

ABSTRACT
Double-stranded RNA (dsRNA)-activated protein kinase (PKR), a major component of the cellular antiviral system, is activated by the binding of either dsRNA or the cellular PKR activator, the PACT protein. The suppression of PKR activation is one of the main strategies that viruses employ to circumvent interferon signaling. Orf virus (ORFV), a parapoxvirus from the Poxviridae family, causes contagious pustular dermatitis in small ruminants. Previous studies have demonstrated that various OV20.0 isoforms, encoded by the OV20.0L gene, are able to inhibit PKR activation both by sequestering dsRNA and by physically interacting with PKR in vitro. Thus, this gene acts as a virulence factor of ORFV when tested using a mouse infection model. In the present study, the regions within OV20.0 that interact with dsRNA and with PKR have been mapped. Furthermore, this study demonstrates for the first time that OV20.0 is also able to interact with the dsRNA binding domain of PACT and that the presence of dsRNA strengthened the interaction of these two molecules. The presence of OV20.0 diminishes PKR phosphorylation when this is stimulated by PACT. Nevertheless, the association of OV20.0 with PKR, rather than with PACT, was found to be essential for reducing PACT-mediated PKR phosphorylation. These observations elucidate a new strategy whereby innate immunity can be evaded by ORFV.
IMPORTANCE Our previous study indicated that ORFV's two OV20.0 isoforms act as a PKR antagonist via sequestering the PKR activator, dsRNA, and by interacting with PKR, leading to an inhibition of PKR activation (Y. Y. Tseng, F. Y. Lin, S. F. Cheng, D. Tscharke, S. Chulakasian, C. C. Chou, Y. F. Liu, W. S. Chang, M. L. Wong, and W. L. Hsu, J Virol 89:4966–4979, 2015, doi:10.1128/JVI.03714-14). In the current study, the possible mechanisms by which OV20.0 protein counteracts PKR activation were studied in depth. OV20.0 is able to bind PKR and its two activators, dsRNA and PACT. In addition, OV20.0 binds directly to the RNA binding domains (RBDs) of PKR, and this interaction does not require dsRNA. Moreover, OV20.0 interacts with or occupies the RBD2 and the kinase domain of PKR, which then prevents PACT binding to PKR. Finally, OV20.0 associates with PACT via the RBDs, which may reduce the ability of PACT to induce PKR activation. The findings in this study provide new concepts in relation to how ORFV modulates PKR activation.
INTRODUCTION
ORFV OV20.0 is the ortholog of the vaccinia virus (VACV) E3 (1). Among all known E3 orthologs, VACV E3 is the most well characterized with respect to innate immune evasion (2). The dsRNA binding domain (RBD) is located at the C terminus (residues 117 to 182) of the VACV E3 protein, and this region also is involved in the protein's association with IFN-stimulating gene 15 (ISG15), which is known to lead to a suppression of the cellular antiviral response (3–5). In addition, the N terminus of VACV E3 interacts with the kinase domain of PKR, which then results in an inhibition of PKR phosphorylation, with the presence of Lys-167 and Arg-168 within the RBD increasing the binding efficiency and inhibition effect (6). Compared with VACV E3, little is known about the function of ORFV OV20.0. Up to the present, only the phenomena of ORFV OV20.0-mediated interferon (IFN) resistance and an inhibition of PKR activation have been reported; however, the underlying mechanism of action of this protein still requires further investigation (7, 8). Based on the sequence alignment, four important amino acid residues responsible for the interaction of VACV E3 with dsRNA are known to be conserved in ORFV OV20.0 (1). Indeed, the dsRNA binding activity of OV20.0 protein has been confirmed in several different experimental systems (1, 8). Furthermore, recent studies have indicated that both the full-length OV20.0 protein and its N-terminal truncated isoform (sh20.0 or a deleted protein described as Δ1-79 here) possess similar dsRNA binding abilities (8). In addition, direct association between PKR and both OV20.0 and the various sh20.0 isoforms has been observed. These findings allow us to identify the approximate region through which OV20.0 interacts with PKR and dsRNA.
PKR is a common target of viruses when they are antagonizing the host innate immune response (6, 9–12). PKR consists of 551 amino acids and is comprised of three functional domains. These consist of a kinase domain in the C terminus and two basic helical domains, with the latter forming two dsRNA binding domains (RBD1 and RBD2) at the N-terminal region of the protein (13). The OH groups of dsRNA interact with the phosphates of the PKR RBD1, and this leads to a conformational change and the formation of the dsRNA-PKR complex, which is further stabilized by RBD2 (14). This process initiates the phosphorylation of PKR and then promotes PKR dimerization; the latter leads to autophosphorylation at residue T446 (14). Sequence specificity within the dsRNA is not required for the induction of PKR activation; however, dsDNA or a DNA/RNA hybrid is unable to trigger the effect (15). The activation of PKR brings about broad cellular responses, including apoptosis, signal transduction, and changes in cell growth (16, 17). Such activation also plays a central role in the antiviral pathway. This occurs via the phosphorylation of the eukaryotic translation initiation factor eIF2α and results in a shutoff of cellular and viral translation initiation (18, 19). In addition, via a regulation of the NF-κB signaling pathways, PKR activation also contributes to the induction of type I interferon production (20). Hence, many viruses have evolved specific mechanisms, such as producing a viral dsRNA-binding protein, the presence of viral dsRNA homologues, and the expression of PKR-binding proteins, in order to counteract this PKR-mediated antiviral effect (6, 10, 11, 21–23).
In addition to dsRNA, an alternative way of bringing about PKR phosphorylation exists, and this involves a cellular protein that acts as a protein activator of PKR, namely, PACT (24). PACT is constitutively expressed in mammalian cells at a low level (24–26). The protein consists of three domains, with the first two domains (I and II) being capable of interacting with the N-terminal dimerization domain (DD) of PKR (25, 26). Domains I and II also have dsRNA-binding ability, which also gives PACT the eligibility to be classified as a member of the dsRNA-binding protein family (25, 26). However, this dsRNA-binding activity is not necessary for PKR activation to occur (24). Furthermore, domain III of PACT, which acts as the effector domain, directly binds to the C-terminal domain of PKR; this facilitates the exposure of the PKR kinase domain and leads to PKR activation in vitro (27–29). Nonetheless, at physiological salt concentrations, one of the other two domains (either I or II) is essential to the strengthening of the weak interactions between the PACT domain III and PKR; this is needed to bring about various further cellular effects, such as enhancing apoptosis (26, 27). PACT-induced PKR phosphorylation is balanced by another cellular protein, named the TAR RNA binding protein (TRBP) (30). Based on the association between PACT and TRBP proteins, free forms of either protein are present at low levels in cells (31). When there is a high expression level, cellular TRBP is prone to form a heterodimer with PACT, which causes PACT to be moved to inactive storage (31–33). In contrast, a low concentration of TRBP results in PACT being more likely to exist as either homodimers or monomers, both of which are able to mediate PKR activation and regulate translation inhibition (31–33).
Recently, we found that various ORFV OV20.0 isoforms act as a PKR antagonist; this occurs via sequestering the PKR activator, dsRNA, as well as by interacting with PKR. Both events lead to an inhibition of PKR activation (8). However, the functional domain of OV20.0 is not known. In the current study, we initially generated systematic deletions of OV20.0 proteins; these then were used to map the regions involved in the interaction of OV20.0 with dsRNA and PKR, as well as the inhibitory effect of PKR activation. Furthermore, experiments were set up to investigate whether cellular PACT, another dsRNA binding protein and PKR activator, also is perturbed by OV20.0. OV20.0 seems to be able to interact with multiple elements involved in PKR activation; therefore, the required hierarchy of the intermolecular interactions that OV20.0 is involved in during the suppression of PKR activation also was explored in more detail. Finally, a potential mechanism of action of these proteins is proposed.
MATERIALS AND METHODS
Cell culture and virus.
The human embryonic kidney cell line (293T) and goat fibroblast cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Life Technologies Corporation, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS; HyClone, Logan, UT) and 1% penicillin-streptomycin (Gibco BRL). Cells were maintained at 37°C with 5% CO2.
Goat fibroblast cells were used for the propagation of recombinant ORFV possessing enhanced green fluorescent protein (EGFP) expression ability (ORFV-GFP) (8). Infection was performed by incubating the indicated multiplicity of infection (MOI) of ORFV with cell monolayers at 80% confluence in infection medium (DMEM without FCS). The infection medium was replaced with 2% FCS DMEM at 1 h postabsorption.
Construction of plasmids.
A series of plasmids harboring various lengths of OV20.0 and PKR were constructed for transient expression in mammalian cells (Table 1). All delineated deletions of OV20.0 and PKR fragments were built by overlap extension PCR (34) with the gene-specific primer sets listed in Table 2. Shortly, two separate rounds of PCRs were performed to generate the upstream or downstream sequences of the deletion region from template containing wild-type genes. Subsequently, products of two separate reactions, with 15-bp overlaps, then served as the template for the next run of PCR using the outermost primer set to generate the final expected fragments.
TABLE 1.
Various protein mutants
| Protein | Description |
|---|---|
| OV200 | Full-length OV20.0 consisting of residues 1 to 184 |
| Δ1-79 | OV20.0 deleted of 1 to 79 residues, consisting of residues 80 to 184 (also referred as sh20 elsewhere) |
| Δ80-129 | OV20.0 deleted of 80 to 129 residues, consisting of residues 1 to 79 and 130 to 184 |
| Δ130-184 | OV20.0 deleted of residues 130 to 184, consisting of residues 1 to 129 |
| Δ80-184 | OV20.0 deleted of residues 80 to 184, consisting of residues 1 to 79 |
| PKR | Full-length PKR, consisting of residues 1 to 551 |
| ΔRBD1 | PKR deleted of residues 10 to 75 (RNA binding domain 1), consisting of residues 1 to 9 and 76 to 551 |
| ΔRBD2 | PKR deleted of residues 101 to 165 (RNA-binding domain 2), consisting of residues 1 to 100 and 166 to 551 |
| Δkinase D | PKR deleted of residues 273 to 551 (kinase domain) |
| PACT | Full-length PACT, consisting of residues 1 to 305 |
| Δ1 | PACT deleted of residues 35 to 99 (domain I), consisting of residues 1 to 34 and 100 to 305 |
| Δ2 | PACT deleted of residues 127 to 192 (domain II), consisting of residues 1 to 126 and 193 to 305 |
| Δ3 | PACT deleted of residues 240 to 305 (domain III), consisting of residues 1 to 239 |
TABLE 2.
Sequences of gene-specific primer sets
| Primer name | Sequencea (5′-3′) |
|---|---|
| OV20.0-HindIII-F | TCAAGCTTATGGCCTGCGAGTG |
| OV20.0-NotI-R | TTAAGCGGCCGCGTAGAAGCTGAT |
| Δ1-79-HindIII-F | GGCCAAGCTTATGGAGACAGAG |
| Δ79-130-F | AACTCCGACACCGAGCCCACGCGTTCTGGCGGCA |
| Δ79-130-R | TGCCGCCAGAACGCGTGGGCTCGGTGTCGGAGTT |
| Δ131-184-NotI-R | TTAAGCGGCCGCCTCGCAGAACT |
| Δ79-184-NotI-R | TTTAAGCGGCCGCGGGCTCGGTGT |
| PKR-BamH I-F | AGCTGGATCCATGGCTGGTGATCTTTC |
| PKR-NotI-R | AATTGCGGCCGCACATGTGTGTCGTTCATT |
| ΔRBD1-BamH I-F | CCGGGGATCCATGGCTGGTGATCTTTCAGCAGGTTTCAATAAGGAAAAGAA |
| ΔRBD2-F | TCCATGGGGAATTTATCAGAAGAAACC |
| ΔRBD2-R | GGTTTCTTCTGATAAATTCCCCATGGA |
| ΔkinaseD-NotI-R | CCGGGCGGCCGCTAATTCTATTTCTTT |
Underlining indicates sequences of restriction enzymes.
For expressing OV20.0 with deletions of different regions, DNA fragments of full-length OV20.0L or with the deletion of residues 1 to 79, 80 to 129, 130 to 184, and 80 to 184 were separately amplified from pcDNA3.1-OV20.0-eGFP (8) by PCR using specific primer sets containing recognition sequences of HindIII and NotI on forward and reverse primers, respectively (sequences of the primers are listed in Table 2). All of these insert fragments were digested by corresponding restriction enzymes and ligated with the linearized p3x FLAG-CMV-14 vector (Sigma-Aldrich).
For the expression of different truncated forms of PKR, inserts deleted of the RNA binding domain (ΔRBD1), ΔRBD2, or Δkinase D were created from pcDNA3.1-PKR-HA plasmid (8) by the described overlap extension PCR with degenerated primers containing BamHI and NotI recognition sequences. The amplicons then were digested with corresponding restriction enzymes, followed by ligation with pcDNA3.1-HA plasmid linearized by BamHI and NotI enzymes.
Transfection and immunoprecipitation.
In general, human 293T cells were seeded in 6-well plates at a density of 1.5 × 106 cells/well. The next day, 5 μg of plasmid was used for transfection by Lipofectamine 2000 (Invitrogen), guided by the manufacturer's instructions.
For conducting immunoprecipitation (IP), constructs expressing various truncated forms of OV20.0, PACT (26), or PKR were transfected to cells seeded 1 day before the experiment. After 24 h of transfection, cells were harvested, resuspended in lysis buffer (50 mM Tris HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA and 1% Triton X-100), and incubated on ice for 10 min, followed by centrifugation at 10,000 × g. Crude cell extract was transferred to a new tube and mixed with anti-FLAG M2 affinity gel (Sigma-Aldrich) at 4°C for 4 h according to the manufacturer's instructions. The gel then was collected by centrifugation at 800 × g and followed by three washes with wash buffer (50 mM Tris-HCl, pH 7.4, with 150 mM NaCl). Lastly, proteins associated with anti-FLAG M2 affinity gel were eluted by SDS sample dye and resolved by SDS-PAGE, followed by Western blotting.
Double-stranded RNA (poly I·C) pulldown assay.
The generation of synthetic dsRNA followed the procedures described previously (8). Briefly, poly(C)-coated agarose beads (Sigma-Aldrich) were incubated with poly(I) (Sigma-Aldrich) in buffer containing 50 mM Tris (pH 7.0) and 150 mM NaCl at 4°C overnight with gentle rotation. Annealed poly(I·C) agarose beads were collected by centrifugation at 800 × g, followed by several washes in buffer containing 50 mM Tris (pH 7.0)–150 mM NaCl, and then were resuspended in the binding buffer A (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1% Nonidet P-40).
To identify the dsRNA-binding domain of OV20.0, cells transfected with various OV20.0 plasmids were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.9, 150 mM NaCl, 0.1% SDS, 1.0% NP-40, 0.5% sodium deoxycholate) on ice followed by centrifugation at 10,000 × g. Supernatants (whole-cell lysate) were mixed with annealed poly(I·C) beads at 4°C. After 1 h of incubation, the beads was collected by centrifugation at 800 × g, followed by washing twice with buffer C (10 mM Tris-HCl, pH 7.8, 6 mM MgCl2, 80 mM KCl, 2 mM dithiothreitol, 250 mM sucrose, and 0.1 mM EDTA). Finally, the poly(I·C) beads were collected by centrifugation and resuspended in SDS sample dye.
Immunofluorescence assay (IFA).
Human 293T cells were individually transfected with plasmids expressing EGFP fused with full-length OV20.0 or deletion of residues 1 to 79 (Δ1-79) or 130 to 184 (Δ130-184) (8). At 24 h posttransfection, cells were fixed by 4% paraformaldehyde for 30 min, followed by permeabilization with 0.1% Triton X-100 for another 10 min. Subsequently, cells were incubated with anti-PKR antibody (Abcam) at a dilution of 1:1,000 for 1 h at room temperature. After six washes, goat anti-rabbit IgG (1:2,000-fold diluted) (Alexa Fluor 594; Invitrogen) was used as the secondary antibody. After 1 h of incubation, nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) for 10 min, followed by confocal microscopy. Between each step, cells were washed at least three times with PBS containing 1% FCS.
Western blot analysis.
Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene difluoride (PVDF) membrane, followed by immune staining procedures described previously (8). In particular, the antibodies (and corresponding dilutions) were the following: FLAG antibody (1:5,000) (F7425; Sigma-Aldrich), PKR antibody (1: 2,000) (ab32052; Abcam), PKR-p antibody (1:1,000) (ab32036; Abcam), β-actin antibody (1:5,000) (GTX26276; Gene Tex), EGFP antibody (1:5,000) (YH80005; Yao-Hong Biotechnology, Taiwan), hemagglutinin (HA) antibody (1:5,000) (YH80008; Yao-Hong Biotechnology, Taiwan), PACT antibody (ab117704; Abcam), and OV20.0 antibody from mice immunized with purified OV20.0 recombinant protein (1:1,000) (8). Goat anti-rabbit or anti-mouse IgG-conjugated horseradish peroxidase (HRP) (1:10,000-fold diluted) (Jackson Laboratory) served as the secondary antibody. After reacting with secondary antibodies for 1 h, the signal was detected by an enzyme-linked chemiluminescence system (ECL; Amersham, GE Healthcare) and revealed by ImageQuant LAS 4000 (GE Healthcare). Washes with PBS-T (PBS with 0.1% Tween) were done between each step.
Sequence analysis.
Sequences of OV20.0 (ABY41265.1), PACT (AAC25672.1), and PKR (XP_011531289.1) proteins were obtained from GenBank. Multiple-sequence alignment was conducted by the ClustalW2 online service (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Fully conserved residues are labeled with an asterisk. Strong similar properties, with a scoring of >0.5 in the Gonnet PAM 250 matrix among groups, are indicated by a colon. A score of <0.5, indicating weak similarity among groups, is labeled with a dot.
RESULTS
The dsRNA binding region of OV20.0.
According to previous research, both OV20.0 and its N-truncated isoform, sh20 (i.e., Δ1-79), are dsRNA binding proteins in vitro. However, the specific residues or domains required for dsRNA binding ability remain uncertain. Sequence alignments based on aligning ORFV OV20.0 with its orthologs from goatpox virus (GPV; a capripoxvirus) and VACV indicate that the dsRNA-binding domain of OV20.0 resides at the C terminus (8). To investigate the minimal region needed for dsRNA binding, several constructs, namely, full-length OV20.0 and various OV20.0 mutants with deletions of residues 1 to 79, 80 to 129, 130 to 184, and 80 to 184, were generated (Fig. 1A). To assess their dsRNA binding ability, lysates of cells transiently expressing each of these OV20.0 proteins were incubated with beads conjugated with annealed poly(I·C), which is a synthetic dsRNA. As shown in the upper portion of Fig. 1B, in addition to full-length OV20.0, only Δ1-79 was able to be pulled down by poly(I·C) beads (Fig. 1B, lanes 1 and 2), while EGFP and the other OV20.0 mutants with various deletions were not detected in the dsRNA-binding fraction. These results indicated that residues 80 to 184 are required for interacting with dsRNA.
FIG 1.
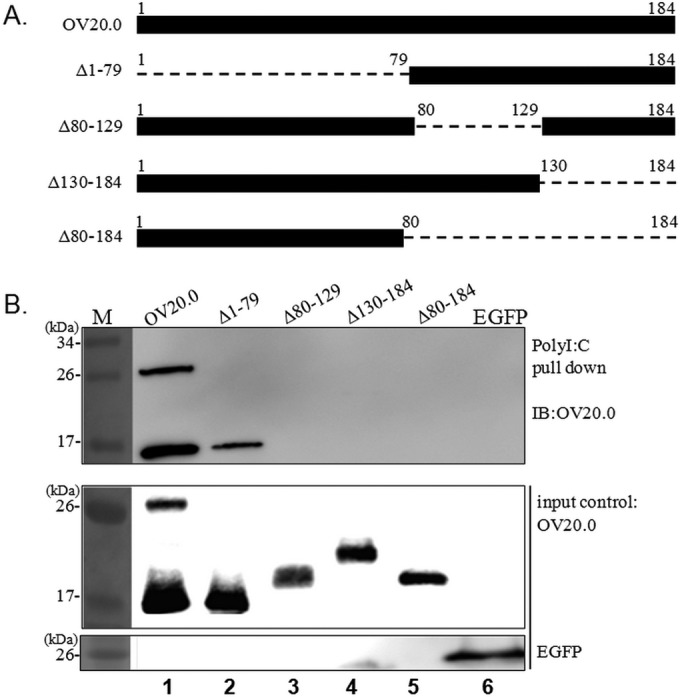
dsRNA binding domain of OV20.0. (A) An illustration showing the constructs expressing the wild type and deletions of ORFV OV20.0 used in this study. The deleted regions are presented as dashes, and the numbers indicate the corresponding amino acids. (B) Plasmids expressing FLAG-tagged wild-type OV20.0 and Δ1-79, Δ80-129, Δ130-184, and Δ79-184 mutants of OV20.0 or EGFP were transfected individually into human 293T cells, which was followed by a poly(I·C) pulldown assay. The collected pellets (upper) as well as 20% of the cell lysate, which was used as the input control (lower), were separately subjected to Western blot analysis using anti-FLAG or anti-EGFP antibodies. The data shown are representative of three independent experiments. Lane M, molecular size marker.
Region of OV20.0 responsible for interaction with PKR.
Interaction between OV20.0 and PKR was reported previously. In this study, we investigated in more detail the binding domain that allows interaction between these two proteins. An earlier study has shown that sh20, a shortened isoform of OV20.0 that consists of residues 80 to 184 (Δ1-79), is sufficient to allow PKR binding; hence, in these experiments, full-length OV20.0 and Δ1-79 were used as positive controls, and the PKR binding ability of the other three mutants was determined. Affinity gel conjugated with FLAG antibody was used to pull down the FLAG-tagged OV20.0 proteins, and the interaction of these proteins with endogenous PKR then was revealed by immunoblot analysis; the results are presented in Fig. 2A. Wild-type OV20.0 and Δ1-79 were able to interact with cellular PKR consistently (Fig. 2A, lanes 1 and 2). Importantly, the deletion mutant lacking residues 80 to 129 (Δ80-129), which has been shown earlier not to bind dsRNA, still is able to interact with PKR (Fig. 2A, lane 3). Nevertheless, the other two mutants (Δ130-184 and Δ80-184), which are missing the last 55 amino acids at the C terminus (residues 130 to 184), are unable to efficiently bind PKR and give only a faint or lack a PKR signal (Fig. 2A, lanes 4 and 5, respectively). These results were confirmed by the immunofluorescent staining (Fig. 2B). Full-length OV20.0 and Δ1-79 fused with EGFP were expressed in the 293T cells and found to colocalize with endogenous PKR. However, when the last 55 amino acids of OV20.0 protein were deleted, this mutant version of OV20.0 (Δ130-184) was found to be present mainly in the nucleus, while PKR remained in the cytoplasm. These findings indicate that OV20.0 uses a domain that contains residues 130 to 184 for PKR binding.
FIG 2.

Regions of OV20.0 involved in PKR interaction. (A) Human 293T cells were individually transfected with plasmids expressing the FLAG-tagged wild type and Δ1-79, Δ80-129, Δ130-184, and Δ79-184 mutants of OV20.0. The next day, the cell lysates were harvested for immunoprecipitation (IP) using anti-FLAG agarose beads. (Lower) An aliquot of total cell lysate (20% of IP) was used as the input control. Proteins eluted from beads then were analyzed by Western blotting using either PKR or FLAG antibodies. (B) Full-length (Kozak20) OV20.0 or the Δ1-79 or Δ130-184 mutant fused with EGFP was transiently expressed in 293T cells. At 24 h after transfection, the expression of endogenous PKR was detected by immunofluorescent assay (IFA) using PKR antibody, which was followed by confocal microscopy. The presence of PKR in nuclei in cells expressing full-length OV20.0 is indicated with an arrow. (C) Map of the constructs expressing the wild type and the deletion of the various individual functional domains of PKR. Dashes and the numbers indicate deleted regions and the residues of PKR, respectively. (D) 293T cells were cotransfected with plasmids expressing FLAG-tagged OV20.0 and one of the HA-tagged PKR proteins or EGFP (as a negative control). Immunoprecipitation was conducted with FLAG antibody-conjugated agarose beads followed by detection of PKR in the pellet fraction (upper) or of OV20.0 and EGFP. (Lower) Proteins in whole-cell lysate, which were used as input controls. Three independent experiments were performed.
To identify the region of PKR interacting with OV20.0, PKR mutants with deletions of each individual functional domain, namely, deletions of RBD1 (ΔRBD1), RBD2 (ΔRBD2), and the kinase domain (Δkinase D), were generated (Fig. 2C). The results of an immunoprecipitation assay showed that there was a strong interaction between wild-type PKR and OV20.0 (Fig. 2D, lane 1); this was used as the positive control, while EGFP acted as a negative control. This approach reduces concerns regarding nonspecific binding in these experiments (Fig. 2D, lane 5). When a similar approach was used with the three deletion mutants, it was found that the absence of the kinase domain (Δkinase D) weakened the association of PKR with OV20.0 (Fig. 2D, lane 4), indicating the contributions of the kinase region to this interaction. However, when ΔRBD1 and ΔRBD2 were used in similar experiments, the binding of PKR to OV20.0 was completely abrogated (Fig. 2D, lanes 2 and 3). This indicates that the interaction between PKR and OV20.0 involves both RBD1 and RBD2. Taking the above-described findings as a whole, the region containing the last 55 amino acids (130 to 184) of OV20.0 protein seems to be critical to the interaction of OV20.0 with PKR via RBD1 and RBD2.
The region of OV20.0 involved in suppression of PKR phosphorylation.
The above-described findings identify the binding sites that allow interaction between OV20.0 and PKR. Based on these findings, we next determined whether OV20.0 binding is responsible for repressing dsRNA-induced PKR activation. Five constructs expressing various regions of OV20.0, namely, OV20.0, Δ1-79, Δ80-129, Δ130-184, and Δ80-184, were transfected individually into cells, and this was followed by poly(I·C) treatment in order to stimulate PKR phosphorylation. As indicated in Fig. 3, the expression level of actin and the basal expression level of PKR were similar across each of the transfection groups; furthermore, poly(I·C) treatment, as a positive control (PC; lane 7), indeed stimulated phosphorylation of PKR compared to that of mock treatment (lane 1). Consistent with our previous results, OV20.0 and Δ1-79 completely ablated PKR phosphorylation (to a level that was below the sensitivity of the assay) (Fig. 3, lanes 2 and 3). Compared with the positive control, a much lower phosphorylation level was observed when Δ80-129 was present (Fig. 3, lane 4). Noticeably, the deletion of residues 130 to 184 and 80 to 184 did not have any significant impact on PKR phosphorylation (Fig. 3, lanes 5 and 6). These findings indicate that a lack of the PKR-interacting sites of OV20.0 (residues 130 to 184; Δ130-184) results in a weakening of the ability to antagonize dsRNA-induced PKR phosphorylation.
FIG 3.

C terminus of OV20.0 suppresses dsRNA-induced PKR phosphorylation. Human 293T cells were mock transfected (lanes 1 and 7) or transfected with different constructs expressing the various OV20.0 mutants (lanes 2 to 6). At 24 h posttransfection, the cells were left untreated (lane 1, mock) or were transfected with 1 μg poly(I·C) (lanes 2 to 7) for a further 4 h. Total proteins were separated by SDS-PAGE, which was followed by Western blotting using antiserum specific to phosphorylated PKR (PKR-p), basal PKR (PKR), or actin (β-actin), which acted as the loading control. Lane PC indicates cells transfected with poly(I·C) (without OV20.0) for PKR activation. Three independent experiments were performed.
OV20.0 inhibition of PKR activation is mediated by PACT.
In addition to dsRNA, PKR also can be activated by cellular PACT, which is also a dsRNA binding protein (24). We next explored whether OV20.0 is able to regulate PKR via this alternative PACT-mediated route. Increased PKR phosphorylation was observed in 293T cells that were transiently expressing PACT alone (Fig. 4A, lane 1), while in mock-transfected cells, PKR phosphorylation was below the detection level (Fig. 4A, lane 6). To identify the effect of OV20.0 on PKR activation mediated by PACT, increasing amounts (1 μg, 3 μg, 5 μg, and 10 μg) of OV20.0 plasmid were cotransfected with PACT into the cells and the PKR activation level was monitored. As shown in Fig. 4A (lanes 2 to 5), PACT-mediated PKR activation was strongly suppressed by OV20.0 in a dose-dependent manner; image quantification of the intensities of the bands indicated that the group transfected with 10 μg OV20.0 showed a significant inhibition of PKR activation compared with that of cells expressing none or lower levels of OV20.0 (Fig. 4B). These findings suggest that OV20.0 is able to block PACT-induced PKR activation.
FIG 4.

OV20.0 inhibits PACT-mediated PKR phosphorylation. (A) Using 293T cells, 5 μg of PACT plasmid was cotransfected with various amounts (0, 1, 3, 5, or 10 μg) of plasmids expressing OV20.0 (lanes 1 to 5) or were mock transfected (lane 6). At 24 h after transfection, total proteins were collected for Western blot analysis using antibodies specific to phosphorylated PKR (PKR-p), basal PKR (PKR), PACT, OV20.0, or actin (β-actin), which acted as the loading control. (B) The intensity of the phosphorylated PKR band was quantified by ImageJ, and the ratio of the various proteins to the basal level of PKR was calculated. The level of PKR phosphorylation in the mock control (sample in lane 1) was arbitrarily set to 1, and the relative inhibition ability of OV20.0 then was estimated. Results are representative of 3 independent experiments.
Interaction of OV20.0 with PACT.
We investigated whether OV20.0 is able to inhibit PACT-mediated PKR phosphorylation through protein-protein interaction. A series of immunoprecipitation reactions were set up to identify the association between endogenous PACT and OV20.0 protein; this involved individually expressing FLAG-tagged wild-type OV20.0 and its various mutants in 293T cells. Wild-type OV20.0, but not empty FLAG vector, strongly bound to cellular PACT (Fig. 5A, lanes 1 and 6, respectively). It appears that PACT is able to interact with the N terminus of the truncated form of OV20.0 (Δ1-79) to a much lesser extent than wild-type OV20.0 (Fig. 5, lane 2). In contrast, the other C-terminal deletion mutants of OV20.0 had completely lost their ability to associate with PACT (Fig. 5A, lanes 3 to 5). This observation suggests that amino acids 80 to 184 of OV20.0 are necessary for its binding to endogenous PACT.
FIG 5.

OV20.0 interacts with PACT. (A) Plasmids expressing FLAG-tagged wild-type OV20.0 (lane 1), various OV20.0 deletion mutants (lanes 2 to 5), or empty vector (lane 6) were separately transfected in 293T cells; this was followed by immunoprecipitation using FLAG antibody-conjugated agarose beads. The FLAG pulldown fractions then were probed using antiserum specific for either PACT or FLAG. (B) Maps of the PACT deletion constructs are shown. The dashes and the numbers identify the truncated regions and corresponding residues, respectively. (C) Plasmids expressing FLAG-tagged wild-type PACT (lane 1) and the various individual PACT deletion mutants (lanes 2 to 4) were cotransfected with HA-tagged OV20.0 into 293T cells for 24 h. The whole-cell lysate next was immunoprecipitated with anti-FLAG agarose beads. The pellet factions (IP: FLAG; upper) and the input control (input; lower) then were separated by SDS-PAGE, which was followed by immunoblot analysis using antibodies specific to HA (for OV20.0), to FLAG (for the PACTs), or to β-actin. Results are representative of 3 independent experiments.
Subsequently, the region of PACT interacting with OV20.0 was determined using three PACT mutants deficient in domain I (Δ1), domain II (Δ2), and domain III (Δ3); domains I and II are responsible for dsRNA binding, while domain III contributes to PKR binding and activation (Fig. 5B). HA-tagged OV20.0 and FLAG-tagged PACT were coexpressed in cells, and FLAG-based immunoprecipitation assays were performed. Consistent with this finding, FLAG-tagged wild-type PACT, which acts as a positive control, interacted with full-length OV20.0 (Fig. 5C, lane 1), while the empty FLAG vector did not pull down HA-OV20.0 (Fig. 5C, lane 5). Furthermore, PACT with a deletion of either domain II or III retained the ability to interact with OV20.0 (Fig. 5C, lanes 3 and 4), while PACT that lacked domain I (Δ1) was found to have lost the ability to allow binding between PACT and OV20.0 (Fig. 5C, lane 2). These findings demonstrated that residues 80 to 184 of OV20.0 and domain I of PACT are required in order to establish the interaction between these two proteins.
OV20.0 represses PKR activation via its association with PKR rather than with PACT.
Since OV20.0 interacts with both PKR and its activator PACT, the question arises as to which association is critical to the OV20.0-dependent inhibition of PKR activation induced by PACT. The problem associated with complicated interactions was simplified by the use of a PACT mutant (Δ1) that fails to interact with OV20.0 but retains the ability to induce PKR phosphorylation. In such a system, if the inhibition effect of OV20.0 still exists, then the physical interaction between PACT and OV20.0 is not absolutely essential for repressing PACT-mediated PKR activation. Human 293T cells were transfected simultaneously with PACT Δ1 and OV20.0, and this was followed by an assay to detect the level of PKR phosphorylation. The results initially revealed that the overexpression of PACT Δ1 in 293T cells led to PKR phosphorylation compared to the level of expression in untreated cells (Fig. 6, lanes 1 and 2). Noticeably, as the doses of OV20.0 increased, the inhibition effect on PKR phosphorylation was gradually elevated (Fig. 6, lanes 3 to 5), while no effect was observed in the cells transfected with the empty vector (Fig. 6, lanes 6 to 8). Thus, it seems that interaction between OV20.0 and PKR is more critical to the inhibition of PACT-induced PKR phosphorylation than is the binding of OV20.0 to PACT.
FIG 6.
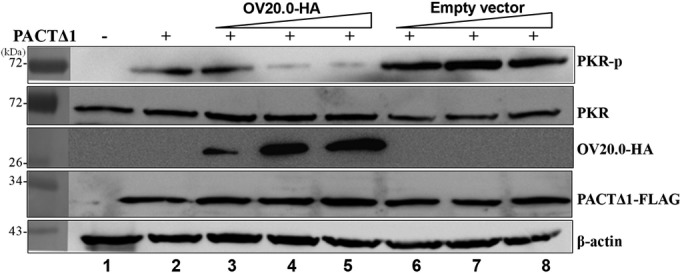
OV20.0 blocks PACT-mediated PKR phosphorylation by interacting with PKR rather than with PACT. A total of 5 μg of plasmid expressing FLAG-tagged PACTΔ1 was transfected alone (lane 2) or cotransfected with 1, 5, or 10 μg of constructs expressing HA-tagged OV20.0 (lanes 3 to 5) or with empty vectors (lanes 6 to 8) into 293T cells. At 24 h after transfection, the individual cell lysates were harvested and then were subjected to Western blot analysis using antibodies specific to phosphorylated PKR (PKR-p), basal PKR (PKR), HA (OV20.0-HA), FLAG (PACTΔ1-FLAG), or β-actin. Results are representative of 3 independent experiments.
OV20.0 interferes with the interaction of PKR with its inducer, PACT.
OV20.0 has the ability to inhibit PACT-induced PKR activation, even after losing its ability to interact with PACT (Fig. 6). A previous report indicated that the existence of domain III alone is not sufficient to allow the association of PACT with PKR, and that there is a requirement for either domain I or domain II in order to stabilize this interaction (28). Hence, it is worth exploring whether OV20.0 blocks PACT-mediated PKR activation by initially occupying PKR, which in turn will prevent the binding of PACT to PKR. By means of immunoprecipitation, the results show that an increase in OV20.0 is associated with a decreased association between PACT and PKR (Fig. 7, lanes 3 to 5). These results suggest the possibility that binding of OV20.0 interferes with the interaction between PACT and PKR, which in turn leads to a repression of PACT-induced PKR activation.
FIG 7.
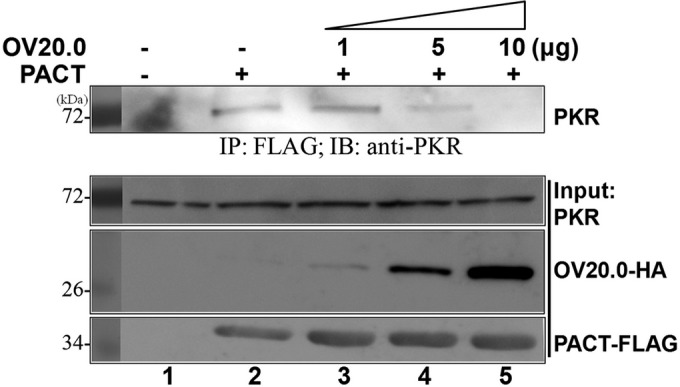
OV20.0 prevents the association of PACT with PKR. Human 293T cells were transfected with 5 μg of construct expressing FLAG-tagged PACT alone (lane 2) or together with 1, 5, or 10 μg of plasmids expressing HA-tagged OV20.0 (lanes 3 to 5) for 24 h. (Lower) Total cell lysates then were harvested individually for immunoprecipitation using anti-FLAG agarose beads; 20% of the cell lysate was kept as the input control. (Upper) The pellet fractions were subjected to Western blot analysis using anti-PKR antibodies. Results are representative of 3 independent experiments.
dsRNA facilitates the association of OV20.0 with PACT but not with PKR.
OV20.0 protein interacts with PKR and its activators PACT and dsRNA. Coincidently, the three proteins all have dsRNA binding ability; therefore, whether dsRNA participates in these associations needs further investigation. To define the role of dsRNA, RNase V1 (Life Technologies Corporation), an RNase that specifically targets dsRNA, was included during the immunoprecipitation reaction. As indicated in Fig. 8A, RNase V1 digestion did not influence the binding of endogenous PKR with OV20.0 in transient expression cells (Fig. 8A, upper, lanes 2 and 3). Nevertheless, the association between OV20.0 and PACT was weakened under RNase V1 treatment (Fig. 8A, middle, lanes 2 and 3).
FIG 8.

Double-stranded RNA facilitates the interaction of OV20.0 with PACT but not between OV20.0 and PKR. (A) A construct expressing FLAG-tagged OV20.0 (lanes 2 and 3) or the vector alone (lane 1) were transfected into 293T cells, or (B) goat fibroblast cells (lane 4) were infected with recombinant ORFV OV20.0-FLAG-GFP virus (lanes 5 and 6) at an MOI of 2 for 24 h. Total cell lysates of these transfections then were immunoprecipitated using anti-FLAG agarose beads with (lanes 2 and 5) or without (lanes 3 and 6) the addition of 0.5 U of RNase V1. The proteins bound to the beads were collected and then subjected to Western blot analysis using antibodies against PKR, PACT, or FLAG (OV20.0-FLAG). Results are representative of two independent experiments.
This finding was further confirmed in cells infected with recombinant ORFV (ORFV-GFP) that also were expressing FLAG-tagged OV20.0 (8). In infected goat fibroblast cells, both PACT and PKR are able to be coprecipitated with viral OV20.0 protein (Fig. 8B, upper and middle). Consistent with this finding, RNase V1 treatment significantly reduced the association between OV20.0 and PACT, while the binding of PKR remained unaffected (Fig. 8B, lanes 2 and 3). These results indicate that the presence of dsRNA increases the interaction and affinity between viral OV20.0 and cellular PACT.
Sequence alignment of the RBD regions of OV20.0, RKR, and PACT.
It is worth noting that sequence alignment of the RBDs of OV20.0 (residues 80 to 184), PACT (domain I and II), and PKR (RBD1 and RBD2) contain several consensus residues that are conserved (Fig. 9), especially residues located at the C terminus. These residues, F148, K167, R168, and K171, have been reported to be involved in the dsRNA binding capacity of these proteins (35, 36). Since members of the dsRNA binding protein family are likely to form homodimers with themselves or heterodimers with other members of the family via their mutual RBDs (37, 38), it is possible that OV20.0 would form heterodimers with PKE and PACT proteins.
FIG 9.

Sequence analysis of RBDs of OV20.0, PACT, and PKR. Sequence of the RBDs of OV20.0, PACT, and PKR were aligned. The dashes represent gaps in the alignment. The asterisks indicate fully conserved residues. The columns (indicated by colons) indicate that one of the following strong residue groups is conserved: FI, FY, KR, KQ, MILV, NQ, or ST. A single dot indicates one of the following groups is conserved: AC, AGS ENS, or HNQR. Boxes mark the conserved residues for dsRNA binding proteins.
DISCUSSION
PKR is an essential host protein that is involved in the innate immune pathway and antiviral responses. Many viruses have developed dedicated mechanisms to counteract PKR activation or that target its downstream signaling pathway (9-11, 39). For example, the E3 and K3 proteins of vaccinia virus (VACV) regulate PKR activity via a range of different mechanisms (21, 40, 41). VACV E3 physiologically interacts with PKR and its activator dsRNA via the protein's N-terminal and C-terminal domains, respectively (6, 35, 42), while K3 mimics the structure of eIF2α, which reduces PKR transphosphorylation of its downstream signaling pathway (43, 44). Unlike VACV, ORFV does not encode a K3 ortholog (45), and as a result, OV20.0 alone plays a role in the modulation of the PKR response by ORFV, which emphasizes this protein's importance to counteracting the host's immune responses. We have previously reported that OV20.0 interacts with PKR and sequesters dsRNA, which leads to the inhibition of PKR activation (8). To explore the mechanism by which this occurs, in this study we identified the dsRNA-binding domain of OV20.0, which is located at the C terminus (residues 80 to 184) (Fig. 1). In addition, interaction sites between OV20.0 and PKR were mapped to residues 130 to 184 of the OV20.0 protein and RBD1 and RBD2 sites of PKR (Fig. 2). These findings imply that the last 55 amino acids at the C terminus of OV20.0 are critical to the inhibition of dsRNA-induced PKR phosphorylation (Fig. 3). Nevertheless, since OV20.0 containing only residues 130 to 184 was not included in this set of experiments, whether these 55 amino acids of OV20.0 is sufficient for PKR binding requires further investigation. Moreover, since our results indicated OV20.0 associates with dsRNA binding regions of PKR, it is possible that in addition to sequestering dsRNA, the association of OV20.0 with PKR would further block dsRNA binding accessibility.
PACT activates PKR by triggering a change in the conformation of PKR that is related to dimerization and is independent of dsRNA binding (24). It has been demonstrated that the existence of domain III is sufficient for PKR phosphorylation in vitro via the protein's constitutive phosphorylation at S246 (27–29). However, the weak interaction between PACT domain III and PKR can be strengthened by either PACT domain I or II (26). In our study, OV20.0 was found, for the first time, to interact with PACT, leading to the abrogation of PACT-mediated PKR activation (Fig. 4). This interaction is brought about via the C-terminal 105 residues of OV20.0 and domain I of PACT (Fig. 5). The C-terminal end of OV20.0 is responsible for the dsRNA binding as well as contributing to the interaction of PKR and PACT, which raises the issue of whether dsRNA mediates the association between these proteins. To clarify this, we used RNase V1 to remove dsRNA, and the results indicate that dsRNA facilitates association between PACT and OV20.0 when the latter is expressed transiently or when it is expressed by ORFV. Nevertheless, OV20.0 is able to directly interact with PKR without the aid of dsRNA, as shown in Fig. 8 of current study and one previous report (8). Consistent with this, a systematic point mutation analysis of the RBD of VACV E3 has revealed that the ability to bind dsRNA binding is not necessary for the inhibition of PKR activation and some other of the protein's biological functions (36).
The inhibition of PACT-mediated PKR activation has been described in two viruses other than ORFV; the proteins involved are Us11 of herpes simplex virus and NS1 of influenza A virus (46–49). As with OV20.0, both Us11 and NS1 are dsRNA binding proteins that are able to block PKR activation when induced by either dsRNA or PACT. The C terminus of Us11 (residues 88 to 155, i.e., the RBD) is used as a target for domains I and II of PACT (47), while the NS1 protein has been shown to associate with domain I of PACT (46). While OV20.0 shares with Us11 and NS1 proteins the same property of interrupting PACT-mediated PKR activation by binding to PKR rather than to PACT (46, 47), we have shown here for the first time that OV20.0 protein is able to prevent PACT binding to PKR (Fig. 7). This property may be explained by the fact that amino acids 130 to 183 of OV20.0 not only interact with RBD1 and RBD2 but also associate with the kinase domain of PKR to a certain extent (Fig. 2D, lane 4). Furthermore, we assume that the binding of the RBDs of PKR by OV20.0 is able to weaken its interaction with domain I/II of PACT. It has been shown that interaction between the PKR RBD and PACT domain I/II is crucial for enhancing the binding affinity and for the partial interaction with the kinase domain of PKR, which in turn will reduce the association between PKR and domain III of PACT (26). These findings imply that OV20.0 is able to suppress PKR activation by preventing PKR from binding to PACT.
Many dsRNA binding proteins form heterodimers with each other via their RBDs (37, 38). For instance, PACT is known to associate with PKR via its RBDs and the C terminus regardless of the presence of dsRNA (24), and with TRBP, a negative regulator of PKR, it is known to form heterodimers with PACT via their RBDs in order to balance PKR functioning (31, 33). In the present study, we have shown that OV20.0 interacts with domain I (the RBD) of PACT (Fig. 5C), and that the sequence similarity of regions involved in the PACT-OV20.0 interaction is 54%, with 35% identity; furthermore, the consensus sequences of the RBD are highly conserved in OV20.0, which implies a high degree of structural similarity between these two proteins. If we consider that the heterodimer of PACT and TRBP shares 40% sequence similarity (31, 33), which is lower than that of OV20.0, it is possible that the RBD of OV20.0 is able to form heterodimers with domain I of PACT.
In conclusion, OV20.0 protein is able to counteract PKR activation by multiple mechanisms, and these are outlined in Fig. 10. First, OV20.0 is able to bind PKR and its two activators, dsRNA and PACT; this finding is supported by our experimental results. In particular, OV20.0 protein is able to sequester dsRNA (as outlined in action 1). Second, OV20.0 is able to bind directly to the RBDs of PKR, which are responsible for the protein's intramolecular conformational change on receiving a suitable substrate (outlined in action 2); importantly, this interaction does not require dsRNA. Third, OV20.0 interacts with or occupies the RBD2 and kinase domain of PKR, which then prevents PACT binding to PKR (28). Finally, another possible mechanism involves OV20.0 forming heterodimers with PACT via their RBDs, which may reduce the ability of PACT to induce PKR activation (as outlined in action 3). The findings in this study provide new concepts in relation to how ORFV modulates PKR activation.
FIG 10.

Mechanisms by which OV20.0 mediates the inhibition of PKR activation. PKR is activated by dsRNA and PACT protein consisting of domains I, II, and III. Binding of the two activators leads to a change in the conformation of PKR that affects the protein's dimerization and autophosphorylation. ORFV OV20.0 inhibits PKR activation via at least three mechanisms: sequestering dsRNA (1), interaction with PKR (2), and binding to PACT (3). RBD, dsRNA-binding domain; kinase D, kinase domain.
ACKNOWLEDGMENTS
The project was supported by the Ministry of Science and Technology, Taiwan (MOST-103–2321-B-005-001), and by the Ministry of Education, Taiwan, Republic of China, under the ATU plan.
REFERENCES
- 1.McInnes CJ, Wood AR, Mercer AA. 1998. Orf virus encodes a homolog of the vaccinia virus interferon-resistance gene E3L. Virus Genes 17:107–115. doi: 10.1023/A:1026431704679. [DOI] [PubMed] [Google Scholar]
- 2.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 3.Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. 2008. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog 4:e1000096. doi: 10.1371/journal.ppat.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eduardo-Correia B, Martinez-Romero C, Garcia-Sastre A, Guerra S. 2014. ISG15 is counteracted by vaccinia virus E3 protein and controls the proinflammatory response against viral infection. J Virol 88:2312–2318. doi: 10.1128/JVI.03293-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang HW, Jacobs BL. 1993. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 194:537–547. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- 6.Romano PR, Zhang F, Tan SL, Garcia-Barrio MT, Katze MG, Dever TE, Hinnebusch AG. 1998. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Mol Cell Biol 18:7304–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haig DM, McInnes CJ, Thomson J, Wood A, Bunyan K, Mercer A. 1998. The orf virus OV20.0L gene product is involved in interferon resistance and inhibits an interferon-inducible, double-stranded RNA-dependent kinase. Immunology 93:335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng YY, Lin FY, Cheng SF, Tscharke D, Chulakasian S, Chou CC, Liu YF, Chang WS, Wong ML, Hsu WL. 2015. Functional analysis of the short isoform of Orf virus protein OV20.0. J Virol 89:4966–4979. doi: 10.1128/JVI.03714-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs BL, Langland JO. 1998. Reovirus sigma 3 protein: dsRNA binding and inhibition of RNA-activated protein kinase. Curr Top Microbiol Immunol 233:185–196. [DOI] [PubMed] [Google Scholar]
- 10.Child SJ, Geballe AP. 2009. Binding and relocalization of protein kinase R by murine cytomegalovirus. J Virol 83:1790–1799. doi: 10.1128/JVI.01484-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergmann M, Garcia-Sastre A, Carnero E, Pehamberger H, Wolff K, Palese P, Muster T. 2000. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J Virol 74:6203–6206. doi: 10.1128/JVI.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langland JO, Jacobs BL. 2004. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology 324:419–429. doi: 10.1016/j.virol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Saunders LR, Barber GN. 2003. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17:961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F, Romano PR, Nagamura-Inoue T, Tian B, Dever TE, Mathews MB, Ozato K, Hinnebusch AG. 2001. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276:24946–24958. doi: 10.1074/jbc.M102108200. [DOI] [PubMed] [Google Scholar]
- 15.Nallagatla SR, Toroney R, Bevilacqua PC. 2011. Regulation of innate immunity through RNA structure and the protein kinase PKR. Curr Opin Struct Biol 21:119–127. doi: 10.1016/j.sbi.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talloczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gil J, Esteban M. 2000. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis 5:107–114. doi: 10.1023/A:1009664109241. [DOI] [PubMed] [Google Scholar]
- 18.Farrell PJ, Balkow K, Hunt T, Jackson RJ, Trachsel H. 1977. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell 11:187–200. doi: 10.1016/0092-8674(77)90330-0. [DOI] [PubMed] [Google Scholar]
- 19.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. 2000. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol Cell Biol 20:4532–4542. doi: 10.1128/MCB.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies MV, Chang HW, Jacobs BL, Kaufman RJ. 1993. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J Virol 67:1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryant KF, Cox JC, Wang H, Hogle JM, Ellington AD, Coen DM. 2005. Binding of herpes simplex virus-1 US11 to specific RNA sequences. Nucleic Acids Res 33:6090–6100. doi: 10.1093/nar/gki919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawagishi-Kobayashi M, Cao C, Lu J, Ozato K, Dever TE. 2000. Pseudosubstrate inhibition of protein kinase PKR by swine pox virus C8L gene product. Virology 276:424–434. doi: 10.1006/viro.2000.0561. [DOI] [PubMed] [Google Scholar]
- 24.Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel CV, Handy I, Goldsmith T, Patel RC. 2000. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem 275:37993–37998. doi: 10.1074/jbc.M004762200. [DOI] [PubMed] [Google Scholar]
- 26.Peters GA, Hartmann R, Qin J, Sen GC. 2001. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol 21:1908–1920. doi: 10.1128/MCB.21.6.1908-1920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters GA, Dickerman B, Sen GC. 2009. Biochemical analysis of PKR activation by PACT. Biochemistry 48:7441–7447. doi: 10.1021/bi900433y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Peters GA, Ding K, Zhang X, Qin J, Sen GC. 2006. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci U S A 103:10005–10010. doi: 10.1073/pnas.0602317103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters GA, Li S, Sen GC. 2006. Phosphorylation of specific serine residues in the PKR activation domain of PACT is essential for its ability to mediate apoptosis. J Biol Chem 281:35129–35136. doi: 10.1074/jbc.M607714200. [DOI] [PubMed] [Google Scholar]
- 30.Gupta V, Huang X, Patel RC. 2003. The carboxy-terminal, M3 motifs of PACT and TRBP have opposite effects on PKR activity. Virology 315:283–291. doi: 10.1016/S0042-6822(03)00589-0. [DOI] [PubMed] [Google Scholar]
- 31.Laraki G, Clerzius G, Daher A, Melendez-Pena C, Daniels S, Gatignol A. 2008. Interactions between the double-stranded RNA-binding proteins TRBP and PACT define the Medipal domain that mediates protein-protein interactions. RNA Biol 5:92–103. doi: 10.4161/rna.5.2.6069. [DOI] [PubMed] [Google Scholar]
- 32.Singh M, Castillo D, Patel CV, Patel RC. 2011. Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry 50:4550–4560. doi: 10.1021/bi200104h. [DOI] [PubMed] [Google Scholar]
- 33.Daher A, Laraki G, Singh M, Melendez-Pena CE, Bannwarth S, Peters AH, Meurs EF, Braun RE, Patel RC, Gatignol A. 2009. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol Cell Biol 29:254–265. doi: 10.1128/MCB.01030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heckman KL, Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc 2:924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 35.Ho CK, Shuman S. 1996. Mutational analysis of the vaccinia virus E3 protein defines amino acid residues involved in E3 binding to double-stranded RNA. J Virol 70:2611–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dueck KJ, Hu YS, Chen P, Deschambault Y, Lee J, Varga J, Cao J. 2015. Mutational analysis of vaccinia E3 protein: the biological functions do not correlate with its biochemical capacity of dsRNA binding. J Virol 89:5382–5394. doi: 10.1128/JVI.03288-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu S, Kaufman RJ. 1997. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J Biol Chem 272:1291–1296. doi: 10.1074/jbc.272.2.1291. [DOI] [PubMed] [Google Scholar]
- 38.Cosentino GP, Venkatesan S, Serluca FC, Green SR, Mathews MB, Sonenberg N. 1995. Double-stranded-RNA-dependent protein kinase and TAR RNA-binding protein form homo- and heterodimers in vivo. Proc Natl Acad Sci U S A 92:9445–9449. doi: 10.1073/pnas.92.21.9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hakki M, Marshall EE, De Niro KL, Geballe AP. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J Virol 80:11817–11826. doi: 10.1128/JVI.00957-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langland JO, Jacobs BL. 2002. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology 299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- 41.Rice AD, Turner PC, Embury JE, Moldawer LL, Baker HV, Moyer RW. 2011. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J Virol 85:550–567. doi: 10.1128/JVI.00254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho CK, Shuman S. 1996. Physical and functional characterization of the double-stranded RNA binding protein encoded by the vaccinia virus E3 gene. Virology 217:272–284. doi: 10.1006/viro.1996.0114. [DOI] [PubMed] [Google Scholar]
- 43.Davies MV, Elroy-Stein O, Jagus R, Moss B, Kaufman RJ. 1992. The vaccinia virus K3L gene product potentiates translation by inhibiting double-stranded-RNA-activated protein kinase and phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J Virol 66:1943–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carroll K, Elroy-Stein O, Moss B, Jagus R. 1993. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J Biol Chem 268:12837–12842. [PubMed] [Google Scholar]
- 45.Bratke KA, McLysaght A, Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. doi: 10.1016/j.meegid.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Min JY, Krug RM, Sen GC. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Peters GA, Khoo D, Mohr I, Sen GC. 2002. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J Virol 76:11054–11064. doi: 10.1128/JVI.76.21.11054-11064.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kew C, Lui PY, Chan CP, Liu X, Au SW, Mohr I, Jin DY, Kok KH. 2013. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol 87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poppers J, Mulvey M, Khoo D, Mohr I. 2000. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 74:11215–11221. doi: 10.1128/JVI.74.23.11215-11221.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]