

Abstract
More effective therapeutic strategies for the prevention and treatment of acute kidney injury (AKI) are needed to improve the high morbidity and mortality associated with this frequently encountered clinical condition. Ischemic and/or hypoxic preconditioning attenuates susceptibility to ischemic injury, which results from both oxygen and nutrient deprivation and accounts for most cases of AKI. While multiple signaling pathways have been implicated in renoprotection, this review will focus on oxygen-regulated cellular and molecular responses that enhance the kidney's tolerance to ischemia and promote renal repair. Central mediators of cellular adaptation to hypoxia are hypoxia-inducible factors (HIFs). HIFs play a crucial role in ischemic/hypoxic preconditioning through the reprogramming of cellular energy metabolism, and by coordinating adenosine and nitric oxide signaling with antiapoptotic, oxidative stress, and immune responses. The therapeutic potential of HIF activation for the treatment and prevention of ischemic injuries will be critically examined in this review.
Keywords: acute kidney injury, ischemic preconditioning, hypoxia, hypoxia-inducible factor, oxygen sensing, HIF prolyl-4-hydroxylases, dioxygenases, erythropoietin, adenosine, inflammation
acute kidney injury (aki) is defined as a sudden reduction in glomerular filtration rate (GFR) (1, 29, 144). Historically, the etiology of AKI is divided into prerenal, intrinsic, and postrenal causes; ischemia, which limits both oxygen and nutrient supply, being one of the major causes (involved in >50% of AKI cases) (97, 144). Although the care of patients with AKI, which is mainly supportive, has seen significant improvements, the clinical outcome of AKI remains poor and is associated with a mortality of ∼40–80% in the intensive care setting (156). While substantial progress has been made toward understanding its pathophysiology, effective therapeutic strategies are urgently needed to lower the increasing prevalence of AKI (3). In addition to the challenges of clinical management, AKI has been shown to accelerate the progression of chronic kidney disease (CKD) to dialysis-dependent end-stage renal disease (ESRD). Recent studies have indicated that ∼6% of AKI patients are likely to develop ESRD within 2 yr of diagnosis (156). This finding furthermore underlines the pressing need for the development of novel therapies that counteract the pathological sequelae associated with AKI.
Despite the large volume of blood that is normally directed toward the kidneys (∼20% of cardiac output), renal Po2 is physiologically low, especially in the medulla, where oxygen tensions of as low as 3 mmHg have been measured (103). Most of renal oxygen is utilized to fuel Na-K-ATPase, which drives tubular sodium reabsorption and other transport processes that are critical for the maintenance of physiological homeostasis. Since these transport processes are load dependent, renal oxygen consumption is directly linked to GFR, which in turn is renal blood flow dependent. Because of this intricate functional relationship, the kidney operates within a narrow range of relatively constant tissue Po2, rendering it susceptible to hypoxic injury (42). Therefore, alterations in renal perfusion from either hypotension and/or vasoconstriction often lead to clinically significant ischemia, a state of inadequate oxygen and nutrient supply. When the duration of ischemia exceeds a certain threshold, intracellular ATP stores become depleted and cells die from either apoptosis or necrosis (32). While reestablishment of adequate renal perfusion is a main therapeutic goal, rapid reoxygenation of the ischemic kidney generates additional tissue injury involving multiple mechanisms, such as increased generation of reactive oxygen species (ROS) (72).
Although somewhat counterintuitive, susceptibility to ischemia-reperfusion injury (IRI) can be attenuated by ischemic preconditioning (IPC), which was first reported in 1986 by Murry and colleagues (114). In their study, anesthetized dogs were subjected to four sequential 5-min-long coronary artery occlusions before the induction of coronary IRI. This intervention reduced the severity of myocardial infarction significantly and protected against myocardial cell death. Subsequently, IPC was shown to be effective in other tissues and across several species, suggesting that the molecular mechanisms, which underlie IPC-induced cytoprotection, are evolutionarily conserved and independent of cell type (20, 46, 54, 73, 86, 124). In the kidney, the degree of IPC-induced cytoprotection depends on the experimental protocol used, such as the duration of ischemia and reperfusion periods (124, 125). By demonstrating that preconditioning with hypoxia is equipotent to preconditioning with ischemia, Shizukuda and colleagues (148) established that acute changes in tissue Po2 alone mediate the cytoprotective effects of IPC (148). Both the perfusion of canine hearts with severely hypoxic blood and a short preinsult period of ischemia resulted in a comparable reduction in infarct size following 60 min of complete coronary artery occlusion (148).
While multiple signaling pathways have been implicated in mediating the protective effects of ischemic/hypoxic preconditioning, this review will focus on oxygen-regulated molecular pathways that enhance tolerance to ischemic injury and have therapeutic potential for the prevention and/or reversal of the pathological sequelae associated with AKI.
Ischemic/Hypoxic Preconditioning: Two Phases of Protection
Preconditioning is characterized by two phases of protection, an acute or early phase, which immediately follows the preconditioning stimulus and lasts no longer than several hours, and a late phase, which starts ∼24 h after the preconditioning stimulus and can last for several days (73, 107, 124). Traditionally, the acute phase has been viewed as being mediated by ion channels and signaling molecules produced during ischemia (61), while the late phase has been thought to depend on transcriptional responses that modulate glucose and mitochondrial metabolism, suppress ROS production and inflammation, and inhibit the expression of proapoptotic molecules. However, recent studies in a model of cardiac ischemia have challenged this concept and have provided experimental evidence that the acute phase of IPC is also transcription factor dependent (139). In mammals, transcriptional responses to hypoxia are primarily mediated by hypoxia-inducible factors (HIFs), oxygen-regulated transcription factors that allow cells to respond and adapt to low tissue Po2.
Oxygen-Dependent Regulation of HIF
HIF-1 and HIF-2 belong to the PER/aryl-hydrocarbon-receptor nuclear translocator (ARNT)/single minded (PAS) family of basic helix-loop-helix transcription factors and consist of an oxygen-sensitive α-subunit and a constitutively expressed β-subunit, also known as ARNT (105, 147). Both transcription factors facilitate oxygen delivery and cellular adaptation to hypoxia by stimulating multiple hypoxia responses including anaerobic glucose metabolism, adenosine and nitric oxide (NO) metabolism, angiogenesis, erythropoiesis, and iron metabolism (59, 167). In the presence of sufficient oxygen, HIF-1α and HIF-2α are targeted for rapid proteasomal degradation by the von Hippel-Lindau (VHL) tumor suppressor, which functions as the substrate recognition component of an E3-ubiquitin ligase complex (109). VHL-mediated targeting of HIF-α requires oxygen- and iron-dependent hydroxylation of specific proline residues located within the oxygen-dependent degradation domain of HIF-α (Fig. 1). HIF prolyl hydroxylation is carried out by three Fe (II)- and 2-oxoglutarate (2OG)-dependent prolyl-4-hydroxylase domain (PHD) proteins, PHD1, 2 and 3 (also known as EGLN2/HPH3, EGLN1/HPH2, and EGLN3/HPH1, respectively) (65, 76), each displaying different expression profiles and specific subcellular localization patterns (for a recent review on this topic, see Ref. 143). PHD enzymes function as oxygen sensors and belong to a large family of 2OG-dependent dioxygenases (∼60 family members have been identified in mammals) that utilize molecular oxygen for catalysis and regulate multiple biological processes (76, 101, 115). Under normoxia, PHD enzymes are catalytically active and HIF-α is degraded, whereas under hypoxia hydroxylation is inhibited and HIF signaling is activated (Fig. 1). PHD catalytic activity is furthermore inhibited by NO and ROS (76), an important consideration for the study of oxygen-sensing mechanisms under conditions where intracellular concentrations of these signaling molecules has changed.
Fig. 1.

Overview of canonical hypoxic signaling via the prolyl-4-hydroxylase domain (PHD)/hypoxia-inducible factor (HIF) axis. Under normoxic conditions, PHD enzymes hydroxylate specific proline residues located within the oxygen-dependent degradation domain of HIF-α subunits. Prolyl-hydroxylation is required for binding to the von Hippel-Lindau (VHL)-E3 ubiquitin ligase complex, which ubiquitylates HIF-α, triggering its proteasomal degradation. When prolyl-4-hydroxylation is inhibited, e.g., in the absence of molecular oxygen or ferrous iron, HIF-α escapes degradation and translocates to the nucleus, where it dimerizes with the aryl-hydrocarbon-receptor nuclear translocator (ARNT), the constitutively expressed HIF-β subunit. HIF-α/ARNT heterodimers bind to hypoxia response element (HRE)-containing regulatory DNA sequences and increase the transcription of oxygen-regulated genes (e.g. EPO, PDK1, LDHα) through recruitment of transcriptional coactivators such as p300/CBP. Factor-inhibiting HIF (FIH), another oxygen- and iron-dependent dioxygenase, hydroxylates a specific asparagine residue located within the HIF-α COOH-terminal transactivation domain, thereby inhibiting transcriptional cofactor recruitment. HIF-α stabilization results in the activation of multiple transcriptional programs, including erythropoiesis, iron metabolism, vascular remodeling, cellular metabolism, and others.
For the purpose of experimental and/or therapeutic IPC, HIF-α stabilization and activation of HIF-dependent signaling pathways can be achieved in several ways: 1) exposure to hypoxia, 2) genetic inactivation of the VHL-ubiquitylation machinery, 3) proteasomal inhibition, and 4) inhibition of HIF-PHDs by genetic or pharmacological means. Pharmacological inactivation of PHD enzymes can be accomplished by exposing cells and/or tissues to either cobalt chloride (CoCl2), iron chelators, such as desferrioxamine, or to small-molecule compounds that reversibly inhibit PHD catalytic activity, several of which are now in clinical trials for the treatment of renal anemia (91). However, pharmacological inhibition of HIF-PHDs may also exert HIF-independent effects, as HIF-PHDs have been shown to hydroxylate targets other than HIF-α. Such non-HIF targets include IκB kinase-β (IKKβ), β2-adrenergic receptor (β2AR), sprouty homolog 2 (SPRY2), and pyruvate kinase muscle isoform 2 (PKM2) (170). To what degree HIF-activating PHD inhibitors, especially those that are in clinical trials, inhibit other non-HIF-related 2OG-dependent dioxygenases remains to be investigated.
Preischemic HIF Activation Protects From Renal Ischemia
In renal cells, the HIF system is activated under conditions of systemic or regional hypoxia, or in response to PHD inhibition. In acute hypoxia, HIF-1α has been detected in glomerular and renal tubular epithelial cells, whereas HIF-2α was detected in glomeruli, peritubular endothelial, and interstitial fibroblast-like cells, the source of renal erythropoietin (EPO) (91, 135). Compared with systemic hypoxia induced by either anemia or exposure to carbon monoxide (CO) (135), the pattern of HIF stabilization in the ischemic mouse kidney appears to be restricted to cells that are predominantly located at the corticomedullary junction (134). With respect to the reperfused rat kidney, significant HIF-1α accumulation was found in proximal tubular cells and felt to be due to increased HIF-1α translation via enhanced mammalian target of rapamycin (mTOR)/AKT activity (28). Consistent with these findings are immunohistochemical studies in human renal postengraftment biopsies, which were obtained between 25 and 40 min after completion of vascular anastomoses, and which revealed significant variability in the extent of HIF-α stabilization. These studies demonstrated a strong positive correlation between HIF-1α expression and allograft function, suggesting a cytoprotective role for HIF-1 in the setting of acute transplant ischemia (136).
Different experimental strategies have been used to test whether enhancement of HIF expression before ischemic kidney injury could afford cytoprotection. Matsumoto and colleagues (108), for example, found that cobalt, a “hypoxia-mimic” and inhibitor of PHD catalysis, conferred significant protection from renal ischemic injury in rats. Similarly, preconditional HIF activation in mice via either administration of CO or administration of PHD inhibitors l-mimosine, dimethyloxaloylglycine (DMOG), or FG-4487 protected the kidney from injury (9, 12, 64, 166). In keeping with this notion, HIF-α stabilization with xenon gas provided morphological and functional renoprotection in a mouse model of IRI. Interestingly, xenon did not activate HIF by inhibiting its degradation, but rather by increasing its translation via the mTOR pathway (104). Similarly, treatment of donor rats with FG-4497 improved tissue injury scores, early mortality rates, as well as long-term graft survival in an allogenic Fisher-Lewis rat kidney transplant model (11).
Strong support for a role of HIF signaling in renal IPC also comes from the study of genetic mouse models. Hill and colleagues (64) found exacerbation of ischemic renal injury in mice that were heterozygously deficient for HIF-1α or HIF-2α and furthermore demonstrated beneficial effects of preischemic HIF activation via the administration of l-mimosine. A cytoprotective role for HIF signaling in ischemic kidney injury is also supported by acute or constitutive deletion of Vhl in mice, which phenocopied the cytoprotective effects of hypoxic preconditioning (68, 142).
Cell Type-Dependent Effects of HIF Activation
A major challenge in the IPC field is the identification of the cell types that mediate HIF-induced renoprotection. Using a genetic approach, our laboratory has shown recently that endothelial cells are critical cellular mediators of ischemic/hypoxic preconditioning in the kidney. We demonstrated that endothelial HIF-2 plays a key role in renoprotection and suppresses IRI-associated inflammation through the modulation of endothelial vascular cell adhesion molecule 1 (VCAM1) expression and inflammatory cell adhesion (79). Specifically, we found that 1) endothelial inactivation of HIF-2α, but not of HIF-1α, was associated with increased expression of renal injury markers and inflammatory cell infiltration in kidneys injured by either ischemia reperfusion or by ureteral obstruction; and 2) the negative effects of HIF-2α deletion were reversed by pharmacological blockade of VCAM1 and its ligand very late antigen 4 (VLA4). In contrast, activation of HIF by either genetic means or via systemic PHD inhibition protected from ischemic kidney injury and inflammation (Fig. 2). In this context, endothelial HIF-2 activation may be useful for the prevention or amelioration of renal fibrogenesis and CKD progression, which are well-recognized, long-term sequelae of AKI. In support of our findings are genetic studies in mice subjected to acute lung injury. In this setting, endothelial HIF-2 was shown to enhance the integrity of adherens junctions and endothelial barrier function (51), supporting the concept that hypoxic signaling in endothelial cells induces a broad cytoprotective response that is directed toward maintaining tissue homeostasis during injury. Therefore, the endothelial PHD/HIF axis represents an attractive therapeutic target for the prevention and treatment of tissue injury associated with ischemia. The renoprotective effects of HIF activation, however, do not appear to be restricted to renal endothelial cells alone, as genetic activation of HIF signaling in other cell types, e.g., epithelial and myeloid cells, has been shown to be renoprotective (43, 87, 142). The role of HIF signaling in immune responses and renal inflammation is discussed in greater detail in a separate section.
Fig. 2.
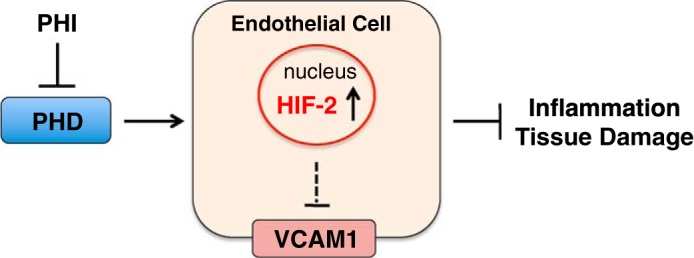
Schematic depicting the role of the endothelial PHD/HIF-2 axis in renoprotection. Pharmacological HIF activation through HIF prolyl-4-hydroxylase inhibition (PHI) promotes recovery from renal ischemic injury via activation of endothelial HIF-2-dependent signaling and suppression of VCAM1.
While renal epithelial HIF-1 had previously been identified as a profibrotic transcription factor in experimental CKD (63, 84), transient global pre- or postischemic HIF stabilization did not promote fibrosis (77). Context, tissue, and cell-type dependence are expected and conceptually supported by several genetic studies in nonrenal injury models. For example, conditional inactivation of Hif1a in neurons resulted in protection from cerebral ischemic injury (62), whereas Baranova and colleagues (7) reported enhanced brain injury in neuron-specific Hif1a−/− mice following transient focal cerebral ischemia. In the heart and kidney, germ line inactivation of one copy of Hif1a worsened both myocardial and renal ischemia (22, 64).
Timing and Duration of HIF Activation Determine Clinical Outcome
In contrast to the robust renoprotection observed with preischemic administration of PHD inhibitors, postischemic HIF-PHD inhibition failed to ameliorate renal injury (77, 164), suggesting that the timing of HIF activation is critical for renoprotection. In contrast, PHD inhibition with DMOG increased HIF-dependent gene expression and reduced injury when administered 30 or 60 min post-IRI in a model of cerebral ischemia (120). Although tissue-specific changes in HIF-mediated gene expression may explain these differences, the regulation of HIF signaling during the actual ischemic-reperfusion phase remains poorly understood and requires further investigation. Studies performed in our laboratory indicate that transcriptional HIF responses are suppressed in the immediate reperfusion phase, when PHD inhibitors are administered postinjury, which may be due to tissue acidification or the increased production of ROS (77).
In addition to timing, the duration of PHD inhibition/HIF activation appears to be critical for disease outcome and may produce adverse effects on tissue homeostasis. For example, cardiac function in mice with cardiomyocyte-specific Phd2 deletion was significantly improved 3 wk after myocardial IRI (66), whereas sustained Phd2 ablation or tissue-specific HIF-1α stabilization resulted in cardiomyopathy (113). In the kidney, chronic HIF activation in proximal tubules induced by Vhl gene loss caused tubular microcysts, which showed evidence of dedifferentiation and increased proliferation (133). In addition to renal cysts, inflammatory and fibrotic lesions were observed after inactivation of Vhl in collecting ducts and a subset of distal tubules (129). Furthermore, expression of a nondegradable form of HIF-2α in renal tubules led to cyst formation and fibrosis (140), while expression of a nondegradable form of HIF-1α resulted in lipid and glycogen deposition and as well as microcyst formation (47), supporting the notion that chronic activation of the renal epithelial HIF system promotes inflammation, fibrosis, cell proliferation, and dysplasia.
HIF Attenuates IRI Through the Coordinated Activation of Renoprotective Metabolic and Signaling Pathways
Although the molecular and cellular mechanisms underlying HIF-mediated cytoprotection in renal IRI are complex and only poorly understood, the concerted transcriptional activation of genes involved in cellular energy metabolism, antioxidant responses, antiapoptotic pathways, and other HIF-regulated biological processes is key to effective and robust cytoprotection (Fig. 3).
Fig. 3.

Molecular mechanisms implicated in PHD/HIF-mediated renoprotection induced by ischemic/hypoxic preconditioning. HIF attenuates ischemia-reperfusion injury through coordinated activation of cytoprotective signaling pathways. Shown are oxygen-regulated biological processes and signaling pathways with key roles in PHD/HIF-mediated renoprotection. NO, nitric oxide.
Hypoxia-induced metabolic reprogramming.
Central to achieving hypoxia tolerance is the shift of cellular metabolism from oxidative phosphorylation to anaerobic glycolysis, which in yeast is referred to as the Pasteur effect (146). HIF-1 mediates this shift by increasing the expression levels of several glycolytic pathway enzymes. Among those are hexokinase (HK1), phosphofructokinase (PFK), aldolase (ALDOA), phosphoglycerate kinase 1 (PGK1), enolase (ENO1), and lactate dehydrogenase-α (LDHα). In addition to increasing glycolytic flux, HIF-1 blocks the conversion of pyruvate to acetyl-CoA by upregulating pyruvate dehydrogenase kinase 1 (PDK1). This suppresses the catalytic activity of pyruvate dehydrogenase (PDH), which is phosphorylated by PDK1, and results in the uncoupling of glycolysis from the tricarboxylic acid cycle (83, 123). The importance of this HIF-1-regulated metabolic response is highlighted by in vitro studies, in which the absence of HIF-1 during hypoxia led to increased ROS production and apoptosis, both of which can be reversed by forced PDK1 expression (83).
To sustain increased glycolytic flux, NAD+ must be regenerated through the conversion of pyruvate to lactate. This is facilitated by HIF-1 through the transcriptional upregulation of LDHα. Inhibition of LDHα in cancer cells results in reduced ATP production, significantly increased oxidative stress and cell death, demonstrating the prosurvival function of LDHα under conditions of increased glycolytic flux (93). To counteract the effects of increased intracellular lactate production and ensuing acidification, HIF-1 facilitates the excretion of lactate and protons by increasing the expression of sodium/hydrogen exchanger (NHE)-1 and monocarboxylic acid transporter (MCT)-4 (126). In fact, intracellular pH is actually kept in a slightly alkaline range as a result of increased expression of the membrane-bound ectoenzyme carbonic anhydrase (CA) 9, which is HIF-1 regulated and converts CO2 to bicarbonate (126).
In addition, HIF-1 activation decreases cellular oxygen consumption through its suppressive effects on mitochondrial biogenesis and metabolism. HIF-1 inhibits c-MYC (177) and improves the efficiency by which mitochondria utilize oxygen. The latter occurs through HIF-1-mediated changes in the subunit composition of mitochondrial complex IV (48). Furthermore, HIF-1 controls mitochondrial mass by regulating mitophagy through B cell lymphoma (BCL)-2 family member BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) (176). The role of individual dioxygenases in cellular metabolism through either HIF-dependent or HIF-independent mechanisms adds additional layers of complexity to the metabolic regulation of hypoxia tolerance. Aragones and colleagues (4) found that inactivation of PHD1 alone lowers oxygen consumption in skeletal muscle by reprogramming glucose metabolism from oxidative to anaerobic ATP production through the activation of peroxisome proliferator-activated receptor-α (PPARA). This metabolic shift, which was HIF-2 dependent, reduced oxidative stress and preserved viability of ischemic myofibers (4). Therefore, activation of the PHD/HIF axis, through its effects on cellular energy metabolism, enhances the capacity of tissues to generate ATP in hypoxic environments, which is predicted to increase resistance to ischemic injury.
Oxidative stress responses.
Intracellular ROS play a key role in the pathophysiology of ischemic renal injury, as signaling pathways that scavenge ROS and/or prevent their formation, promote cell survival, and limit postischemic damage. HIF-2 has been implicated in the regulation of cellular oxidative stress responses, as HIF-2α-deficient mice exhibit impaired ROS clearance and develop multiple organ pathologies (145). HIF-2 plays a crucial role in this regard, as it stimulates the transcription of primary antioxidant enzymes such as catalase (CAT), glutathione peroxidase type 1 (GPX1), copper/zinc superoxide dismutase (SOD1), and manganese superoxide dismutase (SOD2) (145). In support of this notion are studies in mice homozygous for a Hif2a hypomorphic allele, which indicated a protective role for endothelial HIF-2 in renal IRI via regulation of ROS scavenging (89). Further support for a key role of HIF-2 in IPC comes from a study where transient ureteral obstruction was used as a preconditioning maneuver and protected from renal IRI in a HIF-2-dependent manner. The investigators of this study proposed that this occurred via enhanced recovery of intrarenal microvascular perfusion (178).
The hypoxic regulation of renal oxidative stress responses also involves HIF-1. Heme oxygenase 1 (HMOX1), for instance, is HIF-1 regulated, degrades heme, and generates CO and bilirubin, both of which have antioxidant and/or anti-inflammatory effects (137). Induction or overexpression of HMOX1 confers protection from renal IRI, while genetic inactivation of HMOX1 results in exacerbation of injury (14, 117, 128, 169).
Adenosine signaling.
Adenosine, an endogenous purine nucleoside involved in cellular adaptation to hypoxia, is considered by many investigators to be a critical mediator of IPC. The relationship between HIF signaling and adenosine became evident when administration of adenosine rescued Hif1a+/− hearts from IRI (22). This finding indicated that the production of adenosine in IPC required intact HIF-1 signaling. Extracellular adenosine is mainly derived from the enzymatic phosphohydrolysis of ATP and ADP, the first step of this reaction being catalyzed by ectonucleoside triphosphate diphosphohydrolase 1 (CD39), and the final step being the dephosphorylation of AMP, which is catalyzed by 5′-ectonucleotidase (CD73).
A mechanism by which IPC enhances adenosine production involves the hypoxic induction of the CD39 and CD73 genes. While HIF-1 binds directly to the CD73 promoter, the hypoxic regulation of CD39 involves the Sp1 transcription factor (41, 152). Both enzymes have been studied in the context of renal IRI and IPC. The role of CD39 was investigated using a hanging weight model to induce ischemia in mice (56). Remarkably, pharmacological inhibition of CD39 eliminated the protection normally conferred by IPC. Similarly, renoprotection was abolished in Cd39−/− mice. Administration of soluble apyrase, an adenosine diphosphatase derived from Solanum tuberosum recapitulated the protective effects conferred by IPC. In contrast to CD39, the protective role of CD73 in renal IRI and IPC is more controversial. While Grenz and colleagues (55) demonstrated a renoprotective function using similar experimental protocols, others reported that CD73 promoted tissue injury in a model of unilateral renal IRI, which was attributed to excessive accumulation of AMP (102). In addition to increasing adenosine production, HIF also modulates the clearance of extracellular adenosine. Specifically, HIF-1 diminishes uptake of adenosine via transcriptional repression of the equilibrative nucleoside transporter 1 (ENT1) and thereby contributes to the accumulation of extracellular adenosine (40). Accordingly, Ent1−/− mice displayed protection from ischemic AKI via improved restoration of postischemic blood flow (53).
Adenosine signals through its receptors A1, A2a, A2b, and A3, which are G protein-coupled receptors that use cAMP as their second messenger. The adenosine A2a receptor (ADORA2A) and adenosine A2b receptor (ADORA2B) are regulated by HIF-2 and HIF-1, respectively, adding an additional layer of complexity to the hypoxic control of adenosine signaling (2, 90). While all four adenosine receptors appear to have a role in the pathogenesis of AKI, Grenz and colleagues (54) used global knockout mice to investigate the contribution of each individual receptor to IPC-induced renoprotection. In contrast to Adora1−/−, Adora2a−/−, or Adora3−/− mice, IPC failed to induce protective responses in Adora2b−/− mice, which displayed increased renal inflammation following IRI (54). The results from these genetic studies are in concordance with the pharmacological inhibition of ADORA2B with PSB1115, which abrogated IPC-induced renoprotection, while treatment with a selective ADORA2B agonist induced protection and phenocopied the renoprotective effects of IPC, which appeared to be specifically mediated by renal endothelial ADORA2B (54). Furthermore, intact adenosine signaling through endothelial ADORA2B was required for the restoration of postischemic renal blood flow induced by ENT1 blockade (53). Additional support for a central role of ADORA2B in HIF-mediated cytoprotection comes from studies of myocardial ischemia, where the protective effects of pharmacological HIF activation were abrogated by genetic Adora2b deficiency (37).
The exact mechanisms that underlie adenosine-induced cytoprotection in the context of IPC are unclear. While mechanistic insights into adenosine's role in renal IRI are limited, studies in other tissues have suggested that adenosine acts as a critical modulator of postischemic inflammation (8). For example, Khoury and colleagues (80) suggested that stimulation of the ADORA2B receptor suppresses NF-κB activity through deneddylation of Cul-1 (80). Although not studied specifically in the context of IPC, stimulation of ADORA2A also confers beneficial immune-modulatory effects in renal IRI, which include the regulation of natural killer T cell (NKT) activation (96, 158). In addition to the suppression of inflammation, adenosine signaling activates the serine/threonine kinase AKT and reduces glycogen synthase kinase (GSK)-3β activity, which prevents mitochondrial leakage, swelling, and cell death (74, 150). The latter is consistent with studies in the heart, kidney, and other tissues, which demonstrated that inhibition of GSK-3β mimics the cytoprotective effects of IPC (118, 163).
NO signaling, ROS, and the regulation of oxygen sensing.
NO has diverse biological effects, it possesses antiapoptotic, anti-inflammatory, and vasodilative activity, it can both promote and prevent cell injury, and it has been implicated as a mediator of IPC in multiple organs, including the kidney (73, 95, 112, 119). Endogenous NO is synthesized by NO synthase (NOS), an enzyme that converts l-arginine to l-citrulline and NO. Three different NOS isoforms have been identified: neuronal NOS (nNOS or NOS1), inducible NOS (iNOS or NOS2), and endothelial NOS (eNOS or NOS3).
With regard to the HIF pathway, NO has been shown to inhibit the catalytic activity of PHDs and FIH, resulting in HIF-α stabilization. NO-mediated HIF activation is therefore likely to contribute to the cytoprotective effects of NO in IPC (110). Furthermore, HIF-1 induces iNOS levels and thus increases NO production (75, 153), creating a positive feedback loop between HIF and NO-dependent signaling that is predicted to enhance cytoprotection. In support of this notion is the finding that the infarct-limiting effect of CoCl2, an inhibitor of HIF-prolyl-4 hydroxylation, was absent in Nos2−/− mice (171).
Another mechanism by which NO is likely to mediate IPC involves the regulation of mitochondrial respiration and ROS generation, as NO has been shown to inhibit the activity of mitochondrial complex IV (121). A certain sublethal level of ROS, however, seems to be required for IPC to take effect and produce cytoprotection in the setting of cardiac ischemia (for a detailed discussion, the reader is referred to Ref. 21). This concept is supported by findings in the kidney, where pharmacological ROS scavenging with manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin or N-acetylcysteine abrogated cytoprotection that was induced by a single event of ischemia and reperfusion (81). In particular, mitochondrial ROS, via modulation of PHD catalytic activity, has been shown to be required for the hypoxic stabilization of HIF-α subunits (19, 23, 58, 106).
An important question in the field concerns the role of individual NOS isoforms in IPC-induced cytoprotection. Using 30 min of bilateral renal ischemia for preconditioning, Park and colleagues (124) identified iNOS as a key contributor to the protective effects of IPC (up to 12 wk of protection), whereas the role of eNOS in IPC was negligible. Either genetic deletion of iNOS or pharmacological inhibition of NOS enzymes abolished IPC-induced renoprotection, whereas genetic deletion of eNOS had no effect (124). In contrast to these studies, eNOS-mediated NO production was found to be important in a study by Yamasowa and colleagues (173), who applied three 2-min cycles of ischemia followed by 5 min of reperfusion before 45-min ischemia in uninephrectomized mice. Using this model, Yamasowa and colleagues found that eNOS deficiency abrogated IPC-mediated cytoprotection. The discrepant results reported by these studies could be due to the differences in experimental protocols used. Nevertheless, further investigation is required to reconcile these findings and to better understand the role of NO signaling in IPC-induced cytoprotection.
Antiapoptotic pathway regulation.
The degree of programmed cell death in the kidney correlates directly with the severity of histological injury and functional impairment in AKI. While many studies have shown that experimental upregulation of HIF decreases apoptosis of renal cells in AKI, only a few studies have provided evidence for a direct HIF-dependent regulation of apoptotic and/or antiapoptotic signaling pathways in the IPC setting. Yang and colleagues (174) employed a repetitive hypoxic preconditioning protocol, which upregulated HIF-1α transcription and protein levels, subsequently evoking resistance against renal IRI-induced apoptosis. The authors attributed this antiapoptotic response to a HIF-1-dependent induction of BCL-2, which was associated with inhibition of BCL2-associated X protein (BAX) and cytochrome c translocation in the mitochondria. In another study, repetitive hypoxic preconditioning inhibited renal tubular cell apoptosis and autophagy due to activation of HIF-1-dependent HSP70 signaling (175).
It is likely that hypoxic preconditioning regulates antiapoptotic signaling pathways independently of HIF. A recent example is the antiapoptotic protein IAP-2, which is induced by hypoxia in a HIF-1-independent manner and confers protection to cells subjected to apoptosis-inducing agents, such as staurosporine (36). In the context of pharmacological HIF-PHD inhibition, it has been shown that PHDs may also contribute to antiapoptotic responses independently of HIF. For instance, genetic loss of PHD1 decreases epithelial cell apoptosis in a model of inflammatory bowel disease induced by dextran sulfate sodium (DSS), potentially via derepression of NF-κB signaling, as PHD1 been shown to hydroxylate and thus negatively regulate IκB kinase (IKK) (30, 154).
EPO in IPC.
The kidney responds to changes in tissue oxygenation with increased production of EPO, a glycoprotein hormone that prevents apoptosis of erythroid progenitor cells and thus stimulates red blood cell production. While studies in Hep3B cells initially identified HIF-1 as the transcription factor that regulates EPO, ours and other laboratories have established that HIF-2 and not HIF-1 mediates the hypoxic induction of renal and hepatic EPO in vivo (57, 78, 132). The EPO receptor (EPOR), which is also hypoxia inducible, is expressed in multiple nonhematopoietic tissues, including the brain, cardiovascular tissues, liver, and kidney (71), and lacks intrinsic enzymatic function. EPOR homodimers undergo conformational changes upon ligand binding (100) and are associated with the tyrosine kinase JAK2, which phosphorylates EPOR at multiple positions, thus providing docking sites for signal-transducing molecules that contain SRC homology 2 domains. EPOR homodimers signal through multiple pathways, including the signal transduction and activator of transcription (STAT) 5 pathway, the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and the MAPK/ERK pathways, and PKC (71). A low-affinity heterodimeric EPO receptor is also present on the surface of multiple cell types and consists of EPOR and the β-common receptor (βcR), a signal transduction subunit that is shared between the IL-3, IL-5, and granulocyte macrophage colony-stimulating factor receptors (GMCSF) (17).
While EPO is mainly known for its role in hematopoiesis, it is well established that it has multiple nonhematopoietic functions (71). For example, EPO administration reduced ischemic neuronal damage and neurological dysfunction in experimental stroke models, suggesting a role in the regulation of cytoprotective and antiapoptotic pathways (16, 18, 149). These pathways are likely controlled by EPO signaling through the EPOR/βcR heterodimer (17) and involve antiapoptotic proteins of the BCL-2 family, ERK, and PI3K/AKT, and JAK2 regulation of NF-κB signaling and proangiogenic signaling (33, 52, 149). Subsequently, several studies have investigated the potential protective effects of EPO in other injury models, including renal IRI and cisplatin-induced nephrotoxicity (10, 24, 45).
In renal injury, EPO-induced cytoprotection was associated with a reduction in apoptosis and inflammation and an increase in tubular cell proliferation (157). These effects could not be attributed to increased erythropoiesis since the experimental protocols employed did not affect hemoglobin concentrations. While the molecular mechanisms by which EPO exerts cytoprotection in the kidney are ill defined, they are likely to involve multiple cell types as the EPOR is expressed in endothelial and mesangial cells and in cells of the proximal and distal renal tubule and collecting duct (168). Aside from regulating antiapoptotic and cytoprotective signaling pathways, EPO has been shown to mobilize endothelial progenitor cells (5) and to modulate inflammation and ischemia-induced neovascularization, indicating a critical role for EPO in the regulation of renal repair (6). However, when administration of EPO alone was compared with preconditional HIF activation with a PHD inhibitor, cytoprotection was reported to be inferior (164), supporting the concept that the concerted activation of HIF-regulated signaling pathways provides more potent and robust renoprotection than the activation of individual HIF target genes or signaling pathways alone.
The potential use of recombinant human EPO (rhEPO) in the prevention of renal IRI in high-risk patients has recently been studied in the critical care setting. In contrast to preclinical studies in animals and clinical pilot trials in humans, the predicted beneficial effects of perioperative administration of rhEPO to patients who underwent cardiac surgery and who were at high risk for the development of AKI could not be demonstrated (82). This was also the case in patients undergoing kidney transplantation, who were studied in a prospective, randomized, double-blind, placebo-controlled trial to assess the effects of high-dose rhEPO treatment on short- and long-term graft function (60). In patients with myocardial infarction, single intravenous bolus injection of epoetin-α did not reduce infarct size within 4 h of percutaneous coronary intervention (REVEAL trial), whereas subgroup analyses showed an increase in infarct size among older patients (116). These concerns for adverse cardiovascular events in the setting of rhEPO administration are reinforced by the TREAT trial, a randomized, double-blind, placebo-controlled trial of darbepoetin-α in diabetic patients with renal anemia, which reported an increase in stroke rate (127). Interestingly, in contrast to the beneficial effects of rhEPO in patients with acute stroke initially reported in a proof-of-concept, prethrombolysis era clinical trial (38), the German Multicenter EPO Stroke Trial was a negative intent-to-treat study and reported increased death rates particularly in patients receiving systemic thrombolysis (39).
Differential Activation of Stress-Related Kinases
Park and colleagues (125) have evaluated the effect of IPC induced by 30 min of bilateral renal ischemia on the activation pattern of stress-related kinases. They found that the ischemia-related activation of JNK and p38 were markedly diminished by IPC, whereas an effect on postischemic activation of ERK1/2 was not observed. Similarly, the phosphorylation of MKK7, MKK4, and MKK3/6, upstream activators of JNK and p38, was markedly reduced. Given the involvement of JNK and p38 in the expression of adhesion molecules and cytokine production, the authors speculated that these alterations may lessen leucocyte-endothelial interactions and therefore explain the remarkable IPC-induced reduction in outer medullary cellular congestion. A link between enhanced cell survival, decreased JNK, and increased ERK1/2 activation was shown in an in vitro model of endoplasmic reticulum stress preconditioning (67). Joo and colleagues (73), using a different IPC protocol, found rapid phosphorylation of both ERK and AKT, but only the inhibition of PI3K-AKT pathway was able to blunt renoprotection afforded by IPC. Since AKT regulates several cellular survival pathways, it is likely that AKT acts as an important mediator of IPC-induced cytoprotection. This concept is supported by recent pharmacological studies, in which inhibition of AKT signaling with PI3K inhibitor wortmannin abrogated cytoprotection induced by single ischemia-reperfusion preconditioning (69).
microRNAs in IPC
microRNAs (miRs) are small noncoding RNA molecules that in their mature form regulate gene expression, primarily through binding to the 3′-untranslated region (UTR) of target mRNAs, leading either to their degradation or to translational inhibition. Maturation and binding to 3′-UTRs involve several enzyme complexes. Dicer cleaves miR precursors and generates a double-stranded RNA product, which then in association with argonaute endonuclease proteins becomes part of the RNA-induced silencing complex (RISC) that directly binds to target RNAs (13). A subset of miRs is hypoxia regulated and is differentially expressed in ischemic tissues, where they modulate cell survival and tissue repair, and either positively or negatively impact the clinical outcome of IRI. In the heart, for example, miR-499 inhibited apoptosis and improved injury in animal models of myocardial infarction (162), while inhibition of miR-92a promoted angiogenesis and functional recovery from limb and myocardial IRI (15). Interestingly, some miRNAs seem to play a dual role in the recovery from IRI, which appears to be cell type dependent. miR-24, for example, inhibits cardiomyocyte apoptosis through the repression of proapoptotic molecule Bcl-2 interacting mediator of cell death (BIM) (130), while it also promotes apoptosis of cardiac endothelial cells (44).
The relevance of miRs to the pathophysiology of renal IRI is underscored by the proximal renal tubule-specific deletion of dicer, which suppressed miR maturation and made kidneys resistant to IRI (165). Several miRs are differentially expressed following warm IRI (50). These include miR-20a, miR-21, miR-192, miR-194, miR-214, and others. Of those, miR-21 is well characterized and has been shown to be required for the late-phase of IPC in the kidney (172) and IPC in the heart (26). miR-21 is expressed in multiple cell types and is hypoxia inducible in a HIF-1-dependent manner (99); its expression increases after IRI (35, 50, 99). miR-21 reduces the expression of programmed cell death 4 (PDCD4), phosphatase and tensin homolog (PTEN), and other proteins, and has been shown to have antiapoptotic properties (35, 50, 98, 99). However, whether miR-21 represents a suitable therapeutic target for the prevention of ischemic injuries is unclear, as it regulates other signaling pathways that may promote inflammation and fibrosis (179). Several reports indicate the involvement of multiple miRs in IPC of other organs (94, 138); to what degree these miRs contribute to renal IPC is currently under investigation.
While their role as single therapeutic agents for the prevention or treatment of IRI is uncertain, it is clear that miRs regulate and fine-tune cellular hypoxia responses. There are numerous examples of miRs that are hypoxia regulated, either positively or negatively. A prototypical example is miR-210, which is HIF inducible and was initially identified in cancer cells (92). More recently, miR-210 was shown to function as a negative regulator of HIF-1α in T cells (161), illustrating that hypoxia- and also non-hypoxia-regulated miRs operate in complex feedback loops that can amplify or suppress HIF-controlled pathways (31) and thus have the potential to modulate IPC. For example, miR-199a is suppressed under hypoxic conditions and modulates HIF-1-induced preconditioning in cardiomyocytes by directly targeting HIF-1α and SIRT1-induced suppression of PHD2 (131).
The Role of Immune Responses in IPC
Immune cells are key players in tissue injury and repair, and recent studies have established their crucial role in renal IPC. Burne-Taney and colleagues (20) transferred immune cells from ischemic and sham-operated mice into T cell-deficient mice, which then underwent IRI. The investigators found that T cell-deficient mice, which received leukocytes from mice with prior exposure to ischemia, had reduced injury compared with mice which received leukocytes from sham-operated animals. Furthermore, experiments using iNOS-deficient leukocytes demonstrated that immune cell-mediated IPC was not dependent on iNOS. Among lymphocytes, regulatory T cells (Tregs) have been identified as mediators of IPC. Tregs are CD4+CD25highFoxp3+ lymphocytes that are recruited into areas of inflammation and have immune-suppressive functions (111). Kinsey and colleagues (86) found that an ischemic insult caused a significant increase in CD4+CD25highFoxP3+ Tregs in the kidney at day 7 post-IRI, which was furthermore associated with an absolute increase in the number of IL-10-expressing Tregs. Treatment of preconditioned mice with a Treg-depleting anti-CD25 antibody blunted the protective effects of IPC with regard to preserving renal function and morphology and reducing neutrophil infiltration. Conversely, adoptive transfer of Tregs to naive mice before IRI produced cytoprotection and mimicked the protective and anti-inflammatory effects of IPC in the kidney (86). Remarkably, in a follow-up study the same investigators showed that adenosine generation by CD73 and adenosine signaling through ADORA2A were essential in Treg-mediated renoprotection (85). Because CD73 and ADORA2A are both hypoxia regulated (Fig. 1), enhanced adenosine signaling may be central to Treg-mediated IPC. Additional support for a crucial role of immune responses in IPC comes from a study, where systemic CD11c+ macrophage/dendritic cell depletion with clodronate diminished renoprotection associated with IPC (27). However, in a separate study a role for infiltrating macrophages could not be established (70), suggesting that the role of macrophages in IPC may depend on the conditions of the experimental strategies used.
While the molecular mechanisms regulating immune cell function in the context of IPC are not well studied, it is likely that the PHD/HIF axis is critically involved, as it controls metabolic and functional adaptation of immune cells to hypoxic microenvironments (122). For example, inflammatory functions and lifespan of neutrophils under hypoxic conditions are promoted by HIF-1-dependent NF-κB activity, and also require PHD3 expression (159, 160), while HIF-2 regulates neutrophilic apoptosis impacting on the resolution of inflammation (155). The impact of HIF-1-dependent metabolic reprogramming on innate immunity (shift toward anaerobic glycolysis) is exemplified by a recent elegant study showing that myeloid HIF-1 is critical in β-glycan-derived trained immunity responses against bacterial sepsis (25). However, whether IPC has the potential to trigger trained immunity via HIF, and to what degree this impacts renoprotection, is not known. It is also unclear whether IPC in the kidney signals to resident immune cells only or whether renal IPC can also signal and condition immune cells in remote locations such as the bone marrow or spleen. Nevertheless, genetic deletion of Hif1a in myeloid cells prevented functional recovery after renal IRI (141), a finding, which supports the notion that myeloid cell-derived hypoxia responses may have significant positive impact on injury outcomes in the setting of IPC. Accordingly, a protective role for myeloid HIF-1 was shown in a model of renal fibrosis (87). The differential effects of HIF homologs on immune cell function add additional complexity to understanding IPC-induced renoprotection. For instance, in response to different cytokines, HIF-1 and HIF-2 act antagonistically in the polarization of macrophages via the induction of either iNOS or arginase 1 expression, respectively, regulating NO levels differentially (153). With regard to adaptive immune responses, HIF controls T cell activation, the function of Tregs, the balance between Tregs and T helper 17 cells, and enhances the effector responses of CD8+ T cells via modulating the expression of transcription factors, effector molecules, and costimulatory receptors (34, 122). Furthermore, both B cell development and hypoxia-induced cell cycle arrest require intact HIF-1 signaling (49, 88). Given the increasing evidence that hypoxic signaling plays a pivotal role in the regulation of immune cell function (122), additional studies are needed that investigate the contribution of innate and adaptive immune responses to IPC.
Conclusions
Recent advances in understanding the molecular and cellular basis of renal hypoxia responses have identified the PHD/HIF axis as a pivotal oxygen-sensing mechanism that mediates IPC through the coordinated activation of renoprotective signaling pathways. Preischemic activation of these pathways through pharmacological HIF stabilization mimics normal hypoxia responses and would represent a comprehensive physiological approach to maintaining renal tissue homeostasis, protecting from ischemia, and promoting renal tissue repair. Effective HIF-activating compounds are currently being investigated in clinical trials for renal anemia and would be readily available for pilot studies in patients at high risk for development of AKI. However, the physiological consequences of systemic HIF activation in humans are only incompletely understood, and more investigations are needed to establish treatment protocols that permit safe and effective targeting of the HIF oxygen-sensing pathway for therapy of ischemic kidney diseases.
GRANTS
P. P. Kapitsinou is supported by National Institutes of Health (NIH) Grant P20 GM104936 and the Carl Gottschalk Award of the American Society of Nephrology (ASN). V. H. Haase receives support from the Krick-Brooks Chair in Nephrology, from NIH Grants R01-DK080821, R01-DK081646, and R01-DK101791, and from a Department of Veterans Affairs Merit Award (1I01BX002348).
DISCLOSURES
V. H. Haase has received honoraria from AstraZeneca and Daiichi Sankyo, and serves on the Scientific Advisory Board of Akebia Therapeutics, a company that develops prolyl-4-hydroxylase inhibitors for the treatment of renal anemia.
AUTHOR CONTRIBUTIONS
Author contributions: P.P.K. and V.H.H. prepared figures; P.P.K. and V.H.H. drafted manuscript; P.P.K. and V.H.H. edited and revised manuscript; P.P.K. and V.H.H. approved final version of manuscript.
ACKNOWLEDGMENTS
To remain within the scope of this review, the authors have limited the number of references and apologize to those colleagues whose original work was not cited. In those cases where original work could not be cited, the reader is referred to selected review articles.
REFERENCES
- 1.Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med 357: 797–805, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, Franklin WA, Bridges JP, Schaack JB, Colgan SP, White CW. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci USA 106: 10684–10689, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE. Outcomes following diagnosis of acute renal failure in US veterans: focus on acute tubular necrosis. Kidney Int 76: 1089–1097, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Aragonés J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet 40: 170–180, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Bahlmann FH. Erythropoietin regulates endothelial progenitor cells. Blood 103: 921–926, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Bahlmann FH, Song R, Boehm SM, Mengel M, von Wasielewski R, Lindschau C, Kirsch T, de Groot K, Laudeley R, Niemczyk E, Güler F, Menne J, Haller H, Fliser D. Low-dose therapy with the long-acting erythropoietin analogue darbepoetin alpha persistently activates endothelial Akt and attenuates progressive organ failure. Circulation 110: 1006–1012, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci 27: 6320–6332, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol 22: 14–20, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Bernhardt WM, Câmpean V, Kany S, Jürgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Günzler V, Amann K, Willam C, Wiesener MS, Eckardt KU. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17: 1970–1978, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Bernhardt WM, Eckardt KU. Physiological basis for the use of erythropoietin in critically ill patients at risk for acute kidney injury. Curr Opin Crit Care 14: 621–626, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Bernhardt WM, Gottmann U, Doyon F, Buchholz B, Campean V, Schodel J, Reisenbuechler A, Klaus S, Arend M, Flippin L, Willam C, Wiesener MS, Yard B, Warnecke C, Eckardt KU. Donor treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in an allogenic kidney transplant model. Proc Natl Acad Sci USA 106: 21276–21281, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernhardt WM, Warnecke C, Willam C, Tanaka T, Wiesener MS, Eckardt KU. Organ protection by hypoxia and hypoxia-inducible factors. Methods Enzymol 435: 221–245, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Bhatt K, Mi QS, Dong Z. microRNAs in kidneys: biogenesis, regulation, and pathophysiological roles. Am J Physiol Renal Physiol 300: F602–F610, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blydt-Hansen TD, Katori M, Lassman C, Ke B, Coito AJ, Iyer S, Buelow R, Ettenger R, Busuttil RW, Kupiec-Weglinski JW. Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J Am Soc Nephrol 14: 745–754, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324: 1710–1713, 2009. [DOI] [PubMed] [Google Scholar]
- 16.Brines M, Cerami A. Opinion: Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci 6: 484–494, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su-Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA 101: 14907–14912, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 97: 10526–10531, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1: 409–414, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Burne-Taney MJ, Liu M, Baldwin WM, Racusen L, Rabb H. Decreased capacity of immune cells to cause tissue injury mediates kidney ischemic preconditioning. J Immunol 176: 7015–7020, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Cadenas S, Aragones J, Landázuri MO. Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc Res 88: 219, 2010. [DOI] [PubMed] [Google Scholar]
- 22.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc Res 77: 463–470, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95: 11715–11720, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chatterjee PK. Pleiotropic renal actions of erythropoietin. Lancet 365: 1890–1892, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JHA, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJW, van der Veer BMJW, Deen PMT, Logie C, O'Neill LA, Willems P, van de Veerdonk FL, van der Meer JWM, Ng A, Joosten LAB, Wijmenga C, Stunnenberg HG, Xavier RJ, Netea MG. mTOR- and HIF-1 -mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345: 1250684–1250684, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res 87: 431–439, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho WY, Choi HM, Lee SY, Kim MG, Kim HK, Jo SK. The role of Tregs and CD11c+ macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int 78: 981–992, 2010. [DOI] [PubMed] [Google Scholar]
- 28.Conde E, Alegre L, Blanco-Sánchez I, Sáenz-Morales D, Aguado-Fraile E, Ponte B, Ramos E, Sáiz A, Jiménez C, Ordoñez A, López-Cabrera M, del Peso L, de Landázuri MO, Liaño F, Selgas R, Sanchez-Tomero JA, García-Bermejo ML. Hypoxia inducible factor 1-alpha (HIF-1 alpha) is induced during reperfusion after renal ischemia and is critical for proximal tubule cell survival. PLoS One 7: e33258, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz DN, Ricci Z, Ronco C. Clinical review: RIFLE and AKIN—time for reappraisal. Crit Care 13: 211, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 103: 18154–18159, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Lella Ezcurra AL, Bertolin AP, Melani M, Wappner P. Robustness of the hypoxic response: another job for miRNAs? Dev Dyn 241: 1842–1848, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol 17: 1503–1520, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-[kappa]B signalling cascades. Nature 412: 641–647, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat Immunol 14: 1173–1182, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, Wang D, Krall TJ, Delphin ES, Zhang C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem 284: 29514–29525, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong Z, Venkatachalam MA, Wang J, Patel Y, Saikumar P, Semenza GL, Force T, Nishiyama J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia. Hif-1-independent mechanisms. J Biol Chem 276: 18702–18709, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eckle T, Köhler D, Lehmann R, El Kasmi K. Hypoxia-inducible factor-1 is central to cardioprotection. Circulation 118: 166–175, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Rüther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Sirén AL. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8: 495–505, 2002. [PMC free article] [PubMed] [Google Scholar]
- 39.Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jahnig P, Herrmann M, Knauth M, Bahr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, Kastrup A, Bartels C; EPO Stroke Trial Group. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke 40: e647–e656, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schönfeld C, Löffler M, Reyes G, Duszenko M, Karhausen J, Robinson A, Westerman KA, Coe IR, Colgan SP. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 202: 1493–1505, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eltzschig HK, Köhler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 113: 224–232, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Evans RG, Gardiner BS, Smith DW, O'Connor PM. Intrarenal oxygenation: unique challenges and the biophysical basis of homeostasis. Am J Physiol Renal Physiol 295: F1259–F1270, 2008. [DOI] [PubMed] [Google Scholar]
- 43.Fähling M, Mathia S, Paliege A, Koesters R, Mrowka R, Peters H, Persson PB, Neumayer HH, Bachmann S, Rosenberger C. Tubular von Hippel-Lindau knockout protects against rhabdomyolysis-induced AKI. J. Am Soc Nephrol 24: 1806–1819, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiedler J, Jazbutyte V, Kirchmaier BC, Gupta SK, Lorenzen J, Hartmann D, Galuppo P, Kneitz S, Pena JT, Sohn-Lee C, Loyer X, Soutschek J, Brand T, Tuschl T, Heineke J, Martin U, Schulte-Merker S, Ertl G, Engelhardt S, Bauersachs J, Thum T. MicroRNA-24 regulates vascularity after myocardial infarction. Circulation 124: 720–730, 2011. [DOI] [PubMed] [Google Scholar]
- 45.Fliser D, Bahlmann FH, Haller H. EPO: renoprotection beyond anemia correction. Pediatr Nephrol 21: 1785–1789, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Fraisl P, Aragonés J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov 8: 139–152, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Fu L, Wang G, Shevchuk MM, Nanus DM, Gudas LJ. Generation of a mouse model of Von Hippel-Lindau kidney disease leading to renal cancers by expression of a constitutively active mutant of HIF1α. Cancer Res 71: 6848–6856, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukuda R, Zhang H, Kim Jw Shimoda L, Dang Chi V, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129: 111–122, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23: 359–369, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proc Natl Acad Sci USA 107: 14339–14344, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, Zhao YY, Komarova YA, Vogel SM, Malik AB. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125: 1364–1364, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94: 87–96, 1999. [PubMed] [Google Scholar]
- 53.Grenz A, Bauerle JD, Dalton JH, Ridyard D, Badulak A, Tak E, McNamee EN, Clambey E, Moldovan R, Reyes G, Klawitter J, Ambler K, Magee K, Christians U, Brodsky KS, Ravid K, Choi DS, Wen J, Lukashev D, Blackburn MR, Osswald H, Coe IR, Nürnberg B, Haase VH, Xia Y, Sitkovsky M, Eltzschig HK. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest 122: 693–710, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med 5: e137, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grenz A, Zhang H, Eckle T, Mittelbronn M, Wehrmann M, Köhle C, Kloor D, Thompson LF, Osswald H, Eltzschig HK. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol 18: 833–845, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Grenz A, Zhang H, Hermes M, Eckle T, Klingel K, Huang DY, Muller CE, Robson SC, Osswald H, Eltzschig HK. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J 21: 2863–2873, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci USA 104: 2301–2306, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–408, 2005. [DOI] [PubMed] [Google Scholar]
- 59.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol 291: F271–F281, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hafer C, Becker T, Kielstein JT, Bahlmann E, Schwarz A, Grinzoff N, Drzymala D, Bonnard I, Richter N, Lehner F, Klempnauer J, Haller H, Traeder J, Fliser D. High-dose erythropoietin has no effect on short- or long-term graft function following deceased donor kidney transplantation. Kidney Int 81: 314–320, 2011. [DOI] [PubMed] [Google Scholar]
- 61.Hausenloy DJ. Signalling pathways in ischaemic postconditioning. Thromb Haemost 101: 626–634, 2009. [PubMed] [Google Scholar]
- 62.Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S. Brain-specific knock-out of hypoxia-inducible factor-1α reduces rather than increases hypoxic-ischemic damage. J Neurosci 25: 4099, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810–3820, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hill P, Shukla D, Tran MGB, Aragones J, Cook HT, Carmeliet P, Maxwell PH. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 19: 39–46, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem 278: 30772–30780, 2003. [DOI] [PubMed] [Google Scholar]
- 66.Hölscher M, Silter M, Krull S, von Ahlen M, Hesse A, Schwartz P, Wielockx B, Breier G, Katschinski DM, Zieseniss A. Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J Biol Chem 286: 11185–11194, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hung CC, Ichimura T, Stevens JL, Bonventre JV. Protection of renal epithelial cells against oxidative injury by endoplasmic reticulum stress preconditioning is mediated by ERK1/2 activation. J Biol Chem 278: 29317–29326, 2003. [DOI] [PubMed] [Google Scholar]
- 68.Iguchi M, Kakinuma Y, Kurabayashi A, Sato T, Shuin T, Hong SB, Schmidt LS, Furihata M. Acute inactivation of the VHL gene contributes to protective effects of ischemic preconditioning in the mouse kidney. Nephron Exp Nephrol 110: e82–e90, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang HS, Kim J, Kim KY, Kim JI, Cho MH, Park KM. Previous ischemia and reperfusion injury results in resistance of the kidney against subsequent ischemia and reperfusion insult in mice; a role for the Akt signal pathway. Nephrol Dial Transplant 27: 3762–3770, 2012. [DOI] [PubMed] [Google Scholar]
- 70.Jang HS, Kim J, Park YK, Park KM. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation 85: 447–455, 2008. [DOI] [PubMed] [Google Scholar]
- 71.Jelkmann W, Wagner K. Beneficial and ominous aspects of the pleiotropic action of erythropoietin. Ann Hematol 83: 673–686, 2004. [DOI] [PubMed] [Google Scholar]
- 72.Johnson KJ, Weinberg JM. Postischemic renal injury due to oxygen radicals. Curr Opin Nephrol Hypertens 2: 625–635, 1993. [DOI] [PubMed] [Google Scholar]
- 73.Joo JD, Kim M, D'Agati VD, Lee HT. Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J Am Soc Nephrol 17: 3115–3123, 2006. [DOI] [PubMed] [Google Scholar]
- 74.Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res 104: 1240–1252, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jung F, Palmer LA, Zhou N, Johns RA. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ Res 86: 319–325, 2000. [DOI] [PubMed] [Google Scholar]
- 76.Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402, 2008. [DOI] [PubMed] [Google Scholar]
- 77.Kapitsinou PP, Jaffe J, Michael M, Swan CE, Duffy KJ, Erickson-Miller CL, Haase VH. Pre-ischemic targeting of HIF prolyl hydroxylation inhibits fibrosis associated with acute kidney injury. Am J Physiol Renal Physiol 302: F1172–F1179, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kapitsinou PP, Liu Q, Unger TL, Rha J, Davidoff O, Keith B, Epstein JA, Moores SL, Erickson-Miller CL, Haase VH. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 116: 3039–3048, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kapitsinou PP, Sano H, Michael M, Kobayashi H, Davidoff O, Bian A, Yao B, Zhang MZ, Harris RC, Duffy KJ, Erickson-Miller CL, Sutton TA, Haase VH. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest 124: 2396–2409, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest 117: 703–711, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim J, Jang HS, Park KM. Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. Am J Physiol Renal Physiol 298: F158–F166, 2010. [DOI] [PubMed] [Google Scholar]
- 82.Kim JH, Shim JK, Song JW, Song Y, Kim HB, Kwak YL. Effect of erythropoietin on the incidence of acute kidney injury following complex valvular heart surgery: a double blind, randomized clinical trial of efficacy and safety. Crit Care 17: R254, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006. [DOI] [PubMed] [Google Scholar]
- 84.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1α in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol 295: F1023–F1029, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kinsey GR, Huang L, Jaworska K, Khutsishvili K, Becker DA, Ye H, Lobo PI, Okusa MD. Autocrine adenosine signaling promotes regulatory T cell-mediated renal protection. J Am Soc Nephrol 23: 1528–1537, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int 77: 771–780, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kobayashi H, Gilbert V, Liu Q, Kapitsinou PP, Unger TL, Rha J, Rivella S, Schlöndorff D, Haase VH. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol 188: 5106–5115, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kojima H, Gu H, Nomura S, Caldwell CC, Kobata T, Carmeliet P, Semenza GL, Sitkovsky MV. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc Natl Acad Sci USA 99: 2170–2174, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, Ohneda O, Takeda N, Sata M, Miyata T, Fujita T, Nangaku M. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol 18: 1218–1226, 2007. [DOI] [PubMed] [Google Scholar]
- 90.Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J 20: 2242–2250, 2006. [DOI] [PubMed] [Google Scholar]
- 91.Koury MJ, Haase VH. Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nat Rev Nephrol 11: 394–410, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol 27: 1859–1867, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107: 2037–2042, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee ST, Chu K, Jung KH, Yoon HJ, Jeon D, Kang KM, Park KH, Bae EK, Kim M, Lee SK, Roh JK. MicroRNAs induced during ischemic preconditioning. Stroke 41: 1646–1651, 2010. [DOI] [PubMed] [Google Scholar]
- 95.Lei J, Vodovotz Y, Tzeng E, Billiar TR. Nitric oxide, a protective molecule in the cardiovascular system. Nitric Oxide 35: 175–185, 2013. [DOI] [PubMed] [Google Scholar]
- 96.Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, Moser EK, Jaworska K, Kinsey GR, Day YJ, Linden J, Lobo PI, Rosin DL, Okusa MD. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest 122: 3931–3942, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liaño F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 50: 811–818, 1996. [DOI] [PubMed] [Google Scholar]
- 98.Liu LZ, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y, Kung HF, Lai L, Jiang BH. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PLoS One 6: e19139, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Y, Nie H, Zhang K, Ma D, Yang G, Zheng Z, Liu K, Yu B, Zhai C, Yang S. A feedback regulatory loop between HIF-1alpha and miR-21 in response to hypoxia in cardiomyocytes. FEBS Lett 588: 3137–3146, 2014. [DOI] [PubMed] [Google Scholar]
- 100.Livnah O. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science 283: 987–990, 1999. [DOI] [PubMed] [Google Scholar]
- 101.Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci 36: 7–18, 2011. [DOI] [PubMed] [Google Scholar]
- 102.Lu B, Rajakumar SV, Robson SC, Lee EKF, Crikis S, d'Apice AJF, Cowan PJ, Dwyer KM. The impact of purinergic signaling on renal ischemia-reperfusion injury. Transplantation 86: 1707–1712, 2008. [DOI] [PubMed] [Google Scholar]
- 103.Lübbers DW, Baumgärtl H. Heterogeneities and profiles of oxygen pressure in brain and kidney as examples of the Po2 distribution in the living tissue. Kidney Int 51: 372–380, 1997. [DOI] [PubMed] [Google Scholar]
- 104.Ma D, Lim T, Xu J, Tang H, Wan Y, Zhao H, Hossain M, Maxwell PH, Maze M. Xenon preconditioning protects against renal ischemic-reperfusion injury via HIF-1alpha activation. J Am Soc Nephrol 20: 713–720, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40: 294–309, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1: 393–399, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation 88: 1264–1272, 1993. [DOI] [PubMed] [Google Scholar]
- 108.Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, Fujita T, Nangaku M. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol 14: 1825–1832, 2003. [DOI] [PubMed] [Google Scholar]
- 109.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275, 1999. [DOI] [PubMed] [Google Scholar]
- 110.Metzen E, Zhou J, Jelkmann W, Fandrey J, Brüne B. Nitric oxide impairs normoxic degradation of HIF-1α by inhibition of prolyl hydroxylases. Mol Biol Cell 14: 3470–3481, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med 13: 108–116, 2007. [DOI] [PubMed] [Google Scholar]
- 112.Moncada S, Higgs A. The l-arginine-nitric oxide pathway. N Engl J Med 329: 2002–2012, 1993. [DOI] [PubMed] [Google Scholar]
- 113.Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, Signoretti S, Liao R, Kaelin WG. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 122: 1004–1016, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74: 1124–1136, 1986. [DOI] [PubMed] [Google Scholar]
- 115.Myllyharju J. HIF prolyl 4-hydroxylases and their potential as drug targets. Curr Pharm Des 15: 3878–3885, 2009. [DOI] [PubMed] [Google Scholar]
- 116.Najjar SS, Rao SV, Melloni C, Raman SV, Povsic TJ, Melton L, Barsness GW, Prather K, Heitner JF, Kilaru R, Gruberg L, Hasselblad V, Greenbaum AB, Patel M, Kim RJ, Talan M, Ferrucci L, Longo DL, Lakatta EG, Harrington RA, REVEAL Investigators. Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. JAMA 305: 1863–1872, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. [DOI] [PubMed] [Google Scholar]
- 118.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J Mol Cell Cardiol 43: 564–570, 2007. [DOI] [PubMed] [Google Scholar]
- 119.Ogawa T, Nussler AK, Tuzuner E, Neuhaus P, Kaminishi M, Mimura Y, Beger HG. Contribution of nitric oxide to the protective effects of ischemic preconditioning in ischemia-reperfused rat kidneys. J Lab Clin Med 138: 50–58, 2001. [DOI] [PubMed] [Google Scholar]
- 120.Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiol Dis 45: 733–742, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Palacios-Callender M, Quintero M, Hollis VS, Springett RJ, Moncada S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc Natl Acad Sci USA 101: 7630–7635, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity 41: 518–528, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197, 2006. [DOI] [PubMed] [Google Scholar]
- 124.Park KM, Byun JY, Kramers C, Kim JI, Huang PL, Bonventre JV. Inducible nitric-oxide synthase is an important contributor to prolonged protective effects of ischemic preconditioning in the mouse kidney. J Biol Chem 278: 27256–27266, 2003. [DOI] [PubMed] [Google Scholar]
- 125.Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276: 11870–11876, 2001. [DOI] [PubMed] [Google Scholar]
- 126.Parks SK, Chiche J, Pouyssegur J. pH control mechanisms of tumor survival and growth. J Cell Physiol 226: 299–308, 2011. [DOI] [PubMed] [Google Scholar]
- 127.Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, Feyzi JM, Ivanovich P, Kewalramani R, Levey AS, Lewis EF, McGill JB, McMurray JJV, Parfrey P, Parving HH, Remuzzi G, Singh AK, Solomon SD, Toto R. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 361: 2019–2032, 2009. [DOI] [PubMed] [Google Scholar]
- 128.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int 68: 611–622, 2005. [DOI] [PubMed] [Google Scholar]
- 129.Pritchett TL, Bader HL, Henderson J, Hsu T. Conditional inactivation of the mouse von Hippel-Lindau tumor suppressor gene results in wide-spread hyperplastic, inflammatory and fibrotic lesions in the kidney. Oncogene 2014. [DOI] [PubMed]
- 130.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med 208: 549–560, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M. Downregulation of MiR-199a derepresses hypoxia-inducible factor-1 and sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res 104: 879–886, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest 117: 1068–1077, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 66: 2576–2583, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rosenberger C, Heyman SN, Rosen S, Shina A, Goldfarb M, Griethe W, Frei U, Reinke P, Bachmann S, Eckardt KU. Up-regulation of HIF in experimental acute renal failure: evidence for a protective transcriptional response to hypoxia. Kidney Int 67: 531–542, 2005. [DOI] [PubMed] [Google Scholar]
- 135.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13: 1721–1732, 2002. [DOI] [PubMed] [Google Scholar]
- 136.Rosenberger C, Pratschke J, Rudolph B, Heyman SN, Schindler R, Babel N, Eckardt KU, Frei U, Rosen S, Reinke P. Immunohistochemical detection of hypoxia-inducible factor-1alpha in human renal allograft biopsies. J Am Soc Nephrol 18: 343–351, 2007. [DOI] [PubMed] [Google Scholar]
- 137.Ryter SW, Choi AM. Heme oxygenase-1/carbon monoxide: from metabolism to molecular therapy. Am J Respir Cell Mol Biol 41: 251–260, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Salloum FN, Yin C, Kukreja RC. Role of microRNAs in cardiac preconditioning. J Cardiovasc Pharmacol 56: 581–588, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sarkar K, Rey S, Zhang X, Sebastian R, Marti GP, Fox-Talbot K, Cardona AV, Du J, Tan YS, Liu L, Lay F, Gonzalez FJ, Harmon JW, Semenza GL. Tie2-dependent knockout of HIF-1 impairs burn wound vascularization and homing of bone marrow-derived angiogenic cells. Cardiovasc Res 93: 162–169, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schietke RE, Hackenbeck T, Tran M, Günther R, Klanke B, Warnecke CL, Knaup KX, Shukla D, Rosenberger C, Koesters R, Bachmann S, Betz P, Schley G, Schödel J, Willam C, Winkler T, Amann K, Eckardt KU, Maxwell P, Wiesener MS. Renal tubular HIF-2α expression requires VHL inactivation and causes fibrosis and cysts. PLoS One 7: e31034, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schley G, Klanke B, Jantsch J, Olbrich S, Eckardt KU, Weidemann A. Hypoxic response of myeloid cells in renal ischemia-reperfusion injury. ASN, Kidney Week FR-PO1110, 2011. [Google Scholar]
- 142.Schley G, Klanke B, Schödel J, Forstreuter F, Shukla D, Kurtz A, Amann K, Wiesener MS, Rosen S, Eckardt KU, Maxwell PH, Willam C. Hypoxia-inducible transcription factors stabilization in the thick ascending limb protects against ischemic acute kidney injury. J Am Soc Nephrol 22: 2004–2015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5: 343–354, 2004. [DOI] [PubMed] [Google Scholar]
- 144.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest 114: 5–14, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Genet 35: 331–340, 2003. [DOI] [PubMed] [Google Scholar]
- 146.Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, Laderoute K, Johnson RS. Transcription factor HIF-1 is a necessary mediator of the Pasteur effect in mammalian cells. Mol Cell Biol 21: 3436–3444, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shizukuda Y, Mallet RT, Lee SC, Downey HF. Hypoxic preconditioning of ischaemic canine myocardium. Cardiovasc Res 26: 534–542, 1992. [DOI] [PubMed] [Google Scholar]
- 149.Sirén A, Fratelli M, Brines M. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 98: 4044–4049, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Solenkova NV, Solodushko V, Cohen MV, Downey JM. Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol Heart Circ Physiol 290: H441–H449, 2006. [DOI] [PubMed] [Google Scholar]
- 152.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110: 993–1002, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Takeda N, O'Dea EL, Doedens A, Kim Jw Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-alpha isoforms in macrophages are essential for NO homeostasis. Genes Dev 24: 491–501, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, Fraisl P, Lasitschka F, Mollenhauer M, Saunders SP, Maxwell PH, Carmeliet P, Fallon PG, Schneider M, Taylor CT. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 139: 2093–2101, 2010. [DOI] [PubMed] [Google Scholar]
- 155.Thompson AAR, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, Williams L, Parmar S, Shaw G, McGrath EE, Formenti F, Van Eeden FJ, Kinnula VL, Pugh CW, Sabroe I, Dockrell DH, Chilvers ER, Robbins PA, Percy MJ, Simon MC, Johnson RS, Renshaw SA, Whyte MKB, Walmsley SR. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 123: 366–376, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818, 2005. [DOI] [PubMed] [Google Scholar]
- 156a.United States Renal Data System. Survey USRD. Annual data report. USRDS Coordinating Center, 2006.
- 157.Vesey DA, Cheung C, Pat B, Endre Z, Gobé G, Johnson DW. Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant 19: 348–355, 2004. [DOI] [PubMed] [Google Scholar]
- 158.Vincent IS, Okusa MD. Adenosine 2A receptors in acute kidney injury. Acta Physiol (Oxf) 214: 303–310, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Walmsley SR, Chilvers ER, Thompson AA, Vaughan K, Marriott HM, Parker LC, Shaw G, Parmar S, Schneider M, Sabroe I, Dockrell DH, Milo M, Taylor CT, Johnson RS, Pugh CW, Ratcliffe PJ, Maxwell PH, Carmeliet P, Whyte MKB. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest 121: 1053–1063, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, Condliffe AM, Cowburn AS, Johnson N, Chilvers ER. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med 201: 105–115, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol 15: 393–401, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 17: 71–78, 2011. [DOI] [PubMed] [Google Scholar]
- 163.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, Schwartz JH, Borkan SC. GSK3 promotes apoptosis after renal ischemic injury. J Am Soc Nephrol 21: 284–294, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang Z, Schley G, Türkoglu G, Burzlaff N, Amann KU, Willam C, Eckardt KU, Bernhardt WM. The protective effect of prolyl-hydroxylase inhibition against renal ischaemia requires application prior to ischaemia but is superior to EPO treatment. Nephrol Dial Transplant 2011. [DOI] [PubMed]
- 165.Wei Q, Bhatt K, He HZ, Mi QS, Haase VH, Dong Z. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol 21: 756–761, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Weidemann A, Bernhardt WM, Klanke B, Daniel C, Buchholz B, Câmpean V, Amann K, Warnecke C, Wiesener MS, Eckardt KU, Willam C. HIF activation protects from acute kidney injury. J Am Soc Nephrol 19: 486–494, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wenger R, Stiehl D, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005: re12, 2005. [DOI] [PubMed] [Google Scholar]
- 168.Westenfelder C, Biddle DL, Baranowski RL. Human, rat, and mouse kidney cells express functional erythropoietin receptors. Kidney Int 55: 808–820, 1999. [DOI] [PubMed] [Google Scholar]
- 169.Wiesel P, Patel AP, Carvajal IM, Wang ZY, Pellacani A, Maemura K, DiFonzo N, Rennke HG, Layne MD, Yet SF, Lee ME, Perrella MA. Exacerbation of chronic renovascular hypertension and acute renal failure in heme oxygenase-1-deficient mice. Circ Res 88: 1088–1094, 2001. [DOI] [PubMed] [Google Scholar]
- 170.Wong BW, Kuchnio A, Bruning U, Carmeliet P. Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes. Trends Biochem Sci 38: 3–11, 2013. [DOI] [PubMed] [Google Scholar]
- 171.Xi L, Taher M, Yin C, Salloum F, Kukreja RC. Cobalt chloride induces delayed cardiac preconditioning in mice through selective activation of HIF-1α and AP-1 and iNOS signaling. Am J Physiol Heart Circ Physiol 287: H2369–H2375, 2004. [DOI] [PubMed] [Google Scholar]
- 172.Xu X, Kriegel AJ, Liu Y, Usa K, Mladinov D, Liu H, Fang Y, Ding X, Liang M. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int 82: 1167–1175, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yamasowa H, Shimizu S, Inoue T, Takaoka M, Matsumura Y. Endothelial nitric oxide contributes to the renal protective effects of ischemic preconditioning. J Pharmacol Exp Ther 312: 153–159, 2005. [DOI] [PubMed] [Google Scholar]
- 174.Yang CC, Lin LC, Wu MS, Chien CT, Lai MK. Repetitive hypoxic preconditioning attenuates renal ischemia/reperfusion induced oxidative injury via upregulating HIF-1alpha-dependent bcl-2 signaling. Transplantation 88: 1251–1260, 2009. [DOI] [PubMed] [Google Scholar]
- 175.Yeh CH, Hsu SP, Yang CC, Chien CT, Wang NP. Hypoxic preconditioning reinforces HIF-alpha-dependent HSP70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci 86: 115–123, 2010. [DOI] [PubMed] [Google Scholar]
- 176.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 177.Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang Chi V, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11: 407–420, 2007. [DOI] [PubMed] [Google Scholar]
- 178.Zhang S, Han CH, Chen XS, Zhang M, Xu LM, Zhang JJ, Xia Q. Transient ureteral obstruction prevents against kidney ischemia/reperfusion injury via hypoxia-inducible factor (HIF)-2α activation. PLoS One 7: e29876, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol 22: 1668–1681, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]