6/E7, or GFP and examined O-GlcNAc in cell lysates by immunoblot (Fig. 2A and Fig. S3). Consistent with previous reports (21, 22), expression of HPV E6 and E7 reduced p53 and Rb levels in the cells (Fig. 2A), respectively. Importantly, using a pan-GlcNAc antibody, proteins with O-GlcNAc were markedly increased in MEF/HPV E6 or E6/E7 relative to the control, whereas E7 had no effect (Fig. 2A and Fig. S3). In addition, immunoblots confirmed that OGT, but not OGA, was elevated in MEF/HPV16 E6 or E6/E7, but not in MEF/E7 cells (Fig. 2A). Thus, E6 is primarily responsible for the elevation of O-GlcNAc and OGT.
Fig. 2.

O-GlcNAc mediates HPV oncogenic activities. (A and B) MEFs (passage 3) were infected with lentiviruses expressing HPV16 E6, E7, E6/E7, or GFP (control). (A) Whole-cell extracts (WCEs) were analyzed with immunoblots for OGT and OGA as well as with O-GlcNAcylated proteins with respective antibodies. E6, E7, Rb, and p53 were determined for evaluating the HPV16 oncogene expression and function. GAPDH served as a loading control. (B) Percentages of MEFs in senescence at passage 12 were determined after staining with SA–β-gal. Data are average ± SEM of three independent experiments. *P < 0.01. Impact of O-GlcNAcylation on HPV16-induced cell migration (C) and invasion (D) as described in SI Materials and Methods. *P < 0.01 (n = 3). (E) Anchorage-independent cellular growth in soft agar. C33A/GFP and C33A/HPV16-E6/E7 cells were grown on 0.4% agar with 1 μM DON for 2 wk. Colonies were recorded with an Envision light microscope and counted. *P < 0.01 (n = 3). (F–I) O-GlcNAc mediates oncogenic activities of HeLa. HPV18-positive HeLa cells were treated with 50 μM ST045849. Migration (F) and invasion (G) assays and anchorage-independent growth (H) were performed as above described. *P < 0.01 (n = 3). WCEs were used for immunoblot analyses. GAPDH served as a loading control (I).
Fig. S3.

Detection of O-GlcNAc in HPV18-infected cells. MEFs (passage 3) were infected with retroviruses expressing HPV18 E6, E7, E6/E7, or empty vector (control). WCEs were analyzed with immunoblots for the expression of O-GlcNAcylated proteins with an anti–O-GlcNAc antibody (RL2). GAPDH served as a loading control.
To determine whether increased O-GlcNAc might affect the activities of HPV oncogenes, we modulated O-GlcNAc with chemicals in MEF/HPV16 E6/E7 or GFP (Ctrl). Early passage (p3) of MEFs infected with an empty lentivirus vector halted growth and entered senescence at passages 9–11. In contrast, MEF/HPV16 E6/E7 continued to grow, and clusters of spindle-shaped cells were observed as early as passage 6 (Fig. S4). Application of 1.0 µM 6-diazo-5-oxo-l-norleucine (DON), a glutamine antagonist that inhibits O-GlcNAc, did not markedly affect cell viability at passage 6 (Fig. S5A), but strikingly blocked the morphological changes of the E6/E7-transduced MEFs (Fig. S4). In addition, MEF/HPV16 E6/E7 had a reduced senescent cell population, but the addition of 1.0 µM DON continuously from passage 6 increased senescent cells (Fig. 2B). These results suggest that O-GlcNAc could affect the oncogenic activities of HPV16 E6/E7. Similar alterations in the MEFs with or without HPV E6/E7 were also observed after exposure to 50 μM ST045849 [3-(2-adamantanylethyl)-2-[(4-chlorophenyl)azamethylene]-4-oxo-1,3-thiazaperhydroine-6-carboxylic acid], which inhibits OGT specifically (23–25) (Figs. S5B and S6).
Fig. S4.

Inhibition of HPV16 E6/E7-mediated morphological transformation of MEFs upon suppression of O-GlcNAc. Primary MEFs (passage 3) were infected with lentiviruses expressing HPV16 E6/E7 or GFP (control). The cells were then treated with 1 μM DON. Cells were cultured and passaged. Populations of the HPV16 E6/E7-transduced cells were morphologically transformed (at passages 6–9). Representative fields of the cells in passage 9 are shown. (Scale bar, 50 µm.)
Fig. S5.

Viability of MEFs upon treatment of DON and ST045849. Primary MEFs (passage 6) were treated with 1 μM DON (A) or 50 μM ST045849 (B) for 72 h. Cell viability was determined by MTT assay. The value in MEFs treated with PBS (control) was set as 100%. The means ± SEM for three independent experiments are shown.
Fig. S6.

Percentages of MEFs in senescence after treatment with ST045849. MEFs (passage 3) were infected with lentiviruses expressing HPV16 E6/E7 or GFP (control). Beginning at passage 6, the cells were exposed to 50 μM ST045849 continuously. Percentages of MEFs in senescence at passage 12 were determined after staining with SA–β-gal. Data are average ± SEM of three independent experiments. *P < 0.01.
To substantiate this interpretation, we transduced viral oncogenes into C33A cells, a rare cervical cancer cell line devoid of oncogenic HPVs. C33A/HPV16 E6/E7 cells exhibited an elevation in O-GlcNAc (Fig. S7 A and B). We then performed assays in Transwell with or without precoated Matrigel. These cells exhibited increased migration and invasion relative to the parental cells. The effects were abrogated by the treatment with 1.0 µM DON overnight (Fig. 2 C and D), a condition that did not affect cell proliferation and viability. The ability to grow in the absence of anchorage to the extracellular matrix is one of the most important oncogenic properties of HPV-positive cancer cells. We next examined the growth of C33A/HPV16 E6/E7 cells in soft agar in the presence or absence of DON for 2 wk. C33A cells formed few colonies (per 5,000 cells) when plated in soft agar (Fig. 2E). Transduction of HPV16 E6/E7 markedly increased the size and number of the colonies. Application of 1 µM DON substantially reversed the viral effects (Fig. 2E). Similar inhibitory effects on migration, invasion, and soft agar colony formation were also observed in ST045849-treated HeLa cells, an HPV18-positive cell line established from a cervical adenocarcinoma (Fig. 2 F–H). The expression of E6/E7 was not affected (Fig. 2I). Taken together, our data show that O-GlcNAc mediates or promotes the oncogenic activities of the HR HPVs.
Fig. S7.

Reduction of O-GlcNAc by DON. C33A cells transduced with GFP or HPV16 E6/E7 were treated with 1 µM DON for 24 h. WCEs were harvested for immunoblot analyses. GAPDH served as a loading control (A). O-GlcNAc levels with different treatments in A were quantified with the NIH ImageJ software (B).
HPV E6/E7 Enhances the Transcription of OGT.
Three to five percent of the intracellular glucose directly enters into the HBP, where it is converted into UDP-GlcNAc, the donor for the O-GlcNAc modification (5). Thus, it is possible that O-GlcNAc is affected by glucose metabolism. Our recent data showed that MEF/HPV16 E7, but not MEF/HPV16 E6, exhibited appreciably elevated glucose consumption compared with MEFs, consistent with the report that HPV16 E7 promotes glycolytic metabolism (26). Thus, our present results would suggest that the increased O-GlcNAc in E6- or E6/E7-containing cells may not be attributed to an alteration in glucose metabolism.
To examine the mechanism by which HR HPV oncogenes up-regulate OGT, we determined the half-life of OGT. It was not affected by HPV16 oncogene expression (Fig. S8). Rather, the expression of HPV16 E6 or E6/E7 in MEFs increased OGT mRNA levels by about threefold, whereas HPV16 E7 had no significant impact (Fig. 3A). HPV infection influences the expression or activity of multiple transcription factors, including AP-1, SP-1, NF-κB, p53, and c-MYC (21, 22). Interestingly, the promoter region of the OGT gene possesses binding sites for these transcriptional factors. Thus, we cloned human OGT promoter sequences −1010 to +10 into a pGL3 vector and transfected the construct into C33A cells expressing HPV16 oncoproteins (Fig. 3B) or cotransfected the reporter into MEF/HPV16 E6, E7, or E6/E7 (Fig. 3C). The results showed that expression of HPV16 E6 or E6/E7 up-regulated the OGT promoter, whereas E7 had little or no effect (Fig. 3 B and C). We further showed that, in C33A cells, cotransfection with an expression vector of Foxo3a, SP1, NF-κB p65, or c-MYC increased the OGT promoter activity, whereas p53 expression was suppressive and CREB and E2F1 had no significant impact (Fig. S9). Our results are consistent with a report by Nees et al. (27), who examined genes in differentiating cervical keratinocytes infected with retroviruses carrying HPV16 E6 or E7; they found that HPV16 E6 stimulated expression of multiple genes known to be regulated by NF-κB and AP-1, whereas E7 was less effective.
Fig. S8.

Stability of OGT is not affected by HPV16 oncogene expression. MEF/GFP, E6, E7, or E6/E7 were treated with 20 µg/mL cycloheximide (CHX). WCEs were collected at each time point. (A) Immunoblot was conducted to detect the expression of OGT. (B) OGT levels at different time points in (A) were quantified with the NIH ImageJ software.
Fig. 3.

HPV16 E6 or E6/E7 enhances OGT transcription. (A) MEFs were infected with lentiviruses expressing HPV16 E6, E7, E6/E7, or GFP (control). mRNA levels of OGT in the transduced cells were determined by qPCR. The relative mRNA levels were calculated. *P < 0.01 (n = 3). HPV activated the promoter of OGT in C33A cells (B) and in MEFs (C). Human OGT promoter sequences −1010 to +10 were cloned to the pGL3 vector and transfected to the cells expressing HPV16 oncoproteins. The pRL vector expressing wild-type Renilla luciferase was used as a control reporter. Relative luciferase unit (RLU) was the ratio of the OGT promoter-driven luciferase activity to Renilla activity. *P < 0.01 (n = 3).
Fig. S9.

Transcription regulation of the OGT promoter by HPV-targeted transcriptional factors. Human OGT promoter sequences −1010 to +10 were cloned to the pGL3 vector and transfected to C33A cells along with constructs expressing Foxo3a, CREB, SP1, NF-κB p65, p53, c-MYC, E2F1, or GFP (Ctrl). The pRL vector expressing wild-type Renilla luciferase was used as a control reporter. RLU was the ratio of OGT promoter-driven luciferase activity to Renilla activity. The means ± SEM for three independent experiments were presented. *P < 0.01 and **P < 0.05 compared with the control (Ctrl) group.
Knockdown of OGT Reduces O-GlcNAc and Xenograft Growth of HPV-Transformed Cervical Cancer Cells in SCID Mice.
To substantiate the in vitro significance of HPV-mediated up-regulation of OGT with regard to viral oncogenic activities, we knocked down (kd) OGT or OGA in HeLa cells with lentiviruses expressing shRNA-OGT or shRNA-OGA. Cells were selected with puromycin (1 μg/mL). Expression of OGT was reduced by ∼75% (Fig. 4A). OGT knockdown markedly reduced O-GlcNAcylated proteins in the cells (Fig. 4A). Importantly, cell migration, invasion, and anchorage-independent cellular growth in soft agar were markedly reduced following OGT kd (Fig. 4 B–D). In contrast, each of these activities was enhanced in the presence of shRNA-OGA, which severely depleted OGA while elevating O-GlcNAc (Fig. 4 A–D). These observations support the importance of O-GlcNAc in mediating the viral oncogenic activities.
Fig. 4.

Knockdown of OGT reduces O-GlcNAc and tumor growth. HeLa cells were stably transduced with lentiviruses expressing shRNA-Ctrl, shRNA-OGT, or shRNA-OGA. (A) Immunoblots were used to detect O-GlcNAc, OGT, and OGA in cell lysates. GAPDH served as a loading control. (B and C) Migration and invasion assays in HeLa cells in which OGT or OGA expression was stably knocked down. *P < 0.01 (n = 3). (D) Anchorage-independent cellular growth in soft agar. *P < 0.01 (n = 3). (E and F) Depletion of OGT reduced tumor growth in SCID mice, whereas OGA knockdown promoted tumor growth. Two million HeLa/shRNA-Ctrl, /shRNA-OGT, or /shRNA-OGA cells were inoculated s.c. under the flank. (E) Tumor volumes were measured every 3 d. *P < 0.01 and **P < 0.05 compared with shRNA-Ctrl group (n = 10). (F) The tumors were removed from euthanized mice. IHC was used to detect cells positive for active cleaved caspase-3 in the tumor tissues. At least 200 cells were counted in each group for determining the percentage of positive cells. *P < 0.01.
To validate the role of OGT in promoting oncogenic properties in an in vivo system, we performed s.c. inoculation of HeLa/shRNA-Ctrl, shRNA-OGT, and shRNA-OGA cells into SCID mice. Knockdown of OGT substantially reduced the tumor volume (Fig. 4E) and the proportion of cells expressing the proliferating cell nuclear antigen (PCNA) (Fig. S10). Apoptotic tumor cells increased, as detected with an antibody to cleaved caspase-3 (Fig. 4F). In stark contrast, depletion of OGA stimulated s.c. tumor growth (Fig. 4E) and increased PCNA-positive cell number (Fig. S10), but had no effect on the cell population positive for cleaved caspase-3 (Fig. 4F). Taken together, our data support the notion that HPV oncogene-induced elevation of OGT and O-GlcNAc have a significant impact on tumor cell survival and growth.
Fig. S10.
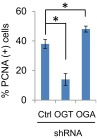
Reduction of tumor growth upon knockdown of OGT. The 2 × 106 HeLa/shRNA-Ctrl, /shRNA-OGT, or /shRNA-OGA cells were inoculated s.c. under the flank. The tumors were removed from euthanized mice at the end of week 3. IHC was used to detect PCNA in the tumor tissues. At least 200 cells were counted in each group for determining the percentage of positive cells. *P < 0.01.
Suppression of O-GlcNAcylation Impedes Tumor Growth and Metastasis of HPV-Transformed TC-1 Cells in Syngeneic Mice.
To verify further the significance of O-GlcNAc in HPV-induced tumor growth, we examined the effects of DON in the TC-1 cell-induced mice tumor model. TC-1 cells were derived from primary lung epithelial cells of C57BL/6 mice transformed by HPV16 E6/E7 and activated K-RAS (28). These cells induce tumors in syngeneic mice and preferentially metastasize to the lungs (28). TC-1 cells were inoculated into the flank of syngeneic mice and treated with DON as described in Materials and Methods. The mouse body weight was stable (Fig. S11), but the s.c. tumor volumes were significantly smaller than those in PBS-treated control mice (Fig. 5 A–C). Immunoblots of tumor tissues showed that DON reduced O-GlcNAc relative to controls (Fig. 5D). IHC of tumor tissues confirmed this result and also revealed that the population of PCNA-positive cells was significantly reduced, whereas cells positive for cleaved caspase-3 were greatly increased in DON-treated mice (Fig. 5E).
Fig. S11.

Unaltered mouse body weights after administration of DON. The 2 × 106 TC-1 cells were inoculated s.c. under the flank of each C57BL6 mouse. The mice were randomly grouped for treatment with PBS or DON (five mice/group). One milligram per kilogram of DON dissolved in 200 µL PBS was administered i.p. on days 1–3 and then repeated every 4 d for a total of seven doses of DON or PBS for each mouse. The body weights of the mice were monitored during the entire treatment process.
Fig. 5.

Suppression of O-GlcNAcylation impedes HPV-positive tumor growth and metastasis in TC-1–induced cancer models in syngeneic mouse. (A–D) The 2 × 106 TC-1 cells were inoculated s.c. under the flank of C57BL6 mouse. Mice were treated with PBS or DON (five mice/group). (A) Tumor volumes for PBS- and DON-treated mice. *P < 0.01, comparison of the tumor volumes between DON and PBS treatment from day 9. (B) Tumors in mice. Arrows indicate the locations of the tumors. (C) Tumors recovered from euthanized mice. (D) O-GlcNAc and c-MYC were reduced in tumors from mice treated with DON. Fresh tumor tissues from the mice were frozen in liquid nitrogen and were kept at −80°. WCEs from the tumor tissues were isolated for immunoblot analyses. GAPDH served as a loading control. (E) IHC detection of O-GlcNAc, PCNA, active cleaved caspase-3, and c-MYC in the tumor tissues. Representative stained tissue sections were presented for each group. (Scale bar, 50 µm.) (F and G) C57BL6 mice were injected with 2 × 105 TC-1 cells via the tail vein. Mice were administered with PBS or DON as in A–D. Lung tissues were collected from euthanized mice, fixed, and stained with H&E. (F) Representative photographs of lung tumor foci. (Scale bar, 100 µm.) Lung metastatic foci in the maximum horizontal layer for each mouse were counted (G). *P < 0.01.
Because inhibition of O-GlcNAc suppresses migration, invasion, and anchorage-independent cell growth of HPV-positive cells (Fig. 2 C–H), O-GlcNAc might also contribute to HPV-promoted tumor metastasis. To test this hypothesis, we used the TC-1 cells in the mouse lung metastasis model (28). TC-1 cells were injected into the tail vein of C57BL/6 mice. Application of DON strikingly reduced the number of TC-1 tumor foci in the lungs as well as the sizes of the foci (Fig. 5 F and G; P < 0.01). No notable toxic side effect (such as leucopenia, lethargy, diarrhea, etc.) was observed. Thus, our results show that suppression of O-GlcNAc reduces HPV-positive tumor growth and metastasis in mouse models.
c-MYC Is O-GlcNAcylated and Stabilized in HPV Infections.
The basis for substrate specificity of O-GlcNAc is unclear, making the identification of key substrates challenging. However, c-MYC, an oncogenic transcriptional factor, is known to be O-GlcNAcylated (12, 13, 25). To examine whether c-MYC and other cellular factors known to be activated by HPVs are O-GlcNAcylated due to the induction of OGT by HPV E6 or E6/E7, we used wheat germ agglutinin affinity purification to pull down O-GlcNAc proteins from MEF/GFP (control) and MEF/HPV16 oncogenes. Immunoblots showed that, relative to the control, both total and O-GlcNAcylated c-MYC were markedly increased in MEF/HPV16 E6 or E6/E7, but not in MEF/HPV16 E7 (Fig. 6A). To confirm the O-GlcNAcylation of c-MYC, we performed a reciprocal pull-down using HeLa cell extracts. Immunoprecipitated c-MYC was detected with a pan-O-GlcNAc antibody, in particular in cells with elevated O-GlcNAc due to OGA kd (Fig. 6B). In our experimental settings, there was no detectible O-GlcNAcylation of GAPDH and S6 (Fig. 6A). However, we cannot rule out the possibility that other host proteins in HPV-infected cells are O-Glycosylated and play a role in mediating HPV oncogenic activities.
Fig. 6.

c-MYC is O-GlcNAcylated and stabilized by HPV oncogene expression. (A) O-GlcNAcylated proteins in MEFs transduced with GFP or with HPV16 E7, E6, or E6/E7 were pulled down with succinylated wheat germ agglutinin beads. c-MYC and O-GlcNAc in the pull-down complexes were detected by immunoblotting. (B) c-MYC was O-GlcNAcylated. c-MYC in HeLa/shRNA-Ctrl, shRNA-OGT, and shRNA-OGA cells was immunoprecipiated with a polyclonal antibody to c-MYC (Upper). The O-GlcNAcylated c-MYC was detected with an O-GlcNAc monoclonal antibody, RL2. (C) HPV16 E6/E7 elevated the O-GlcNAcylated proteins and the c-MYC protein. Both enhancements were abolished by the inhibition of O-GlcNAc with DON. MEFs transduced with GFP or E6/E7 were treated with 10 µM DON overnight. The WCEs were harvested for immunoblot analyses. (D) The amounts of c-MYC correlated with the levels of O-GlcNAc. TC-1 cells were treated with 10 µM DON, 10 mM 2-DG, 10 µM TG, or starved (no glucose, −G) for overnight. The WCEs were collected and subjected to immunoblot analyses. In C and D, GAPDH served as a loading control. (E and F) c-MYC was stabilized in HPV16 E6- and E6/E7-expressing cells. MEF/GFP, E6, E7, and E6/E7 cells were treated with 20 µg/mL CHX for the indicated duration. The WCEs were then collected for immunoblots to detect c-MYC and S6 in the cells (E). c-MYC levels at each time point were quantified with NIH ImageJ software (F). (G) Overexpression of c-MYC partially rescued O-GlcNAc inhibition-induced repression of cell viability. HeLa cells were stably transfected with empty vector or c-MYC expression construct. The cells were then selected with 500 µg/mL G418 for 2 wk. The survived clones were pooled and subjected to cell viability assay in the presence or absence of DON (10 µM) for 72 h. *P < 0.01 (n = 3).
To validate that O-GlcNAc is responsible for elevated c-MYC levels in HPV-infected cells, we exposed MEF/HPV16 E6E7 or MEF/GFP to 10 µM DON for 16 h. As before, the level of c-MYC was elevated upon E6/E7 expression relative to the control (Fig. 6C). However, it was dramatically reduced in the presence of DON regardless of the presence or absence of E6/E7 (Fig. 6C). This O-GlcNAc–mediated regulation of c-MYC levels was further verified in mouse TC-1 cells (Fig. 6D). Suppression of O-GlcNAc with 10 µM DON for 16 h in TC-1 cells markedly reduced the level of c-MYC (Fig. 6D). In contrast, elevating O-GlcNAc with thiamet-G (TG), an inhibitor of OGA, or 2-deoxyglucose (2-DG), a glucose-mimic, correlated with increased levels of c-MYC (Fig. 6D). We noted that depletion of glucose (−G), a means previously reported to promote O-GlcNAc, did not do so in our experimental setting (Fig. 6D). Interestingly, the levels of OGT and OGA changed in opposite directions and correlated with levels of intracellular O-GlcNAc (Fig. 6 C and D), possibly due to a feedback regulation (5). The reduction of c-MYC by DON treatment was also confirmed in TC-1 tumors by IHC (Fig. 5E). Collectively, our data suggest that HPV-up–regulated O-GlcNAcylation is responsible for the elevation of c-MYC protein.
To examine the mechanism by which O-GlcNAc elevated c-MYC protein levels, we determined the protein stability in MEFs transduced with HPV16 E6, E7, E6/E7, or GFP. Cells were exposed to 20 µg/mL cycloheximide (CHX) for up to 8 h, and the levels of c-MYC were determined by immunoblots. The half-life of c-MYC in the control or E7-expressing cells was shorter than 30 min. In contrast, its half-lives in E6- or E6/E7-expressing cells were substantially longer, reaching 60 min (Fig. 6 E and F). c-MYC was reported to be O-GlcNAcylated on Thr58 (8, 12, 13). We observed that O-GlcNAc on his-tagged ectopic c-MYC Thr58A (Thr-to-ala mutation) was dramatically reduced in HeLa cells relative to wild-type his-tagged c-MYC (Fig. S12A). Moreover, unlike endogenous wild-type c-MYC (Figs. 5E and 6 C and D), the his-c-MYC Thr58A level was not affected by O-GlcNAc modulators ST045849 or TG (Fig. S12B). Taken together, our results suggest that c-MYC was stabilized in E6- and E6/E7-expressing cells upon O-GlcNAcylation on Thr58.
Fig. S12.

Reduction of O-GlcNAc on ectopic his-c-MYC Thr58A without alterations in total his-c-MYC levels by O-GlcNAc modulators. (A) HeLa cells were transfected with pD40-His/V5-c-MYC, pD40-His/V5-c-MYC Thr58A, or empty His vector for 48 h. WCEs were isolated and immunoprecipitation was performed with His tag antibody or IgG. O-GlcNAc and c-MYC were then detected in the immune precipitates with RL2 or with His tag antibody. Six hundred micrograms of WCEs was used for the co-immunoprecipitation. (B) The abundance of the his-c-MYC Thr58A protein was not affected by modulators of O-GlcNAc. HeLa cells were transfected with pD40-His/V5-c-MYC Thr58A for 48 h. The cells were then exposed to TG (10 µM), ST045849 (50 µM), or PBS for another 24 h. WCEs were collected for immunoblot assays to detect O-GlcNAc, c-MYC, and GAPDH. Thirty micrograms of WCEs was used for the Western blot.
Overexpression of c-MYC Partially Rescues HPV-Mediated Cell Viability Blocked by a High Concentration of O-GlcNAc Inhibitor.
We then set out to determine the effect of O-GlcNAc on c-MYC function. We genetically manipulated the expression of c-MYC in HeLa cells and determined cell viability under the influence of the O-GlcNAc inhibitor DON. As shown in Fig. 6G, stable expression of c-MYC increased HeLa cell number and partially reversed cellular viability suppressed by 10 µM of DON for 72 h. These results suggest that O-GlcNAc enhances HPV-mediated cell survival and that this effect is at least in part mediated through elevated c-MYC upon O-GlcNAcylation.
Discussion
Although HPV E6 and E7 proteins possess multiple biochemical activities, including inactivating the two major tumor suppressors p53 and pRb, these interactions cannot completely account for HPV carcinogenesis. Therefore, it is of interest to identify cellular factors that contribute to lesion progression to cancer. In this study, we demonstrated that O-GlcNAc and OGT were markedly increased in cervical neoplasms associated with HR HPV infections, whereas OGA was not significantly altered (Fig. 1). We demonstrated in vitro that this increase was due to HR HPV E6-stimulated transcriptional up-regulation of OGT (Figs. 2A and 3 and Fig. S3), and a number of transcription factors were implicated (Fig. S9). Some of these factors, such as NF-κB, AP-1, and p53, are known targets of the viral E6 protein for activation or inactivation (21, 22, 27). We further demonstrated that O-GlcNAc enhanced HPV oncogenic activities, including cell proliferation, migration, invasion, and anchorage-independent cell growth in vitro (Figs. 2 and 4), as well as tumorigenesis and metastasis of HPV-transformed cells in mouse models (Figs. 4 and 5). Consistent with these observations, suppression of O-GlcNAc by the chemical reagents DON and ST045849 or by OGT knockdown substantially inhibited these transformed phenotypes in vitro and in vivo. In contrast, knockdown of OGA elevated O-GlcNAc and enhanced the transformed phenotypes in vitro and in vivo (Figs. 2, 4, and 5). Collectively, our results support the conclusion that O-GlcNAc mediates the oncogenic activity of HPV oncogenes.
Yew et al. previously identified an isoform of mixed lineage leukemia 5 (MLL5β), which regulates HPV E6/E7 oncogene transcription through its interaction with AP-1 at the 5′ segment of the HPV16/18 long control region (LCR) (29). Interestingly, Nin et al. (30) reported that MLL5β was O-GlcNAcylated at T440 residue and this modification is necessary for its interaction with AP-1 and its recruitment to the HPV16/18-LCR. Thus, MLL5β O-GlcNAcylation is an important initiation step in E6/E7 transcription. Inhibition of O-GlcNAcylation by azaserine decreased E6/E7 levels and selectively suppressed the survival of HPV-positive cervical cancer cells (30). Our current data suggest that O-GlcNAcylation also mediates the HPV downstream oncogenic activities. It would seem that there is a feed-forward action for O-GlcNAcylation in HPV infection.
The MYC family proteins are key regulators of cell growth, proliferation, differentiation, and apoptosis by modulating the expression of a significant number of genes (31). MYC also governs events associated with tumor progression, including genetic instability, cell migration, and angiogenesis (31, 32). In particular, c-MYC is a critical mediator for HPV-induced immortalization and transformation. HPV-E6 interacts with c-MYC to reactivate the transcription of human telomerase reverse transcriptase (33–36). However, the effect of HPV oncoproteins on c-MYC protein levels is inconclusive (33–36). Due to the pleiotropic activities of c-MYC, its expression and function must be fine-tuned. In particular, the protein is subject to posttranslational modifications, including phosphorylation, ubiquitinylation, acetylation, and glycosylation (12, 13, 37, 38).
We demonstrated that, in HPV E6- and E6/E7-expressing cells, c-MYC was elevated through protein stabilization via O-GlcNAcylation (Fig. 6). c-MYC has been reported to be O-Glycosylated at Thr58, a known phosphorylation site and a mutational hot spot in lymphomas (12, 13). Our current data indicate that c-MYC Thr58 is targeted by HPV oncogene-induced O-Glycosylation (Fig. S12). We showed that stable expression of c-MYC in HeLa cells increased cell viability and partially restored viable cell populations that were reduced by inhibiting O-GlcNAc with a high concentration of DON (Fig. 6G). Importantly, c-MYC and O-GlcNAc levels were coregulated in the mouse tumor model with regard to cell proliferation and survival (Fig. 5 D and E). Our findings are also consistent with previous reports demonstrating that c-MYC is positively correlated to cell proliferation in cervical tissue specimens (39).
In summary, we have uncovered a mechanism for HPV carcinogenesis in the form of E6-stimulated OGT expression, leading to elevated O-GlcNAcylation. We have identified c-MYC to be one of the OGT substrates, and c-MYC level is highly elevated through this posttranslational protein stabilization. Because of the extensively collaborative activities between O-GlcNAc and HPV oncogenes, we believe that reduction of O-GlcNAc in HPV-infected cells with inhibitors, knockdown of OGT, or overexpression of OGA could be instrumental in reducing HPV oncogenic actions. The O-GlcNAc targeting in mice tumor models described in this study supports this hypothesis and could ultimately prove to be of therapeutic value.
Materials and Methods
The animal protocol was approved by the Institutional Animal Care and Use Committee at Jilin University and the University of Alabama at Birmingham (UAB). Human cervical tissues were obtained from the Department of Pathology at Jilin University and the University of Alabama at Birmingham tissue bank. Institutional review board approvals for the usage of these tissues were obtained from Jilin University and UAB. C33A and MEFs were infected with lentiviruses expressing HPV16 E6, E7, E6/E7, or GFP. After a selection with puromycin (1.0 μg/mL) for 2 wk, resistant stable clones were pooled and passaged. To knock down OGT or OGA stably in HeLa cells, cells were infected with lentiviruses containing the OGT, OGA, or scramble shRNA constructs and selected with puromycin (1 μg/mL) for 2 wk. There were no deleterious effects on the viability of selected cells, as determined by trypan blue exclusion and ATP levels.
Additional information on materials and methods is presented in SI Materials and Methods.
SI Materials and Methods
Plasmids.
The pD40-His/V5-c-MYC and its Thr58A mutation, shRNA-Ctrl, shRNA-OGT, and shRNA-OGA constructs were purchased from Addgene. HPV16/18 E6, E7, and E6/E7 were each in a Moloney murine leukemia virus-based retroviral vector and confirmed by sequencing and function in organotypic cultures of primary human keratinocytes. The HPV16 oncogenes were then cloned into pEF1a lentiviral vector and packaged in 293T cells. The cDNAs of Foxo3a, SP1, NF-κB p65, c-MYC, E2F1, p53, and CREB were cloned in the pcDNA3 vector.
Cell Culture, Gene Transfection, and Reagents.
The human cervical cancer cell lines C33A and HeLa as well as primary MEFs and HPV-16–transformed TC-1 cells were maintained in DMEM supplemented with 10% (vol/vol) FBS (Atlanta Biologicals) at 37 °C in 5% CO2. Gene transfection was performed with Fugene 6 transfection reagent (Roche Diagnostics). Puromycin, G418, DON, and TG were purchased from Sigma-Aldrich. The ST045849 was purchased from TimTec. Dual-Luciferase Reporter Assay kit was purchased from Promega.
Immunoblotting and Immunoprecipitation.
Immunoblotting was performed using 30–50 µg of total proteins from lysed cells. GAPDH was used as loading controls. c-MYC (His) or O-GlcNAc (RL2) antibody was used for immunoprecipitation at 4 °C. Immunoprecipitated complexes were eluted and subjected to immunoblot analysis. Polyclonal antibodies against OGT and OGA were purchased from Sigma-Aldrich. Monoclonal antibodies against His-tag and O-GlcNAc (RL2) were purchased from Fisher-Thermo Scientific. Polyclonal antibody against c-MYC was obtained from Abcam. GAPDH antibody was obtained from Bethyl Laboratories. HPV16/18 E6 and E7 antibodies were purchased from Santa Cruz Biotechnology Inc. S6 antibody was purchased from Cell Signal Technology, Inc.
Anchorage-Independent Cellular Growth in Soft Agar.
C33A cells or HeLa cells that were transduced with shR-OGT, shR-OGA, or shR-Ctrl and exposed to 1 µM DON or 50 µM ST045849 were collected and suspended in the top layer of medium containing 10% FBS, 0.4% select agar (Invitrogen), and 1 µM DON or 50 µM ST045849. Five thousand cells per well were plated in triplicate in six-well plates on the bottom layer of medium containing 10% FBS and 1% select agar. After 2 wk in culture, cells were stained with 0.5 mL 1% crystal violet for 1 h and counted by standard methods.
Transwell Cell Migration/Invasion Matrigel Assays.
C33A, HeLa, and TC-1 cells alone or transduced with shR-OGT, shR-OGA, or shR-Ctrl were placed in the upper chamber of the Transwell Membrane Inserts or precoated Matrigel Chambers from BD Biosciences. O-GlcNAc modulators (1 µM DON or 50 µM ST045849) as a chemoattractant were added to the lower chamber. After 24 h, migrated/invasive cells were stained with 1% crystal violet and quantified under a microscope.
Detection of Senescence.
MEFs were transduced with HPV16 E6/E7 or empty vector. MEFs and transduced MEFs were treated with 1 µM DON, 50 µM ST045849, or PBS continuously from passage 6. At passage 12, cells were washed with PBS and fixed with 4% (vol/vol) paraformaldehyde for 15 min at room temperature. Cells were then incubated in staining solution from an SA–β-gal staining kit (Cell Signaling Technology). Images were taken using an Olympus IX51 inverted microscope. Percentages of senescent MEFs were calculated.
Luciferase Reporter Assay.
Human OGT promoter sequences −1010 to +10 were amplified with PCR and cloned into the pGL3 vector. After verification of the clone by DNA sequencing, the construct was transfected into C33A cells and MEFs along with an expression vector of viral oncogene(s), GFP or a host transcription factor. The pRL vector expressing wild-type Renilla luciferase was used as a control reporter. RLU was the ratio of OGT luciferase activity to Renilla activity.
Cell Viability Assay.
Six thousand cells were seeded into each well of 96-well plates and allowed to adhere overnight. Cell viability was determined 72 h after the treatments with DON or ST045849 using the MTT assay. Plates were read with a Synergy H1 microplate reader (BioTek Instruments) at a wavelength of 530/620 nm.
Tumorigenesis Models.
C57BL/6 and SCID mice were housed in an animal barrier facility on a 12-h light–dark cycle with food and water available ad libitum.
In the xenograft tumor model, early-passage HeLa/shR-OGT, HeLa/shR-OGA, and HeLa/shR-Ctrl cells were harvested and mixed 2:1 in matrigel (BD Biosciences). Two million cells were inoculated s.c. into the flanks of 10 SCID mice. Mice were randomized into treatment (with DON) or control PBS groups (five mice/group). One milligram per kilogram of DON dissolved in 200 µL PBS was applied i.p. on day 1 to day 3 and then administered every 4 d for a total seven doses for each mouse. Xenograft tumors were measured every 3 d with calipers. Tumor volumes were calculated by the following formula: a2 × b × 0.4, where a is the smallest diameter and b is the diameter perpendicular to a. The body weight, feeding behavior, and motor activity of each animal were monitored as indicators of general health. At the end of 3 wk, the mice were euthanized and the tumors were removed for the histological and biochemical analyses.
For the lung metastasis model, 2 × 105 TC-1 cells were injected into the tail vein of C57BL/6 mice. After 3 d of recovery, the mice were randomized into DON treatment and control groups with five mice in each group as described above. At the end of 3 wk, the mice were euthanized and lungs were fixed to determine the metastatic nodules and for microscopy analysis.
Human Cervical Tissues and Immunohistochemistry.
Human cervical tissues were obtained from the Department of Pathology at Jilin University and the University of Alabama at Birmingham tissue bank. Cervical carcinoma tissue microarray slides (#CR2088, #CR602, and #CR242) were ordered from US Biomax. Tissues from healthy controls (n = 22; 4 were HPV16-positive, and 2 were HPV18-positive), intraepithelial neoplasia (n = 43; 34 were HPV16-positive), and carcinoma (n = 229; 223 squamous cell carcinomas were HPV16-positive, and 1 adenosquamous carcinoma and 5 adenocarcinomas were HPV18 positive) were used for the analysis. IHC was performed using the streptavidin–peroxidase–biotin (SP) IHC method. The staining index was based on the staining intensity, which was graded as “−,” no staining; “+,” weak staining; “++,” moderate staining; and “+++,” strong staining. Samples that scored as “−” or “+” were considered as low expression and those scored as “++” or “+++” as high expression. All stained slides were observed and scored by two pathologists. If the staining interpretation differed between the two investigators, the data for the slide were discarded. For determining the H score, stained tissues were scored by calculating the product of the intensity level and the percentage of cells staining at that level (0, negative; 1, weak; 2, moderate; 3, strong). An H score was then calculated by summing the individual intensity level scores.
Statistical Analysis.
The Pearson’s χ2-test was used to analyze the distribution difference of O-GlcNAc and OGT staining among human cervical tissues. Student’s t test was used for additional statistical analyses in the study. P < 0.05 was considered significant.
Acknowledgments
We thank Dr. T.-C. Wu (Johns Hopkins University) for providing TC-1 cells. The study was supported by NIH Grants R01CA133053, R01CA83679, P50CA098252, and U19AI113212 and National Natural Science Foundation of China Grants 81271853, 81272243, and 81573087.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1606801113/-/DCSupplemental.
References
- 1.Walboomers JM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.zur Hausen H. Papillomaviruses in the causation of human cancers: A brief historical account. Virology. 2009;384(2):260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 3.Lajer CB, von Buchwald C. The role of human papillomavirus in head and neck cancer. APMIS. 2010;118(6-7):510–519. doi: 10.1111/j.1600-0463.2010.02624.x. [DOI] [PubMed] [Google Scholar]
- 4.White EA, Howley PM. Proteomic approaches to the study of papillomavirus-host interactions. Virology. 2013;435(1):57–69. doi: 10.1016/j.virol.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanover JA, Krause MW, Love DC. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol. 2012;13(5):312–321. doi: 10.1038/nrm3334. [DOI] [PubMed] [Google Scholar]
- 7.Stine ZE, Dang CV. Stress eating and tuning out: Cancer cells re-wire metabolism to counter stress. Crit Rev Biochem Mol Biol. 2013;48(6):609–619. doi: 10.3109/10409238.2013.844093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slawson C, Hart GW. O-GlcNAc signalling: Implications for cancer cell biology. Nat Rev Cancer. 2011;11(9):678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Q, Chen Y, Bian C, Fujiki R, Yu X. TET2 promotes histone O-GlcNAcylation during gene transcription. Nature. 2013;493(7433):561–564. doi: 10.1038/nature11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson JR, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502(7471):372–376. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang X, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451(7181):964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- 12.Chou TY, Dang CV, Hart GW. Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci USA. 1995;92(10):4417–4421. doi: 10.1073/pnas.92.10.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou TY, Hart GW, Dang CV. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem. 1995;270(32):18961–18965. doi: 10.1074/jbc.270.32.18961. [DOI] [PubMed] [Google Scholar]
- 14.Jang H, et al. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell. 2012;11(1):62–74. doi: 10.1016/j.stem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Ha JR, et al. β-catenin is O-GlcNAc glycosylated at Serine 23: Implications for β-catenin’s subcellular localization and transactivator function. Exp Cell Res. 2014;321(2):153–166. doi: 10.1016/j.yexcr.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J Biol Chem. 2013;288(21):15121–15130. doi: 10.1074/jbc.M113.470047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang WH, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8(10):1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 18.Fujiki R, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480(7378):557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: A New Cancer Hallmark? Front Endocrinol (Lausanne) 2013;4:99. doi: 10.3389/fendo.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrer CM, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54(5):820–831. doi: 10.1016/j.molcel.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLaughlin-Drubin ME, Münger K. Oncogenic activities of human papillomaviruses. Virus Res. 2009;143(2):195–208. doi: 10.1016/j.virusres.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moody CA, Laimins LA. Human papillomavirus oncoproteins: Pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 23.Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc. 2005;127(42):14588–14589. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- 24.Filhoulaud G, Guillemain G, Scharfmann R. The hexosamine biosynthesis pathway is essential for pancreatic beta cell development. J Biol Chem. 2009;284(36):24583–24594. doi: 10.1074/jbc.M109.025288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itkonen HM, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013;73(16):5277–5287. doi: 10.1158/0008-5472.CAN-13-0549. [DOI] [PubMed] [Google Scholar]
- 26.Mazurek S, Zwerschke W, Jansen-Dürr P, Eigenbrodt E. Effects of the human papilloma virus HPV-16 E7 oncoprotein on glycolysis and glutaminolysis: Role of pyruvate kinase type M2 and the glycolytic-enzyme complex. Biochem J. 2001;356(Pt 1):247–256. doi: 10.1042/0264-6021:3560247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nees M, et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol. 2001;75(9):4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji H, et al. Antigen-specific immunotherapy for murine lung metastatic tumors expressing human papillomavirus type 16 E7 oncoprotein. Int J Cancer. 1998;78(1):41–45. doi: 10.1002/(sici)1097-0215(19980925)78:1<41::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 29.Yew CW, et al. A novel MLL5 isoform that is essential to activate E6 and E7 transcription in HPV16/18-associated cervical cancers. Cancer Res. 2011;71(21):6696–6707. doi: 10.1158/0008-5472.CAN-11-1271. [DOI] [PubMed] [Google Scholar]
- 30.Nin DS, et al. O-GlcNAcylation of MLL5β is essential for MLL5β-AP-1 transcription complex assembly at the HPV16/18-long control region. J Mol Cell Biol. 2015;7(2):180–183. doi: 10.1093/jmcb/mjv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4(6):a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuzyk A, Mai S. c-MYC-induced genomic instability. Cold Spring Harb Perspect Med. 2014;4(4):a014373. doi: 10.1101/cshperspect.a014373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gewin L, Galloway DA. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J Virol. 2001;75(15):7198–7201. doi: 10.1128/JVI.75.15.7198-7201.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Veldman T, Horikawa I, Barrett JC, Schlegel R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol. 2001;75(9):4467–4472. doi: 10.1128/JVI.75.9.4467-4472.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMurray HR, McCance DJ. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J Virol. 2003;77(18):9852–9861. doi: 10.1128/JVI.77.18.9852-9861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, et al. Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J Virol. 2008;82(23):11568–11576. doi: 10.1128/JVI.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vervoorts J, Lüscher-Firzlaff J, Lüscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem. 2006;281(46):34725–34729. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- 38.Yeh E, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6(4):308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 39.Dellas A, Schultheiss E, Leivas MR, Moch H, Torhorst J. Association of p27Kip1, cyclin E and c-myc expression with progression and prognosis in HPV-positive cervical neoplasms. Anticancer Res. 1998;18(6A):3991–3998. [PubMed] [Google Scholar]