

ABSTRACT
Despite the identification of horseshoe bats as the reservoir of severe acute respiratory syndrome (SARS)-related coronaviruses (SARSr-CoVs), the origin of SARS-CoV ORF8, which contains the 29-nucleotide signature deletion among human strains, remains obscure. Although two SARS-related Rhinolophus sinicus bat CoVs (SARSr-Rs-BatCoVs) previously detected in Chinese horseshoe bats (Rhinolophus sinicus) in Yunnan, RsSHC014 and Rs3367, possessed 95% genome identities to human and civet SARSr-CoVs, their ORF8 protein exhibited only 32.2 to 33% amino acid identities to that of human/civet SARSr-CoVs. To elucidate the origin of SARS-CoV ORF8, we sampled 348 bats of various species in Yunnan, among which diverse alphacoronaviruses and betacoronaviruses, including potentially novel CoVs, were identified, with some showing potential interspecies transmission. The genomes of two betacoronaviruses, SARSr-Rf-BatCoV YNLF_31C and YNLF_34C, from greater horseshoe bats (Rhinolophus ferrumequinum), possessed 93% nucleotide identities to human/civet SARSr-CoV genomes. Although these two betacoronaviruses displayed lower similarities than SARSr-Rs-BatCoV RsSHC014 and Rs3367 in S protein to civet SARSr-CoVs, their ORF8 proteins demonstrated exceptionally high (80.4 to 81.3%) amino acid identities to that of human/civet SARSr-CoVs, compared to SARSr-BatCoVs from other horseshoe bats (23.2 to 37.3%). Potential recombination events were identified around ORF8 between SARSr-Rf-BatCoVs and SARSr-Rs-BatCoVs, leading to the generation of civet SARSr-CoVs. The expression of ORF8 subgenomic mRNA suggested that the ORF8 protein may be functional in SARSr-Rf-BatCoVs. The high Ka/Ks ratio among human SARS-CoVs compared to that among SARSr-BatCoVs supported that ORF8 is under strong positive selection during animal-to-human transmission. Molecular clock analysis using ORF1ab showed that SARSr-Rf-BatCoV YNLF_31C and YNLF_34C diverged from civet/human SARSr-CoVs in approximately 1990. SARS-CoV ORF8 originated from SARSr-CoVs of greater horseshoe bats through recombination, which may be important for animal-to-human transmission.
IMPORTANCE Although horseshoe bats are the primary reservoir of SARS-related coronaviruses (SARSr-CoVs), it is still unclear how these bat viruses have evolved to cross the species barrier to infect civets and humans. Most human SARS-CoV epidemic strains contain a signature 29-nucleotide deletion in ORF8, compared to civet SARSr-CoVs, suggesting that ORF8 may be important for interspecies transmission. However, the origin of SARS-CoV ORF8 remains obscure. In particular, SARSr-Rs-BatCoVs from Chinese horseshoe bats (Rhinolophus sinicus) exhibited <40% amino acid identities to human/civet SARS-CoV in the ORF8 protein. We detected diverse alphacoronaviruses and betacoronaviruses among various bat species in Yunnan, China, including two SARSr-Rf-BatCoVs from greater horseshoe bats that possessed ORF8 proteins with exceptionally high amino acid identities to that of human/civet SARSr-CoVs. We demonstrated recombination events around ORF8 between SARSr-Rf-BatCoVs and SARSr-Rs-BatCoVs, leading to the generation of civet SARSr-CoVs. Our findings offer insight into the evolutionary origin of SARS-CoV ORF8 protein, which was likely acquired from SARSr-CoVs of greater horseshoe bats through recombination.
INTRODUCTION
Coronaviruses (CoVs) are known to cause respiratory, enteric, hepatic, and neurological diseases of varying severity in a variety of animals. They are currently classified into four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus, replacing the traditional three groups, groups 1 to 3 (1–4). The genus Betacoronavirus is further classified into lineages A to D (3, 5, 6). Among CoVs that infect humans, human CoV 229E (HCoV 229E) and human CoV NL63 (HCoV NL63) belong to Alphacoronavirus; human CoV OC43 (HCoV OC43) and human CoV HKU1 (HCoV HKU1) belong to Betacoronavirus lineage A; Severe Acute Respiratory Syndrome-related CoV (SARSr-CoV) belongs to Betacoronavirus lineage B; and the recently emerged Middle East Respiratory Syndrome CoV (MERS-CoV) belongs to Betacoronavirus lineage C (7–16). The high recombination rate, coupled with the infidelity of the RNA-dependent RNA polymerase (RdRp), may have facilitated CoVs to adapt to new hosts and ecological niches, causing epidemics in animals and humans (5, 17–24).
The SARS epidemic and identification of SARSr-CoVs from palm civet and horseshoe bats in China have boosted interest in the discovery of novel CoVs in both humans and animals, especially bats (25–29). With the exception of lineage A betacoronaviruses, bats are now known to be an important reservoir of diverse alphacoronaviruses and of lineage B, C, and D betacoronaviruses (30–38), with bat CoVs being the gene source for other mammalian CoVs (4). In particular, the findings of bat CoVs related to SARS-CoV and MERS-CoV suggest that bats may be the animal origin of both SARS and MERS epidemics, while other animals have served as the intermediate or amplifying hosts for animal-to-human transmission, palm civets in the case of SARS and dromedary camels in the case of MERS (25, 27, 28, 39–41). However, the evolutionary paths from bat CoVs to CoVs capable of infecting intermediate hosts and humans are not fully understood.
SARSr-CoVs have been detected in at least 11 different species of horseshoe bats (genus Rhinolophus) from various countries in Asia, Africa, and Europe (27, 28, 35, 37, 38, 42, 43). Related viruses have also been reported in bats of other genera, such as Chaerephon and Hipposideros, from Africa and China (43–45). However, it is still unclear how these bat CoVs have evolved to generate the ancestor of civet/human SARSr-CoVs capable of crossing the species barrier. The genome organization of SARSr-CoVs, similar to that of other CoVs, possesses the characteristic gene order from 5′ to 3′ of open reading frame 1ab (ORF1ab), spike (S), ORF3, envelope (E), membrane (M), ORF6 to ORF8, and nucleocapsid (N). It is known that most human SARS-CoVs during the epidemic contained a signature 29-nucleotide (nt) deletion in ORF8, compared to civet SARSr-CoVs (25), suggesting that this genomic region may be important for interspecies transmission. However, the origin of SARS-CoV ORF8 remains obscure. Genomes of SARS-related Rhinolophus sinicus bat CoVs (SARSr-Rs-BatCoVs), previously designated SARSr-Rh-BatCoVs, from Chinese horseshoe bats (Rhinolophus sinicus) in Hong Kong and the Guangdong Province shared only 87 to 92% nucleotide identities to human/civet SARSr-CoV genomes (22, 27, 28). A subsequent study identified two SARSr-Rs-BatCoVs, RsSHC014 and Rs3367, in Yunnan Province which were more closely related to human/civet SARSr-CoVs (with 95% genome sequence identities) than any other SARSr-BatCoVs (42). The S proteins of these two SARSr-Rs-BatCoVs from Yunnan shared 90.1 to 92.3% amino acid identities to those of human/civet SARSr-CoVs, compared to 79 to 80% amino acid identities between SARSr-Rs-BatCoVs from Hong Kong and human/civet SARSr-CoVs (27, 42). Moreover, a highly similar virus, SARSr-Rs-BatCoV WIV1, isolated in Vero E6 cells, was able to use angiotensin converting enzyme II (ACE2) from humans, civets, and Chinese horseshoe bats as a receptor for cell entry, suggesting that intermediate hosts between bats and humans/civets may not be necessary for interspecies transmission (42). However, considerable genetic distance still exists between the two SARSr-Rs-BatCoVs from Yunnan and human/civet SARSr-CoVs, especially in the ORF8 region, with only 32.2 to 33% amino acid identities.
To elucidate the evolutionary origin of SARS-CoV ORF8 and search for even closer bat CoV ancestors of SARS-CoV, we conducted a 3-month study (May to July 2013) on CoVs among various bats from different regions of Yunnan Province. Diverse CoVs were detected, including two SARS-related Rhinolophus ferrumequinum BatCoVs (SARSr-Rf-BatCoVs) from greater horseshoe bats (R. ferrumequinum), which possessed an expressed ORF8 much more closely related to human/civet SARSr-CoVs than CoVs detected from other bat species. Recombination and molecular clock analysis were also performed to elucidate the evolutionary paths and time of interspecies transmission of SARSr-CoVs.
MATERIALS AND METHODS
Ethics statement.
The collection of bat samples was approved and performed by the Yunnan Institute of Endemic Diseases Control and Prevention, Dali, Yunnan, China. All bats were maintained and handled using standard procedures approved by the Medical Ethical Committee of Yunnan Institute of Endemic Diseases Control and Prevention, China.
Sample collection.
Bats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013 (Fig. 1). Samples were collected using procedures described previously (27, 46). All samples were placed in viral transport medium (Earle's balanced salt solution, 0.09% glucose, 0.03% sodium bicarbonate, 0.45% bovine serum albumin, 50 mg/ml amikacin, 50 mg/ml vancomycin, 40 U/ml nystatin) and stored at −80°C before RNA extraction.
FIG 1.

Map showing five locations of bat sampling in four autonomous prefectures in Yunnan Province, China. Sampling locations in Yunnan are in red. The location of SARSr-Rs-BatCoV strains Rs3367 and RsSHC014, detected in a previous study (42), is in blue.
RNA extraction.
Viral RNA was extracted from alimentary samples using a QIAamp viral RNA minikit (Qiagen, Hilden, Germany). The RNA was eluted in 50 μl of AVE buffer and used as the template for reverse transcription (RT)-PCR.
RT-PCR for CoVs and DNA sequencing.
CoV screening was performed by amplifying a 440-bp fragment of the RdRp gene of CoVs using conserved primers (5′-GGTTGGGACTATCCTAAGTGTGA-3′ and 5′-ACCATCATCNGANARDATCATNA-3′) targeted to RdRp genes of CoVs (12). Reverse transcription was performed using the SuperScript III kit (Invitrogen, Life Technologies, Grand Island, NY, USA). The PCR mixture (25 μl) contained cDNA, PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 3 mM MgCl2, 0.01% gelatin), 200 μM (each) deoxynucleoside triphosphates (dNTPs), and 1.0 U Taq polymerase (Applied Biosystems, Life Technologies, Grand Island, NY, USA). The mixtures were amplified for 40 cycles of 94°C for 1 min, 48°C for 1 min, and 72°C for 1 min and a final extension at 72°C for 10 min in an automated thermal cycler (Applied Biosystems). Standard precautions were taken to avoid PCR contamination, and no false-positive result was observed in the negative controls.
The PCR products were gel purified using the QIAquick gel extraction kit (Qiagen). Both strands of the PCR products were sequenced twice with an ABI Prism 3700 DNA analyzer (Applied Biosystems), using the two PCR primers. The sequences of the PCR products were compared with known sequences of the RdRp genes of CoVs in the GenBank database. A phylogenetic tree was constructed using 266-bp fragments of the RdRp gene with the maximum likelihood method using the substitution model of the general time reversible model with gamma distribution as well as allowance of evolutionarily invariable sites (GTR+G+I) by MEGA 5.0 (47).
Viral culture.
The two samples positive for SARSr-Rf-BatCoVs were subjected to virus isolation in Vero E6 (African green monkey kidney) and primary R. sinicus lung cells as described previously (21).
Complete genome sequencing and analysis of SARSr-Rf-BatCoVs.
Two complete genomes of SARSr-Rf-BatCoVs were amplified and sequenced using RNA extracted from the alimentary samples as templates. RNA was converted to cDNA by a combined random-priming and oligo(dT) priming strategy. The cDNA was amplified by using degenerate primers as described previously (27). A total of 75 sets of primers, available on request, were used for PCR. The 5′ end of the viral genome was confirmed by rapid amplification of cDNA ends (RACE) using the 5′/3′ SMARTer RACE cDNA amplification kit (Clontech, USA). Sequences were assembled and manually edited to produce the final sequences. The nucleotide sequences of the genomes and the deduced amino acid sequences of the ORFs were compared to those of other CoVs using the coronavirus database CoVDB (48). Phylogenetic tree construction was performed using the maximum likelihood method with MEGA 6.0.
Recombination analysis.
To detect possible recombination between different SARSr-BatCoVs and civet SARSr-CoVs, sliding window analysis was performed using nucleotide alignment of the available genome sequences generated by ClustalX version 1.83 and edited manually with BioEdit version 7.1.3. Similarity plot analysis and Bootscan analysis were performed using Simplot version 3.5.1 (49) (F84 model; window size, 1,000 bp; step, 200 bp) with civet SARSr-CoV SZ3 as the query.
Estimation of synonymous and nonsynonymous substitution rates.
The number of synonymous substitutions per synonymous site, Ks, and the number of nonsynonymous substitutions per nonsynonymous site, Ka, for each coding region were calculated for all available SARSr-Rf-BatCoV, SARSr-Rs-BatCoV, civet SARSr-CoV, and human SARSr-CoV genomes using the Nei-Gojobori method (Jukes-Cantor) in MEGA 5.0.
Estimation of divergence dates.
The time to most recent common ancestor (tMRCA) was estimated based on an alignment of ORF1ab and nsp5 sequences, using the uncorrelated exponential distributed relaxed clock model (UCED) in BEAST version 1.8 (http://beast.bio.ed.ac.uk/) (50). Under this model, the rates were allowed to vary at each branch drawn independently from an exponential distribution. The sampling dates of all strains were collected from the literature or from the present study and were used as calibration points. Depending on the data set, Markov chain Monte Carlo (MCMC) sample chains were run for 2 × 108 states, with sampling every 1,000 generations under the GTR nucleotide substitution model, determined by MODELTEST and allowing γ-rate heterogeneity for all data sets. A constant population coalescent prior was assumed for all data sets. The median and highest posterior density (HPD) were calculated for each of these parameters from two identical but independent MCMC chains using TRACER 1.3 (http://beast.bio.ed.ac.uk). The tree was annotated by TreeAnnotator, a program of BEAST, and displayed by using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Expression of ORF8 and determination of leader-body junction sequence.
The leader-body junction site and flanking sequences of the ORF8 subgenomic mRNA in SARSr-Rf-BatCoV YNLF_31C were determined using RT-PCR as described previously (21, 51). Briefly, RNA was extracted directly from the bat samples using TRIzol reagent (Invitrogen). Reverse transcription was performed using random hexamers and the SuperScript III kit (Invitrogen). cDNA was PCR amplified with a forward primer (5′-CTACCCAGGAAAAGCCAAC-3′) located in the leader sequence and a reverse primer (5′-TGAACCATAGTGTGCCATCT-3′) located in the body of the ORF8 mRNA. The PCR mixture (25 μl) contained cDNA, PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 2 mM MgCl2, 0.01% gelatin), 200 μM (each) dNTPs, and 1.0 U Taq polymerase (Applied Biosystems). The mixtures were amplified for 60 cycles of 94°C for 1 min, 50°C for 1 min, and 72°C for 1 min and a final extension at 72°C for 10 min in an automated thermal cycler (Applied Biosystems). RT-PCR products were subjected to agarose gel electrophoresis, gel purified using a QIAquick gel extraction kit (Qiagen), and sequenced to obtain the leader-body junction sequences for the ORF8 subgenomic mRNA.
Nucleotide sequence accession numbers.
The nucleotide and genome sequences of the CoVs detected in this study have been deposited in the GenBank sequence database under accession numbers KP886808, KP886809, and KP895482 to KP895525.
RESULTS
Detection of CoVs in bats.
A total of 348 alimentary samples from bats belonging to five different genera were obtained from various regions of Yunnan Province. RT-PCR for a 440-bp fragment of the RdRp gene of CoVs was positive in alimentary samples from 46 bats of five species belonging to four genera (Table 1; Fig. 1). Sequence analysis showed that 35 samples contained diverse alphacoronaviruses, while 11 samples contained betacoronaviruses, including two lineage B betacoronaviruses and nine lineage D betacoronaviruses.
TABLE 1.
Detection of CoVs in different bat species by RT-PCR of the 440-bp fragment of the RdRp gene
| Bat species | Common name | No. of bats tested | No. of bats positive for CoV | CoV detected or closest match in GenBank (no. of bats) | % Nucleotide identity to closest match | Sampling location of positive bats |
|---|---|---|---|---|---|---|
| Rhinolophus luctus | Woolly horseshoe bat | 32 | 0 | |||
| Rhinolophus affinis | Intermediate horseshoe bat | 22 | 0 | |||
| Rhinolophus ferrumequinum | Greater horseshoe bat | 11 | 2 | SARS-CoV (2) | 100 | Lufeng |
| Rhinolophus stheno | Lesser brown horseshoe bat | 34 | 1 | Rs-BatCoV HKU2 (1) | 92 | Mojiang |
| Hipposideros pomona | Pomona roundleaf bat | 17 | 2 | Hi-BatCoV HKU10 (2) | 81–87 | Mojiang |
| Myotis daubentonii | Daubenton's bat | 98 | 32 | My-BatCoV HKU6 (24) | 78–99 | Xiangyun |
| Rs-BatCoV HKU2 (1) | 93 | Mojiang | ||||
| Rs-BatCoV HKU2 (4) | 80–81 | Xiangyun | ||||
| Mi-BatCoV HKU7 (2) | 96 | Mojiang | ||||
| Mi-BatCoV HKU8 (1) | 96 | Mojiang | ||||
| Rousettus leschenaulti | Leschenault's rousette | 115 | 9 | Ro-BatCoV HKU9 (9) | 75–79 | Mengla |
| Unknown | 19 | 0 |
Detection of diverse bat alphacoronaviruses.
Phylogenetic analysis of the 440-bp fragments of the RdRp gene of alphacoronaviruses detected in 35 bat samples showed that two sequences from one Rhinolophus stheno bat and one Myotis daubentonii bat captured in Mojiang possessed 92 to 93% nucleotide identities to Rhinolophus bat CoV HKU2 (Rh-BatCoV HKU2) (GenBank accession no. NC_009988.1) (Table 1; Fig. 2). Four sequences from M. daubentonii in Xiangyun possessed 81% nucleotide identity to Rh-BatCoV HKU2 (GenBank accession no. NC_009988.1). Twenty-four sequences from M. daubentonii in Xiangyun possessed 78 to 99% nucleotide identities to Myotis bat CoV HKU6 (My-BatCoV HKU6) (GenBank accession no. DQ249224.1). Two sequences from M. daubentonii in Mojiang possessed 96% nucleotide identities to Miniopterus bat CoV HKU7 (Mi-BatCoV HKU7) (GenBank accession no. DQ249226.1). One sequence from M. daubentonii in Mojiang possessed 96% nucleotide identities to Miniopterus bat CoV HKU8 (GenBank accession no. NC_010438.1). Two sequences from Hipposideros pomona in Mojiang possessed 81 to 87% nucleotide identities to Hipposideros bat CoV HKU10 (Hi-BatCoV HKU10) (GenBank accession no. JQ989267.1).
FIG 2.
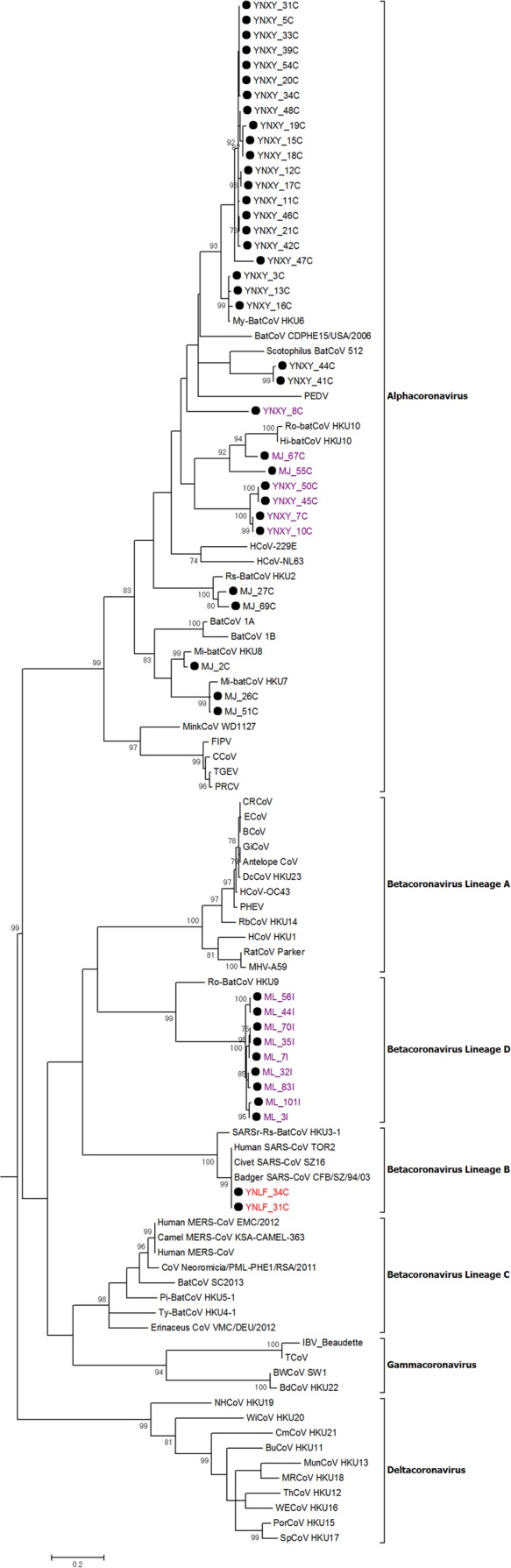
Phylogenetic analysis of the nucleotide sequences of the 267-nt fragment of RdRp gene regions of the 46 positive samples identified in bats in Yunnan in this study. The tree was constructed by the maximum likelihood method with the model GTR+G. Bootstrap values were calculated from 1,000 trees, and only values of >700 are shown and given at the nodes. The scale bar represents 5 nucleotide substitutions per site. The two SARSr-Rf-BatCoV strains, YNLF_31C and YNLF_34C, are in red. The potentially novel bat CoVs are in purple. AntelopeCoV, sable antelope coronavirus (EF424621); BatCoV CDPHE15/USA/2006, bat coronavirus CDPHE15/USA/2006 (NC_022103.1); BatCoV/SC2013, betacoronavirus/SC2013 (KJ473821.1); Erinaceus CoV/VMC/DEU/2012, betacoronavirus Erinaceus/VMC/DEU/2012 (NC_022643); BCoV, bovine coronavirus (NC_003045); BdHKU22, bottlenose dolphin coronavirus HKU22 (KF793826); BuCoV HKU11, bulbul coronavirus HKU11 (FJ376619); BWCoV SW1, beluga whale coronavirus SW1 (NC_010646); CCoV, canine coronavirus strain CCoV/NTU336/F/2008 (GQ477367.1); CRCoV, canine respiratory coronavirus strain K37 (JX860640.1); CmCoV HKU21, common moorhen coronavirus HKU21 (NC_016996); CoV Neoromicia/PML-PHE1/RSA/2011, coronavirus Neoromicia/PML-PHE1/RSA/2011 (KC869678); DcCoV HKU23, dromedary camel coronavirus HKU23 (KF906251); ECoV, equine coronavirus (NC_010327); FIPV, feline infectious peritonitis virus (AY994055); GiCoV, giraffe coronavirus US/OH3-TC/2006 (EF424622.1); HCoV-229E, human coronavirus 229E (NC_002645); HCoV-HKU1, human coronavirus HKU1 (NC_006577); HCoV-NL63, human coronavirus NL63 (NC_005831); HCoV-OC43, human coronavirus OC43(NC_005147); Hi-BatCoV HKU10, Hipposideros bat coronavirus HKU10 (JQ989269); IBV-beaudette, Beaudette coronavirus (AY692454); Human MERS-CoV, Middle East respiratory syndrome coronavirus (NC_019843.3); Human MERS-CoV EMC/2012, human betacoronavirus 2c_EMC/2012 (JX869059.2); Camel MERS-CoV KSA-CAMEL-363, Middle East respiratory syndrome coronavirus isolate KSA-CAMEL-363 (KJ713298); MRCoV HKU18, magpie robin coronavirus HKU18(NC_016993); BatCoV 1A, Miniopterus bat coronavirus 1A (NC_010437); BatCoV 1B, Miniopterus bat coronavirus 1B(NC_010436); Mi-BatCoV HKU7, Miniopterus bat coronavirus HKU7 (DQ249226); Mi-BatCoV HKU8, Miniopterus bat coronavirus HKU8 (NC_010438); Mink CoV strain WD1127, mink coronavirus strain WD1127 (NC_023760.1); MunCoV HKU13, munia coronavirus HKU13 (FJ376622); MHV-A59, murine hepatitis virus (NC_001846); My-BatCoV HKU6, Myotis bat coronavirus HKU6 (DQ249224); NH CoV HKU19, night heron coronavirus HKU19 (NC_016994); PEDV, porcine epidemic diarrhea virus (NC_003436); PHEV, porcine hemagglutinating encephalomyelitis virus (NC_007732); Pi-BatCoV-HKU5-1, Pipistrellus bat coronavirus HKU5 (NC_009020); PorCoV HKU15, porcine coronavirus HKU15 (NC_016990); PRCV, porcine respiratory coronavirus (DQ811787); RbCoV HKU14, rabbit coronavirus HKU14 (NC_017083); RatCoV parker, rat coronavirus Parker (NC_012936); Rs-BatCoV HKU2, Rhinolophus bat coronavirus HKU2 (EF203064); Ro-BatCoV-HKU9, Rousettus bat coronavirus HKU9 (NC_009021); Ro-BatCoV HKU10, Rousettus bat coronavirus HKU10 (JQ989270); Human SARS-CoV TOR2, SARS-related human coronavirus (NC_004718); Civet SARS-CoV SZ16, SARS-related palm civet coronavirus (AY304488); Badger SARS-CoV, SARS-related badger coronavirus CFB/SZ/94/03 (AY545919.1); SARSr-Rs-BatCoV HKU3, SARS-related Rhinolophus bat coronavirus HKU3 (DQ022305); Scotophilus BatCoV 512, Scotophilus bat coronavirus 512 (NC_009657); SpCoV HKU17, sparrow coronavirus HKU17 (NC_016992); TCoV, turkey coronavirus (NC_010800); TGEV, transmissible gastroenteritis virus (DQ443743); ThCoV HKU12, thrush coronavirus HKU12 (FJ376621); Ty-BatCoV-HKU4-1, Tylonycteris bat coronavirus HKU4 (NC_009019); WECoV HKU16, white-eye coronavirus HKU16 (NC_016991); WiCoV HKU20, widgeon coronavirus HKU20 (NC_016995).
Detection of lineage B and D bat betacoronaviruses.
Phylogenetic analysis of the 440-bp fragments of the RdRp gene of betacoronaviruses detected in two bat samples, YNLF_31C and YNLF_34C, showed that they belonged to Betacoronavirus lineage B, with 100% nucleotide identities to human SARS-CoV TOR2 (GenBank accession no. AY274119.3) and 90% nucleotide identities to SARSr-Rs-BatCoV HKU3 (GenBank accession no. DQ022305), thus representing SARSr-Rf-BatCoVs (Table 1; Fig. 2). Both samples were collected from greater horseshoe bats (Rhinolophus ferrumequinum) captured in Lufeng County, Chuxiong Yi Autonomous Prefecture (Fig. 1). Phylogenetic analysis of the 440-bp fragments of the RdRp gene of betacoronaviruses detected in nine other bat samples showed that they belonged to Betacoronavirus lineage D, with 75 to 79% nucleotide identities to Rousettus bat coronavirus HKU9 (Ro-BatCoV HKU9) (GenBank accession no. NC_009021.1). These nine samples were collected from Leschenault's rousettes (Rousettus leschenaulti) in Mengla County, Xishuangbanna Dai Autonomous Prefecture. Attempts to passage SARSr-Rs-BatCoV YNLF_31C and YNLF_34C in various cell lines were not successful, with no cytopathic effect or viral replication being detected.
Genome comparison between SARSr-Rf-BatCoV and other SARSr-CoVs.
The complete genome sequences of the two SARSr-Rf-BatCoV strains, YNLF_31C and YNLF_34C, were obtained by assembly of the sequences of RT-PCR products obtained directly from alimentary samples. Their genome sizes were 29,723 bases, with a G+C content of 40.7%, comparable to those of most other SARSr-CoVs (27, 28). They were closely related to each other, with 99.9% overall nucleotide identities, while they possessed 88.2% nucleotide identities to the genomes of SARSr-Rs-BatCoV HKU3 and 93% nucleotide identities to the genomes of human/civet SARSr-CoVs. SARSr-Rf-BatCoV strains share similar genome organization with other SARSr-CoV strains, containing the putative transcription regulatory sequence (TRS) motif, 5′-ACGAAC-3′, at the 3′ end of the 5′ leader sequence and preceding each ORF except ORF7b. Similar to most other SARSr-BatCoVs, SARSr-Rf-BatCoV strains YNLF_31C and YNLF_34C contained a single long ORF8.
The nsp3, S, ORF3, and ORF8 regions are known to be the most rapidly evolving regions among SARSr-CoV genomes (27, 28, 52, 53). Pairwise comparison of amino acid sequences between civet SARSr-CoV SZ3 and other SARSr-CoVs showed that the S and ORF3a of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C displayed relatively low sequence identities to civet SARSr-CoV (Table 2). However, the nsp3 protein of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C exhibited 97.1% amino acid identity to civet SARSr-CoV, which is comparable to the high sequence identity of 96.8 to 97.5% between civet SARSr-CoV and SARSr-BatCoVs Rs3367, RsSHC014, WIV1, and BtCoV-Cp/2011, reported previously from Yunnan (42). Furthermore, an exceptionally high sequence identity (80.4 to 81.3% amino acid identity) was observed in the ORF8 protein between SARSr-Rf-BatCoVs and human/civet SARSr-CoVs, much higher than that between human/civet SARSr-CoVs and other SARSr-BatCoVs (23.2 to 37.3% amino acid identity). Therefore, civet SARSr-CoV SZ3 was most closely related to SARSr-Rs-BatCoV Rs3367 and WIV1 in S and ORF3a but was most closely related to SARSr-Rf-BatCoVs in ORF8.
TABLE 2.
Percentage amino acid identities of the selected predicted gene products of SARSr-CoVs to civet SARSr-CoV strain SZ3
| CoV | % Amino acid identitya |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| nsp2 | nsp3 | nsp5 | nsp12 | S | ORF3 | E | M | ORF8 | N | |
| Civet SARSr-CoV civet007 | 99.5 | 99.5 | 100.0 | 99.7 | 98.6 | 98.1 | 100.0 | 100.0 | 98.3 | 99.5 |
| Civet SARSr-CoV SZ16 | 100.0 | 99.9 | 100.0 | 99.9 | 99.9 | 100.0 | 100.0 | 100.0 | 98.3 | 100.0 |
| Human SARS-CoV BJ01 | 99.8 | 99.6 | 100.0 | 99.9 | 98.8 | 98.1 | 100.0 | 99.5 | 38.2 | 100.0 |
| Human SARS-CoV GZ02 | 99.8 | 99.8 | 100.0 | 99.9 | 99.0 | 97.8 | 100.0 | 99.5 | 98.3 | 100.0 |
| Human SARS-CoV Tor2 | 99.8 | 99.6 | 100.0 | 99.9 | 98.6 | 98.1 | 100.0 | 99.5 | 37.3 | 100.0 |
| SARSr-Rs-BatCoV Rs3367 | 97.8 | 96.8 | 100.0 | 99.6 | 92.3 | 96.7 | 99.1 | 97.7 | 32.2 | 100.0 |
| SARSr-Rs-BatCoV RsSHC014 | 98.3 | 96.8 | 99.7 | 99.6 | 90.1 | 96.7 | 99.1 | 97.7 | 33.0 | 99.5 |
| SARSr-Rs-BatCoV WIV1 | 97.8 | 96.8 | 99.7 | 99.5 | 92.3 | 96.3 | 99.6 | 97.7 | 32.2 | 99.8 |
| SARSr-Rs-BatCoV HKU3-1 | 90.6 | 91.7 | 99.3 | 98.6 | 77.9 | 81.3 | 97.4 | 98.2 | 31.4 | 96.4 |
| SARSr-Rs-BatCoV HKU3-2 | 90.6 | 91.7 | 99.3 | 98.6 | 77.8 | 81.3 | 96.5 | 98.2 | 31.4 | 96.7 |
| SARSr-Rs-BatCoV HKU3-3 | 90.6 | 91.7 | 99.3 | 98.6 | 77.9 | 81.3 | 96.1 | 98.2 | 31.4 | 96.4 |
| SARSr-Rs-BatCoV HKU3-6 | 90.6 | 91.7 | 99.3 | 98.5 | 78.0 | 81.3 | 97.4 | 98.2 | 31.4 | 96.4 |
| SARSr-Rs-BatCoV HKU3-8 | 90.0 | 91.7 | 99.0 | 98.8 | 78.1 | 81.7 | 97.4 | 96.4 | 23.2 | 98.1 |
| SARSr-Rs-BatCoV HKU3-12 | 90.4 | 91.7 | 99.3 | 98.9 | 78.1 | 81.7 | 97.4 | 98.2 | 31.4 | 96.2 |
| SARSr-Rs-BatCoV HKU3-13 | 90.6 | 91.2 | 99.3 | 98.6 | 78.0 | 81.0 | 97.4 | 98.2 | 31.4 | 96.4 |
| SARSr-Rs-BatCoV Rs672/2006 | 98.3 | 87.1 | 99.3 | 99.7 | 78.0 | 89.4 | 98.7 | 98.2 | 32.2 | 98.6 |
| SARSr-Rb-BatCoV BM48-31/BGR | 70.8 | 75.9 | 94.4 | 97.7 | 74.8 | 69.4 | 96.5 | 89.4 | 87.2 | |
| SARSr-Rm-BatCoV 279/2005 | 89.6 | 90.3 | 99.7 | 99.1 | 78.6 | 83.2 | 97.4 | 96.8 | 31.7 | 96.9 |
| SARSr-Rm-BatCoV Rm1 | 89.5 | 90.0 | 99.3 | 92.4 | 78.7 | 83.2 | 97.8 | 96.8 | 33.0 | 97.4 |
| SARSr-Rp-BatCoV Rp3 | 96.7 | 95.1 | 99.7 | 92.8 | 78.4 | 83.2 | 99.6 | 96.8 | 33.0 | 97.9 |
| SARSr-Rp-BatCoV Rp/Shaanxi2011 | 93.6 | 93.0 | 100.0 | 92.3 | 79.0 | 82.1 | 90.0 | 96.4 | 33.0 | 97.9 |
| SARSr-Cp-BatCoV Cp/Yunnan2011 | 90.8 | 97.5 | 100.0 | 92.2 | 78.9 | 89.4 | 97.0 | 98.6 | 31.4 | 98.1 |
| SARSr-Rf-BatCoV Rf1 | 90.1 | 92.0 | 99.7 | 91.6 | 76.5 | 85.7 | 96.1 | 97.3 | 80.4 | 95.5 |
| SARSr-Rf-BatCoV 273/2005 | 89.8 | 92.3 | 99.7 | 98.4 | 76.6 | 85.7 | 98.7 | 97.3 | 80.4 | 96.2 |
| SARSr-Rf-BatCoV YNLF_31C | 95.0 | 97.1 | 99.7 | 99.5 | 77.3 | 86.8 | 97.4 | 98.2 | 81.3 | 98.1 |
| SARSr-Rf-BatCoV YNLF_34C | 95.0 | 97.1 | 99.7 | 99.0 | 77.3 | 86.8 | 99.1 | 98.2 | 81.3 | 97.9 |
The high amino acid identities in nsp3 and ORF8 between SARSr-Rf-BatCoVs and civet SARSr-CoV are in bold.
The predicted receptor binding domain (RBD) of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C possessed 89% and 68.1% amino acid identities to that of SARSr-Rs-BatCoV HKU3-1 and civet SARSr-CoV SZ3, respectively. Previous studies have identified five critical residues (residues 442, 472, 479, 487, and 491) for ACE2 binding in human and civet SARSr-CoVs (54). In particular, residues 479 and 487 are the two key residues that are different between human and civet SARSr-CoV strains, with an S→T substitution at residue 487 resulting in a 20-fold reduction in human ACE2 binding affinity (54). In SARSr-Rs-BatCoV Rs3367, two (residues 479 and 491) of the five critical residues were conserved. In SARSr-Rf-BatCoVs and most other SARSr-Rs-BatCoVs, only residue 491 was conserved (Fig. 3). Compared to human/civet SARSr-CoVs and SARSr-Rs-BatCoV Rs3367, WIV1, and RsSHC014, the RBD of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C, similar to some SARSr-BatCoV strains, contained two deletions of 5 and 12 amino acids (aa), respectively.
FIG 3.

Multiple alignment of the amino acid sequences of the receptor-binding motifs of the spike proteins of human and civet SARSr-CoV and the corresponding sequences of SARSr-BatCoVs in different Rhinolophus species. Asterisks indicate positions that have fully conserved residues. Amino acid deletions among some SARSr-BatCoVs are highlighted in yellow. The five critical residues for receptor binding in human SARS-CoV, at positions 442,472,479,487,491, are highlighted in pink.
Phylogenetic analysis.
Phylogenetic trees were constructed using nsp2, nsp3, nsp5, nsp12 (RdRp), S, ORF3a, ORF8, and N genes of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C and other SARSr-CoVs (Fig. 4). These regions were selected because they were commonly used in phylogenetic analysis of CoVs (RdRp, S, N), represent regions of rapid evolution in SARSr-CoVs (nsp3, ORF3, ORF8), or are free from recombination upon subsequent analysis (nsp2, nsp5). In nsp2, nsp3, nsp5, RdRp, and N genes, SARSr-Rf-BatCoV YNLF_31C and YNLF_34C were more closely related to other SARSr-BatCoVs than to two other SARSr-Rf-BatCoV strains, Rf1 and BtCoV/273/2005, previously detected from greater horseshoe bats in Hubei (28, 37). However, in S, ORF3, and ORF8 genes, SARSr-Rf-BatCoV YNLF_31C and YNLF_34C were most closely related to SARSr-Rf-BatCoV Rf1 and BtCoV/273/2005, forming a distinct cluster among other SARSr-BatCoVs.
FIG 4.

Phylogenetic analyses of nsp2, nsp3, nsp5, RdRp, S, ORF3, ORF8, and N gene nucleotide sequences of SARSr-BatCoVs from different bat species. The trees were constructed by the maximum likelihood method using GTR+G (A), GTR+G (B), GTR+G+I (C), TN93+G (D), GTR+G (E), TN93+G (F), T92 +G (G), and GTR+G (H) substitution models, and bootstrap values were calculated from 1,000 trees. Except for ORF3 and ORF8, all trees were rooted using the corresponding sequences of HCoV HKU1 (GenBank accession number NC_006577). Only bootstrap values of >70% are shown. Nucleotide positions 1736 (A), 5019 (B), 908 (C), 2777 (D), 3638 (E), 804 (F), 345 (G), and 1222 (H) were included in the analyses. The scale bars represent 50 (A), 10 (B), 20 (C), 20 (D), 10 (E), 20 (F), 10 (G), and 200 (H) substitutions per site. Human and civet SARSr-CoVs are in green, SARSr-Rs-BatCoVs from R. sinicus are in blue, and SARSr-Rs-BatCoVs from R. ferrumequinum are in red. The two SARSr-Rf-BatCoV strains YNLF_31C and YNLF_34C detected in this study are in bold.
In S and ORF3 regions, human/civet SARSr-CoVs were most closely related to SARSr-Rs-BatCoV Rs3367, WIV1, and RsSHC014 previously detected in Yunnan bats (42). This is in line with the ability of SARSr-Rs-BatCoV WIV1 to replicate in Vero E6 cells and to use ACE2 as a receptor (42). In the nsp3 region, human/civet SARSr-CoVs were most closely related to SARSr-Rf-BatCoV YNLF_31C and YNLF_34C as well as SARSr-Rs-BatCoV Rs3367, WIV1, and RsSHC014. Furthermore, in ORF8, SARSr-Rf-BatCoV strains were clustered with human and civet SARSr-CoV strains with a high bootstrap value of 990, whereas all SARSr-Rs-BatCoV strains, including Rs3367, WIV1, and RsSHC014, formed another cluster. This concurs with results from a pairwise amino acid sequence comparison and suggests that the ORF8 of civet and human SARSr-CoV originated from SARSr-Rf-BatCoVs from greater horseshoe bats instead of SARSr-Rs-BatCoV from Chinese horseshoe bats.
Recombination analysis.
Since the ORF8 of SARSr-Rf-BatCoVs showed high sequence identity to those of human/civet SARSr-CoVs, we hypothesize that the ancestor of civet SARSr-CoVs acquired its ORF8 from SARSr-Rf-BatCoVs through recombination between SARSr-Rf-BatCoVs from greater horseshoe bats and SARSr-Rs-BatCoVs from Chinese horseshoe bats. When civet SARSr-CoV SZ3 was used as the query for sliding window analysis with SARSr-Rf-BatCoV YNLF_31C and SARSr-Rs-BatCoV Rs3367 and HKU3 as potential parents, several recombination breakpoints were observed. In particular, two breakpoints, between which ORF8 was located, were identified (Fig. 5). Downstream to the first breakpoint at position 27128 and upstream to the second breakpoint at position 28635, an abrupt change in clustering occurred with high bootstrap support for clustering of civet SARSr-CoV SZ3 with SARSr-Rf-BatCoV YNLF_31C. This is in line with results from phylogenetic and similarity plot analyses. Moreover, using multiple alignments, civet SARSr-CoV SZ3 was shown to possess much higher sequence similarities to SARSr-Rf-BatCoVs than to SARSr-Rs-BatCoVs within ORF8, which includes the region corresponding to the 29-nt deletion found in human SARS-CoVs (Fig. 5).
FIG 5.


(A) Bootscan (upper panel) and Simplot (lower panel) analysis using the genome sequence of civet SARSr-CoV strain SZ03 as the query sequence. Bootscanning was conducted with Simplot version 3.5.1 (F84 model; window size, 1,000 bp; step, 200 bp) on a gapless nucleotide alignment, generated with ClustalX. The red line denotes SARSr-Rf-BatCoV strain YNLF_31C, the blue line denotes SARSr-Rs-BatCoV strain Rs3367, and the black line denotes SARSr-Rs-BatCoV strain HKU3-1. The ORF8 region with potential recombination is highlighted in yellow. (B) Multiple alignment of nucleotide sequences from genome positions 27000 to 28700. Bases conserved between civet SARSr-CoV SZ03 and SARSr-Rf-BatCoVs (strains YNLF_31C and Rf1) are marked in yellow boxes. Bases conserved between civet SARSr-CoV SZ03 and SARSr-Rs-BatCoVs (strains Rs3367 and HKU3-1) are marked in green boxes. The 29-nt deletion in human SARS coronavirus TOR2 is highlighted in orange. The start codon and stop codon of ORF8 are labeled with black boxes.
Besides ORF8, another region of interest was S, which was situated between two breakpoints at positions 20900 and 26100 (Fig. 5). Downstream to position 20900 and upstream to position 26100, an abrupt change in clustering occurred with high bootstrap support for clustering of civet SARSr-CoV SZ3 with SARSr-Rs-BatCoV Rs3367. This is in line with phylogenetic analysis and the ability of strain Rs3367 to use ACE2 as a receptor for cellular entry (42). However, similarity plot analysis still showed a substantial difference between the S of civet SARSr-CoV SZ3 and that of SARSr-Rs-BatCoV Rs3367, especially in the S1 region.
Estimation of synonymous and nonsynonymous substitution rates.
Using all available SARSr-BatCoV genome sequences for analysis, the Ka/Ks ratios for various coding regions, compared to those of civet SARSr-CoVs and human SARS-CoVs, were determined and are shown in Table 3. Notably, the Ka/Ks ratios for most coding regions of SARSr-BatCoVs, including ORF8 of SARS-Rf-BatCoVs, were low, supporting purifying selection. In contrast, many regions of civet SARSr-CoVs and human SARS-CoVs exhibited relatively high Ka/Ks ratios that are suggestive of positive selection. Positive selection was particularly strong at the S (Ka/Ks = 3) and ORF3 (Ka/Ks = 2) regions of civet SARSr-CoVs and the M (Ka/Ks = 2) and ORF8 (Ka/Ks = 3.5) regions of human SARS-CoVs.
TABLE 3.
Nonsynonymous and synonymous substitution rates in the coding regions of SARSr-CoVs among different hostsa
| Gene | SARSr-Rf-BatCoV (n = 4) |
SARSr-Rs-BatCoV (n = 17) |
Civet SARSr-CoV (n = 18)b |
Human SARS-CoV (n = 122)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | Ka | Ks | Ka/Ks | |
| nsp1 | 0.013 | 0.081 | 0.161 | 0.003 | 0.108 | 0.028 | 0.000 | 0.000 | 0.000 | 0.000 | ||
| nsp2 | 0.036 | 0.349 | 0.103 | 0.023 | 0.230 | 0.100 | 0.001 | 0.003 | 0.333 | 0.000 | 0.001 | 0.000 |
| nsp3 | 0.030 | 0.414 | 0.073 | 0.018 | 0.288 | 0.063 | 0.001 | 0.002 | 0.500 | 0.004 | 0.005 | 0.800 |
| nsp4 | 0.012 | 0.391 | 0.031 | 0.010 | 0.222 | 0.045 | 0.001 | 0.002 | 0.500 | 0.002 | 0.002 | 1.000 |
| nsp5 | 0.003 | 0.442 | 0.007 | 0.004 | 0.244 | 0.016 | 0.001 | 0.000 | 0.000 | 0.001 | 0.000 | |
| nsp6 | 0.009 | 0.331 | 0.027 | 0.005 | 0.178 | 0.028 | 0.000 | 0.002 | 0.000 | 0.002 | 0.001 | 2.000 |
| nsp7 | 0.018 | 0.549 | 0.033 | 0.000 | 0.181 | 0.000 | 0.002 | 0.000 | 0.000 | 0.001 | 0.000 | |
| nsp8 | 0.004 | 0.249 | 0.016 | 0.003 | 0.175 | 0.017 | 0.001 | 0.000 | 0.000 | 0.000 | ||
| nsp9 | 0.000 | 0.199 | 0.000 | 0.003 | 0.199 | 0.015 | 0.001 | 0.000 | 0.001 | 0.000 | ||
| nsp10 | 0.011 | 0.355 | 0.031 | 0.000 | 0.158 | 0.000 | 0.000 | 0.000 | 0.002 | 0.002 | 1.000 | |
| nsp12 | 0.038 | 0.109 | 0.349 | 0.026 | 0.076 | 0.342 | 0.000 | 0.003 | 0.000 | 0.001 | 0.001 | 1.000 |
| nsp13 | 0.002 | 0.347 | 0.006 | 0.002 | 0.199 | 0.010 | 0.000 | 0.003 | 0.000 | 0.001 | 0.001 | 1.000 |
| nsp14 | 0.006 | 0.485 | 0.012 | 0.005 | 0.270 | 0.019 | 0.001 | 0.003 | 0.333 | 0.001 | 0.001 | 1.000 |
| nsp15 | 0.016 | 0.452 | 0.035 | 0.012 | 0.275 | 0.044 | 0.000 | 0.000 | 0.000 | 0.001 | 0.000 | |
| nsp16 | 0.008 | 0.306 | 0.026 | 0.005 | 0.277 | 0.018 | 0.002 | 0.002 | 1.000 | 0.002 | 0.003 | 0.667 |
| S | 0.012 | 0.174 | 0.070 | 0.049 | 0.412 | 0.119 | 0.003 | 0.001 | 3.000 | 0.001 | 0.002 | 0.500 |
| ORF3 | 0.012 | 0.065 | 0.185 | 0.041 | 0.220 | 0.186 | 0.002 | 0.001 | 2.000 | 0.072 | 0.386 | 0.187 |
| E | 0.015 | 0.070 | 0.214 | 0.003 | 0.037 | 0.081 | 0.000 | 0.000 | 0.001 | 0.002 | 0.500 | |
| M | 0.003 | 0.096 | 0.313 | 0.007 | 0.097 | 0.072 | 0.001 | 0.002 | 0.500 | 0.002 | 0.001 | 2.000 |
| ORF8 | 0.021 | 0.110 | 0.190 | 0.035 | 0.197 | 0.178 | 0.004 | 0.000 | 0.007 | 0.002 | 3.500 | |
| N | 0.015 | 0.143 | 0.105 | 0.008 | 0.069 | 0.116 | 0.002 | 0.005 | 0.400 | 0.000 | 0.001 | 0.000 |
Ka/Ks ratios of ≥0.5 are in bold.
Only sequences without deletions were included in the analysis of the ORF8 gene for these CoVs.
Estimation of divergence dates.
Using the uncorrelated relaxed clock model on ORF1ab, the time of the most recent common ancestor (tMRCA) of all SARSr-CoVs was estimated to be 1960.1 (highest posterior density regions at 95% [HPDs], 1899.1 to 1988.6). The tMRCA of human and civet SARSr-CoVs was estimated to be 2001.5 (HPDs, 1999.1 to 2002.5), approximately 2 years before the SARS epidemic. The tMRCA of human/civet SARSr-CoVs, SARSr-Rp-BatCoV Rp3/2004, and SARSr-Rs-BatCoV RsSHC014/2011, Rs3367/2012, and WIV1/2012 was estimated to be 1995.3 (HPDs, 1984.5 to 2001), while that of human/civet SARSr-CoVs and SARSr-Rf-BatCoVs was estimated to be 1990.6 (HPDs, 1973.2 to 1999.6) (Fig. 6).
FIG 6.

Estimation of tMRCA of SARSr-CoVs based on ORF1ab (A) and nsp5 (B). The mean estimated dates are indicated. The taxa are labeled with their sampling dates.
Since some regions in ORF1ab may be involved in recombination (Fig. 5), nsp5, which was free from recombination, was also used for analysis and showed similar tree topology. Using the uncorrelated relaxed clock model on nsp5, the time of the most recent common ancestor (tMRCA) of all SARSr-CoVs was estimated to be 1961.5 (highest posterior density regions at 95% [HPDs], 1898.9 to 1991.5). The tMRCA of human and civet SARSr-CoVs was estimated to be 2000.7 (HPDs, 1996.7 to 2002.6), approximately 2 years before the SARS epidemic. The tMRCA of human/civet SARSr-CoVs, SARSr-Rp-BatCoV Rp3/2004, and SARSr-Rs-BatCoV RsSHC014/2011, Rs3367/2012, and WIV1/2012 was estimated to be 1996.3 (HPDs, 1985.2 to 2001.7), while that of human/civet SARSr-CoVs and SARSr-Rf-BatCoVs was estimated to be 1989.9 (HPDs, 1969.6 to 2000.3) (Fig. 6) The estimated mean substitution rates of the ORF1ab and nsp5 data set under the uncorrelated exponentially distributed relaxed clock model (UCED) were 2.00 ×10−3 and 1.36 ×10−3 substitutions per site per year, respectively, which are comparable to those of other CoVs and RNA viruses (55, 56).
Expression of ORF8 and determination of leader-body junction sequence.
CoVs are characterized by a unique mechanism of discontinuous transcription with the synthesis of a nested set of subgenomic mRNAs (1, 2). To determine if ORF8 is expressed in SARSr-Rf-BatCoV and the location of the leader and body TRS used for mRNA synthesis, the leader-body junction sites and flanking sequences of ORF8 subgenomic mRNA were determined. The obtained subgenomic mRNA sequence was aligned to the leader sequence, which confirmed the core sequence of the TRS motifs as 5′-ACGAAC-3′ (Fig. 7), as in other SARSr-CoVs. The leader TRS and the ORF8 subgenomic mRNA exactly matched each other. The SARSr-Rf-BatCoV leader was confirmed as the first 66 nt of the genome.
FIG 7.

SARSr-Rf-BatCoV YNLF_31C mRNA leader-body junction and flanking sequences. The subgenomic ORF8 mRNA sequences are shown in alignment with the leader and genomic sequences. The start codon AUG in subgenomic RNA is depicted in red. The putative TRS is depicted in boldface type and underlined. Identical bases between leader sequence and subgenomic mRNA sequence are in blue. Identical bases between genome and subgenomic mRNA sequences are in green.
DISCUSSION
The ORF8 of civet SARSr-CoV is likely to have been acquired from SARSr-Rf-BatCoVs in greater horseshoe bats (R. ferrumequinum) through recombination. In this study, two SARSr-Rf-BatCoV strains, YNLF_31C and YNLF_34C, were identified from greater horseshoe bats. Although their genomes possessed only 93% nucleotide identities to the genomes of human/civet SARSr-CoVs, which is lower than the 95% nucleotide identities between human/civet SARSr-CoV and SARSr-Rs-BatCoV strains Rs3367 and RsSHC014, from Chinese horseshoe bats in Yunnan, the nsp3 and ORF8 protein of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C exhibited the highest amino acid identities among all SARSr-BatCoVs to that of civet SARSr-CoV SZ3. In particular, their ORF8 proteins demonstrated much higher amino acid identities (81.3%) to ORF8 protein of civet SARSr-CoV SZ3 than of SARSr-BatCoVs from other horseshoe bats (23.2% to 37.3%). Phylogenetic analysis of the ORF8 revealed a distinct clade formed by human/civet SARSr-CoVs and SARSr-Rf-BatCoVs separate from other SARSr-BatCoVs. This is in line with a previous report showing that the ORF8 of SARSr-Rf-BatCoV Rf1 was clustered with human/civet SARSr-CoVs but not SARSr-BatCoV Rm1 and Rp3 upon phylogenetic analysis, although only one SARSr-Rf-BatCoV strain was available for analysis (28). Moreover, potential recombination sites were identified between SARSr-Rf-BatCoVs and SARSr-Rs-BatCoVs around the ORF8 region, leading to the generation of civet SARSr-CoV SZ3 with the ORF8 acquired from SARSr-Rf-BatCoVs. Similar to other regions of the genome, the ORF8 region of SARSr-Rf-BatCoVs has been under purifying selection, which supports greater horseshoe bats as a reservoir for SARSr-Rf-BatCoVs. In contrast, the ORF8 of human SARS-CoVs was under strong positive selection, which reflects the rapid evolution soon after interspecies jumping. These findings support that recombination is the key mechanism involved in the acquisition of ORF8 by the ancestor of civet SARSr-CoVs. In fact, previous studies have demonstrated frequent recombination events between SARSr-Rs-BatCoV strains from different bat species of different geographical locations in China (22, 55). Moreover, a recombination breakpoint at the nsp16-S gene intergenic region was detected between SARSr-Rp-BatCoV Rp3 from Pearson's horseshoe bats (Rhinolophus pearsoni) and SARSr-Rf-BatCoV Rf1 during the evolution of SARSr-BatCoVs to civet SARSr-CoV (22). On the other hand, some genomic regions of SARSr-Rf-BatCoV YNLF_31C and YNLF_34C, such as those of the nsp3, RdRp, and N genes, were evolutionarily distinct from two previously reported SARSr-Rf-BatCoV strains, Rf1 and 273/2005, upon phylogenetic analysis. This suggests that SARSr-Rf-BatCoVs from different geographical locations in China may have evolved separately through other recombination events. The present findings offer new insights into the origin and evolution of SARS-CoV by showing that the ancestor of civet SARSr-CoV is a likely recombinant virus, with ORF8 originating from SARSr-Rf-BatCoVs in greater horseshoe bats and other genome regions originating from different horseshoe bats.
Although SARSr-Rs-BatCoV Rs3367 and RsSHC014 represented the closest bat CoVs to SARS-CoV in terms of genome identity, they were unlikely to be the immediate ancestor of civet SARSr-CoVs. Previous molecular dating studies estimated that the time of divergence between human/civet and bat SARSr-CoVs ranged from 4 to 17 years before the SARS epidemic (22, 55, 57). SARSr-CoVs were also shown to be a newly emerged subgroup of Betacoronavirus, with the median date of their MRCA estimated to be from 1961 to 1982 (55, 57). The present results are in line with such estimations, with the tMRCA between human/civet and the closest bat strains estimated to be approximately 1995 (8 years before the SARS epidemic) and that among all SARSr-CoVs estimated to be approximately 1960, using ORF1ab. Similar results were also obtained when using the nsp5 region, which was recombination-free. Moreover, we demonstrated that SARSs-Rf-BatCoV YNLF_31C and YNLF_34C diverged from civet/human SARSr-CoVs only around approximately 1990. This is in contrast to previous studies that showed SARSr-Rp-BatCoV Rp3 as the only recently diverged strain (55, 57). Together with the evidence on the acquisition of ORF8, it is likely that civet SARSr-CoV originated from recombination between SARS-Rs-BatCoVs and SARS-Rf-BatCoVs from different horseshoe bat species within several years before the SARS epidemic.
The overlapping habitats and geographical distributions of different horseshoe bats may have fostered recombination between different SARSr-BatCoVs and the emergence of SARS-CoV. Chinese horseshoe bats are widely distributed throughout China, including in Yunnan, Guangdong, and Hong Kong. While greater horseshoe bats are also widely distributed across different provinces in China, including Yunnan, they are not found in Guangdong (58). The two bat species share similar diet and habits, such as the ability to roost in man-made structures, suggesting that they may cohabit in similar environments in Yunnan, the province with the highest biodiversity in China. In fact, SARSr-Rf-BatCoV YNLF_31C and YNLF_34C and SARSr-Rs-BatCoV Rs3367 and RsSHC014 were detected in Lufeng and Kunming of Yunnan Province, respectively, which are only ∼80 km apart and within the migration distances of horseshoe bats (Fig. 1) (22, 59, 60). Since greater horseshoe bats are not found in Guangdong, recombination between SARSr-Rf-BatCoVs and SARS-Rs-BatCoVs with the generation of the ancestor of civet SARSr-CoVs may have occurred in yet unidentified bats in Yunnan or nearby provinces, which were then transported to wildlife markets in Guangdong and infected civets. Alternatively, recombination may have occurred in civets or other animals within wildlife farms or markets, where many different wild animal species are often housed together (61). A possible scenario is that the animals were coinfected with SARSr-Rf-BatCoVs and SARSr-Rs-BatCoVs from different horseshoe bats, which was followed by recombination events. More extensive surveillance in bats from Yunnan and neighboring provinces, as well as wildlife markets in Guangdong, may reveal the immediate ancestor of civet SARSr-CoVs.
The ORF8 region, unique to SARSr-CoVs, is prone to mutations or deletions during interspecies transmission. One of the most striking genomic changes observed in SARS-CoV soon after its zoonotic transmission to humans was the acquisition of a characteristic 29-nt deletion which splits ORF8 into two ORFs, ORF8a and ORF8b (25, 62). While SARS-CoVs isolated from the later human cases of the epidemic contained this 29-nt deletion, isolates from civets and some early human cases possessed a single continuous ORF8 (25, 63). Besides, some early human strains and a farmed civet strain from Hubei possessed an alternative 82-nt deletion in ORF8 (63). On the other hand, four late human isolates possessed a 415-nt deletion, resulting in the loss of the entire ORF8 (63). Although studies using reverse genetics showed that the ORF8 is not essential for virus replication in vitro and in vivo (64, 65), the full-length 8ab protein is a functional protein that is delivered by a cleavable signal sequence to the lumen of the endoplasmic reticulum, where it becomes N-glycosylated (62). Different subcellular localizations and functions have also been reported for 8ab, 8a, and 8b proteins (66–69). Inside the endoplasmic reticulum, 8ab activates the ATF6 branch of unfolded-protein response (70). The 8a protein enhances SARS-CoV replication and induces caspase-dependent apoptosis through a mitochondrion-dependent pathway (66). Moreover, antibodies against 8a protein have been detected in the sera of SARS patients (66). The 8b protein downregulates the expression of the E protein, which supports a modulatory role in viral replication (68). Moreover, overexpression of the 8b protein induces DNA synthesis (67). The 8b and 8ab proteins also play a role in the host ubiquitin-proteasome system (71). In this study, the expression of ORF8 subgenomic mRNA in SARSr-Rf-BatCoV YNLF_31C suggested that the ORF8 protein may also be functional in SARSr-BatCoVs. Moreover, the high Ka/Ks ratio among human SARS-CoVs compared to SARSr-BatCoVs supports that ORF8 is subject to rapid evolution under strong positive selection during animal-to-human transmission. Further studies may help clarify the importance of ORF8 evolution for interspecies transmission of SARSr-CoVs.
Besides SARSr-BatCoVs, diverse alphacoronaviruses and betacoronaviruses, including potentially novel CoVs, with potential interspecies transmission events were identified in this study. Bats are known important reservoirs of lineage B, C, and D betacoronaviruses, while rodents are likely the reservoir of lineage A betacoronaviruses (30). Nine samples belonging to lineage D betacoronaviruses were detected in Leschenault's rousettes (R. leschenaulti), a known reservoir of Ro-BatCoV HKU9 (24). However, the partial RdRp sequences of these nine samples possessed only 75 to 79% nucleotide identities to sequences of Ro-BatCoV HKU9, suggesting that they may represent either novel CoV species or a novel genotype of Ro-BatCoV HKU9. As for alphacoronaviruses, 24 samples from Daubenton's bats (M. daubentonii) contained viruses most closely related to My-BatCoV HKU6, with 78 to 99% nucleotide identities in the partial RdRp region, which may represent My-BatCoV HKU6 or related viruses previously reported for the same bat species (38). Six samples contained alphacoronaviruses most closely related to Rh-BatCoV HKU2. However, four samples (YNXY_7C, YNXY_10C, YNXY_45, and YNXY_50C) from Daubenton's bats possessed partial RdRp sequences of only 80 to 80% nucleotide identities to that of Rh-BatCoV HKU2, suggesting that they may represent novel CoVs. Although the other two samples (MJ_27C and MJ_69C) possessed RdRp sequences with 92 to 93% identities to that of Rh-BatCoV HKU2, they were detected from Daubenton's bats and lesser brown horseshoe bats (R. stheno) instead of Chinese horseshoe bats (R. sinicus), which were previously reported to carry Rh-BatCoV HKU2 (34). This may suggest interspecies transmission of Rh-BatCoV HKU2 among different bat species. Two samples from Pomona roundleaf bats (Hipposideros pomona) contained alphacoronaviruses most closely related to Hi-BatCoV HKU10. However, the partial RdRp sequences possessed only 81 to 87% nucleotide identity to the latter. We have previously described recent interspecies transmission of BatCoV HKU10 between Leschenault's rousettes (R. leschenaulti) and Pomona roundleaf bats, two very different bats belonging to different families, through the rapid evolution of the S protein (72). Further studies are warranted to determine if the two samples from Pomona roundleaf bats contained potentially novel CoVs that are closely related to BatCoV HKU10 or variants of BatCoV HKU10 due to interspecies transmission.
ACKNOWLEDGMENTS
We thank Wing-Man Ko, Secretary for Food and Health Bureau, and Constance Hon-Yee Chan, Director of Health, for facilitation and support. We are grateful to the generous support of Carol Yu, Richard Yu, Hui Hoy, and Hui Ming in the genomic sequencing platform.
This work was partly supported by the Health and Medical Research Fund of the Food and Health Bureau of HKSAR (13121102); the Theme-Based Research Scheme and Research Grant Council Grant, University Grant Council; Croucher Senior Medical Research Fellowships, Committee for Research and Conference Grant, Strategic Research Theme Fund, University Development Fund and Special Research Achievement Award, The University of Hong Kong; and the Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease for the HKSAR Department of Health.
REFERENCES
- 1.Brian DA, Baric RS. 2005. Coronavirus genome structure and replication. Curr Top Microbiol Immunol 287:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai MM, Cavanagh D. 1997. The molecular biology of coronaviruses. Adv Virus Res 48:1–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya A, Holmes KV, Perlman S, Poon L, Rottier PJ, Talbot PJ, Woo PC, Ziebuhr J. 2011. Coronaviridae, p 806–828. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy, classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses, International Union of Microbiological Societies, Virology Division. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 4.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol 86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Wong BH, Gao K, Tsoi HW, Huang Y, Li KS, Lam CS, Chan KH, Zheng BJ, Yuen KY. 2007. Comparative analysis of 12 genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol 81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorbalenya AE, Snijder EJ, Spaan WJ. 2004. Severe acute respiratory syndrome coronavirus phylogeny: towards consensus. J Virol 78:7863–7866. doi: 10.1128/JVI.78.15.7863-7866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguière AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Müller S, Rickerts V, Stürmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 8.Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC, Simon JH, Osterhaus AD. 2004. A previously undescribed coronavirus associated with respiratory disease in humans. Proc Natl Acad Sci U S A 101:6212–6216. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ, SARS Working Group. 2003. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 10.Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. 2004. Identification of a new human coronavirus. Nat Med 10:368–373. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo PC, Lau SK, Tsoi HW, Huang Y, Poon RW, Chu CM, Lee RA, Luk WK, Wong GK, Wong BH, Cheng VC, Tang BS, Wu AK, Yung RW, Chen H, Guan Y, Chan KH, Yuen KY. 2005. Clinical and molecular epidemiological features of coronavirus HKU1-associated community-acquired pneumonia. J Infect Dis 192:1898–1907. doi: 10.1086/497151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 15.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e004773–12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish ZA, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr J. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J Virol 87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J Virol 72:4508–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Yuen KY. 2006. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80:7136–7145. doi: 10.1128/JVI.00509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keck JG, Matsushima GK, Makino S, Fleming JO, Vannier DM, Stohlman SA, Lai MM. 1988. In vivo RNA-RNA recombination of coronavirus in mouse brain. J Virol 62:1810–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kottier SA, Cavanagh D, Britton P. 1995. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology 213:569–580. doi: 10.1006/viro.1995.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lau SK, Woo PC, Yip CC, Fan RY, Huang Y, Wang M, Guo R, Lam CS, Tsang AK, Lai KK, Chan KH, Che XY, Zheng BJ, Yuen KY. 2012. Isolation and characterization of a novel Betacoronavirus subgroup A coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J Virol 86:5481–5496. doi: 10.1128/JVI.06927-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau SK, Li KS, Huang Y, Shek CT, Tse H, Wang M, Choi GK, Xu H, Lam CS, Guo R, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J Virol 84:2808–2819. doi: 10.1128/JVI.02219-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau SK, Lee P, Tsang AK, Yip CC, Tse H, Lee RA, So LY, Lau YL, Chan KH, Woo PC, Yuen KY. 2011. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol 85:11325–11337. doi: 10.1128/JVI.05512-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, Guo R, Li KS, Gao K, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2010. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol 84:11385–11394. doi: 10.1128/JVI.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 26.Yang ZY, Werner HC, Kong WP, Leung K, Traggiai E, Lanzavecchia A, Nabel GJ. 2005. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci U S A 102:797–801. doi: 10.1073/pnas.0409065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A 102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 29.Woo PC, Lau SK, Li KS, Poon RW, Wong BH, Tsoi HW, Yip BC, Huang Y, Chan KH, Yuen KY. 2006. Molecular diversity of coronaviruses in bats. Virology 351:180–187. doi: 10.1016/j.virol.2006.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau SK, Woo PC, Li KS, Tsang AK, Fan RY, Luk HK, Cai JP, Chan KH, Zheng BJ, Wang M, Yuen KY. 2015. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports murine origin of Betacoronavirus 1 with implications on the ancestor of Betacoronavirus lineage A. J Virol 89:3076–3092. doi: 10.1128/JVI.02420-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandão PE, Scheffer K, Villarreal LY, Achkar S, Oliveira RDN, Fahl WDO, Castilho JG, Kotait I, Richtzenhain LJ. 2008. A coronavirus detected in the vampire bat Desmodus rotundus. Braz J Infect Dis 12:466–468. doi: 10.1590/S1413-86702008000600003. [DOI] [PubMed] [Google Scholar]
- 32.Dominguez SR, O'Shea TJ, Oko LM, Holmes KV. 2007. Detection of group 1 coronaviruses in bats in North America. Emerg Infect Dis 13:1295–1300. doi: 10.3201/eid1309.070491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gloza-Rausch F, Ipsen A, Seebens A, Gottsche M, Panning M, Felix Drexler J, Petersen N, Annan A, Grywna K, Muller M, Pfefferle S, Drosten C. 2008. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis 14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, Xu H, Guo R, Chan KH, Zheng BJ, Yuen KY. 2007. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology 367:428–439. doi: 10.1016/j.virol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pfefferle S, Oppong S, Drexler JF, Gloza-Rausch F, Ipsen A, Seebens A, Muller MA, Annan A, Vallo P, Adu-Sarkodie Y, Kruppa TF, Drosten C. 2009. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg Infect Dis 15:1377–1384. doi: 10.3201/eid1509.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poon LL, Chu DK, Chan KH, Wong OK, Ellis TM, Leung YH, Lau SK, Woo PC, Suen KY, Yuen KY, Guan Y, Peiris JS. 2005. Identification of a novel coronavirus in bats. J Virol 79:2001–2009. doi: 10.1128/JVI.79.4.2001-2009.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang XC, Zhang JX, Zhang SY, Wang P, Fan XH, Li LF, Li G, Dong BQ, Liu W, Cheung CL, Xu KM, Song WJ, Vijaykrishna D, Poon LL, Peiris JS, Smith GJ, Chen H, Guan Y. 2006. Prevalence and genetic diversity of coronaviruses in bats from China. J Virol 80:7481–7490. doi: 10.1128/JVI.00697-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He B, Zhang Y, Xu L, Yang W, Yang F, Feng Y, Xia L, Zhou J, Zhen W, Feng Y, Guo H, Zhang H, Tu C. 2014. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J Virol 88:7070–7082. doi: 10.1128/JVI.00631-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reusken CB, Haagmans BL, Muller MA, Gutierrez C, Godeke GJ, Meyer B, Muth D, Raj VS, Smits-De Vries L, Corman VM, Drexler JF, Smits SL, El Tahir YE, De Sousa R, van Beek J, Nowotny N, van Maanen K, Hidalgo-Hermoso E, Bosch BJ, Rottier P, Osterhaus A, Gortazar-Schmidt C, Drosten C, Koopmans MP. 2013. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect Dis 13:859–866. doi: 10.1016/S1473-3099(13)70164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haagmans BL, Al Dhahiry SH, Reusken CB, Raj VS, Galiano M, Myers R, Godeke GJ, Jonges M, Farag E, Diab A, Ghobashy H, Alhajri F, Al-Thani M, Al-Marri SA, Al Romaihi HE, Al Khal A, Bermingham A, Osterhaus AD, AlHajri MM, Koopmans MP. 2014. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect Dis 14:140–145. doi: 10.1016/S1473-3099(13)70690-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan JF, Lau SK, To KK, Cheng VC, Woo PC, Yuen KY. 2015. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin Microbiol Rev 28:465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, Wu Z, Ren X, Yang F, He G, Zhang J, Dong J, Sun L, Zhu Y, Du J, Zhang S, Jin Q. 2013. Novel SARS-like betacoronaviruses in bats, China, 2011. Emerg Infect Dis 19:989–991. doi: 10.3201/eid1906.121648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwand B, Breiman RF, Anderson LJ, Rupprecht CE. 2009. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis 15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quan PL, Firth C, Street C, Henriquez JA, Petrosov A, Tashmukhamedova A, Hutchison SK, Egholm M, Osinubi MO, Niezgoda M, Ogunkoya AB, Briese T, Rupprecht CE, Lipkin WI. 2010. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio 1(4):e00208–10. doi: 10.1128/mBio.00208-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, Ksiazek T. 2001. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg Infect Dis 7:439–441. doi: 10.3201/eid0703.017312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lau SK, Li KS, Tsang AK, Lam CS, Ahmed S, Chen H, Chan KH, Woo PC, Yuen KY. 2013. Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J Virol 87:8638–8650. doi: 10.1128/JVI.01055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Y, Lau SK, Woo PC, Yuen KY. 2008. CoVDB: a comprehensive database for comparative analysis of coronavirus genes and genomes. Nucleic Acids Res 36:D504–D511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pyrc K, Jebbink MF, Berkhout B, van der Hoek L. 2004. Genome structure and transcriptional regulation of human coronavirus NL63. Virol J 1:7. doi: 10.1186/1743-422X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ren W, Li W, Yu M, Hao P, Zhang Y, Zhou P, Zhang S, Zhao G, Zhong Y, Wang S, Wang LF, Shi Z. 2006. Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis. J Gen Virol 87:3355–3359. doi: 10.1099/vir.0.82220-0. [DOI] [PubMed] [Google Scholar]
- 53.Song HD, Tu CC, Zhang GW, Wang SY, Zheng K, Lei LC, Chen QX, Gao YW, Zhou HQ, Xiang H, Zheng HJ, Chern SW, Cheng F, Pan CM, Xuan H, Chen SJ, Luo HM, Zhou DH, Liu YF, He JF, Qin PZ, Li LH, Ren YQ, Liang WJ, Yu YD, Anderson L, Wang M, Xu RH, Wu XW, Zheng HY, Chen JD, Liang G, Gao Y, Liao M, Fang L, Jiang LY, Li H, Chen F, Di B, He LJ, Lin JY, Tong S, Kong X, Du L, Hao P, Tang H, Bernini A, Yu XJ, Spiga O, Guo ZM, Pan HY, He WZ, Manuguerra JC, Fontanet A, Danchin A, Niccolai N, Li YX, Wu CI, Zhao GP. 2005. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci U S A 102:2430–2435. doi: 10.1073/pnas.0409608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, Wong SK, Huang IC, Xu K, Vasilieva N, Murakami A, He Y, Marasco WA, Guan Y, Choe H, Farzan M. 2005. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J 24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hon CC, Lam TY, Shi ZL, Drummond AJ, Yip CW, Zeng F, Lam PY, Leung FC. 2008. Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus. J Virol 82:1819–1826. doi: 10.1128/JVI.01926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jenkins GM, Rambaut A, Pybus OG, Holmes EC. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J Mol Evol 54:156–165. doi: 10.1007/s00239-001-0064-3. [DOI] [PubMed] [Google Scholar]
- 57.Vijaykrishna D, Smith GJ, Zhang JX, Peiris JS, Chen H, Guan Y. 2007. Evolutionary insights into the ecology of coronaviruses. J Virol 81:4012–4020. doi: 10.1128/JVI.02605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flanders J, Wei L, Rossiter SJ, Zhang S. 2011. Identifying the effects of the greater horseshoe bat, Rhinolophus ferrumequinum, in East Asia using ecological niche modelling and phylogenetic analyses. J Biogeogr 38:439–452. doi: 10.1111/j.1365-2699.2010.02411.x. [DOI] [Google Scholar]
- 59.Neuweiler G. 2000. The biology of bats, p 266 Oxford University Press, New York, NY. [Google Scholar]
- 60.Nowak RM, Paradiso JL. 1983. Walker's mammals of the world, p 230 The Johns Hopkins University Press, Baltimore, MD. [Google Scholar]
- 61.Woo PC, Lau SK, Yuen KY. 2006. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr Opin Infect Dis 19:401–407. doi: 10.1097/01.qco.0000244043.08264.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oostra M, de Haan CA, Rottier PJ. 2007. The 29-nucleotide deletion present in human but not in animal severe acute respiratory syndrome coronaviruses disrupts the functional expression of open reading frame 8. J Virol 81:13876–13888. doi: 10.1128/JVI.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chinese SARS Molecular Epidemiology Consortium. 2004. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 64.Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, Denison MR, Davis N, Baric RS. 2005. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol 79:14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dediego ML, Pewe L, Alvarez E, Rejas MT, Perlman S, Enjuanes L. 2008. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology 376:379–389. doi: 10.1016/j.virol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen CY, Ping YH, Lee HC, Chen KH, Lee YM, Chan YJ, Lien TC, Jap TS, Lin CH, Kao LS, Chen YM. 2007. Open reading frame 8a of the human severe acute respiratory syndrome coronavirus not only promotes viral replication but also induces apoptosis. J Infect Dis 196:405–415. doi: 10.1086/519166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Law PY, Liu YM, Geng H, Kwan KH, Waye MM, Ho YY. 2006. Expression and functional characterization of the putative protein 8b of the severe acute respiratory syndrome-associated coronavirus. FEBS Lett 580:3643–3648. doi: 10.1016/j.febslet.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keng CT, Choi YW, Welkers MR, Chan DZ, Shen S, Gee Lim S, Hong W, Tan YJ. 2006. The human severe acute respiratory syndrome coronavirus (SARS-CoV) 8b protein is distinct from its counterpart in animal SARS-CoV and down-regulates the expression of the envelope protein in infected cells. Virology 354:132–142. doi: 10.1016/j.virol.2006.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. 2014. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res 109:97–109. doi: 10.1016/j.antiviral.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sung SC, Chao CY, Jeng KS, Yang JY, Lai MM. 2009. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 387:402–413. doi: 10.1016/j.virol.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le TM, Wong HH, Tay FP, Fang S, Keng CT, Tan YJ, Liu DX. 2007. Expression, post-translational modification and biochemical characterization of proteins encoded by subgenomic mRNA8 of the severe acute respiratory syndrome coronavirus. FEBS J 274:4211–4222. doi: 10.1111/j.1742-4658.2007.05947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lau SK, Li KS, Tsang AK, Shek CT, Wang M, Choi GK, Guo R, Wong BH, Poon RW, Lam CS, Wang SY, Fan RY, Chan KH, Zheng BJ, Woo PC, Yuen KY. 2012. Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's rousettes to pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J Virol 86:11906–11918. doi: 10.1128/JVI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]