

Abstract
Along with the canonical miRNA, distinct miRNA-like sequences called sibling miRNAs (sib-miRs) are generated from the same pre-miRNA. Among them, isomeric sequences featuring slight variations at the terminals, relative to the canonical miRNA, constitute a pool of isomeric sibling miRNAs (isomiRs). Despite the high prevalence of isomiRs in eukaryotes, their features and relevance remain elusive. In this study, we performed a comprehensive analysis of mature precursor miRNA (pre-miRNA) sequences from Arabidopsis to understand their features and regulatory targets. The influence of isomiR terminal heterogeneity in target binding was examined comprehensively. Our comprehensive analyses suggested a novel computational strategy that utilizes miRNA and its isomiRs to enhance the accuracy of their regulatory target prediction in Arabidopsis. A few targets are shared by several members of isomiRs; however, this phenomenon was not typical. Gene Ontology (GO) enrichment analysis showed that commonly targeted mRNAs were enriched for certain GO terms. Moreover, comparison of these commonly targeted genes with validated targets from published data demonstrated that the validated targets are bound by most isomiRs and not only the canonical miRNA. Furthermore, the biological role of isomiRs in target cleavage was supported by degradome data. Incorporating this finding, we predicted potential target genes of several miRNAs and confirmed them by experimental assays. This study proposes a novel strategy to improve the accuracy of predicting miRNA targets through combined use of miRNA with its isomiRs.
Keywords: arabidopsis, bioinformatics, isomiRs, miRNA, miRNA target prediction, RNA-Seq
Introduction
MicroRNAs (miRNAs) constitute a class of short RNA molecules found in eukaryotic cells. Approximately 21 nucleotides (nt) long, miRNAs are derived from hairpin structures embedded in their primary miRNA transcripts.1,2 The biogenesis of plant miRNAs and their biological functions in gene regulation are well understood.3,4 In a typical plant cell, the Dicer-Like 1 (DCL1) enzyme recognizes the hairpin-loop structure of a primary miRNA (pri-miRNA) and cleaves it to generate a long hairpin of about 100 nt called precursor miRNA (pre-miRNA).5,6 The pre-miRNA is then processed by DCL1 at the terminal loop, leading to generation of a miRNA:miRNA* duplex which is methylated at the 3′-end by RNA methylase HUA ENHANCER 1 (HEN1) and transported to the cytoplasm.7 Next, the guide strand miRNA is incorporated into protein Argonaute (AGO) to form the RNA-induced silencing complex (RISC), which binds to target gene(s) for gene silencing.8-10
Extensive experimental and computational studies have been conducted to understand the role of miRNAs and miRNA-target gene (mRNA transcript) binding for gene silencing. These studies have led to the discovery of a myriad of miRNAs and their targets in various organisms, as well as the creation of different public databases (e.g., miRBase,11 miRTarBase,12 TarBase,13 and miRecords 14) for cataloging them. Originally, research suggested that a pre-miRNA produced only one mature miRNA sequence; however, later studies have revealed that some cells contain low levels of miRNA* for mediating gene silencing.15,16 Further studies indicate that the abundance of miRNA and miRNA* are not strictly static and may vary across cell types and developmental stages.17 Higher level of miRNA* compared to its partner miRNA has been reported in animal and plant tissues, implying a critical biological function for miRNA*.15,16,18–20
Initially, it was believed that DCL cleaves the hairpin at a precise position to liberate the mature miRNA from the hairpin. However, recent advancements in high-throughput sequencing technology have enabled large-scale profiling of miRNAs within cells. These studies demonstrated that, in addition to the canonical miRNA (miRBase 18: http://www.mirbase.org/), distinct species of miRNA-like RNAs called sibling miRNAs (sib-miRs) are also generated from the same hairpin.21,22 Some sib-miR sequences are highly similar to the canonical miRNA with only slight variations at their termini. These molecules are known as isomeric miRNAs (isomiRs).23-26 The termini of an isomiR are heterogeneous compared to its canonical miRNA, and may arise due to variations in DCL cleavage sites on precursor molecules.27,28 Therefore, a single pre-miRNA can generate distinct sib-miRs, which include isomiRs and the miRNA (Fig. 1, Table S1). Depending on the nature of the terminal nucleotide, isomiRs can be divided into 2 major classes:29 (1) templated, in which each nucleotide in the isomiRs matches to the genome; and (2) un-templated, in which an extra nucleotide is added at the isomiR terminus, thereby rendering all but the last nucleotide identical to the genome.23,28,30 Base modifications from adenine to inosine in miRNAs have also been reported as sources of isomiRs28,31-33
Figure 1.

Diverse sib-miRs generated from the pre-miRNA ath-MIR156a. The canonical mature miRNA sequence, ath-miR156a, is represented in red. The isomiRs are enclosed into blue lines within 5 nt upstream or downstream of the terminal canonical miRNA. The first column indicates normalized counts as RPM, while the second column lists normalized counts within pre-miRNA (for details, see Materials and Methods).
Much research has been focused recently on characterizing isomiRs and elucidating their roles in biological processes. These molecules are present at much lower levels than the corresponding canonical miRNA. However, isomiR levels may change with time and tissue type, thus impacting gene regulation.20,25,34,35 Templated isomiRs in Drosophila melanogaster are dynamically regulated, indicating their relevance in gene silencing.20 Recent studies in human and fish suggest that isomiR biogenesis is not random, but is instead highly regulated. Moreover, isomiRs have been shown to play numerous biological roles,17,26,34 and the identification of novel miRNA/miRNA* and development of a systematic method for annotating them was reported recently.35 Studies have also revealed several advantages to un-templated extensions, including increased stability of miRNAs with addition of a nucleotide at the 3′-end in D. melanogaster and Populus trichocarpa20,36 and improved miRNA loading into RISC.28 Interestingly, a very low prevalence of un-templated isomiRs was found in humans.34
In spite of the prevalence of sib-miRs/isomiRs, a systematic and comprehensive analysis of their sequence features and the functional consequences of variation in their terminal nucleotide on target binding remains lacking in animals and plants.25 Therefore, in this study, we systematically analyzed sib-miRs using 51 small RNA (sRNA) deep-sequencing datasets (http://mpss.udel.edu/at_sbs/) from the model plant Arabidopsis thaliana.37 Our computational analysis revealed an important role for isomiRs in gene regulation via their terminal heterogeneity, which enhances the specificity of target gene silencing. Moreover, our study revealed, for the first time, that the group of genes targeted by several members of isomiRs are enriched in Gene Ontology (GO) terms and also identified as validated targets. Furthermore, the isomiRs mediated target cleavage was supported by degradome data. Applying our approach we predicted several novel miRNA target genes, of which some were experimentally validated. Therefore, our study recommended using the sequences of miRNA and its isomiRs for accurate target prediction. In addition, the knowledge gained from this study enhances our understanding of the features that characterize mature miRNA sequences generated from pre-miRNAs, thereby enabling the design of improved artificial miRNAs/small interfering RNA (siRNA) for efficient gene silencing.
Results
We began our investigation by examining the propensity of each arm of a pre-miRNA to generate sib-miRs. Then, we focused on identifying the characteristic length of sib-miRs, their composition patterns at both terminals, and loading specificity toward different AGO proteins. We also studied the sib-miRs generated from conserved and non-conserved pre-miRNAs. Finally, the heterogeneity of terminal sequences was calculated to understand the cleavage accuracy of DCL in generating isomiRs (see Supplemental Results and Discussion for more details).
Identification of targets of miRNAs and their corresponding isomiRs
We became particularly interested in how miRNA terminal heterogeneity impacts its binding to target transcripts. Imprecise cleavage by DCL generates heterogeneous termini that can alter the miRNA seed region (2–8 nt from the 5′-end) and lead to different mRNA targets. However, to the best of our knowledge, no systematic study has been conducted yet to determine the effect of miRNA terminal heterogeneity on target mRNA selection. To understand the biological role of isomiRs, we predicted isomiR targets using psRNATarget.38 miRNAs, and thus their isomiRs, were selected for this study if (1) experimental validation of target mRNAs was published, and (2) at least 8 known isomiRs are produced from the miRNA (Tables 1A and 1B). The isomiRs were mapped to the 5′-end of the canonical miRNA, with at most a 5-nucleotide shift in the upstream or downstream direction.
Table 1A.
miRNAs generated from the 5p-arm of hairpins and their experimentally validated target genes.
| miR | #isomiR | Arm | #species | miFam | Targets | ||
|---|---|---|---|---|---|---|---|
| ath-miR156a | 17 | 5p | 34 | MIPF0000008 | AT1G27370 | AT1G53160 | AT1G69170 |
| AT2G33810 | AT5G43270 | ||||||
| ath-miR160 | 13 | 5p | 25 | MIPF0000032 | AT1G77850 | AT2G28350 | AT4G30080 |
| ath-miR161 | 28 | 5p | 2 | MIPF0000455 | AT1G06580 | AT1G62910 | AT1G63080 |
| AT1G63130 | AT1G63150 | AT1G63230 | |||||
| AT1G63400 | AT5G41170 | ||||||
| ath-miR164 | 11 | 5p | 21 | MIPF0000045 | AT1G56010 | AT3G15170 | AT5G07680 |
| AT5G39610 | AT5G53950 | AT5G61430 | |||||
| ath-miR167 | 16 | 5p | 23 | MIPF0000023 | AT1G30330 | AT5G37020 | |
| ath-miR168 | 29 | 5p | 20 | MIPF0000081 | AT1G48410 | ||
| ath-miR169 | 13 | 5p | 16 | MIPF0000037 | AT1G17590 | AT1G54160 | AT1G72830 |
| AT3G05690 | AT3G20910 | AT5G06510 | |||||
| AT5G12840 | |||||||
| ath-miR173 | 16 | 5p | 2 | MIPF0001175 | AT1G50055 | AT2G27400 | AT2G39675 |
| AT2G39681 | AT4G36920 | ||||||
| ath-miR391 | 14 | 5p | 2 | MIPF0001151 | AT3G17185 | AT5G49615 | AT5G57735 |
| ath-miR393 | 9 | 5p | 15 | MIPF0000083 | AT1G12820 | AT3G23690 | AT3G26810 |
| AT3G62980 | AT4G03190 | ||||||
| ath-miR394 | 13 | 5p | 14 | MIPF0000100 | AT1G27340 | ||
| ath-miR396a | 27 | 5p | 29 | MIPF0000047 | AT1G10120 | AT2G06200 | AT2G22840 |
| AT2G36400 | AT2G45480 | AT3G13960 | |||||
| AT3G52910 | AT4G24150 | AT4G37740 | |||||
| AT5G53660 | |||||||
| ath-miR400 | 9 | 5p | 2 | MIPF0001157 | AT1G06580 | ||
| ath-miR778 | 14 | 5p | 1 | NO_FAM | AT2G22740 | ||
| ath-miR824 | 10 | 5p | 5 | MIPF0000442 | AT3G57230 | ||
| ath-miR858 | 10 | 5p | 2 | MIPF0001138 | AT1G06180 | AT1G66230 | AT2G47460 |
| AT3G08500 | AT5G49330 | ||||||
| ath-miR859 | 8 | 5p | 2 | MIPF0001191 | AT3G49510 |
isomiRs: number of distinct mature sequences of isomiRs (including miRNA) found in the data set; #species: number of species (including A. thaliana) in which the pre-miRNA is conserved; miFam: accession number of the pre-miRNA. NO_FAM indicates that the precursor was not assigned in miFam and that the molecule is evolutionarily young; arm indicates relative to miRNA hairpin from which canonical miRNA and isomiRs generate. The genes highlighted in green were predicted by the psRNATarget tool.
All predicted targets were further analyzed to understand the varied patterns that arise due to terminal heterogeneity of the miRNA. To accomplish this, a binary matrix was created in which each row indicated the predicted mRNA transcript (TAIR10, http://www.arabidopsis.org/) and each column specified an isomiR sequence. Values of “1” or “0” signified the presence or absence, respectively, of an isomiR target site within the transcript. The isomiRs were arranged based on their 5′-end position and length. Gitools (version 1.6.1, http://www.gitools.org/) was then used to generate a heat map of the matrix for easier visualization. Moreover, we clustered mRNAs based on the number of predicted isomiRs that may target them.
Intriguingly, we observed that most of the validated mRNAs could be targeted by the majority of isomiRs and the miRNA, indicating that “authentic” targets have binding sites for multiple isomiRs and not only by the “canonical” miRNA (Fig. 2, S1, and S2). We also identified several isomiRs with specificity toward distinct mRNAs. Figure S1 shows that ath-miR156a has a total of 17 isomiRs (including the miRNA) targeting 47 mRNA transcripts. However, all of the experimentally validated mRNAs are targeted by 15 or more (88–100%) isomiRs. In the case of ath-miR164a, 11 isomiRs were found to target 69 mRNAs. Each of the experimentally validated mRNAs is targeted by all of the isomiRs (Figure S2). Similarly, analysis of ath-miR172a produced 20 isomiRs that could target 90 mRNAs (Fig. 2). Ten of 14 experimentally validated target mRNAs were predicted to be recognized by more than 18 isomiRs (90–100%). Our analysis predicted that one mRNA (i.e., AT3G14770.1) could be targeted by 10 isomiRs, while 3 mRNAs (i.e., AT1G53780.1, AT1G53780.2, and AT1G53780.3) are targeted by one isomiR.
Figure 2.
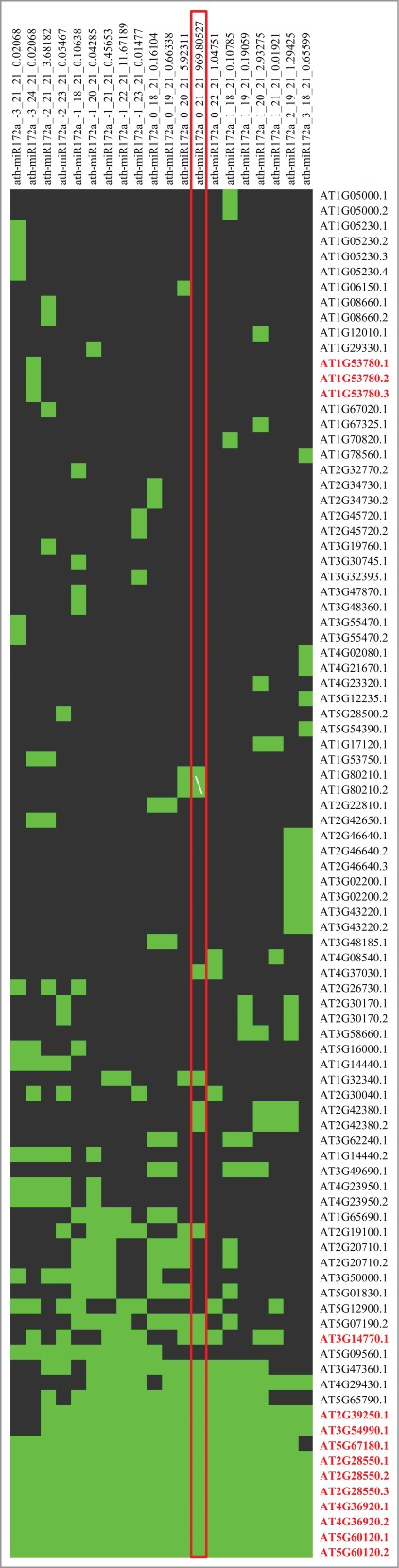
Effect of terminal heterogeneity of ath-miR172a-derived isomiRs on target mRNAs. Green and black in the heatmap indicates the presence and absence of the isomiR target, respectively. The canonical miRNA, ath-miR172a, is denoted with a red box while experimentally validated targets are written in red bold.
To understand the significance of isomiRs in target selection, we calculated the percentage of isomiRs that can target experimentally validated mRNAs. Here we only considered targets predicted by psRNATarget (highlighted in green in Tables 1A and 1B). This analysis revealed 31 miRNAs (and their isomiRs) capable of targeting 2,163 mRNAs, of which 2,075 are unique and 88 are targeted by multiple miRNAs. Table 2 demonstrates that if we increase the percentage of isomiRs targeting an mRNA, there is an increased likelihood of the mRNA being an experimentally validated target. The mRNA targeted by miRNA and its all isomiRs can cover ∼57% of validated targets (Table 2). This result indicates that “authentic” target mRNAs are recognized by multiple isomiRs, and not only by the canonical miRNA. Furthermore, we analyzed the recognition sites within a few transcripts and found that isomiRs can target the same site as the canonical miRNA with slight variations in a few nucleotides (Fig. 3A, 3B, and S3). Therefore, the possibility exists that isomiRs can sort into different AGO proteins to target the same transcript through different RNA interference (RNAi) mechanisms. Our computational models support this hypothesis (Tables 3 and S2). In addition, we checked the pattern of miRNA:target binding in humans. Like Arabidopsis, we observed that experimentally verified target mRNAs can be recognized by several human miRNA/isomiRs (see Supplemental Results and Discussion).
Table 1B.
miRNAs generated from the 3p-arm of hairpins and their experimentally validated target genes.
| miR | #isomiR | Arm | #species | miFam | Targets | ||
|---|---|---|---|---|---|---|---|
| ath-miR159a | 24 | 3p | 30 | MIPF0000010 | AT1G30210 | AT1G53230 | AT2G26960 |
| AT2G31070 | AT2G32460 | AT2G34010 | |||||
| AT3G11440 | AT3G15030 | AT3G60460 | |||||
| AT4G18390 | AT4G30440 | AT4G37770 | |||||
| AT5G06100 | AT5G18100 | AT5G55020 | |||||
| AT5G55930 | |||||||
| ath-miR162 | 10 | 3p | 14 | MIPF0000127 | AT1G01040 | ||
| ath-miR163 | 19 | 3p | 2 | MIPF0001116 | AT1G66690 | AT1G66700 | AT1G66720 |
| AT3G44860 | AT3G44870 | ||||||
| ath-miR165 | 10 | 3p | 26 | MIPF0000004 | AT1G30490 | AT1G52150 | AT2G34710 |
| AT4G32880 | AT5G60690 | ||||||
| ath-miR172a | 20 | 3p | 22 | MIPF0000035 | AT2G28550 | AT2G39250 | AT3G54990 |
| AT4G36920 | AT5G60120 | AT5G67180 | |||||
| AT1G53780 | AT3G14770 | AT3G54990 | |||||
| AT4G36920 | AT5G60120 | ||||||
| ath-miR403 | 9 | 3p | 8 | MIPF0000290 | AT1G31280 | ||
| ath-miR408 | 16 | 3p | 18 | MIPF0000102 | AT1G72230 | AT2G02850 | AT2G30210 |
| AT2G44790 | AT2G47020 | AT3G02200 | |||||
| AT5G05390 | AT5G07130 | ||||||
| ath-miR447 | 14 | 3p | 1 | MIPF0000170 | AT5G60760 | ||
| ath-miR773 | 8 | 3p | 2 | MIPF0001106 | AT4G14140 | ||
| ath-miR775 | 16 | 3p | 1 | NO_FAM | AT1G23390 | AT1G53290 | |
| ath-miR780 | 23 | 3p | 1 | NO_FAM | AT5G41610 | ||
| ath-miR823 | 8 | 3p | 2 | MIPF0001135 | AT1G69770 | ||
| ath-miR827 | 11 | 3p | 3 | MIPF0001113 | AT1G02860 | ||
| ath-miR857 | 11 | 3p | 2 | MIPF0001180 | AT3G09220 |
Table 2.
Performance of miRNA target prediction using their isomiRs.
| % of isomiRs targeting (% equal or more than value) | Total predicted target mRNA (A) | Experimentally validated target mRNA (B) | Coverage (%) of validated target mRNA in predicted (Sensitivity=B *100/A) |
|---|---|---|---|
| 0 | 2163 | 125 | 5.78 |
| 10 | 1430 | 114 | 7.97 |
| 20 | 903 | 111 | 12.29 |
| 30 | 628 | 110 | 17.52 |
| 40 | 460 | 106 | 23.04 |
| 50 | 397 | 104 | 26.20 |
| 60 | 323 | 101 | 31.27 |
| 70 | 265 | 97 | 36.60 |
| 80 | 206 | 87 | 42.23 |
| 90 | 139 | 69 | 49.64 |
| 100 | 93 | 53 | 56.99 |
Figure 3.
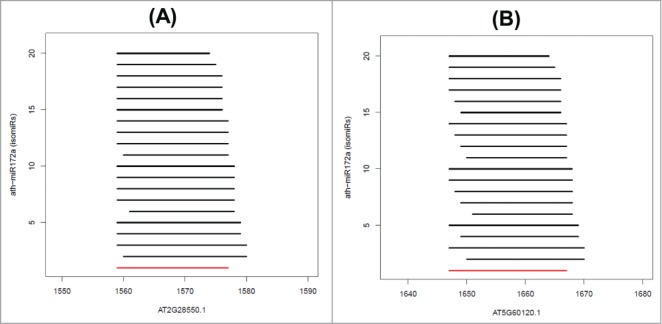
Position-specific binding of ath-miR172a isomiRs on (A) AT2G28550.1 and (B) AT5G60120.1. Numbering on the x-axis denotes the nucleotide position on the transcript while the y-axis denotes the isomiR. isomiR sequences are provided in Table 3. The numbering of isomiRs on the y-axis corresponds to Serial Number (SN) in Table 3. The canonical miRNA, ath-miR172a, is denoted in red.
Table 3.
Sorting of ath-miR172a-derived isomiRs into different Argonaute (AGO) proteins. SN 1 in bold is the canonical miRNA. Exp denotes experimentally validated; Pred denotes predicted by SVM models.
| SN | isomiRs of ath-miR172a | Sequence | Argonaute | Evidence |
|---|---|---|---|---|
| 1 | ath-miR172a_0_21_21_969.80527 | —AGAAUCUUGAUGAUGCUGCAU- | AGO1,AGO2,AGO4,AGO5 | Exp |
| 2 | ath-miR172a_-3_21_21_0.02068 | AUGAGAAUCUUGAUGAUGCUG—- | AGO2,AGO4 | Pred |
| 3 | ath-miR172a_-3_24_21_0.02068 | AUGAGAAUCUUGAUGAUGCUGCAU- | AGO4 | Exp |
| 4 | ath-miR172a_-2_21_21_3.68182 | -UGAGAAUCUUGAUGAUGCUGC— | AGO1,AGO2,AGO4 | Exp |
| 5 | ath-miR172a_-2_23_21_0.05467 | -UGAGAAUCUUGAUGAUGCUGCAU- | AGO1 | Pred |
| 6 | ath-miR172a_-1_18_21_0.10638 | –GAGAAUCUUGAUGAUGCU—– | AGO4 | Pred |
| 7 | ath-miR172a_-1_20_21_0.04285 | –GAGAAUCUUGAUGAUGCUGC— | AGO1,AGO4 | Exp |
| 8 | ath-miR172a_-1_21_21_0.45653 | –GAGAAUCUUGAUGAUGCUGCA– | AGO1,AGO4,AGO5 | Exp |
| 9 | ath-miR172a_-1_22_21_11.67189 | –GAGAAUCUUGAUGAUGCUGCAU- | AGO1,AGO4 | Exp |
| 10 | ath-miR172a_-1_23_21_0.01477 | –GAGAAUCUUGAUGAUGCUGCAUC | AGO4 | Pred |
| 11 | ath-miR172a_0_18_21_0.16104 | —AGAAUCUUGAUGAUGCUG—- | AGO2 | Pred |
| 12 | ath-miR172a_0_19_21_0.66338 | —AGAAUCUUGAUGAUGCUGC— | AGO1,AGO2,AGO4,AGO5 | Exp |
| 13 | ath-miR172a_0_20_21_5.92311 | —AGAAUCUUGAUGAUGCUGCA– | AGO1,AGO2,AGO4,AGO5 | Exp |
| 14 | ath-miR172a_0_22_21_1.04751 | —AGAAUCUUGAUGAUGCUGCAUC | AGO1,AGO2,AGO4 | Exp |
| 15 | ath-miR172a_1_18_21_0.10785 | —-GAAUCUUGAUGAUGCUGC— | AGO4 | Pred |
| 16 | ath-miR172a_1_19_21_0.19059 | —-GAAUCUUGAUGAUGCUGCA– | AGO1,AGO2,AGO4,AGO5 | Exp |
| 17 | ath-miR172a_1_20_21_2.93275 | —-GAAUCUUGAUGAUGCUGCAU- | AGO1,AGO2,AGO4,AGO5 | Exp |
| 18 | ath-miR172a_1_21_21_0.01921 | —-GAAUCUUGAUGAUGCUGCAUC | AGO4 | Pred |
| 19 | ath-miR172a_2_19_21_1.29425 | —–AAUCUUGAUGAUGCUGCAU- | AGO1,AGO2,AGO4 | Exp |
| 20 | ath-miR172a_3_18_21_0.65599 | ——AUCUUGAUGAUGCUGCAU- | AGO2,AGO4 | Pred |
Gene ontology analysis of targets
Transcripts targeted by Arabidopsis isomiRs were further subjected to Gene Ontology (GO) analysis using the agriGO tool (http://bioinfo.cau.edu.cn/agriGO/).39 We hypothesized that mRNAs targeted by a single miRNA and its isomiRs are natural targets involved in a common, specific biological function. Our results show that mRNAs targeted by more than 50% of isomiRs derived from ath-miR156a are significantly enriched for 23 GO terms, while another group of test transcripts targeted by up to 50% of isomiRs had no significant terms in common (Table 4 and Table S3). We also observed similar patterns of GO term enrichment for other test sets. For example, 5 and 4 GO terms were uncovered for ath-miR164a and ath-miR172a (Table 4 and Table S3), respectively. Moreover, we searched the significance of GO terms in all predicted targets of 31 miRNAs and their isomiRs. This analysis can further help us to understand the significance of isomiRs in target finding, and overall functional characteristics of miRNA target genes. Among 2,163 mRNAs, 1,792 mRNA are targeted by up to 50% of isomiRs, while only 371 mRNA are targeted by more than 50% of isomiRs. Interestingly, we found significantly enriched 96 GO terms in a group of mRNA targeted by more than 50% of isomiRs, while other genes targeted by up to 50% of isomiRs does not show significant GO terms (Table 4 and Table S3). Among them, the terms “transcription factor activity” and “transcription regulator activity” appeared regularly. This result further supports our observation that authentic miRNA target genes are also targeted by various isomiRs instead of by only the canonical miRNA, and that they most likely participate in transcription factor activity. Figure 4 shows interactive GO graphs according to biological process. Supplementary Figure S4 depicts results from the GO analysis according to cellular component and molecular function.
Table 4.
Predicted target and Gene Ontology (GO) of isomiRs and canonical miRNA.
| Targets with more than 50% of isomiRs |
Targets with up to 50% of isomiRs |
Targets with canonical miRNA |
Targets with isomiR but not with canonical miRNA |
|||||
|---|---|---|---|---|---|---|---|---|
| miRNA | #mRNA | # GO | #mRNA | # GO | #mRNA | # GO | #mRNA | # GO |
| ath-miR156a | 21(10) | 23 | 26(0) | 0 | 25(10) | 10 | 22(0) | 0 |
| ath-miR164a | 12(8) | 5 | 57(0) | 0 | 20(8) | 2 | 49(0) | 0 |
| ath-miR172a | 13(10) | 4 | 77(4) | 0 | 22(11) | 0 | 68(3) | 0 |
| 31 miRNAs | 371(102) | 96 | 1792(22) | 0 | 569(108) | 64 | 1594(16) | 0 |
| Computational isomiRs of ath-miR172a (-5/+5 nt) | 11(10) | 8$ — | 169(4) | 0 | 22(11) | 8 | 158(3) | 0 |
| Computational isomiRs of ath-miR172a (-3/+3 nt) | 11(10) | 14$ | 110(4) | 0 | 22(11) | 2 | 100(3) | 0 |
mRNA column: number of experimentally validated target mRNA is given in bracket.
number of input mRNA was not sufficient to map with GO, so 2 more mRNAs was taken.
Figure 4.
The interactive Gene Ontology graph of biological processes.
In addition, we studied how isomiRs and canonical miRNA influenced the performance of target predictions. Our result showed that targets predicted with only canonical miRNA have significant GO terms, while targets predicted with isomiRs without canonical miRNA does not have significant GO term (Table 4 and Table S3). Our result showed that 31 miRNAs targeting 569 mRNAs in the class of canonical miRNA, and among them 108 mRNAs are experimentally known (Table 4). On the other hand 371 mRNAs targeted by class of more than 50% of isomiRs, and among them 102 mRNAs are experimentally known. Therefore, sensitivity of target prediction increases from 18.98% to 27.49%, and enrichment of GO terms become stronger if isomiRs were combined with the canonical miRNA (Table 4). Though, complete targets of miRNAs were not experimentally known, but if we consider the available list of experimentally known targets, the true positive and true negative targets would be 108 and 1578, respectively for class of canonical miRNA. While true positive and true negative targets would be 102 and 1770, respectively for class of more than 50% of isomiRs. Therefore accuracy of target prediction increase from 77.9% to 86% if isomiRs were combined with the canonical miRNA (Table 4). This further supports that combination of isomiRs with miRNA can predict the target with high accuracy.
Furthermore, we assessed the significance of isomiRs in target prediction using computationally generated all possible isomiRs of ath-miR172a from its precursor sequence with length varies from 18 to 28 nt (see Length diversity of sib-miRs of Supplemental Results and Discussion). Two sets of isomiRs were generated having heterogeneous termini of: (I) -3/+3 nt, and (II) -5/+5 nt, relative to 5′-end of canonical miRNA. These isomiRs along with canonical miRNA were used for target prediction by psRNATarget, and compared with experimentally validated targets (Table 4 and Table S4). Compared to natural isomRs, randomly generated isomiRs can increase the sensitivity from 77% (10/13) to 91% (10/11) and give strong GO enrichment (Table 4). We observed that mRNA targeted by up to 50% of isomiRs does not show any GO terms, while mRNA targeted by more than 50% of isomiRs showed the GO enrichments which is similar to the target of natural isomiRs (Table 4). It was interesting that we observed that targets predicted with only canonical miRNA had prediction sensitivity of 50% (11/22) and targets predicted by more than 50% of isomiRs (including canonical miRNA) had significantly increased prediction sensitivity, which was 91% (10/11) (Table 4).
Evidence of target cleavage by isomiRs
Concentration level of isomiRs are considered to be lower compared to canonical miRNA, but still can cleave the target gene and have a biological impact on gene regulation. In order to prove this hypothesis, we investigated the isomiRs directed target cleavage through publically available degradome data of polyadenylated transcripts using a computational pipeline CleaveLand.40 Experimental studies showed that miRNA cleaves the target mRNA precisely between the 10th and 11th nucleotides relative to the 5′-end of miRNA.40-42 The resulting downstream fragment of the cleaved mRNA is uncapped and polyadenylated, called as degradome,42 and has been exploited to identifying miRNA guided cleavage site accurately in the mRNA transcripts 40,43-45 Through degradome data analysis, we have detected the cleaved targets by both isomiRs and canonical miRNAs (Figure S5A–L). Our analysis also showed that some isomiRs are more effective in target cleavage than their canonical miRNA (Figure S5P–Q). Interestingly, we also detected the targets which are cleaved by isomiRs but not by its canonical miRNA (Figure S5M-N), and thus support the finding of other study showing significance of isomiRs in gene regulation.46
Prediction of novel targets using miRNA and isomiRs
We observed that a “true” target is suppressed by various isomiRs and the canonical miRNA in our genome-wide bioinformatics analysis. Furthermore, GO analysis showed that target mRNAs of these isomiRs are enriched for a particular biological function, which indicates that natural miRNA targets may be recognized by several isomiRs. Based on these observations, we extended and applied our new strategy to predict novel miRNA targets. In this analysis, a transcript was considered a probable target if it contained a target site for 80% or more isomiRs (including the miRNA). We also lowered the threshold if targets were not found using this criterion. Therefore, the highest number of isomiRs associated with a target indicates a real target gene. Tables 5A and 5B list the miRNA targets predicted with this method.
Table 5A.
Predicted miRNA targets generated from the 5p-arm of the hairpin in Arabidopsis based on psRNATarget with the highest number of isomiRs.
| miRNA | locusID | isomiRs targeting mRNA | Total isomiRs | %isomiRs targeting mRNA |
|---|---|---|---|---|
| ath-miR390a | AT3G17185.1 | 19 | 25 | 76.00 |
| AT3G17185.2 | 19 | 25 | 76.00 | |
| AT5G57735.1 | 22 | 25 | 88.00 | |
| AT5G49615.1 | 23 | 25 | 92.00 | |
| ath-miR822 | AT1G44020.1 | 14 | 17 | 82.35 |
| AT1G66440.1 | 14 | 17 | 82.35 | |
| AT1G66450.1 | 14 | 17 | 82.35 | |
| AT2G02620.1 | 16 | 17 | 94.12 | |
| AT2G13900.1 | 17 | 17 | 100.00 | |
| AT5G02330.1 | 17 | 17 | 100.00 | |
| AT5G02350.1 | 17 | 17 | 100.00 | |
| ath-miR833–5p | AT2G36890.1 | 12 | 12 | 100.00 |
| ath-miR839 | AT2G27350.1 | 10 | 12 | 83.33 |
| AT2G27350.2 | 10 | 12 | 83.33 | |
| AT2G27350.3 | 10 | 12 | 83.33 | |
| AT2G27350.4 | 10 | 12 | 83.33 | |
| AT2G27350.5 | 10 | 12 | 83.33 | |
| AT2G27350.6 | 10 | 12 | 83.33 | |
| AT5G38383.1 | 10 | 12 | 83.33 | |
| ath-miR840 | AT2G02750.1 | 8 | 9 | 88.89 |
| ath-miR843 | AT1G14780.1 | 7 | 8 | 87.50 |
| AT4G31340.1 | 7 | 8 | 87.50 | |
| AT4G31340.2 | 7 | 8 | 87.50 | |
| AT1G11810.1 | 8 | 8 | 100.00 | |
| AT3G13830.1 | 8 | 8 | 100.00 | |
| ath-miR854a | AT5G05100.1 | 16 | 19 | 84.21 |
| ath-miR864–5p | AT2G06880.1 | 9 | 10 | 90.00 |
| AT1G02840.1 | 10 | 10 | 100.00 | |
| AT1G02840.2 | 10 | 10 | 100.00 | |
| AT1G02840.3 | 10 | 10 | 100.00 | |
| ath-miR2936 | AT1G15690.1 | 9 | 11 | 81.82 |
| AT1G15690.2 | 9 | 11 | 81.82 | |
| AT1G76720.1 | 9 | 11 | 81.82 | |
| AT3G49430.2 | 9 | 11 | 81.82 | |
| AT3G52770.1 | 9 | 11 | 81.82 | |
| ath-miR3933 | AT1G72310.1 | 6 | 10 | 60.00 |
| AT4G22235.1 | 6 | 10 | 60.00 | |
| AT4G22235.2 | 6 | 10 | 60.00 | |
| ath-miR5028 | AT1G68540.1 | 8 | 9 | 88.89 |
| AT1G68540.2 | 8 | 9 | 88.89 | |
| AT3G10010.1 | 9 | 9 | 100.00 |
Biological validation of predicted targets
The predicted targets of ath-miR158a were validated experimentally to confirm the accuracy of our method. Our analysis predicted 4 targets, namely, AT1G49910, AT1G64100, AT3G03580, and AT1G62860 (Table 5B), for ath-miR158a and its isomiRs. Therefore, we hypothesized that, in Arabidopsis, the absence of ath-miR158a and its isomiRs leads to increased expression of these 4 target genes. To test this hypothesis, we obtained 2 T-DNA knockout mutants for the ath-miR158a gene (Figure S6), namely SALK_025691 and SALK_083227, from ABRC (https://abrc.osu.edu/). The expression levels of miR158 and its isomiRs were quantified by RT-qPCR in plants that were homozygous for the respective T-DNA insertions. RT-qPCR was performed using forward primers against miR158 and an isomiR predicted to be generated toward the 5′-end of miR158. Wild-type (Col-0) plants showed amplification of both miR158 and isomiR158, indicating the expression of both sRNAs in Arabidopsis (Fig. 5). Our data also indicated that the expressions of the mature miR158a and one of its isomiRs were significantly reduced in both mutant plants (Fig. 5).
Table 5B.
Predicted miRNA targets generated from the 3p-arm of the hairpin in Arabidopsis based on psRNATarget with the highest number of isomiRs.
| miRNA | locusID | isomiRs targeting mRNA | Total isomiRs | %isomiRs targeting mRNA |
|---|---|---|---|---|
| ath-miR158a | AT1G49910.1 | 16 | 21 | 76.19 |
| AT1G64100.1 | 17 | 21 | 80.95 | |
| AT1G64100.2 | 17 | 21 | 80.95 | |
| AT3G03580.1 | 17 | 21 | 80.95 | |
| AT1G62860.1 | 18 | 21 | 85.71 | |
| ath-miR776 | AT3G44240.1 | 7 | 8 | 87.50 |
| AT5G56360.1 | 7 | 8 | 87.50 | |
| AT2G28260.1 | 8 | 8 | 100.00 | |
| AT5G62310.1 | 8 | 8 | 100.00 | |
| ath-miR783 | AT4G01090.1 | 13 | 20 | 65.00 |
| ath-miR846 | AT5G49870.1 | 17 | 19 | 89.47 |
| AT1G61230.1 | 18 | 19 | 94.74 | |
| AT1G33790.2 | 19 | 19 | 100.00 | |
| AT1G52050.1 | 19 | 19 | 100.00 | |
| AT1G52060.1 | 19 | 19 | 100.00 | |
| AT1G52110.1 | 19 | 19 | 100.00 | |
| AT1G52130.1 | 19 | 19 | 100.00 | |
| AT1G57570.1 | 19 | 19 | 100.00 | |
| AT2G25980.1 | 19 | 19 | 100.00 | |
| AT5G28520.1 | 19 | 19 | 100.00 | |
| AT5G49850.1 | 19 | 19 | 100.00 | |
| ath-miR848 | AT5G60490.1 | 5 | 11 | 45.45 |
| ath-miR851–3p | AT2G02110.1 | 7 | 10 | 70.00 |
| ath-miR866–3p | AT1G57835.1 | 8 | 9 | 88.89 |
| AT1G20230.1 | 9 | 9 | 100.00 | |
| AT2G22070.1 | 9 | 9 | 100.00 | |
| AT4G21400.1 | 9 | 9 | 100.00 | |
| ath-miR869.1 | AT4G08210.1 | 11 | 22 | 50.00 |
| ath-miR5026 | AT5G14180.1 | 12 | 16 | 75.00 |
Figure 5.
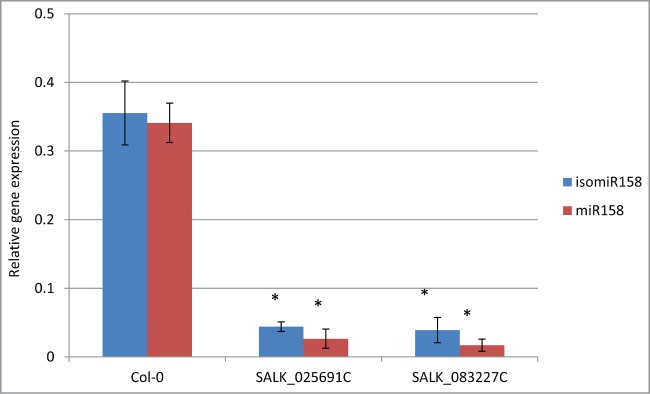
Quantification of endogenous mature ath-miR158a and an isomiR158. Total RNA was isolated from 5- week-old Arabidopsis mutant (SALK_025691 and SALK_083227) and wild-type (Col-0) plant leaves. Levels of miR158 and an isomiR158 were quantified using RT-qPCR. Values represented are relative gene expression over 2 internal controls, miR159 and UBQ5. Error bars represent the standard deviation. Asterisks indicate values that were considered statistically significant (p < 0.05) compared to wild-type according to the Student's t-test. Experiment was performed with 2 biological and 3 technical replications.
Endogenous levels of AT1G49910, AT1G64100, AT3G03580, and AT1G62860 mRNA were also assessed by RT-qPCR in the leaf tissues of mutant and wild-type plants. The SALK_025691 mutant plant exhibited an approximately 4-fold increase in AT3G03580 mRNA compared to wild-type (Fig. 6A). This mutant also showed significantly higher levels of AT1G64100, and AT1G62860 mRNA. Degradome data also support our finding that AT1G62860 mRNA is the target of miR158a (Figure S5O). Similarly, SALK_083227 mutant plants showed a 3-fold increase in AT3G03580 mRNA level (Fig. 6A). This increase in expression of 3 out of 4 predicted targets in ath-miR158a-deficient plants demonstrated that these mRNAs are regulated by ath-miR158a in wild-type plants. These mutants also showed significantly decreased level of expression of miR158-derived isomiRs (Fig. 5). However, not all predicted targets were confirmed. For example, AT1G49910 mRNA was expressed similarly in wild-type and mutant plant leaves (Fig. 6A). Also, unlike SALK_025691 mutants, SALK_083227 mutant plant leaves did not display an increase in AT1G49910, AT1G64100, and AT1G62860 mRNA levels.
Figure 6.
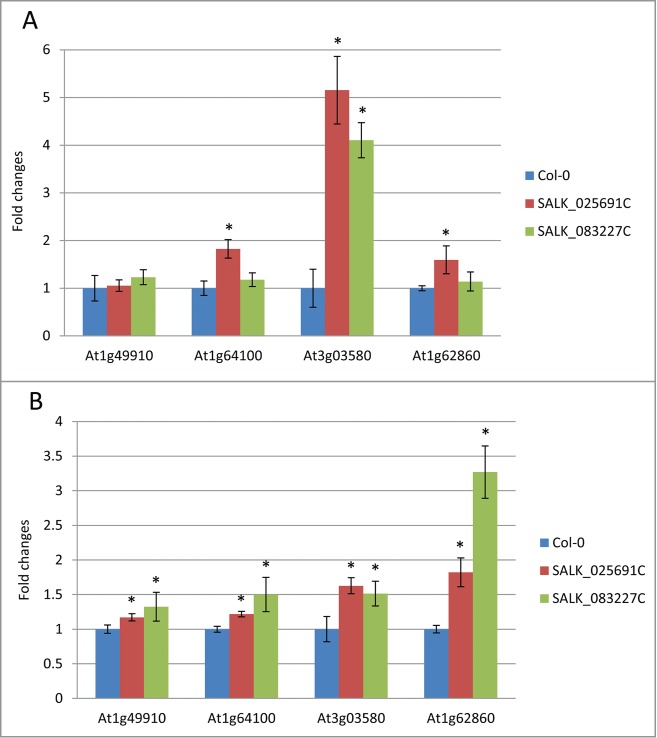
Quantification of endogenous levels of mRNAs predicted to be targeted by ath-miR158a. (A) Expression levels of 4 target mRNAs of ath-miR158a (At1g49910, At1g64100, At3g03580, and At1g62860) were determined in total RNA isolated from Arabidopsis mutant and wild-type leaves. (B) Expression levels of 4 target mRNAs of ath-miR158a (At1g49910, At1g64100, At3g03580, and At1g62860) were determined in total RNA isolated from Arabidopsis mutant and wild-type flowers. Error bars represent the standard deviation. Asterisks indicate values were considered statistically significant (p < 0.05) compared to wild-type according to the Student's t-test.
To understand the reason for these variations, we investigated the expression pattern of these 4 mRNAs in different plant parts across different growth stages using Arabidopsis eFP Browser, a publicly available database (http://bar.utoronto.ca/efp/cgi-bin/efpWeb.cgi).47 Data from the Arabidopsis eFP Browser indicates that all 4 mRNAs are highly expressed in reproductive tissues or meristems, especially flower tissues (Figure S7), but not in leaves. Thus, we further analyzed the endogenous expression level of all 4 mRNAs in the flowers of mutant and wild-type plants by (Col-0) RT-qPCR. Results from this experiment demonstrated that all 4 transcripts are significantly up-regulated in the mutant flower tissues compared to wild-type (Fig. 6B). Convincingly, AT1G62860 mRNA was highly expressed in the flower tissues of both mutant plants.
Through bioinformatics analysis, we also predicted that ath-miR869 can target AT4G08210 mRNA (Table 5B). To verify this, we analyzed the expression of AT4G08210 mRNA in an Arabidopsis mutant, SALK_052117, which contains a T-DNA insertion in the ath-miR869 gene (i.e., AT5G39693) by RT-qPCR. Our results showed that this mRNA was expressed more than 10-fold higher in mutant plant leaves compared to wild-type (Figure S8A). Similarly, expression of the ath-miR5028 target, namely AT1G68540, was analyzed (Table 5A) in SALK_025566 and SALK_108642 mutants, which possess a mutation in the ath-miR5028 gene. Consistent with the previous results, AT1G68540 mRNA was significantly increased in both the SALK mutants when compared to wild-type plant leaves (Figure S8B). These experimental results validated the predicted target genes, and thus demonstrated that our bioinformatics approach of combining the sequences of canonical miRNA and its isomiRs can accurately predict the target of miRNA in Arabidopsis.
Discussion
Cells have developed precise mechanisms to regulate gene expression using miRNA and other cis-regulatory elements present on genes and mRNA.48 High-throughput sequencing data of small RNA has been an important source for understanding the characteristic features of miRNA, their biogenesis, and their role in gene regulation. The functional role of isomer sequences has been established for protein-coding genes, where distinct isoforms of mRNAs can be generated from a single gene; however, the role of isomiRs in pre-miRNAs has yet to be fully deciphered.49,50 Therefore, in this study, a comprehensive global analysis of small RNA high-throughput data from 51 libraries was carried out using bioinformatics approach to understand the properties of annotated miRNAs and their un-annotated sRNAs (sibling and isomeric miRNAs). These analyses are described and discussed in detail in the Supplemental Results and Discussion.
miRNAs target several thousand genes in Arabidopsis to regulate various biological processes. However, predicting miRNA targets remains a challenging task because the features governing miRNA:mRNA hybrid formation are still poorly understood, thereby hindering the development of a more accurate model for target prediction that produces few false positives.51 Some studies showed that a consensus approach using several prediction tools can increase miRNA target specificity; however, this approach also compromises sensitivity. Therefore, in this study, we applied a novel strategy to accurately identify miRNA targets. First, we predicted the targets of miRNAs and their isomiRs using psRNATarget. We did not consider the mature sequences generated from the other arm of the hairpin since they generally target a different set of genes.52 We systematically analyzed and compared these predicted targets to experimentally verified ones, and found that most of the validated targets have a binding site for distinct isomiRs instead of only the canonical miRNA (Fig. 2, S1, and S2). Moreover, common target mRNAs of isomiRs (including the canonical miRNA) are significantly enriched for a diversity of GO terms, indicating that the range of miRNA regulation is much wider (Table 4, S3, and S4) yet consistent with a previous report.53 Moreover, the genes targeted by up to 50% of isomiRs did not show significant GO term enrichment. Sensitivity (or true positive rate) of a method measures the probability to identify correct miRNA target out of total predicted miRNA targets. Our analysis showed that the probability of predicted miRNA targets as validated target increase as we increase the percent of isomiRs targeted gene (Table 2 and Table 4). Interestingly, we found that combination of isomiRs with canonical miRNA drastically increase the miRNA prediction sensitivity from 18.98% to 27.49% and accuracy from 77.9% to 86% (Target with more than 50% of isomiRs in Table 4). But we also noticed that our method missed some true targets while we increase the percent of isomiRs targeted genes (Table 2). Furthermore, computationally generated all possible isomiRs of ath-miR172a can increase the sensitivity of target prediction, and strong GO enrichment, which indicate that virtually generated isomiRs can also be used to improve miRNA target prediction (Table 4, Table S4).
Published data has demonstrated that miRNA and siRNA can down-regulate several unintended target genes, which limits the specificity of RNAi machinery.54-56 We conjecture that, to accomplish more specific gene silencing, the RNAi machinery generates several isomiRs along with the canonical miRNA against a bona fide target gene (Table 2). Like the canonical miRNA, isomiRs also load into AGO proteins, form an active RISC complex, and bind to mRNA through base pair complementarity to cause gene silencing.17,25
Two different modes of gene silencing by RISC have been described. Perfect complementarity between miRNA:mRNA results in RISC-catalyzed endonucleolytic mRNA cleavage, which occurs between the 10th and 11th nucleotide of complementarity, relative to the miRNA 5′-end.57,58 Whereas central mismatches in the complementary region of miRNA:mRNA promote translational repression.59 Silencing through both modes has been reported in plants. However, unlike in animals, most plant miRNAs show mRNA cleavage.59 Each miRNA and its isomiRs can target distinct sets of transcripts; however, due to high sequence similarity, they have many targets in common. We hypothesized that a miRNA and its isomiRs increases the number of active RISC complexes, thus increasing the chances of binding and silencing the common “bona fide” target gene (Fig. 7). Meanwhile, each miRNA and its isomiRs may have unique targets, but the likelihood of binding and silencing the uncommon (un-intended) target is very low. This strategy can increase the specificity and efficiency of silencing true targets while minimizing off-target silencing. Our study also supports a previous study that showed cooperation in hsa-miR10a, hsa-miR-10b, and their 2 isomiRs in increasing the specificity of silencing target genes involved in similar biological pathways.17 Moreover, our GO enrichment analysis is consistent with other studies indicating that miRNA targets are mostly transcription factors.53,60-62
Figure 7.
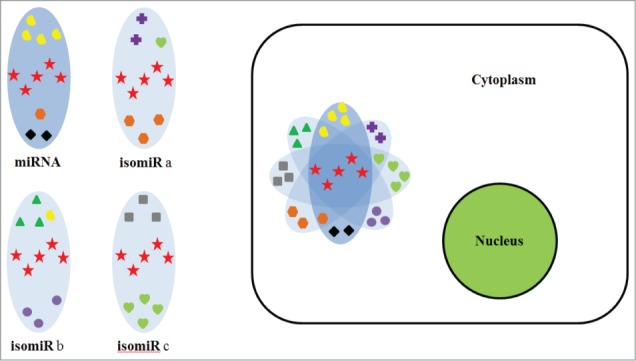
Schematic depicting how miRNA and its isomiRs increase the specificity and efficacy of gene silencing. (A) Each miRNA/isomiR targeting to different group of mRNAs is illustrated as 4 different ovals. Each group contains different genes, as indicated by different symbols within the oval. However, due to high sequence similarity between isomiRs and the miRNA, they also have some common (bona fide) targets. Generally, the abundance of the miRNA is higher than its isomiRs, thus providing a greater degree of silencing, as indicated by the intensity of color in the oval. (B) The cytoplasm contains a myriad of mRNAs. Expression of miRNA and its isomiRs in the cell increases their chance of binding to common “bona fide” targets (star symbol) with a low chance of binding to off-targets. This leads to a greater silencing effect of bona fide target genes and a reduced silencing effect for off-targets, as depicted by the intensity of color in the oval. Common target mRNAs are enriched for Gene Ontology terms.
miRNA and its isomiRs possess a different seed sequence, 2–8 nt from 5′-end, that can dramatically affect target selection. Previous studies showed that miRNA and their 5′-end-derived isomiRs target different mRNAs.46,63,64 Interestingly, we also observed that a slight variation in the terminus of isomiRs derive from a miRNA can alter their targets (Figure S5M-N). In this study, we identified and validated miRNA targets that would have been missed if only the canonical miRNA had been used for its target prediction. For instance, 3 mRNAs (AT1G53780.1, AT1G53780.2, and AT1G53780.3) were predicted to be targeted by one isomiR (i.e., ath-miR172a_-3_24_21_0.02068) instead of ath-miR172a (Fig. 2). Several studies reported that the expression patterns of isomiRs change during development and differ between cell types.17,20,26 Study also identified highly expressed isomiRs with similar abundance as their canonical miRNA.65 Our analysis on degradome data clearly showed that isomiRs mediated target cleavage (Figure S5). Though, isomiRs mediated target cleavage may be less significant. It is difficult to determine the contribution of each isomiRs of a miRNA in target cleavage, because isomiRs having the same 5′-end but heterogeneous 3′-end have the same cleavage site. These published reports that isomiR biogenesis is not random, but is instead highly regulated to modulate specific targets missed by the annotated miRNA. In addition, consistent with other studies, our bioinformatics analysis suggested that a miRNA and its isomiRs can sort into a diverse class of AGO proteins (Tables 3 and S2).17,66–68 Therefore, our study indicates that a miRNA and its isomiRs may regulate a more complex repertoire of target genes.
Finally, new insights about miRNA/isomiRs were incorporated into a strategy to identify novel miRNA targets (Tables 5A and 5B). We predicted 5 potential targets for ath-miR158a, namely AT1G49910.1, AT1G64100.1, AT1G64100.2, AT3G03580.1, and AT1G62860.1, belonging to the Transducin/WD40 repeat-like superfamily protein, Pentatricopeptide (PPR), and Tetratricopeptide (TPR) repeat-containing super family protein. Other studies have also predicted AT3G03580.1 to be a target of ath-miR158a.53,69 RT-qPCR was performed to validate the predicted results for ath-miR158a, as well as ath-miR869 and ath-miR5028, in T-DNA insertion mutants in which expression of the 3 miRNA and their isomiRs was disrupted (Table 5A and 5B). Our analysis demonstrated increased target mRNA levels in all 3 mutant plants, thereby verifying our computational predictions (Fig. 6 and S7). For example, AT3G03580, one of the genes targeted by ath-miR158a showed higher expression in both SALK_025691 and SALK_083227 mutant plant leaves (Fig. 6A) and flower (Fig. 6B) when compared to wild-type. Further, AT1G62860 gene transcripts were higher in mutant plants lacking ath-miR158a expression compared to wild-type. It should be noted that both AT3G03580 and AT1G62860 encode repeat-containing proteins (tetra and penta, respectively) and that AT1G62860 is associated with miRNA-mediated gene regulation.70
More interestingly, these experiments indirectly reveal that the target mRNA is best silenced in wild-type plants when their endogenous expression is at its highest levels. This miRNA-mediated transcript silencing may be a necessary regulatory mechanism for the plant cell in order to effectively achieve the developmental phase changes or stress responses. Supporting this notion, AT1G49910, which is targeted by ath-miR158a, is highly down-regulated following mutation of hub1, a gene encoding RING E3 ligase proteins that function in organ growth.71 The ubiquitin-protein ligase BRE1-like 1 (hub1) mutant exhibits developmental defects, especially in leaf morphology. Taken together, we conclude that the regulation of target mRNA expression by miRNAs and their isomiRs may depend on their endogenous expression pattern. Nevertheless, our data validate the mRNA targets predicted for 3 selected miRNAs, thus demonstrating the robustness of our new method. Furthermore, analysis of target binding patterns of miRNA/isomiRs in humans (see Supplemental Results and Discussion) also showed that validated targets can have binding potentiality with several miRNA/isomiRs and suggests its applicability beyond plants.
Conclusion
Important factors that greatly impact RNAi include the expression levels of miRNA and its target mRNA, its complementarity with target mRNA, and the type of AGO protein into which the miRNA is loaded. However, studies suggest that all of these factors may be dependent on uncharacterized features of miRNA/sib-miRs. Our study identified some of these features of mature miRNA sequences, and revealed the effect of terminal heterogeneity on target mRNA selection. These features may help to sort miRNA/sib-miRs into suitable AGOs and lead to more extensive gene silencing. Altogether, these features of miRNAs/sib-miRs/isomiRs could be incorporated into designing a hairpin that acts as a better DCL substrate, thus further improving gene silencing technology. Moreover, we have developed a robust, novel approach for target prediction by using sequences of miRNA and isomiRs in Arabidopsis that could be also applied to identifying miRNA/isomiR targets in other species.
Materials and Methods
Data sets
We retrieved sRNAs of Arabidopsis from Plant MPSS databases (http://mpss.udel.edu/) released on November 25, 2011.37 The database contains information about sRNAs that are perfectly mapped to 231 Arabidopsis pre-miRNAs taken from 51 sRNA high-throughput Sequencing-by-Synthesis (SBS) sequencing libraries (104,576,308 sequences in total) covering a diversity of tissues and cell lines. We retrieved sequences mapped to each miRNA precursor with their normalized read number (read per million; RPM) from various libraries along with their genomic coordinates (TAIR10).
According to miRBase 18 (release November 2011), Arabidopsis possesses 291 miRNA precursors that generate 328 mature sequences. Among them, 200 were placed into 97 different families based upon their sequence similarity (miRFam.dat of miRBase). To achieve more accurate sequence analysis, a clean and non-redundant dataset was prepared according to 2 criteria: (A) taking one precursor from each family, and (B) taking all precursors not assigned into any family. Among the 231 precursors obtained from the Massively Parallel Signature Sequencing (MPSS) database, 90 belonged to category (A) while 55 were from category (B). Moreover, we removed sRNAs whose total read number was less than 10 RPM in all 51 sRNA libraries, and mapped to opposite strands of the genome relative to annotated pre-miRNA. Thus, the final data set (termed pre-miR_data) contained 132 precursor sequences and their perfectly matched sRNAs, which we called sib-miRs. The number of sib-miRs and their expression level across 51 sRNA libraries was given in the Table S1.
Furthermore, we constructed a dataset of 132 precursors and the number of species in which these precursors are conserved. We also included the number of unique sib-miRs and its read counts (RPM). The data set (called as conserve_pre-miR.txt) was used to study the relationship between miRNA gene conservation and their expression.
To study the cleavage accuracy of DCL on pre-miRNA and the effect of terminal heterogeneity of mature mRNA on targeting genes, a dataset of isomiRs, isomiR_data, was created. For this, sib-miRs mapping within 5 nucleotides upstream and downstream relative to end (5′-end or 3′-end) of canonical miRNAs were taken. Here we considered canonical miRNAs as annotated in miRBase 18.
We also compiled data of experimentally validated targets of miRNAs from miRTarBase, SeqTar, and the published literature into a data set called exp_valid_miR_target.txt.12,43,53,72 We used an in-house-developed Perl script, along with careful manual inspection, to select and analyze the final data.
Data normalization
Datasets were normalized using 2 different methods. The first method involved normalizing the counts as RPM. This method involved calculating the read counts of an sRNA divided by the total number of read counts in that library and multiplying by 106. The data set contains only “genome-matched reads" excluding t/r/sn/snoRNAs. Throughout this manuscript, we have used “read counts” to indicate “normalized read counts” unless otherwise mentioned. The second method involved normalizing within the pre-miRNA. Here, the read count of a sRNA was divided by the total number of sRNA read counts generated from the same pre-miRNA and multiplying by 103. This calculation normalized the sib-miRs at the single pre-miRNA level. We considered read counts of sib-miRs in RPM, derived from 51 libraries, and mapped to a pre-miRNA.
Sorting into AGO proteins
All unique sib-miRs were first exactly matched with experimentally known sequences with AGO (Gene Expression Omnibus (GEO):GSE10036) and assigned a specificity for each AGO protein.73 The remaining sequences were predicted for AGO specificity using SVM models developed through standard protocols.9,74,75 These models were developed using features of nucleotide frequency of sRNA and binary patterns of 6 nt from both terminals of sRNA9 of AGO-specific experimental sequences (GEO:GSE10036). The binary models of AGO1, AGO2, AGO4, and AGO5 achieved an accuracy of 85%, 78%, 88%, and 93%, respectively, on threshold “0” using 5-fold cross validation techniques (Table S5). The performance of these models as assessed on independent datasets showed high accuracy (Table S5).
Analysis of DCL cutting accuracy
Terminal heterogeneity showed absolute distances between the observed 5′- or 3′-ends of sRNA and the reference miRNA (annotated in miRBase 18). We adopted a previously described method to calculate the terminal heterogeneity of each miRNA.27,28 In brief, to calculate 5′-end heterogeneity, sequences mapped within 5 nucleotides upstream or downstream of the 5′-end of a mature miRNA was used. A heterogeneity value was calculated for each canonical miRNA using the total shift in cleavage site divided by the total copy of sequences.
For example, the 6 sRNA sequences mapped to the 5′-arm of ath-miR164a as follows:
ath-miR164a|0|21nt|50
ath-miR164a|-1|22nt|25
ath-miR164a|1|21nt|20
ath-miR164a|2|19nt|13
ath-miR164a|-3|23nt|7
ath-miR164a|3|22nt|5
where the format of ath-miR164a|-1|22nt|25: miRNA, -1 is (1 nt upstream) the shifted nucleotide position in the sequence compared to the 5′-end of the canonical ath-miR164a, 22 nt is the sequence length, and 25 is the read count in RPM. Total copy of sequences = 50+25+20+13+7+5 = 120 Total shift in cleavage site = 50*abs(0)+25*abs(-1)+20*abs(1)+13*abs(2)+7*abs(3) +5*abs(3) = 107 Heterogeneity value for ath-miR164a is : Total shift in cleavage site / Total copy of sequences =107/120= 0.89.
Finally, the mean heterogeneity of the 5′-end terminal was calculated as the average heterogeneity of all studied canonical mature miRNA.
miRNA/isomiRs and their target identification
The computational tool, psRNATarget (http://plantgrn.noble.org/psRNATarget/), was utilized to identify isomiR target mRNAs.38 We used TAIR10 as a genomic library with default parameters except the hpsize, which we set to 15. We named each sequence of isomiRs in a format which gives complete information about the sequence relative to canonical miRNA. For instance, “ath-miR164a_-1_21_21_0.11359” indicates ath-miR164a as the canonical miRNA, -1 represents shift in one nucleotide upstream compared to the 5′-end of canonical ath-miR164a, 21 is the length of the isomiR, 21 is the length of canonical the ath-miR164a, 0.11359 is the normalized value of the isomiR within the pre-miRNA. Therefore, “ath-miR164a_0_21_21_985.78476” would be the name of canonical miRNA expressed in great abundance.
Gene Ontology (GO) analysis
The Singular Enrichment Analysis (SEA) of agriGO (v 1.2) tool was used for GO term analysis.39 To accomplish this, we divided the predicted target transcripts of a miRNA and its isomiRs into 2 groups: (1) mRNA targeted by up to 50% of all isomiRs, and (2) mRNA targeted by more than 50% of all isomiRs. We then analyzed both groups for Gene Ontology enrichment by comparing all targets as “Customized reference." The Fisher statistical test, Bonferroni correction, a significance level of 0.01, and a minimum number of mapping entries equal to 5 were selected as additional parameters.
Degradome data analysis
We downloaded 7 libraries of degradome data of Arabidopsis from Plant MPSS databases (https://mpss.udel.edu/at_pare/): (1) AxIDT [GSM278334], (2) AxIRP [GSM278335], (3) AxSRP [GSM278370], (4) Col [GSM284751], (5) ein5 [GSM284752], (6) TWF [GSM280226], and (7) Tx4F [GSM2802267]. These files were already normalized, where information of each sequence was given in the form of sequence and its normalized copy number value. In order to use in CleaveLand, each sequence was represented with multiple copy of sequence according to its copy number, and all files were merged into a single file. Sliced targets of isomiRs and miRNA were identified and classified using CleaveLand 4.3 pipeline. For analysis, we used 3 different input files: (1) degradome data, (2) Our's list of isomiRs and miRNA sequences, and (3) Transcript database of Arabidopsis previously used in psRNATarget analysis (http://plantgrn.noble.org/psRNATarget/).
Plant material and growth conditions
Arabidopsis T-DNA insertion mutants of the ath-miR158a, ath-miR869, and ath-miR5028 genes were obtained from the Arabidopsis Biological Resource Center in Columbus, Ohio, USA. Arabidopsis ecotype Col-0 was used as the wild-type control. All mutant plants used in this study were confirmed to be homozygous for their respective T-DNA insertion (described below). Detailed information about the mutant plants and the respective gene IDs used in this study is described in Supplementary Table S6. Wild-type Col-0 and mutant seeds were cold-treated for 3 d at 4°C and germinated in Metro Mix Professional Growing Mix (SUNGRO) in a growth chamber maintained at 20° to 22°C. The plants were grown at this temperature under short day conditions (8 h of light).
Zygosity test for mutant plants
Arabidopsis wild-type Col-0 and respective mutant plants were grown for 3 weeks and leaf samples were collected to test for zygosity using the method recommended by SALK (http://signal.salk.edu/tdnaprimers.2.html). Genomic DNA was isolated by using the REDExtract-N-Amp Plant PCR Kit (Sigma-Aldrich Co. St. Louis MO) according to the manufacturer's recommended protocol. Two paired PCR reactions using primer combinations, namely LP+RP and LB+RP (primer details are provided in Supplementary Table S7), were performed. Reactions that showed amplification of the expected PCR product with the LB+RP primers and no amplification in the LP+RP reaction were considered homozygous for the respective mutation.
Quantification of endogenous miRNAs
The homozygous mutant plants for miR158, namely SALK_025691 and SALK_083227, were confirmed to be deficient in the endogenous mature miRNA transcript using primers as described in Supplementary Table S8. Total RNA was extracted from 5-week-old Arabidopsis plant leaves using the RNeasy plant mini kit (Qiagen Valencia, CA, USA) according to the manufacturer's instructions. The High-specificity miRNA RT-qPCR detection kit and protocol (Agilent Technologies Inc.. Clara, CA USA) were used to quantify miRNAs and isomiRs. This kit has been optimized for the detection and quantification of miRNA from 15–250 ng of total RNA. RT-qPCR (BioRad-CFX connect real-time system, Hercules, CA, USA) was performed using a miRNA-specific forward primer (Supplementary Table S8) and universal reverse primer provided in the kit. The forward primer for isomiR158 was designed using the miRNA primer design tool at http://www.astridbio.com/ and synthesized by Integrated DNA Technologies (IDT, Inc.., Coralville, IA, USA). Forward primer design and RT-qPCR were performed as described previously76 with modifications. Relative expression as fold increase was calculated using the ΔCP method77 with comparison to a reference (i.e., AtUbiquitin 5 and miR159).
Real-time PCR for quantification of endogenous mRNA expression
Total RNA was extracted from the leaf or floral tissues as described above. RNA samples were treated with RNAse-free DNAse prior to reverse transcription using the SuperscriptTM First Strand Synthesis System for RT-PCR (Invitrogen, Life Technologies, Carlsbad, CA, USA). First strand cDNA was synthesized from 2 μg of total RNA. Quantitative real-time PCR (RT-qPCR) was conducted with a SYBR green (Sigma Aldrich Co. St. Louis MO) reaction mix using a 1:20 dilution of Cdna.78 Using the Ct values, the relative expression of target transcripts was calculated by the ΔCP method77 with comparison to the reference genes AtEF1α, and AtUbiquitin 5. Primers for the respective endogenous transcripts were designed using Primer Quest software (IDT, Inc..) and the primer sequences provided in Supplementary Table S9.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Dr. Nitish Kumar Mishra for his assistance in analyzing the loading potential of plant small RNAs into different AGOs, and to Dr. Hee-Kyung Lee for zygosity testing of T-DNA mutants and RNA extraction.
Funding
Project funding support: National Science Foundation (Grant No. ABI-0960897); Oklahoma Center for the Advancement of Science and Technology (OCAST Project No. PSB11–004); Samuel Roberts Noble Foundation.
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
Authors' Contributions
PZ conceived the study and oversaw the analyses. FA, XD, and PZ performed the feature analysis of the miRNAs and isomiRs, and identified their regulatory targets in Arabidopsis. MSK, SL, and KSM designed and performed experiments to validate the predicted miRNA targets. FA, MSK, and PZ wrote the manuscript. All authors read and approved the final manuscript.
References
- 1. Ul Hussain M. Micro-RNAs (miRNAs): genomic organisation, biogenesis and mode of action. Cell Tissue Res 2012; 349:405-13; PMID:22622804; http://dx.doi.org/ 10.1007/s00441-012-1438-0 [DOI] [PubMed] [Google Scholar]
- 2. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 2009; 10:126-39; PMID:19165215; http://dx.doi.org/ 10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- 3. Meng Y, Shao C, Wang H, Chen M. The regulatory activities of plant microRNAs: a more dynamic perspective. Plant Physiol 2011; 157:1583-95; PMID:22003084; http://dx.doi.org/ 10.1104/pp.111.187088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell 2009; 136:642-55; PMID:19239886; http://dx.doi.org/ 10.1016/j.cell.2009.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 2009; 25:21-44; PMID:19575669; http://dx.doi.org/ 10.1146/annurev.cellbio.042308.113417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell 2011; 23:431-42; PMID:21317375; http://dx.doi.org/ 10.1105/tpc.110.082784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X. Methylation as a crucial step in plant microRNA biogenesis. Science 2005; 307:932-5; PMID:15705854; http://dx.doi.org/ 10.1126/science.1107130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim Y, Kim VN. MicroRNA factory: RISC assembly from precursor microRNAs. Mol Cell 2012; 46:384-6; PMID:22633486; http://dx.doi.org/ 10.1016/j.molcel.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 9. Ahmed F, Ansari HR, Raghava GP. Prediction of guide strand of microRNAs from its sequence and secondary structure. BMC Bioinformat 2009; 10:105; PMID:19358699; http://dx.doi.org/ 10.1186/1471-2105-10-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kawamata T, Tomari Y. Making RISC. Trends Biochem Sci 2010; 35:368-76; PMID:20395147; http://dx.doi.org/ 10.1016/j.tibs.2010.03.009 [DOI] [PubMed] [Google Scholar]
- 11. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008; 36:D154-8; PMID:17991681; http://dx.doi.org/ 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res 2011; 39:D163-9; PMID:21071411; http://dx.doi.org/ 10.1093/nar/gkq1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N, Dalamagas T, Hatzigeorgiou AG. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res 2012; 40:D222-9; PMID:22135297; http://dx.doi.org/ 10.1093/nar/gkr1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res 2009; 37:D105-10; PMID:18996891; http://dx.doi.org/ 10.1093/nar/gkn851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA* species has substantial influence on microRNA and 3' UTR evolution. Nat Struct Mol Biol 2008; 15:354-63; PMID:18376413; http://dx.doi.org/ 10.1038/nsmb.1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC. Widespread regulatory activity of vertebrate microRNA* species. RNA 2011; 17:312-26; PMID:21177881; http://dx.doi.org/ 10.1261/rna.2537911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cloonan N, Wani S, Xu Q, Gu J, Lea K, Heater S, Barbacioru C,Steptoe AL, Martin HC, Nourbakhsh E, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol 2011; 12:R126; PMID:22208850; http://dx.doi.org/ 10.1186/gb-2011-12-12-r126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo L, Lu Z. The fate of miRNA* strand through evolutionary analysis: implication for degradation as merely carrier strand or potential regulatory molecule? PLoS One 2010; 5:e11387; PMID:20613982; http://dx.doi.org/ 10.1371/journal.pone.0011387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Devers EA, Branscheid A, May P, Krajinski F. Stars and symbiosis: microRNA- and microRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol 2011; 156:1990-2010 PMID:21571671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fernandez-Valverde SL, Taft RJ, Mattick JS. Dynamic isomiR regulation in drosophila development. RNA 2010; 16:1881-8; PMID:20805289; http://dx.doi.org/ 10.1261/rna.2379610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W, Gao S, Zhou X, Xia J, Chellappan P, Zhang X, Zhang X, Jin H. Multiple distinct small RNAs originate from the same microRNA precursors. Genome Biol 2010; 11:R81; PMID:20696037; http://dx.doi.org/ 10.1186/gb-2010-11-8-r81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in arabidopsis thaliana. Genes Dev 2006; 20:3407-25; PMID:17182867; http://dx.doi.org/ 10.1101/gad.1476406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu AL, Zhao Y, McDonald H, Zeng T, Hirst M, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 2008; 18:610-21; PMID:18285502; http://dx.doi.org/ 10.1101/gr.7179508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer 2010; 10:389-402; PMID:20495573; http://dx.doi.org/ 10.1038/nrc2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neilsen CT, Goodall GJ, Bracken CP. IsomiRs - the overlooked repertoire in the dynamic microRNAome. Trends Genet 2012; 28:544-9; PMID:22883467; http://dx.doi.org/ 10.1016/j.tig.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 26. Bizuayehu T, Lanes C, Furmanek T, Karlsen B, Fernandes J, Johansen S, Babiak I. Differential expression patterns of conserved miRNAs and isomiRs during atlantic halibut development. BMC Genomics 2012; 13:11; PMID:22233483; http://dx.doi.org/ 10.1186/1471-2164-13-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Zhou YH, Chen W, Khaitovich P. Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics 2009; 10:413; PMID:19732433; http://dx.doi.org/ 10.1186/1471-2164-10-413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seitz H, Ghildiyal M, Zamore PD. Argonaute loading improves the 5' precision of both microRNAs and their miRNA strands in flies. Curr Biol 2008; 18:147-51; PMID:18207740; http://dx.doi.org/ 10.1016/j.cub.2007.12.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, Manjunath N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One 2007; 2:e1020; PMID:17925868; http://dx.doi.org/ 10.1371/journal.pone.0001020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. A comprehensive survey of 3' animal miRNA modification events and a possible role for 3' adenylation in modulating miRNA targeting effectiveness. Genome Res 2010; 20:1398-410; PMID:20719920; http://dx.doi.org/ 10.1101/gr.106054.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li J, Yang Z, Yu B, Liu J, Chen X. Methylation protects miRNAs and siRNAs from a 3'-end uridylation activity in arabidopsis. Curr Biol 2005; 15:1501-7; PMID:16111943; http://dx.doi.org/ 10.1016/j.cub.2005.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reid JG, Nagaraja AK, Lynn FC, Drabek RB, Muzny DM, Shaw CA, Weiss MK, Naghavi AO, Khan M, Zhu H, et al. Mouse let-7 miRNA populations exhibit RNA editing that is constrained in the 5'-seed/ cleavage/anchor regions and stabilize predicted mmu-let-7a:mRNA duplexes. Genome Res 2008; 18:1571-81; PMID:18614752; http://dx.doi.org/ 10.1101/gr.078246.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007; 315:1137-40; PMID:17322061; http://dx.doi.org/ 10.1126/science.1138050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo L, Yang Q, Lu J, Li H, Ge Q, Gu W, Bai Y, Lu Z. A comprehensive survey of miRNA repertoire and 3' addition events in the placentas of patients with pre-eclampsia from high-throughput sequencing. PLoS One 2011; 6:e21072; PMID:21731650; http://dx.doi.org/ 10.1371/journal.pone.0021072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X, Liu S. Systematic curation of miRBase annotation using integrated small RNA high-throughput sequencing data for C. elegans and drosophila. Front Genet 2011; 2; PMID:22303321; 10.3389/fgene.2011.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu S, Sun YH, Chiang VL. Adenylation of plant miRNAs. Nucleic Acids Res 2009; 37:1878-85; PMID:19188256; http://dx.doi.org/ 10.1093/nar/gkp031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakano M, Nobuta K, Vemaraju K, Tej SS, Skogen JW, Meyers BC. Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res 2006; 34:D731-5; PMID:16381968; http://dx.doi.org/ 10.1093/nar/gkj077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 2011; 39 2:W155-9; PMID:21622958; http://dx.doi.org/ 10.1093/nar/gkr319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 2010; 38:W64-70; PMID:20435677; http://dx.doi.org/ 10.1093/nar/gkq310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009; 25:130-1; PMID:19017659; http://dx.doi.org/ 10.1093/bioinformatics/btn604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the arabidopsis degradome. Curr Biol 2008; 18:758-62; PMID:18472421; http://dx.doi.org/ 10.1016/j.cub.2008.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of scarecrow-like mRNA targets directed by a class of arabidopsis miRNA. Science 2002; 297:2053-6; PMID:12242443; http://dx.doi.org/ 10.1126/science.1076311 [DOI] [PubMed] [Google Scholar]
- 43. Zheng Y, Li YF, Sunkar R, Zhang W. Seqtar: an effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res 2012; 40:e28; PMID:22140118; http://dx.doi.org/ 10.1093/nar/gkr1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang W, Sunkar R. et al. Transcriptome-wide identification of microRNA targets in rice. Plant J 2010; 62:742-59; PMID:20202174; http://dx.doi.org/ 10.1111/j.1365-313X.2010.04187.x [DOI] [PubMed] [Google Scholar]
- 45. Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of brassica juncea. BMC Genomics 2013; 14:9; PMID:23324572; http://dx.doi.org/ 10.1186/1471-2164-14-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tan GC, Chan E, Sarkar R, Cui W, Molnar A, Meister G, et al. Canonical microRNAs and their isomiRs have different target genes. Abstract Presented at: Biogenesis and turnover of small RNAs, Royal Society 2013 January 15—17; Edinburgh, UK 2013; http://www.biochemistry.org/ conference. http://www.biochemistry.org/Portals/0/Conferences/abstracts/SA144/SA144P014.pdf [Google Scholar]
- 47. Winter D, Vinegar B, Nahal H, Ammar R, Wilson GV, Provart NJ. An "electronic fluorescent pictograph" browser for exploring and analyzing large-scale biological data sets. PloS one 2007; 2:e718; PMID:17684564; http://dx.doi.org/ 10.1371/journal.pone.0000718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahmed F, Benedito VA, Zhao PX. Mining functional elements in messenger RNAs: overview, challenges, and perspectives. Front Plant Sci 2011; 2; PMID:22639614; http://dx.doi.org/ 10.3389/fpls.2011.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/ 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ahmed F, Zhao P. A comprehensive analysis of isomirs and their targets using high-throughput sequencing data for arabidopsis thaliana. J Nat Sci, Biol Med 2011; 2:32 [Google Scholar]
- 51. Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol 2009; 10:141-8; PMID:19145236; http://dx.doi.org/ 10.1038/nrm2619 [DOI] [PubMed] [Google Scholar]
- 52. Marco A, Macpherson JI, Ronshaugen M, Griffiths-Jones S. MicroRNAs from the same precursor have different targeting properties. Silence 2012; 3:8; PMID:23016695; http://dx.doi.org/ 10.1186/1758-907X-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alves L, Jr., Niemeier S, Hauenschild A, Rehmsmeier M, Merkle T. Comprehensive prediction of novel microRNA targets in arabidopsis thaliana. Nucleic Acids Res 2009; 37:4010-21; PMID:19417064; http://dx.doi.org/ 10.1093/nar/gkp272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burchard J, Jackson AL, Malkov V, Needham RH, Tan Y, Bartz SR, Dai H, Sachs AB, Linsley PS. MicroRNA-like off-target transcript regulation by siRNAs is species specific. RNA 2009; 15:308-15; PMID:19144911; http://dx.doi.org/ 10.1261/rna.1326809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 2003; 21:635-7; PMID:12754523; http://dx.doi.org/ 10.1038/nbt831 [DOI] [PubMed] [Google Scholar]
- 56. Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA "off-target" transcript silencing mediated by seed region sequence complementarity. RNA 2006; 12:1179-87; PMID:16682560; http://dx.doi.org/ 10.1261/rna.25706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004; 304:594-6; PMID:15105502; http://dx.doi.org/ 10.1126/science.1097434 [DOI] [PubMed] [Google Scholar]
- 58. Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002; 297:2056-60; PMID:12154197; http://dx.doi.org/ 10.1126/science.1073827 [DOI] [PubMed] [Google Scholar]
- 59. Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008; 320:1185-90; PMID:18483398; http://dx.doi.org/ 10.1126/science.1159151 [DOI] [PubMed] [Google Scholar]
- 60. Guo AY, Sun J, Jia P, Zhao Z. A novel microRNA and transcription factor mediated regulatory network in schizophrenia. BMC Syst Biol 2010; 4:10; PMID:20156358; http://dx.doi.org/ 10.1186/1752-0509-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 2006; 57:19-53; PMID:16669754; http://dx.doi.org/ 10.1146/annurev.arplant.57.032905.105218 [DOI] [PubMed] [Google Scholar]
- 62. Dugas DV, Bartel B. MicroRNA regulation of gene expression in plants. Curr Opin Plant Biol 2004; 7:512-20; PMID:15337093; http://dx.doi.org/ 10.1016/j.pbi.2004.07.011 [DOI] [PubMed] [Google Scholar]
- 63. Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, Siomi MC. Characterization of endogenous human argonautes and their miRNA partners in RNA silencing. Proc Natl Acad Sci U S A 2008; 105:7964-9; PMID:18524951; http://dx.doi.org/ 10.1073/pnas.0800334105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Humphreys DT, Hynes CJ, Patel HR, Wei GH, Cannon L, Fatkin D, Suter CM, Clancy JL, Preiss T. Complexity of murine cardiomyocyte miRNA biogenesis, sequence variant expression and function. PloS one 2012; 7:e30933; PMID:22319597; http://dx.doi.org/ 10.1371/journal.pone.0030933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Baran-Gale J, Fannin EE, Kurtz CL, Sethupathy P. Beta cell 5'-shifted isomiRs are candidate regulatory hubs in type 2 diabetes. PLoS One 2013; 8:e73240; PMID:24039891; http://dx.doi.org/ 10.1371/journal.pone.0073240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burroughs AM, Ando Y, de Hoon MJ, Tomaru Y, Suzuki H, Hayashizaki Y, Daub CO. Deep-sequencing of human argonaute-associated small RNAs provides insight into miRNA sorting and reveals argonaute association with RNA fragments of diverse origin. RNA Biol 2011; 8:158-77; PMID:21282978; http://dx.doi.org/ 10.4161/rna.8.1.14300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res 2009; 37:2461-70; PMID:19255090; http://dx.doi.org/ 10.1093/nar/gkp093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ebhardt HA, Fedynak A, Fahlman RP. Naturally occurring variations in sequence length creates microRNA isoforms that differ in argonaute effector complex specificity. Silence 2010; 1:12; PMID:20534119; http://dx.doi.org/ 10.1186/1758-907X-1-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005; 121:207-21; PMID:15851028; http://dx.doi.org/ 10.1016/j.cell.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 70. Howell MD, Fahlgren N, Chapman EJ, Cumbie JS, Sullivan CM, Givan SA, Kasschau KD, Carrington JC. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant cell 2007; 19:926-42; PMID:17400893; http://dx.doi.org/ 10.1105/tpc.107.050062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fleury D, Himanen K, Cnops G, Nelissen H, Boccardi TM, Maere S, Beemster GT, Neyt P, Anami S, Robles P, et al. The Arabidopsis thaliana homolog of yeast BRE1 has a function in cell cycle regulation during early leaf and root growth. Plant Cell 2007; 19:417-32; PMID:17329565; http://dx.doi.org/ 10.1105/tpc.106.041319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moldovan D, Spriggs A, Yang J, Pogson BJ, Dennis ES, Wilson IW. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in arabidopsis. J Exp Bot 2010; 61:165-77; PMID:19815687; http://dx.doi.org/ 10.1093/jxb/erp296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, et al. Sorting of small RNAs into arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell 2008; 133:116-27; PMID:18342361; http://dx.doi.org/ 10.1016/j.cell.2008.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ahmed F, Kaundal R, Raghava GP. PHDcleav: a SVM based method for predicting human dicer cleavage sites using sequence and secondary structure of miRNA precursors. BMC Bioinformat 2013; 14 14:S9; PMID:24267009; http://dx.doi.org/ 10.1186/1471-2105-14-S14-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ahmed F, Kumar M, Raghava GP. Prediction of polyadenylation signals in human DNA sequences using nucleotide frequencies. Silico Biol 2009; 9:135-48; PMID:19795571 [PubMed] [Google Scholar]
- 76. Varkonyi-Gasic E, Wu R, Wood M, Walton EF, Hellens RP. Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 2007; 3:12; PMID:17931426; http://dx.doi.org/ 10.1186/1746-4811-3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:6; PMID:11328886; http://dx.doi.org/ 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Senthil-Kumar M, Mysore KS. Ornithine-delta-aminotransferase and proline dehydrogenase genes play a role in non-host disease resistance by regulating pyrroline-5-carboxylate metabolism-induced hypersensitive response. Plant Cell Environment 2012; 35:1329-43; PMID:22321246; http://dx.doi.org/ 10.1111/j.1365-3040.2012.02492.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.