

Abstract
Potassium Kir4.1/5.1 channels are abundantly expressed at the basolateral membrane of principal cells in the cortical collecting duct (CCD), where they are thought to modulate transport rates by controlling transepithelial voltage. Insulin and insulin-like growth factor-1 (IGF-1) stimulate apically localized epithelial sodium channels (ENaC) to augment sodium reabsorption in the CCD. However, little is known about their actions on potassium channels localized at the basolateral membrane. In this study, we implemented patch-clamp analysis in freshly isolated murine CCD to assess the effect of these hormones on Kir4.1/5.1 at both single channel and cellular levels. We demonstrated that K+-selective conductance via Kir4.1/5.1 is the major contributor to the macroscopic current recorded from the basolateral side in principal cells. Acute treatment with 10 μM amiloride (ENaC blocker), 100 nM tertiapin-Q (TPNQ; ROMK inhibitor), and 100 μM ouabain (Na+-K+-ATPase blocker) failed to produce a measurable effect on the macroscopic current. In contrast, Kir4.1 inhibitor nortriptyline (100 μM), but not fluoxetine (100 μM), virtually abolished whole cell K+-selective conductance. Insulin (100 nM) markedly increased the open probability of Kir4.1/5.1 and nortriptyline-sensitive whole cell current, leading to significant hyperpolarization of the basolateral membrane. Inhibition of the phosphatidylinositol 3-kinase cascade with LY294002 (20 μM) abolished action of insulin on Kir4.1/5.1. IGF-1 had similar stimulatory actions on Kir4.1/5.1-mediated conductance only when applied at a higher (500 nM) concentration and was ineffective at 100 nM. We concluded that both insulin and, to a lesser extent, IGF-1 activate Kir4.1/5.1 channel activity and open probability to hyperpolarize the basolateral membrane, thereby facilitating Na+ reabsorption in the CCD.
Keywords: sodium reabsorption, distal nephron, PI3-kinase, transepithelial transport, nortriptyline
the cortical collecting duct (CCD) plays a critical role in adaptation of water and electrolyte transport in response to dietary and endocrine stimuli to achieve systemic homeostasis (34). In this tubular segment, the dominant cell type, principal cells, exerts epithelial Na+ channel (ENaC)-mediated sodium reabsorption coupled to renal outer medullary potassium (ROMK)-dependent potassium secretion, whereas intercalated cells are mainly involved in regulation of acid-base balance and Cl− transport (34, 39). Inward rectifying Kir4.1 (KCNJ10) and Kir5.1 (KCNJ16) potassium channels are prominently expressed on the basolateral membrane of CCD principal cells (20, 45). Kir5.1, despite being unable to produce measurable current when expressed in heterologous systems (4), coassembles with Kir4.1 to form a heteromer with distinct biophysical properties (37). In fact, previous studies demonstrate that the Kir4.1/5.1 heteromer is the prevailing functional potassium channel at the basolateral membrane of the CCD principal cells (20, 45). Through coordinated actions with the Na+-K+-ATPase, this channel mediates potassium recycling across the basolateral membrane thereby modulating sodium reabsorption in distal parts of the renal nephron with the most established role in the distal convoluted tubule (14). The importance of Kir4.1/5.1 activity for Na+ transport in the connecting tubule (CNT) and CCD is less defined. Loss-of-function mutations in gene encoding Kir4.1 causes SeSAME/EAST syndrome in humans, leading to complex renal-based electrolyte imbalance, including salt wasting, hypocalciuria, hypomagnesemia, and hypokalemic metabolic alkalosis (3, 33). It is also thought that Kir4.1/5.1-mediated K+ efflux contributes to setting the resting basolateral membrane potential to establish a favorable electrochemical driving force for transport of electrolytes (40). However, the importance of the channel for electrical properties of CCD principal cells is not completely understood.
Apart from their actions on metabolism, insulin and structurally related insulin-like growth factor-1 (IGF-1) are known to regulate urinary excretion of electrolytes affecting tubular transport in different segments (1, 12, 13). Insulin and IGF-1 stimulate ENaC-mediated sodium reabsorption in native and immortalized CCD principal cells (16, 30, 35). The signaling mechanism involves activation of the respective insulin and IGF-1 receptors belonging to the receptor tyrosine kinase family and subsequent activation of phosphatidylinositol 3-kinase (PI3-kinase) (30, 35). Genetic deletion of insulin receptors decreases ENaC open probability (Po) without notable changes in ENaC protein abundance (27). Systemic administration of both insulin and IGF-1 in humans reduces urinary sodium excretion (5, 9). However, little is known about whether, in addition to their actions on ENaC, insulin and IGF-1 also affect basolateral membrane conductance and transepithelial voltage to modulate electrolyte transport.
In the current study, we report that the activity of the Kir4.1/5.1 channel is the major contributor of the macroscopic K+ current recorded from the basolateral side in principal cells from freshly isolated murine CCDs. Insulin and less efficiently IGF-1 acutely stimulate single-channel Kir4.1/5.1 Po, leading to a respective increase in whole cell K+ currents. PI3-kinase blockade abolishes actions of insulin and IGF-1 on Kir4.1/5.1. We also demonstrate that this regulation is capable of controlling basolateral membrane voltage, subsequently increasing the electrochemical driving force for sodium reabsorption in the CCD.
MATERIALS AND METHODS
Reagents and animals.
All chemicals and materials were from Sigma (St. Louis, MO), VWR (Radnor, PA), and Tocris (Ellisville, MO) unless noted otherwise and were at least of reagent grade. Animal use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Animal Care and Use Committees of the University of Texas Health Science Center at Houston and the Medical College of Wisconsin. For experiments, male C57BL/6J mice (Charles River Laboratories, Wilmington, MA), 6–10 wk old, were used. Animals were maintained on a standard rodent regimen and had free access to tap water.
Tissue isolation.
The procedure for isolation of the CCD suitable for electrophysiology is a modification from the protocols described previously (20–22, 44). Mice were euthanized by CO2 administration followed by cervical dislocation, and the kidneys were removed immediately. Kidneys were cut into thin slices (<1 mm) with slices placed into ice-cold physiological saline solution (PSS) containing (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35). Straight cortical-to-medullary sectors, containing ∼30–50 renal tubules, were isolated by microdissection using watchmaker forceps under a stereomicroscope. Isolated sectors were further incubated in PSS containing 0.8 mg/ml collagenase type I (Alfa Aesar, Ward Hill, MA) and 5 mg/ml of dispase II (Roche Diagnostics, Mannheim, Germany) for 20 min at 37°C followed by extensive washout with PSS. Individual CCDs were visually identified by their morphological features (pale color, coarse surface, and, in some cases, bifurcations) and were mechanically isolated from the sectors by microdissection. Isolated CCDs were attached to a 5 × 5-mm coverglass coated with poly-l-lysine. A coverglass containing a CCD was placed in a perfusion chamber mounted on an inverted Nikon Eclipse Ti microscope and perfused with PSS at room temperature. The tubules were used within 1–2 h after isolation.
Immunohistochemistry.
Kidneys were fixed in 10% formalin and processed for paraffin embedding. Kidney sections (4 μm) were double-labeled with rabbit anti-Kir5.1 (SAB4501636, Sigma) and aquaporin-2 water channel (AQP2; sc-28629, Santa Cruz Biotechnology, Dallas, TX) or rabbit anti-Kir4.1 (APC-035, Alomone Labs) and thiazide-sensitive Na+-Cl− cotransporter (NCC; AB3553, Millipore, Billerica, MA) antibodies. Bindings were revealed with Alexa Fluor 488 or 643 with goat anti-rabbit biotinylated IgG (Molecular Probes, Waltham, MA). Immunostaining was performed in tissues from at least four different kidneys. All tissue sections were examined by TCS SP5 confocal laser-scanning microscopy (Leica, Buffalo Grove, IL).
Whole cell currents and membrane potential.
Whole cell currents in CCD cells were measured under voltage-clamp conditions in the perforated-patch mode with GΩ seals formed on the basolateral membrane. All patch-clamp data were acquired with an Axopatch 200B (Molecular Devices, Sunnyvale, CA) patch-clamp amplifier interfaced via a Digidata 1440 (Molecular Devices) to a computer running pClamp 10.4 (Molecular Devices). The bath solution was (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35). Freshly made amphotericin-B (400 μM, Enzo Life Sciences, Farmingdale, NY) was dissolved in the pipette solution containing 150 mM K acetate, 5 mM KCl, 2 mM MgCl2, and 10 mM HEPES (pH 7.35) by ultrasonication. Electrical recordings were made once the access resistance from the pipette to the cell interior fell to <15 MΩ, usually 5–10 min after achievement of a pipette-to-membrane seal resistance of 5–10 GΩ. Capacity of individual principal cells was on average 15 pF and was manually compensated. Real-time changes in membrane voltage in CCD cells were studied under current-clamp mode using the perforated-patch technique as was described previously (43, 45).
Single-channel recordings.
Single-channel activity of Kir4.1/5.1 in CCD cells was determined in cell-attached patches on the basolateral membrane made under voltage-clamp conditions. Recording pipettes had resistances of 8–10 MΩ. Bath and pipette solutions were (in mM) 150 NaCl, 5 mM KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.35) and 150 mM KCl, 2 mM MgCl2, and 10 mM HEPES (pH 7.35), respectively. In the cell-attached configuration, the actual voltage applied to a membrane patch (Vpatch) is a sum of the pipette voltage and the resting basolateral membrane potential (Vbasolateral) of principal cells, which is close to −70 mV for principal and −15 mV for intercalated cells (see Fig. 1).
Fig. 1.
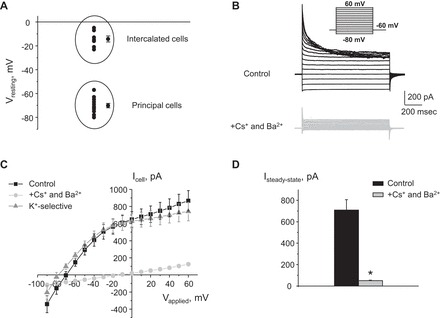
Characterization of K+-selective conductance in cortical collecting duct (CCD) principal cells. A: summary graph of resting membrane potentials in cells from freshly isolated CCDs. Two cell populations representing principal (with high negative resting membrane voltage) and intercalated (with low negative resting membrane voltage) are highlighted in circles. B: representative macroscopic currents in individual principal cells in response to voltage steps from −80 to +60 (shown in inset) from the holding potential of −60 mV in the control (black) and upon equimolar substitution of intracellular K+ with Cs+ and in the presence of 30 μM Ba2+ in extracellular medium (light grey). C: current-voltage (I–V) relationships obtained from voltage-step protocols, as shown in B in control (black) and in the presence of Cs+ and Ba2+ (light grey). K+-selective current (grey) was calculated as the difference in the aforementioned conditions. D: summary graph of amplitude of the steady-state current estimated as the absolute current values at the applied voltage of +20 mV. *Significant decrease vs. control.
For paired patch-clamp experiments, pipette voltage was −Vp = −40 mV. Current recordings were made in a permanently perfused bath (1.5 ml/min). Drug administration times are shown with bars on the top of representative traces. All applied reagents were prepared as 1,000× stock solutions in water or dimethyl sulfoxide (DMSO) as instructed. There were no changes in single-channel activity and macroscopic current amplitude during vehicle application. For each experimental condition, CCDs from at least three different mice were assayed. Currents were low-pass filtered at 1 kHz with an eight-pole Bessel filter (Warner Instruments, Hamden, CT). Events were inspected visually before acceptance. In paired experiments, Kir4.1/5.1 activity was analyzed over a span of 60–120 s for each experimental condition after reaching a new steady state in response to a treatment. Channel activity (NPo) and Po were assessed using Clampfit 10.4. For calculating Po in paired experiments, N was fixed as the greatest number of active channels observed in control or experimental conditions. For representation, current traces were filtered at 200 Hz and corrected for a slow baseline drift as necessary.
Data analysis.
All summarized data are reported as means ± SE. In paired experiments, data from before and after treatment were compared using the paired t-test. Data from unpaired experiments were compared with a Student's (two-tailed) t-test or a one-way ANOVA as appropriate. P < 0.05 was considered significant.
RESULTS
Basolateral K+ conductance via Kir4.1 and Kir4.1/5.1 is the major contributor to the whole cell current recorded from the basolateral side in CCD principal cells.
The CCD is known to contain principal (∼70% from total cell population) and intercalated (the remaining 30%) cells, which are presumably not electrically coupled (41). Indeed, we detected two independent cell populations maintaining resting membrane potentials of −70.2 ± 1.9 mV (n = 12) and −14.3 ± 2.5 mV (n = 7), as assessed in current clamp mode in patches on the basolateral membrane of freshly isolated CCDs (Fig. 1A). These values are close to the estimated reversal potentials for K+ (first group) and Cl− (second group) assuming physiological distribution of the ions. These groups are consistent with principal and intercalated cells known to have predominantly potassium and chloride basolateral conductance, respectively (23). In further studies, we focused exclusively on principal cells. A typical whole cell current in response to a voltage-step protocol is shown in Fig. 1B. As is clear from the respective current-voltage relationship (I–V) in Fig. 1C, whole cell current demonstrates a noticeable rectification at negative voltages. Extracellular application of 30 μM Ba2+ and equimolar substitution of intracellular K+ by Cs+ to block K+ channels drastically reduce the amplitude of the steady-state current, directly indicating its K+-dependent nature (Fig. 1, B and D).
The basolateral membrane of principal cells is known to contain Kir4.1/5.1 potassium channels and electrogenic Na+-K+-ATPase (40). Indeed, Fig. 2 demonstrates the immunolocalization of Kir4.1 and Kir5.1 channels on the basolateral membrane in mouse kidney cortex. Double immunofluorescence staining studies using antibodies against Kir5.1 and the collecting duct-specific marker, water channel AQP2, revealed colocalization within the CCD. In addition, there is a clear staining in some tubules not positive to AQP2 (Fig. 2A). Double immunofluorescence staining with the specific marker of the distal convoluted tubule (DCT), thiazide-sensitive NCC, and Kir4.1 revealed colocalization of Kir4.1 with NCC in the DCT. Similarly, there is staining in tubules not positive for NCC, which most likely represents expression in the CCD (Fig. 2B). No positive staining was observed in glomeruli and proximal tubules. Therefore, both Kir4.1 and Kir5.1 proteins appeared localized in the basolateral membranes of CCD and DCT, as it was reported previously (20).
Fig. 2.

Immunolocalization of Kir4.1 and Kir5.1 channels in the renal cortex. A: representative micrograph of double-labeled kidney cortex sections stained for Kir5.1 (red) and aquaporin-2 (AQP2; green) demonstrating colocalization of Kir5.1 with AQP2 on the basolateral membrane of CCD. Staining for Kir5.1 in distal convoluted tubules (DCT) is also apparent. B: representative micrograph of double-labeled kidney cortex sections stained for Kir4.1 (red) and sodium-chloride cotransporter (NCC; green) showing colocalization of Kir4.1 protein with NCC in the DCT. Staining for Kir4.1 in the CCD is also present. G, glomeruli; PT, proximal tubules. Scale bars = 20 μM.
Both electrical currents across the apical and basolateral membranes can contribute to the whole cell current. We next determined their relative contribution upon formation of a patch on the basolateral membrane. Amiloride is known to inhibit ENaCs, thereby eliminating the driving force for the apically localized ROMK channels. However, treatment with amiloride (10 μM for 3 min) has no apparent effect on the amplitude of the whole cell current (Fig. 3, A and B), suggesting a minor contribution of the apical membrane, and the recorded whole cell current represents chiefly basolateral conductance. Consistently, inhibition of the apically localized ROMK channels with a selective antagonist, tertiapin-Q (TPNQ; 100 nM for 3 min), also does not inhibit the whole cell current (Fig. 3, A and C). These results support the concept that the basolateral membrane has much higher conductance than the apical and therefore the monitored whole cell current flows predominantly across the basolateral membrane.
Fig. 3.
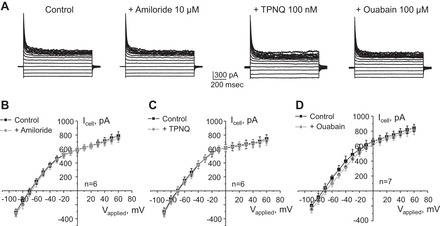
The basolateral Kir4.1/5.1 channel is the major contributor to the macroscopic current in CCD principal cells. A: representative current traces in response to voltage-step protocol as shown in Fig. 1B in control and after 3-min pretreatment with epithelial sodium channel (ENaC) blocker amiloride, renal outer medullary potassium (ROMK) inhibitor tertiapin-Q (TPNQ), and Na+-K+-ATPase blocker ouabain. Also shown are I–V relationships in control and upon blockade of ENaC (B), ROMK (C), and Na+-K+ ATPase (D). Numbers of individual experiments are also shown.
To further explore the molecular identity underlying basolateral K+ conductance, we treated freshly isolated CCDs with the pump blocker ouabain (100 μM for 3 min). We detected just a minor trend toward a reduction of the whole cell current and depolarization, which was not statistically significant (Fig. 3, A and D). Prolonged treatment with ouabain has detrimental consequences for cellular health, which precluded patch-clamp analysis of this condition. This is consistent with the view that the activity of Kir4.1 and Kir5.1 potassium channels on the basolateral membrane is the chief contributor to the whole cell current in PC of freshly isolated CCDs.
There are no specific commercially available inhibitors of Kir4.1/5.1 channels, although it was previously reported that fluoxetine (serotonin reuptake blocker) and nortriptyline (tricyclic antidepressant) are potent inhibitors of homomeric Kir4.1 channels heterologously expressed in HEK293T cells (25, 36). Whereas fluoxetine (100 μM for 4 min) had only a minor effect (Fig. 4A), nortriptyline (100 μM for 4 min) virtually abolished macroscopic whole cell current in CCD principal cells (Fig. 4B). These results are consistent with the concept that fluoxetine affects only monomeric Kir4.1 and nortriptyline can also target Kir4.1/5.1 channels. Furthermore, our observations strongly argue that activity of the heteromeric Kir4.1/5.1 is the major contributor to the K+-selective whole cell current recorded from the basolateral side in CCD principal cells.
Fig. 4.
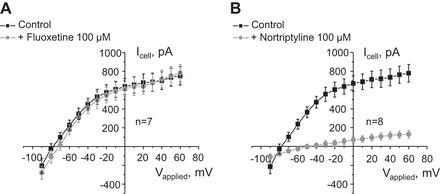
Nortriptyline but not fluoxetine drastically decreases the macroscopic K+-selective conductance in CCD principal cells. Shown are average I–V relationships of macroscopic whole cell current in CCD principal cells in control and upon application of Kir4.1 inhibitors 100 μM fluoxetine (A) and 100 μM nortriptyline (B) for 4 min. Numbers of individual experiments are shown.
Insulin acutely increases activity of basolateral Kir4.1/5.1 in CCD principal cells in a PI3-kinase-dependent manner.
Insulin, by acting on its basolateral receptors, is known to stimulate ENaC activity in principal cells to promote Na+ reabsorption (35). We next asked whether insulin also modulates activity of basolateral K+ channels to affect transepithelial voltage. Acute insulin application significantly increases the amplitude of whole cell current from 688 ± 119 to 1,096 ± 52 pA during the steady state (Fig. 5, A and B). Insulin washout returned the current to control values of 729 ± 77 pA, indicating a reversible nature of this stimulation. Figure 5C shows the average I–V relationships of the whole cell current of PCs in control and during insulin treatment. Of note, insulin also causes a shift of the reversal potential toward more negative values (Fig. 5C), which is consistent with hyperpolarization of the basolateral membrane. Importantly, stimulatory actions of insulin were precluded in the presence of nortriptyline (Fig. 6, A and B). This suggests that activation of Kir4.1/5.1 plays a central role in augmentations of whole cell current in response to insulin.
Fig. 5.
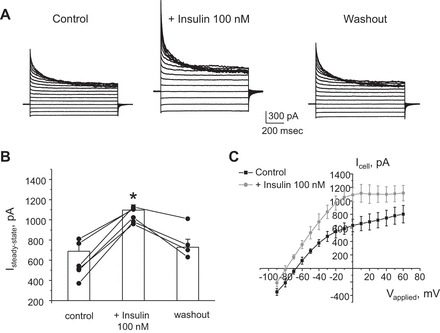
Insulin reversibly augments macroscopic currents in CCD principal cells. A: representative whole cell current recordings in response to voltage-step protocol as shown in Fig. 1B in control, after 3-min treatment with insulin (100 nM), and followed washout with regular medium. B: summary graph of changes in amplitude of the steady-state current estimated as the absolute current values at applied voltage of +20 mV in paired experiments similar to that shown in A. *Significant increase vs. control. C: average I–V relationships from individual CCD principal cells in control (black) and after treatment with insulin (light grey). Of note, insulin treatment induces a leftward shift of the reversal potential.
Fig. 6.
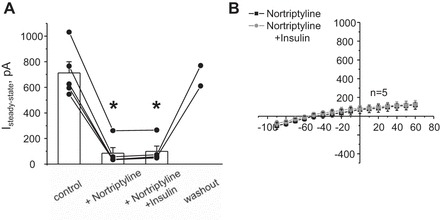
Inhibition of Kir4.1/5.1 channels with nortriptyline abolishes stimulatory actions of insulin on the macroscopic whole cell current in CCD principal cells. A: summary graph of changes in amplitude of the steady-state current estimated as the absolute current values at applied voltage of +20 mV in control, after pretreatment with 100 μM nortriptyline, simultaneous application of 100 nM insulin and 100 μM nortriptyline, then washout with control media. *Significant decrease vs. control. B: average I–V relationships from individual CCD principal cells pretreated with 100 μM nortriptyline at baseline and in the presence of insulin (light grey).
We next directly monitored insulin actions on this channel using a cell-attached configuration in freshly isolated CCDs. Similarly as we and other described previously (20, 45), heteromeric Kir4.1/5.1 channels were identified by their fast kinetics, estimated single-channel conductance of ∼40 pS, and high selectivity for potassium. As demonstrated in a representative continuous experiment (Fig. 7A) and the summary graph (Fig. 7B), insulin (100 nM) reversibly increases channel Po from 0.34 ± 0.03 to 0.45 ± 0.03. Of note, we have also detected a small insulin-induced increase (∼10%) in the unitary current amplitude (Fig. 7A, regions 1 and 2), indicating hyperpolarization of the basolateral membrane. Pretreatment with the PI3-kinase inhibitor LY294002 (20 μM for 3 min) has no effect on basal Kir4.1/5.1 activity but abolishes the stimulatory effect of insulin (Fig. 7C), suggesting that insulin increases Kir4.1/5.1 activity in a PI3-kinase dependent manner.
Fig. 7.
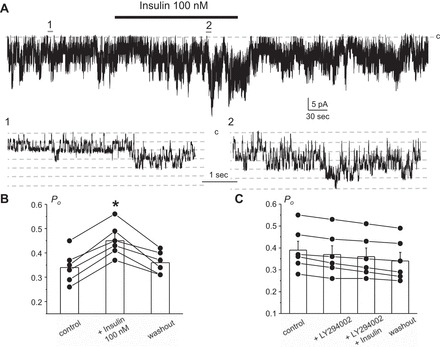
Insulin acutely increases open probability (Po) of Kir4.1/5.1 channel via activation of PI3-kinase. A: representative continuous current trace from a cell-attached patch monitoring activity of basolateral Kir4.1/5.1 channels in CCD principal cells in control, after application of 100 nM insulin, and followed washout with the control medium. The patch was clamped to −Vp = −40 mV. Insulin application time is shown by the bar on top. Areas 1 and 2 are shown below at an expanded timescale. Insulin treatment mildly increases the unitary current amplitude, indicative of basolateral membrane hyperpolarization. B: summary graph of changes in Kir4.1/5.1 Po upon treatment with 100 nM insulin from paired patch-clamp experiments similar to that shown in A. *Significant increase vs. control. C: summary graph of changes in Kir4.1/5.1 Po in paired cell-attached patch-clamp experiments in control, upon treatment with phosphatidylinositol 3-kinase (PI3)-K inhibitor LY294002 (20 μM), insulin (100 nM) in the continued presence of the inhibitor, and following washout.
IGF-1 is less potent to stimulate Kir4.1/5.1 activity in CCD cells.
In addition to insulin, IGF-1 also has a stimulatory effect on ENaC-mediated Na+ reabsorption in the CCD (35). However, application of 100 nM IGF has no effect on the whole cell current and Kir4.1/5.1 activity (data not shown). In contrast, a higher concentration of IGF-1 (500 nM) is capable of reversibly stimulating the amplitude of whole cell current from 694 ± 79 to 1,110 ± 80 pA during the steady state (Fig. 8, A and B). The respective I–V relationships for whole cell current in control and during IGF-1 treatment are shown in Fig. 8C. A consistent stimulatory effect of a high IGF-1 concentration (500 nM) is also apparent at the single-channel level, increasing Kir4.1/5.1 Po from 0.31 ± 0.03 to 0.44 ± 0.03 (Fig. 9, A and B). Again, pretreatment with PI3-kinase inhibitor LY294002 (20 μM for 3 min) abolishes the effect of IGF-1 (Fig. 9C). Overall, we concluded that IGF-1, while at a higher concentration, produces a similar stimulatory action on Kir4.1/5.1-mediated current in the CCD cells as insulin.
Fig. 8.
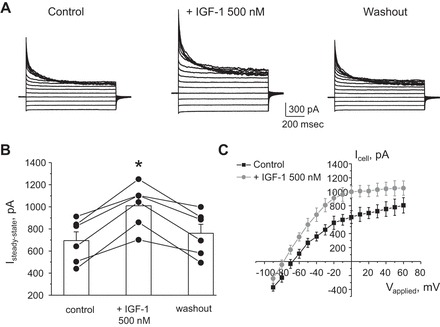
High levels of IGF-1 stimulate macroscopic currents in CCD principal cells. A: representative whole cell current recordings in response to voltage-step protocol as shown in Fig. 1B in control, after 3 min treatment with IGF-1 (500 nM), and followed washout with regular medium. B: summary graph of changes in amplitude of the steady-state current estimated as the absolute current values at applied voltage of +20 mV in paired experiments similar to that shown in A. *Significant increase vs. control. C: average I–V relationships from individual CCD principal cells in control (black) and after treatment with IGF-1 (light grey). Similarly to the effect of insulin in Fig. 5C, IGF-1 application shifts the reversal potential to more negative values.
Fig. 9.
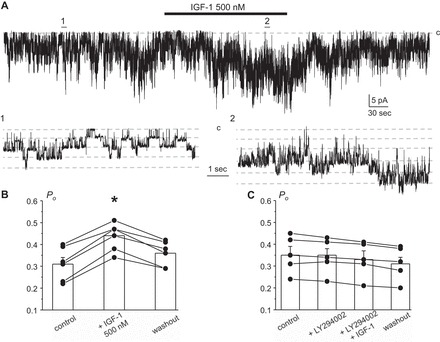
High concentration of IGF-1 augments Kir4.1/5.1 single-channel activity in a PI3-kinase dependent manner. A: representative continuous current trace from a cell-attached patch monitoring activity of basolateral Kir4.1/5.1 channels in CCD principal cells in control, after application of 500 nM IGF-1, and followed washout with the control medium. The patch was clamped to −Vp = −40 mV. IGF-1 application time is shown with the bar on top. Areas 1 and 2 are shown below at an expanded timescale. IGF-1 treatment mildly increases the unitary current amplitude, indicative of basolateral membrane hyperpolarization. B: summary graph of changes in Kir4.1/5.1 Po upon treatment with 500 nM IGF-1 from paired patch-clamp experiments similar to that shown in A. *Significant increase vs. control. C: summary graph of changes in Kir4.1/5.1 Po in paired cell-attached patch-clamp experiments in control, upon treatment with PI3-K inhibitor LY294002 (20 μM), IGF-1 (500 nM) in the continued presence of the inhibitor, and following washout.
Insulin and IGF-1 hyperpolarize the basolateral membrane of PC in the CCD.
Our results (Figs. 5C and 8C) demonstrate that insulin and IGF-1 elicit a leftward shift of the whole cell current reversal potential. Furthermore, we detected a small increase in the Kir4.1/5.1 unitary current amplitude during treatment with both hormones (Figs. 7A and 9A). This is consistent with an effect of insulin and IGF-1 on the basolateral membrane voltage in CCD principal cells. To directly probe this, we continuously monitored real-time change in the resting membrane potential using patch-clamp electrophysiology in current-clamp mode. Insulin (Fig. 10A) and IGF-1 (Fig. 10B) elicit an acute hyperpolarization of the basolateral membrane from −67.6 ± 1.7 to −76.0 ± 2.2 and −73.8 ± 2.2 mV, respectively (Fig. 10C). Short applications of 50 mM KCl at the end of experiments were used to test viability of cells and to verify that K+ conductance is the major contributor of the resting membrane potential.
Fig. 10.
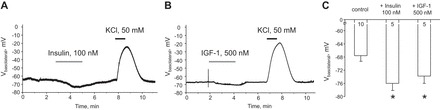
Insulin and IGF-1 hyperpolarize basolateral membrane in CCD principal cells. Representative continuous voltage traces monitoring basolateral membrane potential in individual cells in control and after application of 100 nM insulin (A) and 500 nM IGF-1 (B). Short applications of 50 mM KCl were used to assess cell viability. Drug application times are shown with respective bars on the top. C: summary graph of the resting basolateral membrane potential in control and after acute application of insulin and IGF-1. *Significant increase vs. control.
DISCUSSION
We have implemented patch-clamp analysis at both cellular and single-channel levels to reveal a novel molecular target, the basolateral Kir4.1/5.1 channels, in the insulin and IGF-1 signaling network in the CCD. Using native tissue preparations, we demonstrate that insulin dynamically regulates Kir4.1/5.1 channel Po in principal cells and the process depends on activation of PI3-kinase. At the cellular level, this transforms into increased K+ conductance due to augmented basolateral K+ recycling and hyperpolarization of the basolateral membrane. IGF-1, while at a higher concentration, produces a similar effect. We propose that, in addition to their well-described direct effects on ENaC, both hormones also increase transepithelial voltage through stimulation of Kir4.1/5.1 channels to further promote Na+ reabsorption and set up a favorable driving force for K+ secretion and Cl− reabsorption.
In this study, we were able to establish how acute changes in Kir4.1/5.1 channel activity contribute to altered electrical properties of CCD principal cells. In contrast to current recordings at the single-channel level, whole cell analysis is harder to interpret due to anatomical and architectural features of the CCD. This segment contains two different functional cell populations: the majority (∼70%) of them are principal and the remaining (∼30%) are intercalated cells. CCD cells are connected to each other by tight junctions. Previous studies argue against electrical coupling between different cells in the CCD (23, 41). Consistently, we can reliably distinguish two cell types based on their basolateral membrane potential: one of them (principal cells) has a resting potential close to the equilibrium potential for K+ (∼ −70 mV), whereas in the other group (intercalated cells) resting potential seems to be determined primarily by Cl− (−15 mV; see Fig. 1A). This is in a good agreement with earlier observations that basolateral membranes of principal and intercalated cells have predominantly potassium and chloride conductance, respectively (23). Polarization of CCD cells is another technical difficulty to overcome. It was assessed that potassium conductance of the basolateral membrane in principal cells is greater than that of the apical membrane by a factor of 10 (11). Thus the measured whole cell current supposedly reflects ion fluxes across the basolateral membrane, and the contribution of the apical membrane should be minimal. Indeed, we did not observe noticeable changes in the amplitude of the whole cell current in principal cells upon inhibition of ENaC and ROMK channels (Fig. 3). One may argue that limited diffusion into the collapsed lumen of a nonopened CCD may explain inefficiency of the antagonists to affect the recorded macroscopic current. However, we isolated CCDs of only several millimeters in length and we did not observe a decrease in current in cells located close to the tubular sides upon inhibition of the apical conductance. Interestingly, Palmer and colleagues (11) did see a moderate decrease in whole cell current amplitude in response to the ROMK inhibitor TPNQ (100 nM) when patches were made on the apical side. Thus it is possible that the presence of the basal lamina, which needs to be otherwise enzymatically digested to make a seal on the basolateral side, blunts conductive properties of the basolateral membrane. In support of this assumption, the same group did not detect an effect of TPNQ in non-split-open CCDs (8). In any case, it does not seem accurate to precisely quantify changes in the apical conductance due to the apparent shunting effect of the basolateral membrane.
We also did not detect any significant contribution of the electrogenic Na+-K+ ATPase to the electrical conductance in CCD principal cells (Fig. 3). A high concentration of ouabain acutely produced only a tendency to reduce whole cell current and did not elicit an apparent depolarization. Our results are coherent with previous studies estimating the current generated by the pump in CCD as ∼35 pA/cell (26), suggesting that Na+-K+-ATPase activity is essential for establishing ion gradients but not for dynamic ion fluxes. Indeed, we did observe cell swelling and decay if ouabain was applied for >30 min. We and others reported that heteromeric Kir4.1/5.1 is the most commonly observed K+ channel in the basolateral membrane of CCD principal cells (20, 45). While there are no specific pharmacological tools targeting Kir4.1/5.1 activity, previous studies documented that the serotonin reuptake blocker fluoxetine and an antidepressant, nortriptyline, inhibited homomeric Kir4.1 channels overexpressed in HEK293T cells, while having no effect on Kir1.1 (ROMK1) channels (25, 36). Intriguingly, we observed discrete effects of these drugs on the macroscopic K+-selective current in CCD principal cells (Fig. 4). While fluoxetine failed to significantly affect current, nortriptyline drastically inhibited macroscopic K+ conductance by >80%, and this action was at least partially reversible (Fig. 6B). The most likely explanation is that fluoxetine targets only homomeric Kir4.1, while nortriptyline can also block heteromeric Kir4.1/5.1. This also suggests that homomeric Kir4.1 has a minor contribution to whole cell conductance, supporting the view that Kir4.1/5.1 is the most abundant K+ channel in CCD principal cells (20, 45). Of course, these drugs cannot be effectively used as Kir4.1/5.1 inhibitors in in vivo settings due to their low potency (100-μmol range). This likely explains why no apparent renal-wasting phenotype, similar to that during SeSAME/EAST syndrome, has been observed upon systemic administration of nortriptyline and fluoxetine to treat depression (2). Moreover, the adverse effects associated with liver and kidney toxicity upon nortriptyline overdose were reported during treatment of depression in humans (10, 28). It is also worth mentioning that structural differences between fluoxetine and nortriptyline might be taken into consideration upon development of future pharmacological inhibitors discriminating between Kir4.1 and Kir4.1/5.1.
We found that insulin and IGF-1 stimulated Kir4.1/5.1 channel Po (Figs. 7, 9) and augmented the macroscopic K+-selective current (Figs. 5 and 8) in CCD principal cells. The observation that higher concentrations of IGF-1 are necessary to stimulate the channel likely indicates that IGF-1 targets insulin receptors to which it has lower affinity (18, 32). The situation might be even more complex assuming that hybrid receptors also exist with their own binding profiles, making insulin and IGF-1 signaling interconnected. Consistently, a functional cross talk between insulin and IGF-1 receptors in regulation of Na+ reabsorption in immortalized mpkCCDc14 cells was recently demonstrated (16). A possibility exists that activation of a component which is not active at baseline conditions might also contribute to the increased whole cell current. Insulin was also reported to increase ouabain-sensitive Rb+ uptake, suggesting a direct action on Na+-K+-ATPase via an unknown mechanism (6). In these studies, insulin induced up to 50% an increase in activity of the pump after 15 min of incubation. Thus it is unlikely that this effect would substantially contribute to the augmented whole cell current due to a minor ability of Na+-K+-ATPase to generate current, as was discussed in the previous paragraph. Whether IGF-1 has a similar effect on pump activity was not tested. Importantly, we found that insulin failed to significantly increase the amplitude of whole cell current in the presence of the Kir4.1/5.1 inhibitor nortriptyline (Fig. 6). This unequivocally suggests that activation of Kir4.1/5.1 is the dominant component in insulin-induced increases in basolateral membrane conductance. Furthermore, we found a strong correlation between the amplitude of the stimulatory effect of insulin and IGF-1 on single-channel Kir4.1/5.1 activity (Figs. 7 and 9) and macroscopic K+ current (Figs. 5 and 8).
We revealed that activation of PI3-kinase mediates actions of insulin and IGF-1 (Figs. 7 and 9). Insulin and IGF-1 receptors are known to possess tyrosine kinase activity and trigger activation of PI3-kinase and MAPK signaling cascades in kidney cells (13). Both hormones were reported to increase ENaC activity in split-open CCDs via a mechanism involving PI3-kinase activation and subsequent generation of phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] (35). It is currently not known whether a similar molecular mechanism is involved in a PI3-kinase-dependent increase in Kir4.1/5.1 Po in response to insulin and IGF-1. While a direct effect of PI(3,4,5)P3 on Kir4.1/5.1 has not been tested, a similar phosphoinositide, PI(4,5)P2, is known to be a key regulator of gating properties of inwardly rectifying potassium channels, such as Kir4.1/5.1 (29). In addition, increased reactive oxygen species (ROS) production was suggested to mediate stimulatory effects of insulin and IGF-1 on ENaC-mediated sodium reabsorption in the CCD (17). Again, it is not clear whether ROS directly modulate Kir4.1/5.1 activity. Increased ROS production in retinal Müller cells due to chronic exposure to high glucose to mimic diabetic conditions was actually accompanied by decreased Kir4.1 expression (42), but the relationship is probably not direct. Thus further studies are necessary to precisely determine the molecular mechanism of PI3-kinase-dependent stimulation of Kir4.1/5.1 activity in CCD principal cells.
An important physiological consequence of insulin and IGF-1 action on Kir4.1/5.1 activity is that it dynamically controls basolateral membrane resting potential and, by extension, transepithelial voltage (Fig. 10). Increased driving force will further facilitate ENaC-mediated sodium reabsorption as well as coordinated Cl− and K+ transport in the CCD. We have recently documented that IGF-1 stimulates whereas insulin inhibits activity of the basolateral ClC-K2 chloride channel in intercalated cells (43). The channel is proposed to be involved in Cl− reabsorption in the CCD (19, 24, 38). Therefore, increased insulin levels will stimulate Na+/K+ exchange, leading to kaliuresis (15, 31). Of interest, a recent report suggested a stimulatory effect of insulin on ROMK (7). In contrast, IGF-1 will favor NaCl retention and reduction of fractional K+ excretion (9). Thus understanding mechanisms controlling basolateral ion channels, such as Kir4.1/5.1 and ClC-K2, in the CCD would allow uncoupling of NaCl reabsorption from potassium secretion in the distal renal tubule. Furthermore, it is conceivable to propose that development of pharmacological tools targeting Kir4.1/5.1 activity might be beneficial to preventing renal sodium retention in clinically relevant cases associated with elevated insulin (such as type II diabetes) and IGF-1 (acromegaly) levels.
GRANTS
This research was supported by National Institutes of Health Grants DK095029 (to O. Pochynyuk) and HL108880 (to A. Staruschenko), American Heart Association Grant 15SDG25550150 (to M. Mamenko), and MCW Neuroscience Research Center/Advancing a Healthier Wisconsin pilot grant 9520217 (to O. Palygin).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: O.Z., O. Palygin, V.T., and M.M. performed experiments; O.Z., O. Palygin, M.M., and O. Pochynyuk analyzed data; O.Z., O. Palygin, V.T., A.S., and O. Pochynyuk interpreted results of experiments; O.Z., O. Palygin, A.S., and O. Pochynyuk prepared figures; O.Z., O. Palygin, V.T., M.M., A.S., and O. Pochynyuk edited and revised manuscript; O.Z., O. Palygin, V.T., M.M., A.S., and O. Pochynyuk approved final version of manuscript; A.S. and O. Pochynyuk provided conception and design of research; A.S. and O. Pochynyuk drafted manuscript.
ACKNOWLEDGMENTS
Christine Duris (Children's Hospital of Wisconsin) is recognized for excellent technical assistance with immunostaining.
REFERENCES
- 1.Bach LA, Hale LJ. Insulin-like growth factors and kidney disease. Am J Kidney Dis 65: 327–336, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Bergstrom RF, Beasley CM Jr, Levy NB, Blumenfield M, Lemberger L. The effects of renal and hepatic disease on the pharmacokinetics, renal tolerance, and risk-benefit profile of fluoxetine. Int Clin Psychopharmacol 8: 261–266, 1993. [DOI] [PubMed] [Google Scholar]
- 3.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van't Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bond CT, Pessia M, Xia XM, Lagrutta A, Kavanaugh MP, Adelman JP. Cloning and expression of a family of inward rectifier potassium channels. Receptors Channels 2: 183–191, 1994. [PubMed] [Google Scholar]
- 5.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest 55: 845–855, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feraille E, Rousselot M, Rajerison R, Favre H. Effect of insulin on Na+,K+-ATPase in rat collecting duct. J Physiol 488: 171–180, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frindt G, Palmer LG. Effects of insulin on Na and K transporters in the rat CCD. Am J Physiol Renal Physiol 302: F1227–F1233, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frindt G, Shah A, Edvinsson J, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am J Physiol Renal Physiol 296: F347–F354, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giordano M, DeFronzo RA. Acute effect of human recombinant insulin-like growth factor I on renal function in humans. Nephron 71: 10–15, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Glazener CM, Evans JH, Peto RE. Tricyclic and related drugs for nocturnal enuresis in children. Cochrane Database Syst Rev: CD002117, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Gray DA, Frindt G, Zhang YY, Palmer LG. Basolateral K+ conductance in principal cells of rat CCD. Am J Physiol Renal Physiol 288: F493–F504, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Hale LJ, Coward RJ. The insulin receptor and the kidney. Curr Opin Nephrol Hypertens 22: 100–106, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Hale LJ, Coward RJ. Insulin signalling to the kidney in health and disease. Clin Sci 124: 351–370, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton KL, Devor DC. Basolateral membrane K+ channels in renal epithelial cells. Am J Physiol Renal Physiol 302: F1069–F1081, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoekstra M, Yeh L, Lansink AO, Vogelzang M, Stegeman CA, Rodgers MG, van der Horst IC, Wietasch G, Zijlstra F, Nijsten MW. Determinants of renal potassium excretion in critically ill patients: the role of insulin therapy. Crit Care Med 40: 762–765, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Ilatovskaya DV, Levchenko V, Brands MW, Pavlov TS, Staruschenko A. Cross-talk between insulin and IGF-1 receptors in the cortical collecting duct principal cells: implication for ENaC-mediated Na+ reabsorption. Am J Physiol Renal Physiol 308: F713–F719, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilatovskaya DV, Pavlov TS, Levchenko V, Staruschenko A. ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am J Physiol Cell Physiol 304: C102–C111, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JJ, Accili D. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res 12: 84–90, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol 12: 1327–1334, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294: F1398–F1407, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Mamenko M, Zaika O, Doris PA, Pochynyuk O. Salt-dependent inhibition of epithelial Na+ channel-mediated sodium reabsorption in the aldosterone-sensitive distal nephron by bradykinin. Hypertension 60: 1234–1241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muto S, Yasoshima K, Yoshitomi K, Imai M, Asano Y. Electrophysiological identification of alpha- and beta-intercalated cells and their distribution along the rabbit distal nephron segments. J Clin Invest 86: 1829–1839, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nissant A, Paulais M, Lachheb S, Lourdel S, Teulon J. Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am J Physiol Renal Physiol 290: F1421–F1429, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Ohno Y, Hibino H, Lossin C, Inanobe A, Kurachi Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res 1178: 44–51, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Palmer LG, Antonian L, Frindt G. Regulation of the Na-K pump of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102: 43–57, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, Staruschenko A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J 27: 2723–2732, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prince JB, Wilens TE, Biederman J, Spencer TJ, Millstein R, Polisner DA, Bostic JQ. A controlled study of nortriptyline in children and adolescents with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol 10: 193–204, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Rapedius M, Paynter JJ, Fowler PW, Shang L, Sansom MS, Tucker SJ, Baukrowitz T. Control of pH and PIP2 gating in heteromeric Kir4.1/Kir51 channels by H-bonding at the helix-bundle crossing. Channels 1: 327–330, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Record RD, Froelich LL, Vlahos CJ, Blazer-Yost BL. Phosphatidylinositol 3-kinase activation is required for insulin-stimulated sodium transport in A6 cells. Am J Physiol Endocrinol Metab 274: E611–E617, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Rossetti L, Klein-Robbenhaar G, Giebisch G, Smith D, DeFronzo R. Effect of insulin on renal potassium metabolism. Am J Physiol Renal Fluid Electrolyte Physiol 252: F60–F64, 1987. [DOI] [PubMed] [Google Scholar]
- 32.Roth J, Kahn CR, Lesniak MA, Gorden P, De Meyts P, Megyesi K, Neville DM Jr, Gavin JR 3rd, Soll AH, Freychet P, Goldfine ID, Bar RS, and Archer JA. Receptors for insulin, NSILA-s, and growth hormone: applications to disease states in man. Recent Prog Horm Res 31: 95–139, 1975. [DOI] [PubMed] [Google Scholar]
- 33.Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staruschenko A. Regulation of transport in the connecting tubule and cortical collecting duct. Compr Physiol 2: 1541–1584, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Su S, Ohno Y, Lossin C, Hibino H, Inanobe A, Kurachi Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J Pharmacol Exp Ther 320: 573–580, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir41. J Physiol 525: 587–592, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchida S, Sasaki S. Function of chloride channels in the kidney. Annu Rev Physiol 67: 759–778, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Wall SM, Weinstein AM. Cortical distal nephron Cl− transport in volume homeostasis and blood pressure regulation. Am J Physiol Renal Physiol 305: F427–F438, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang WH, Yue P, Sun P, Lin DH. Regulation and function of potassium channels in aldosterone-sensitive distal nephron. Curr Opin Nephrol Hypertens 19: 463–470, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woda CB, Leite M Jr, Rohatgi R, Satlin LM. Effects of luminal flow and nucleotides on [Ca2+]i in rabbit cortical collecting duct. Am J Physiol Renal Physiol 283: F437–F446, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Xie B, Jiao Q, Cheng Y, Zhong Y, Shen X. Effect of pigment epithelium-derived factor on glutamate uptake in retinal Muller cells under high-glucose conditions. Invest Ophthalmol Vis Sci 53: 1023–1032, 2012. [DOI] [PubMed] [Google Scholar]
- 43.Zaika O, Mamenko M, Boukelmoune N, Pochynyuk O. IGF-1 and insulin exert opposite actions on ClC-K2 activity in the cortical collecting ducts. Am J Physiol Renal Physiol 308: F39–F48, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaika O, Mamenko M, O'Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol 300: F1105–F1115, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/51 and Kir41 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol 305: F1277–F1287, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]