

To the editor:
The WHO/OIE/FAO H5N1 Evolution Working Group has previously proposed a nomenclature system designed to classify the A/goose/Guangdong/1/1996 lineage of Eurasian highly pathogenic H5N1 avian influenza viruses. To date the nomenclature system has identified 10 distinct clades of viruses based primarily on the phylogenetic characterization and sequence homology of the hemagglutinin (HA) gene. 1 Clades were strictly defined using criteria based on sharing of a common node and monophyletic grouping with a bootstrap value of ≥60 at the clade defining node (after 1000 neighbor‐joining bootstrap replicates) and maintaining average percentage pairwise nucleotide distances between and within clades of >1·5% and <1·5% respectively. However, as viruses within these clades continue to evolve, new H5N1 clades (defined by the same specific criteria) can be expected to periodically emerge from within established clades. These new clades are defined as second (or third, etc.) order clades and assigned a numerical ‘address’ which links them to their original clade. In this progressive system, the WHO/OIE/FAO H5N1 Evolution Working Group examines the available data and updates the associated phylogenetic trees and clade nomenclature on an ongoing basis.
Divergence of clade 2·2 H5N1 viruses
The continuing circulation of highly pathogenic avian influenza (HPAI) H5N1 viruses in poultry and/or wild birds of Asia, the Middle East, Europe, and Africa has led to an increase in the cumulative number of isolates available for sequence analysis. The H5 HA from these viruses has continued to evolve rapidly. Molecular phylogenetic analyses of the clade 2·2 HA genes are of particular importance due to the wide geographic spread of viruses of this clade and their proven ability to cause human disease. Several recent independent reports in the scientific literature describe the evolution of clade 2·2 HPAI viruses 2 , 3 , 4 , 5 , 6 , 7 and identify emerging lineages using either novel naming conventions or modifications of the criteria proposed by the WHO/OIE/FAO H5N1 Evolution Working Group to classify HPAI H5N1 viruses.
In order to understand how these new designations are related to the H5 HA phylogenetic tree previously published by the WHO Evolution Working Group, a comprehensive phylogenetic analysis of the HA of clade 2·2 viruses was performed using these new data, as well as other sequences more recently added to public databases. In this letter, we consider the further delineation of this specific clade by applying the unified nomenclature criteria used previously to classify HPAI H5N1 virus lineages. 1
To address the question of whether the clade 2·2 viruses have evolved sufficiently to require subdivision into third‐order clades, as has been done with other clades (i.e., clade 2·1 and 2·3), we initially constructed a phylogenetic tree composed of more than 300 nearly complete HA sequences available from GenBank. The neighbor‐joining tree (Figure 1) constructed using a GTR + I + G model in PAUP with 1000 bootstraps was rooted to clade 2·5 viruses and its topology analyzed. Using this approach, we show that all previously described clade 2·2 viruses remain in a single monophyletic group comprising isolates from many regions of Asia, the Middle East, Europe, and Africa. As expected, many isolates cluster geographically and temporally, but there are many exceptions, which could be attributed to previously identified mechanisms of viral spread (i.e., poultry trade and bird migration). Despite the apparent divergence of isolates into specific groups, when classified according to the unified nomenclature criteria and using a wide variety of available strains, clade 2·2 H5 HA genes remain an extremely closely related group at the nucleotide level with short branch lengths (note the 0·003 nucleotide substitution scale of the tree). Additionally, there is low bootstrap support at the primary nodes separating the major viral groups (only bootstrap support ≥60 is shown on tree), such that the overall topology of the tree is not robust at these primary nodes.
Figure 1.
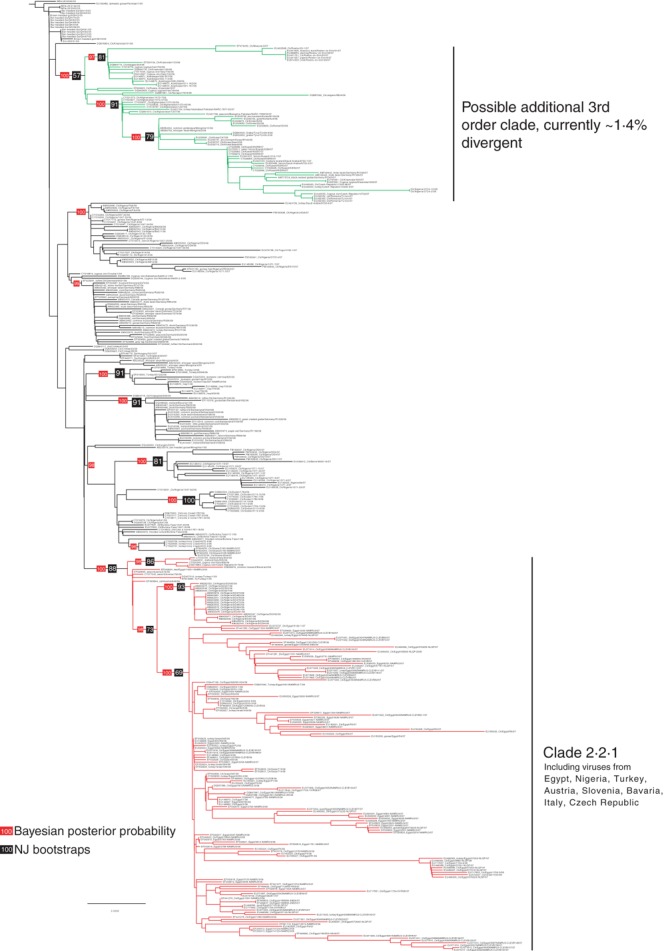
Neighbor‐joining (NJ) tree of clade 2·2 H5N1 hemagglutinin gene sequences descending from A/goose/Guangdong/1/96‐like viruses. The tree is rooted at the clade 2·5 node. Bayesian posterior probabilities and NJ bootstrap values (1000 replicates) are shown in red and black, respectively.
In addition to neighbor‐joining analysis, Bayesian analysis was conducted with MRBAYES 3·0 (Tallahassee, FL, USA) by using two replicates of 3 million generations sampling every 100 generations. Bayesian posterior probabilities (BPPs) were calculated from the consensus of 54 000 trees after excluding the first 6000 trees as burnin. BPPs are calculated taking into account the specified nucleotide model, while the bootstrap resamples from the nucleotide dataset allowing the possibility that the resampled alignment in each bootstrap replicate does not fit the nucleotide model of the actual dataset. In contrast, the BPPs are calculated from >10 000 trees that were generated from the original dataset and taking into account the model. As such, BPPs provide an alternative method for the estimation of phylogenetic support in these analyses and an additional layer of statistical support when interpreting phylogenetic relationships (Figure 1).
Nucleotide similarity analyses between most groups of sequences in clade 2·2 reveal a nucleotide divergence of <1·5%. However, a highly divergent group of viruses (at the bottom of the tree in Figure 1) comprising isolates primarily from Egypt, Israel, the Gaza Strip, Nigeria, and Europe are characterized by strong statistical support (bootstrap 88%, BPP 100%) and ‘inter‐group’ nucleotide divergence >1·5% between this group and all other clade 2·2 viruses. In addition, pairwise comparison of the ‘within‐group’ nucleotide distances is <1·5%. Based on these previously defined criteria, this group of viruses can be classified as a third‐order clade.
Thus, the WHO/OIE/FAO H5N1 Evolution Working Group recommends describing clade 2·2 as having a third‐order clade: clade 2·2·1. The remaining viruses in clade 2·2 from Asia, the Middle East, Europe, and Africa will continue to be classified as belonging to clade 2·2. Because this analysis has only considered the ‘global’ evolution of clade 2·2, the working group has not attempted to address the evidence that additional distinct sublineages within clade 2·2 have emerged recently. However, the working group recommends that newly described sublineages of H5N1 not be classified using the decimal‐numbering system until that sublineage has met each of the specific criteria for H5 HA clade designation. Of note is a sublineage of clade 2·2 comprised of isolates from Europe, Russia, Mongolia, Korea, India, the Middle East, and Nigeria, which has evolved extensively in recent years (marked in green in Figure 1). Because this group falls slightly below the >1·5% divergence threshold it has not been assigned a third‐order clade status but is likely to meet this criteria soon assuming further divergence occurs. The emergence of additional third‐order clades will be monitored as clade 2·2 viruses continue to evolve.
Acknowledgements
We thank Ian H. Brown, Giovanni Cattoli, C. Todd Davis, Ruben O. Donis, Ron A. M. Fouchier, Elizabeth Mumford, and Gavin J. D. Smith for drafting the manuscript on behalf of the H5N1 Evolution Working Group. We also thank Todd Davis and Gavin Smith for performing sequence homology comparisons and phylogenetic analyses. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
List of H5N1 Evolution Working Group
The working group was established by request of the World Health Organization’s Global Influenza Programme, Department of Epidemic and Pandemic Alert and Response (WHO, GIP, and EPR), the World Organisation for Animal Health (OIE), and the Food and Agriculture Organization (FAO). It consisted of the following persons:
Ian H. Brown, Veterinary Laboratories Agency, Addlestone, England, United Kingdom; Ilaria Capua, Instituto Zooprofilattico Sperimentale delle Venezie, Padova, Italy; Giovanni Cattoli, Instituto Zooprofilattico Sperimentale delle Venezie, Padova, Italy; Hualan Chen, Harbin Veterinary Research Institute, CAAS, China; Nancy Cox, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; C. Todd Davis, Centers for Disease Control and Prevention, Atlanta, Georgia; Ruben O. Donis, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; Ron A. M. Fouchier, Erasmus University, Netherlands; Rebecca Garten, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; Yi Guan, The University of Hong Kong, HK SAR, China; Alan Hay, MRC National Institute for Medical Research, London, UK; Yoshihiro Kawaoka, University of Wisconsin‐Madison, Madison, Wisconsin, USA and Institute of Medical Science, University of Tokyo, Tokyo, Japan; John Mackenzie, Curtin University of Technology, Perth, Australia; John McCauley, MRC National Institute for Medical Research, London, UK; Elizabeth Mumford, WHO, GIP, EPR, Geneva, Switzerland; Christopher Olsen, University of Wisconsin‐Madison, Madison, Wisconsin, USA; Michael L. Perdue, HHS, Washington, DC, USA; Colin A. Russell, Department of Zoology, University of Cambridge, England, United Kingdom; Catherine Smith, Centers for Disease Control and Prevention, Atlanta, Georgia, USA; Derek Smith, Department of Zoology, University of Cambridge, England, United Kingdom; Gavin J. D. Smith, The University of Hong Kong, Hong Kong SAR, China; Yuelong Shu, Chinese Center for Disease Control and Prevention, Beijing, China; Masato Tashiro, National Institute of Infectious Diseases, Tokyo, Japan; Dhanasekaran Vijaykrishna, The University of Hong Kong, HK SAR, China; Robert Webster, St. Jude Children’s Research Hospital, Memphis, USA.
References
- 1. World Health Organization/World Organisation for Animal Health/Food and Agriculture Organization H5N1 Evolution Working Group . Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg Infect Dis 2008. Available from http://www.cdc.gov/EID/content/14/7/e1.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ducatez MF, Olinger CM, Owoade AA et al. Molecular and antigenic evolution and geographical spread of H5N1 highly pathogenic avian influenza viruses in western Africa. J Gen Virol 2007; 88:2297–2306. [DOI] [PubMed] [Google Scholar]
- 3. Kiss I, Gyarmati P, Zohari S et al. Molecular characterization of highly pathogenic H5N1 avian influenza viruses isolated in Sweden in 2006. Virol J 2008; 5:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Monne I, Fusaro A, Al‐Blowi MH et al. Co‐circulation of two sublineages of HPAI H5N1 virus in the Kingdom of Saudi Arabia with unique molecular signatures suggesting separate introductions into the commercial poultry and falconry sectors. J Gen Virol 2008; 89:2691–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Salzberg SL, Kingsford C, Cattoli G et al. Genome analysis linking recent European and African influenza (H5N1) viruses. Emerg Infect Dis 2007; 5:713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Starick E, Beer M, Hoffmann B et al. Phylogenetic analyses of highly pathogenic avian influenza virus isolates from Germany in 2006 and 2007 suggest at least three separate introductions of H5N1 virus. Vet Microbiol 2008; 128:243–252. [DOI] [PubMed] [Google Scholar]
- 7. Chen H, Smith GJD, Zhang SY et al. H5N1 virus outbreak in migratory waterfowl. Nature 2005; 436:191–192. [DOI] [PubMed] [Google Scholar]