

Background: Finerenone is a novel nonsteroidal mineralocorticoid antagonist, currently in clinical phase IIb trials.
Results: Finerenone delays mineralocorticoid receptor nuclear import and inhibits its binding and transcriptional coactivator recruitment onto target gene promoters.
Conclusion: Finerenone impedes three critical steps of the mineralocorticoid receptor signaling pathway.
Significance: Finerenone, which behaves differently from currently available mineralocorticoid antagonists, is potentially a promising molecule to treat cardiorenal diseases.
Keywords: aldosterone, cardiovascular disease, drug action, heart, inhibition mechanism, inhibitor, mineralocorticoid receptor, steroid hormone receptor, antagonist, mechanism of action
Abstract
Aldosterone regulates sodium homeostasis by activating the mineralocorticoid receptor (MR), a member of the nuclear receptor superfamily. Hyperaldosteronism leads todeleterious effects on the kidney, blood vessels, and heart. Although steroidal antagonists such as spironolactone and eplerenone are clinically useful for the treatment of cardiovascular diseases, they are associated with several side effects. Finerenone, a novel nonsteroidal MR antagonist, is presently being evaluated in two clinical phase IIb trials. Here, we characterized the molecular mechanisms of action of finerenone and spironolactone at several key steps of the MR signaling pathway. Molecular modeling and mutagenesis approaches allowed identification of Ser-810 and Ala-773 as key residues for the high MR selectivity of finerenone. Moreover, we showed that, in contrast to spironolactone, which activates the S810L mutant MR responsible for a severe form of early onset hypertension, finerenone displays strict antagonistic properties. Aldosterone-dependent phosphorylation and degradation of MR are inhibited by both finerenone and spironolactone. However, automated quantification of MR subcellular distribution demonstrated that finerenone delays aldosterone-induced nuclear accumulation of MR more efficiently than spironolactone. Finally, chromatin immunoprecipitation assays revealed that, as opposed to spironolactone, finerenone inhibits MR, steroid receptor coactivator-1, and RNA polymerase II binding at the regulatory sequence of the SCNN1A gene and also remarkably reduces basal MR and steroid receptor coactivator-1 recruitment, unraveling a specific and unrecognized inactivating mechanism on MR signaling. Overall, our data demonstrate that the highly potent and selective MR antagonist finerenone specifically impairs several critical steps of the MR signaling pathway and therefore represents a promising new generation MR antagonist.
Introduction
The mineralocorticoid receptor (MR)5 and its physiological ligand aldosterone are main effectors of the renin-angiotensin-aldosterone system (1, 2). MR, a ligand-induced transcription factor belonging to the nuclear receptor family, plays a critical physiological role in water-electrolyte homeostasis and regulation of blood pressure. However, the past two decades have brought compelling evidence that pathophysiological overactivation of MR signaling may also induce disorders like heart failure and chronic kidney disease (3, 4). All of these new insights have spurred intense investigative interest in the use of MR antagonists as a new therapeutic strategy in controlling blood pressure, chronic renal disease progression, and cardiovascular remodeling and morbidity (5). Spironolactone is known as the first generation of potent MR antagonists (6). It has been shown to enhance survival in patients with heart failure and to prevent the development of cardiac fibrosis (7). Although highly efficient, this molecule lacks selectivity because it is able to antagonize androgen receptor (8) and to act as a progesterone receptor agonist (9). Consequently, prolonged use of spironolactone is associated with gynecological side effects such as gynecomastia and menstrual irregularities (10). More importantly, another known side effect of spironolactone administration is the induction of potentially life-threatening hyperkalemia in patients with diminished kidney function (11). Therefore, considerable efforts were made to develop a next generation of nonsteroidal antagonists that would overcome the inherent limitation of steroidal antagonists by combining the high potency of spironolactone with better selectivity and less adverse secondary effects (12). Recently, a novel nonsteroidal MR antagonist, finerenone, has been developed that combines potency and high selectivity and demonstrated a more efficient protection from cardiorenal injuries in comparison with steroidal antagonists at equinatriuretic doses in preclinical models (13, 14). These remarkable properties, together with the nonsteroidal nature of finerenone, raise the question of the mechanisms by which this molecule interferes with aldosterone in the MR signaling.
Aldosterone binding to MR, a ligand-induced transcription factor, promotes a receptor conformational change that allows dissociation from chaperone heterocomplexes and a critical hyperphosphorylation that are associated with a rapid translocation into the nuclear compartment. Once in the nucleus, MR binds as a dimer to hormone response elements and recruits transcriptional coregulators, allowing the transcription or repression of its target genes (15). Modulation of MR activity upon antagonist binding may thus occur at different steps of the receptor signaling pathway such as at nucleocytoplasmic shuttling (16, 17), its post-translational modifications (phosphorylation, ubiquitinylation), and its interaction with the transcriptional machinery (18). In the present study, we investigated the molecular mechanism of action of finerenone by using structural, biochemical, and cellular approaches and highlighted that finerenone impairs MR signaling at the levels of nucleocytoplasmic shuttling, post-translational modification, turnover, and coregulator recruitment.
Experimental Procedures
Compounds
Finerenone was synthesized as previously described (13). Aldosterone and spironolactone were purchased from Sigma-Aldrich. 18-Oxo-18-vinylprogesterone was a gift from A. Marquet (19).
Synthesis
A mixture of 3.83 mg (10.1 μmol) finerenone and 9.39 mg of (1,5-cyclooctadien)-(pyridine)-(tricyclohexylphosphine)-iridium(I)-hexafluorophosphate dissolved in 1.0 ml of dichloromethane was stirred with tritium gas at a partial pressure of ∼80.6 kPa (806 mbar) for 60 min in a reaction vessel that was connected to a tritium labeling apparatus. After freezing of the reaction mixture with liquid nitrogen, the nonreacted tritium and the solvent were adsorbed in a trap filled with platinum oxide and charcoal at the temperature of liquid nitrogen. To remove the labile tritium, the dry residue was dissolved in 1.0 ml of acetonitrile and 0.5 ml of ethanol, and the solvent was evaporated in the vacuum. This process of removing labile tritium was repeated four times. The labeling position is in the methylene group of the ethoxy moiety, which was determined by [3H]NMR spectroscopy. No other signals were detected. The labeling degree was determined by LC MS, and the specific activity was calculated as 312 MBq/mg (3.2 Ci/mmol) for [3H]finerenone. Radiochemical purity was determined as being >98.1%.
Expression Vectors
The expression vectors pchMR, pchMRN770A, pchMRA773G, pchMRQ776A, pchMRS810A, pchMRS810L, pchMRS810M, pchMRR817A, pchMRM852A, pchMRC942A, and pchMRT945A encode for the human MR, MRN770A, MRA773G, MRQ776A, MRS810A, MRS810L, MRS810M, MRR817A MRM852A, MRC942A, and MRT945A, respectively (20–24). The plasmid pFC31Luc contains the mouse mammary tumor virus promoter that drives the luciferase gene (25).
Cell Culture
Cells from the human cortical collecting duct HK-GFP-MR cell line (human kidney GFP-MR, clone 20) (26) were routinely cultured at 37 °C, in an humidified incubator gassed with 5% CO2. Cells were seeded on Petri dishes with 10 ml of DMEM high glucose medium supplemented with l-glutamine (Life Technologies), 2.5% FBS (Biowest, Courtaboeuf, France), 1% penicillin/streptomycin (GE Healthcare), and 200 μg/ml Geneticin (Life Technologies). The murin cortical collecting duct KC3AC1 cells (27) were seeded on collagen I-coated Petri dishes and cultured in a mixture of DMEM/Ham's F-12 (1:1) supplemented with 2 mm glutamine, 50 nm dexamethasone (Sigma), 50 nm sodium selenite (Sigma), 5 μg/ml transferrin, 5 μg/ml insulin (Sigma), 10 ng/ml EGF (Tebu, Le Perray en Yvelines, France), 2 nm T3 (Sigma), 100 units/ml penicillin/streptomycin, 20 mm HEPES, pH 7.4, and 5% dextran charcoal-treated serum. HEK 293T cells (12,000 cells/well) were cultured in high glucose Dulbecco's minimal essential medium (Invitrogen) containing 2 mm glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10% FBS at 37 °C.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 30 min at room temperature and permeabilized for 30 min with a 0.5% solution of PBS-Triton X-100. Cells were then washed with PBS and incubated for 1 h at room temperature in PBS-Tween buffer containing 5% nonfat dry milk (PBS, 5% nonfat dry milk, 0.1% (v/v) Tween 20) before incubation with primary antibody. Cells were incubated with a rabbit anti-GFP antibody overnight at 4 °C (Millipore SAS, Guyancourt, France), followed by an Alexa Fluor 555 anti-rabbit secondary antibody for 30 min at room temperature (Life Technologies). After antibody labeling, cells were postfixed 10 min with 4% paraformaldehyde and nuclear counterstaining was performed with 0.5 μg/ml DAPI.
Automated High Throughput Microscopy
HK-GFP-MR cells were seeded (6000 cells/well) on a 24-well tissue culture plate in DMEM containing 2.5% dextran charcoal-stripped FBS. Forty-eight hours later, a subsequent hormone withdrawal was achieved by rinsing the cells three times with DMEM containing 2.5% dextran charcoal-stripped FBS prior to further incubation of the cells for 48 h within this medium. Ligand treatment was then performed at the indicated concentrations with ligands alone (aldosterone, finerenone, or spironolactone) or with aldosterone plus a 100-fold excess of finerenone or spironolactone. Control vehicle condition was run in parallel with DMEM containing 0.01% DMSO and 0.1% ethanol. Sequential images were acquired (20×/0.4 NA), analyzed and quantified by the ArrayScan VTI imaging platform (Thermo Fisher Scientific). DAPI and GFP-hMR fluorescence were captured using sequential acquisition to give separate image files for each. The Molecular Translocation V4 Bioapplication algorithm (vHCS Scan, version 6.3.1, Build 6586) was used to quantify the relative distribution of GFP-hMR. Briefly, the DAPI staining identifying the nuclear region was used for focusing during acquisition and to define a binary nuclear mask. This mask (CIRC mask) was used to quantify the amount of target channel fluorescence within the nucleus (CircAvgIntensity or nuclear average intensity, NI). The nuclear mask was then dilated to cover the maximum cytoplasmic region as possible without exceeding cell boundaries (RINGWidth = 5 pixels). Subtraction of the nuclear mask from this dilated mask creates a binary cytoplasmic mask covering the cytoplasmic region. An automatic cut-off threshold was applied to exclude pixels of the outside cell boundary background signal (fixed threshold = 55). The cytoplasmic mask (RING mask) was used to quantify the amount of target channel fluorescence within the cytoplasm (RingAvgIntensity or cytoplasmic average intensity, CI). The translocation index measurement (CircAvgInten/RingAvgIntensity or NI/CI) represents the average value ratio of the nuclear intensity to the cytoplasmic intensity calculated for each selected cell per well (n = 3,000–12,000 cells) (28).
Western Blot Analysis
Twenty-four h before the experiment, HK-GFP-MR cells were incubated in DMEM containing 2.5% dextran-coated charcoal-treated FBS. Cells were then incubated for 1, 4, or 24 h at 37 °C with either 10−8 m aldosterone, 10−6 m finerenone or spironolactone, or vehicle. Total protein extracts were then prepared using the protein extraction buffer (150 mm NaCl; 50 mm Tris-HCl, pH 7.5; 0.5 mm EDTA; 30 mm sodium pyrophosphate; 50 mm NaF), extemporaneously supplemented with 1% Triton X-100, 1% protease inhibitor cocktail 100× (Sigma), and 0.1% SDS. Samples were kept at 4 °C for 30 min and then centrifuged at 16,000 × g for 20 min at 4 °C. Protein concentrations were quantified using the bicinchoninic acid assay. After migration on a 6% polyacrylamide gel in denaturing conditions, total proteins (50 μg) were transferred onto a nitrocellulose membrane and revealed by the 39N anti-MR (27) (1:1000 dilution) or the anti-α-tubulin (1:10,000 dilution) antibodies.
Chromatin Immunoprecipitation
Geneticin was removed from cell medium 8 days before experiments. Twenty-four hours before ChIP, HK-GFP-MR cells were incubated in DMEM containing 2.5% dextran charcoal-stripped FBS. Cells were then incubated for 1 h at 37 °C with either 10−7 m aldosterone, 10−6 m finerenone or spironolactone, vehicle, or a combination of 10−7 m aldosterone plus 10−6 m spironolactone or finerenone, all in DMEM supplemented by 2.5% dextran charcoal-stripped FBS. Cells were then fixed for 10 min at room temperature by adding 1% paraformaldehyde to the medium. After 8 min of neutralization of the medium by adding 0.125 m glycine, cells were washed twice with ice-cold PBS. Cell lysis and chromatin shearing were carried out using the #HighCellChIP kit buffers (Diagenode, Seraing, Belgium), according to the manufacturer's recommendations, with the following modifications. Chromatin shearing was performed in 500 μl of shearing buffer S1 + protease inhibitor mixture (Sigma-Aldrich), applying 2 runs of 10 cycles at high intensity (Bioruptor®; Diagenode). Each cycle was composed of 30 s with effective application of ultrasounds (ON) and 30 s without (OFF). Between each run, heated water had to be removed and replaced by ice-cold water and ice. The total quantity of sheared chromatin protein was measured, using the bicinchoninic acid assay (Interchim, Montluçon, France) and the spectrophotometer Victor3® (Perkin Elmer). Protein A-coated magnetic beads (25 μl of the stock suspension) were washed three times in buffer C1 and resuspended in C1 buffer (100 μl of final volume). Sheared chromatin (1.3 mg of proteins) and 7 μg of one of the tested antibodies were then added to the beads, and the final volume was adjusted to 500 μl with C1 buffer supplemented with 1% protease inhibitor mixture and 0.1% BSA (final concentrations). Samples were incubated overnight at 4 °C using a rotary incubator. Magnetic beads were then isolated and washed three times with C1 buffer and once with W1 buffer. DNA fragments from the immunoprecipitated chromatin were eluted with the DNA isolating buffer supplemented with 1% Proteinase K, for 15 min at 55 °C and finally for 15 min at 100 °C and quantified by real time quantitative PCR.
Real Time Quantitative PCR
ChIP-resulting DNA samples were amplified by real time quantitative PCR, using the QuantStudio 6Flex System (Life Technologies) and 96-well plates. In each well were mixed 5 μl of a DNA sample, 6 μl of Fast SYBR Green master Mix (Applied Biosystems, Saint-Aubin, France), and 0.5 μl of forward (5′-TTCCTGCAACTCTGTGAC-3′) and reverse (5′-GCCCTAGGACATTCTGTT-3′) primers (300 nm final concentration) (SCNN1A promoter). After a 10-min preincubation at 95 °C, 40 cycles were performed. Each cycle was composed of 15-s denaturation phase at 95 °C, followed by 1-min hybridization and amplification phase at 60 °C. Each DNA sample was analyzed in triplicate. The results are expressed as percentages of input and are the mean ± S.E. of three independent measurements.
Reverse Transcription
KC3AC1 cells were cultured for 48 h in minimal medium that has the same composition than the epithelial medium but that lacks dexamethasone, EGF and dextran charcoal-treated serum. Cells were then treated for 4 h with vehicle, 10−8 m aldosterone, or 10−6 m finerenone alone or in combination with 10−8 m aldosterone. After twice washing of cells with ice-cold PBS, total RNA was extracted by the phenol-chloroform method, using the TRI reagent (Molecular Research Center, Euromedex, Mundolsheim, France) following the manufacturer's recommendations, then recovered in 20 μl of water, and quantified with Nanodrop spectrophotometer (Thermo Scientific, Villebon, France). Total RNA (1 μg) was first digested for 10 min at 37 °C with 0.5 μl of DNase I (Biolabs, Evry, France) in a total volume of 10 μl to remove potential genomic DNA and then incubated for 10 min at 75 °C with 1 μl of 25 mm EDTA (Life Technologies). For each sample, 9 μl of the reverse transcription mix was incubated in the presence of Superscript reverse transcriptase (1 μl), random primers (2 μl), 10 mm dNTP (0.8 μl), 10× reverse transcriptase buffer (2 μl) and water (3.2 μl). After 10 min of annealing at 25 °C, the reverse transcription was carried out for 2 h at 37 °C, followed by rapid denaturation of the enzyme at 85 °C. The real time quantitative PCR was performed on the complementary DNA (cDNA), as described above. Specific primer sequences are 5′-CTGCACCTTTGGCATGATGTA and 5′-TTCCGGGTACCTGTAGGGATT. A 1:10 dilution standard range was run in parallel to quantify cDNA (attomol amount = f(Ct value)). All samples were quantified in duplicate. Relative expression in a given sample was normalized to the internal reference 18S rRNA values where the control condition value was arbitrarily set at 1. The results are expressed as means ± S.E.
Transactivation Assays in Transiently Transfected Cells
HEK 293T cells were cultured and transfected using the calcium phosphate method with the vectors allowing the expression of the wild type or mutant MRs (MRWT, MRN770A, MRA773G, MRQ776A, MRS810A, MRR817A MRM852A, MRC942A, and MRT945A), the reporter vector pFC31Luc, and the pcβgal vector as previously described (29) or with Lipofectamine 2000 as recommended by the manufacturer (Invitrogen) (MRWT, MRA773G, MRS810L, and MRS810M) and the pMIR-REPORT β-gal (Applied Biosystems). Twenty-four hours after transfection, finerenone was added (10−9 to 10−6 m) together with an agonist at the concentration able to maximally activate each receptor (10−9 m aldosterone for MR, MRA773G, MRS810A, MRS810L, and MRS810M; 10−7 m aldosterone for MRQ776A, MRC942A, and MRT945A; 10−7 m 18-oxo-18-vinylprogesterone for MRN770A; and 10−8 m spironolactone for MRM852A), and after a 16-h incubation, cell extracts were assayed for luciferase and β-galactosidase activities (29). The GraphPad Prism software (version 6; GraphPad Software Inc., San Diego, CA) was used for curve fitting and calculation of the IC50 values. The IC50 values were determined from at least three independent experiments performed in triplicate. The IC50 values are given as means ± S.E.
Finerenone Binding Characteristics at Equilibrium and Kinetic Experiments
The human MR was expressed in vitro in the rabbit reticulocyte lysate system as previously described (21). Lysates were diluted 4-fold with TEGWM buffer (20 mm Tris-HCl, 1 mm EDTA, 20 mm sodium tungstate, 1 mm β-mercaptoethanol, and 10% glycerol (v/v), pH 7.4) and incubated with 0.3 to 300 nm [3H]finerenone for 4 h at 4 °C. Bound and Unbound ligands were separated by the dextran-charcoal method (23). The change in bound/unbound as a function of bound was analyzed, and the Kd value was calculated as previously described (30).
Dissociation kinetics studies were performed using 4-fold diluted hMR-containing lysates incubated with 10−8 m [3H]finerenone for 4 h at 4 °C. One-half of the labeled lysate was kept at 4 °C and used to determine the stability of the [3H]finerenone/MR complexes, whereas the other half was incubated with 10−6 m finerenone for various periods of time. Bound and free ligands were separated using charcoal-dextran treatment. The data were corrected for receptor stability and were expressed as a percentage of the binding measured at time 0.
Protein Modeling and Ligand Docking
The x-ray structure of the wild type MR ligand-binding domain complexed with deoxycorticosterone (Protein Data Bank code 2ABI) (24) served as a model for docking of finerenone. In contrast to other x-ray structures, it is missing the C808S mutation, leading to a slightly more natural side chain arrangement. The structure was complemented for unresolved loops by superimposing it with 2A3I (31) and transferring the loops Arg-904–Gln-919 and Gly-753–Asp-760. Helix 12 (Pro-957–His-982) was deleted. The resulting protein structure including crystallographic water molecules was protonated. Likely protonation states and hydrogen bonding networks were predicted using the “protein preparation workflow” in Maestro (Schrödinger). Initial docking was carried out by manually placing the small molecule x-ray-derived conformation of BR-4628 into the binding site and varying the relative orientation of the ligand (29). The conformation of Met-852, Ser-811, and Thr-945 side chains were altered to enlarge the volume of the binding pocket. The program Glide SP version 3.5 (32) (Schrödinger) was used to dock finerenone into the binding cleft. The ligand was placed at the center of a 22 Å box to calculate the interaction grid. The van der Waals scaling factor was set at 0.8, and the partial charge cutoff was set to 0.15. The ligands were docked flexibly, and nonplanar amide bonds were penalized; 10,000 poses per docking run were sampled. The complex with the most probable binding pose was energy minimized using the OPLS2.1 force field (33) (dielectric constant, 1.0; constant dielectric, solvent water, Polak-Ribier conjugate gradient; convergence threshold, 0.5) in MacroModel (Schrödinger).
Statistical Analysis
Experimental data are presented as means ± S.E. Statistical analyses were performed using the Prism 6 software (GraphPad Software). Statistical significance was calculated with the Mann-Whitney nonparametric U test with unpaired data and two-tailed calculations.
Results
Finerenone Is a Potent Mineralocorticoid Antagonist That Displays a High Affinity for MR
Finerenone was initially reported to strongly inhibit aldosterone-induced transcriptional activity of the MR ligand-binding domain (LBD) fused to the GAL4 DNA-binding domain (13). We investigated the antagonistic properties of finerenone on the wild type full-length human MR in comparison with the steroidal MR antagonist spironolactone. HEK 293T cells transiently expressing human MR and a reporter vector in which the luciferase gene was placed under the control of the mouse mammary tumor virus promoter were treated by aldosterone in the presence of increasing concentrations of finerenone or spironolactone. Luciferase activity was measured, and IC50 values of 58 ± 9 and 74.0 ± 15 nm were determined for finerenone and spironolactone, respectively (Table 1). These results indicate that finerenone is slightly more potent than spironolactone in vitro. We then evaluated the binding properties of [3H]finerenone on the recombinant MR by performing binding experiments at equilibrium. Scatchard plot analysis revealed that finerenone binds the in vitro translated full-length MR with a high affinity, as determined by the Kd value of 1.52 ± 0.01 nm (Fig. 1A), which is in the same range as that of aldosterone (0.5 nm) and the mineralocorticoid antagonist progesterone (1.0 nm) (22). We then evaluated the dynamics of the finerenone-MR interaction. Dissociation kinetic experiments, performed at 4 °C revealed a half-life time of 50 min for the finerenone-MR complex (Fig. 1B), which is in the same order of magnitude as that for the complex between MR and the antagonist RU26752 (22). Altogether, these results indicate that finerenone has a high affinity for MR combined with a rapid receptor off rate.
TABLE 1.
Half-maximally inhibitory concentrations of finerenone for the wild-type and mutant MRs
HEK 293T cells were cultured and transfected. Twenty-four hours after transfection, finerenone was added (10−9 to 10−6 m) together with an agonist at the concentration able to maximally activate each receptor (10−9 m aldosterone for MR, MRA773G, MRS810A, MRS810L and MRS810M; 10−7 m aldosterone for MRQ776A, MRC942A, and MRT945A; 10−7 m 18OVP for MRN770A; and 10−8 m spironolactone for MRM852A), and after a 16-h incubation, cell extracts were assayed for luciferase and β-galactosidase activities (29). The IC50 values were calculated using the GraphPad Prism Software and are the means ± S.E. of three independent experiments performed in triplicate. NA, nonapplicable.
| Finerenone |
Spironolactonea |
||||
|---|---|---|---|---|---|
| IC50 | Fold | IC50 | Fold | ED50 | |
| nm | nm | nm | |||
| MRWTb | 58 ± 9 | 1 | 74.0 ± 15 | 1 | |
| MRN770Ab | 1925 ± 185 | 33.2 | 1313 ± 167 | 17.7 | |
| MRQ776Ab | 74 ± 8 | 1.3 | 301 ± 77 | 4.1 | |
| MRS810Ab | 524 ± 50 | 9.0 | 109 ± 24 | 1.5 | |
| MRR817Ab | 155 ± 12 | 2.7 | 331 ± 20 | 4.5 | |
| MRC942Ab | 321 ± 49 | 5.5 | 506 ± 55 | 6.8 | |
| MRT945Ab | 119 ± 19 | 2.1 | 444 ± 43 | 6.0 | |
| MRWTc | 31.8 ± 1.8 | 1 | 74.0 ± 15 | 1 | |
| MRA773Gc | 1370 ± 280 | 43.1 | 84 ± 15 | 17.7 | |
| MRS810Mc | 5100 ± 800 | 160.4 | NA | NA | 6 |
a According to Ref. 29.
b Transfected using the calcium phosphate method.
c Transfected using Lipofectamine 2000.
FIGURE 1.
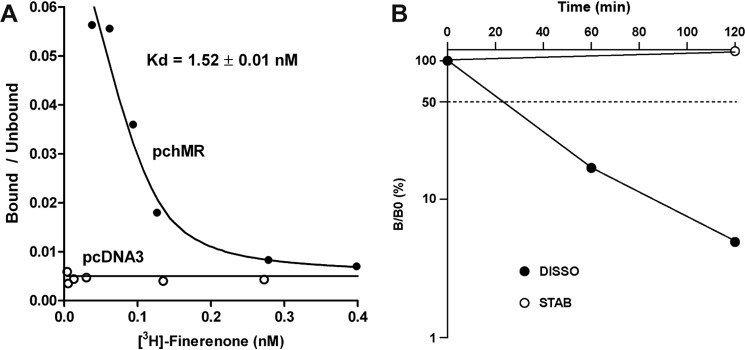
Finerenone binding characteristics at equilibrium and kinetic experiments. A, the human MR was expressed in vitro in the rabbit reticulocyte lysate system. Lysates were diluted 4-fold with TEGWM buffer (20 mm Tris-HCl, 1 mm EDTA, 20 mm sodium tungstate, 1 mm β-mercaptoethanol, and 10% glycerol (v/v), pH 7.4) and incubated with 3 × 10−10 to 3 × 10−7 [3H]finerenone for 4 h at 4 °C. Bound (B) and Unbound (U) ligands were separated by the dextran-charcoal method. The change in B/U as a function of B was analyzed, and the Kd value was calculated. B, dissociation kinetics studies were performed using 4-fold diluted hMR containing lysates incubated with 10−8 m [3H]finerenone for 4 h at 4 °C. One-half of the labeled lysate was kept at 4 °C and used to determine the stability of the [3H]finerenone-MR complexes, whereas the other half was incubated with 10−6 m finerenone for various periods of time. Bound and free ligands were separated using charcoal-dextran treatment. The data were corrected for receptor stability and were expressed as percentages of the binding measured at time 0.
The Ligand-induced Nuclear Import of MR Is Differentially Delayed by Finerenone and Spironolactone
Nuclear translocation is one of the critical steps that convert steroid receptors into their transcriptionally active state. It was previously shown that steroidal MR antagonists drastically affected intracellular localization of MR as opposed to aldosterone that rapidly triggered MR nuclear translocation within one hour (16, 17). We thus investigated the impact of finerenone and spironolactone on the subcellular trafficking of MR to determine whether both antagonists could exert differential actions on MR cytoplasmic sequestration and/or inhibition of aldosterone-induced MR nuclear import. A human renal cortical collecting duct GFP-MR cell line (HK-GFP-MR) stably expressing low levels of GFP-hMR was selected for our experiments (26) to avoid nonspecific mislocalization of the receptor. Control experiments indicate that GFP-hMR was not detected in control parental cell line (data not shown). Cells were treated for 1 h-24 h with each ligand alone or by combining aldosterone and one of the antagonists. Results show that 1 h aldosterone treatment induces the complete accumulation of GFP-hMR into the nuclear compartment (Fig. 2A). In contrast, in the presence of finerenone or spironolactone, hMR was only partially imported into the nucleus, because a large fraction of GFP-hMR still remains detectable in the cytoplasmic compartment (Fig. 2A).
FIGURE 2.

Finerenone and spironolactone delay the ligand-induced nuclear translocation of the mineralocorticoid receptor. A, HK-GFP-MR cells were cultured for 48 h in medium containing 2.5% charcoal-stripped fetal bovine serum, followed by 1 h of incubation with either 10−8 m aldosterone, 10−6 m finerenone, or spironolactone. Cells were processed for immunocytochemistry using an anti-GFP antibody, and fluorescence was captured with an automated ArrayScan VTI fluorescent microscope. DAPI staining delineates the nuclei. The left panel shows the raw images acquired in the GFP channel. The right panel shows the DAPI acquisition. B, automated quantification of GFP-hMR nuclear translocation kinetics by automated high throughput microscopy. HK-GFP-MR cells were cultured in medium containing 2.5% charcoal-stripped fetal bovine serum. Forty-eight hours after, cells were treated with either 10−8 m aldosterone, 10−6 m finerenone, or 10−6 m spironolactone or incubated with a mix of aldosterone 10−8 m and antagonist at 10−6 m for 1, 4, or 24 h as indicated. The panel shows the translocation index that represents the ratio of the average nuclear intensity to the average cytoplasmic intensity (NI/CI) calculated in their respective binary mask (see “Experimental Procedures”). C, the panel shows the average nuclear fluorescence intensity (NI) values calculated for each indicated condition (CircAvgInten). Statistical significance was calculated with the Mann-Whitney nonparametric U test with unpaired data and two-tailed calculations. Because of the very large number of cells (3,000–12,000), p values < 0.0001 were obtained, with some exceptions. For the NI/CI, p values < 0.001 were obtained for S versus A+F (1 h), S versus F (24 h), S versus A+S (24 h), and F versus A+F (24 h), p values < 0.01 for F versus A+F (1 h) and A versus S (4 h); p values < 0.05 for F versus A+S (1 h) and A+F versus A+S (24 h), and nonsignificant values were obtained for A+F versus A+S (1 h), S versus A+S (4 h), and S versus A+F (24 h). For the NI, p values < 0.001 were obtained for S versus A+S (24 h), and p values < 0.01 were obtained for A versus S (4 h) and F versus A+F (24 h); nonsignificant values were obtained for F versus A+S (1 h), A versus V (24 h), and S versus F (24 h), F versus A+S (24 h), and A+S versus A+F (24 h).
Precise quantification of the subcellular localization of the receptor after 1–24 h of treatment with various ligands was made possible by the use of an automated high throughput microscope, an analysis we previously developed for the characterization of progesterone receptor antagonists (34). As shown in Fig. 2B, the translocation index of GFP-hMR as defined by the ratio between the averaged intensities of the nuclei and cytoplasmic rings (NI/CI) (see “Experimental Procedures”) had a value of 1.33 ± 0.05 (n = 2993) in the absence of aldosterone, indicating a localization of the fusion protein within the whole cell compartments. Upon 1 h of incubation with 10 nm aldosterone, the translocation index of GFP-hMR drastically increased and reached a maximum value of 3.11 ± 0.02 (n = 3940; p < 0.0001). No significant additional increase of the index value was observed after prolonged incubation time with aldosterone (4–24 h), indicating that the aldosterone-dependent MR nuclear accumulation reaches its maximum after 1 h treatment. In sharp contrast, the translocation index only increases moderately in cells treated 1h with antagonists alone (10−6 m) or with a mix of aldosterone (10−8 m) and a 100-fold excess of each antagonist (1.84 ± 0.01 (n = 2872), 1.76 ± 0.01 (n = 3746), 1.75 ± 0.01 (n = 4823) and 1.78 ± 0.01 (n = 2379)), for spironolactone, finerenone, aldosterone plus spironolactone, and aldosterone plus finerenone, respectively (p < 0.0001 versus vehicle, and versus aldosterone). These data are consistent with previous observations (Fig. 2A) and demonstrate that both antagonists are able to compete with aldosterone by inhibiting the hormone-dependent nuclear accumulation of GFP-hMR. However, a prolonged exposure to antagonists (4–24 h) further increases MR nuclear import (translocation index 2.71 ± 0.01 (n = 9095), 2.71 ± 0.01 (n = 7972), 2.82 ± 0.01 (n = 7901) and 2.82 ± 0.02 (n = 2333)) for spironolactone, finerenone, aldosterone plus spironolactone, and aldosterone plus finerenone, respectively (p < 0.0001 versus vehicle, and versus aldosterone; Fig. 2B), indicating that both antagonists do not exert their inhibitory effects by sequestering MR into the cytoplasm but rather by delaying its nuclear import kinetics. To confirm these observations, we precisely quantified the average fluorescence intensity within the nucleus for each condition (i.e. NI parameter; Fig. 2C). As expected, the graph shows that the nuclear intensity increases dramatically after 1 h of treatment with aldosterone (NI = 390 ± 3 (n = 3940) versus vehicle 250 ± 3 (n = 2293), p < 0.0001 versus vehicle; Fig. 2C). Strikingly, the nuclear fluorescence intensity rapidly declines after 4 and 24 h of aldosterone exposure (NI = 315 ± 2 (4 h) (n = 12272) and 275 ± 2 (24 h) (n = 6370); Fig. 2C), suggesting that a ligand-induced down-regulation of MR occurs in the presence of the agonist. In contrast, 1 h of finerenone exposure alone or in combination with aldosterone only moderately increases the nuclear fluorescence intensity (NI = 275 ± 3, n = 3746) at a much lower extend than that observed with spironolactone (NI = 313 ± 3, n = 2872), indicating that finerenone is more potent to inhibit the nuclear import of GFP-hMR than spironolactone. However, upon 4 h of exposure with antagonist ligands (alone or in combination with aldosterone), NI values slightly increased to reach their maximum value (311 ± 2 (n = 9095), 329 ± 2 (n = 7972), 344 ± 2 (n = 7901), and 351 ± 4 (n = 2333) for spironolactone, finerenone, aldosterone plus spironolactone, and aldosterone plus finerenone, respectively). Thereafter, nuclear intensities decreased by ∼50% after 24 h of antagonist exposure, suggesting that antagonist-induced MR degradation might also occur in a delayed manner with a diminished efficacy. Taken together, our findings demonstrate that finerenone impedes MR nuclear translocation more efficiently than spironolactone and indicate that MR degradation might be differentially regulated by agonist and antagonist ligands.
Finerenone and Spironolactone Impair Aldosterone-dependent Down-regulation of the Mineralocorticoid Receptor
To better understand how ligands modulate MR nuclear fluorescence intensity, we investigated whether MR protein levels could be differentially affected after incubation with aldosterone or MR antagonists. Steroid receptor transcriptional activation has been shown to be directly coupled to the receptor degradation by the ubiquitin-proteasome pathway, and it has been also shown that gene transactivation could not occur when the receptor is not down-regulated (35–37). To analyze the effect of ligands on MR expression level, HK-GFP-MR cells were incubated for various periods of time (1–24 h) in the presence of each molecule. An antibody directed against MR (27) was used to detect the 150-kDa band corresponding specifically to GFP-hMR. Western blot analyses of whole cell extracts revealed that GFP-hMR expression level did not significantly change upon 1 h of ligand treatment (agonist or antagonist) (Fig. 3, A and D). Of particular interest, an upper shift of the GFP-hMR band was observed after 1, 4, and 24 h of aldosterone exposure, indicating a ligand-induced phosphorylation of the receptor. This upper shift was neither observed in the vehicle nor in antagonist-treated conditions, suggesting that the receptor phosphorylation is an agonist-dependent process. In addition, although MR level significantly decreased after 4 h of aldosterone treatment (Fig. 3, B, lanes 3 and 4 versus lanes 1 and 2, and D), the receptor level remained stable in the presence of both antagonists (Fig. 3, B, lanes 6 and 7 and lines 8 and 9 versus lanes 1 and 2, and D). This result is consistent with the data obtained from the in situ nuclear fluorescence intensity quantification (Fig. 2C). Interestingly, GFP-hMR level dramatically decreased after 24 h of exposure to aldosterone (Fig. 3, C, lanes 4 and 5 versus lanes 2 and 3, and D), whereas no significant reduction of the receptor level was observed after both antagonists treatment (Fig. 3C, lanes 7 and 8 and lanes 9 and 10 versus lanes 2 and 3, and D). Overall, these data clearly indicate that MR expression is reduced in an aldosterone-dependent manner and that both antagonists are unable to promote such receptor degradation.
FIGURE 3.

GFP-MR degradation upon ligand binding. A, Western blot analysis of the ligand-induced degradation of the GFP-hMR fusion protein. A–C, HK-GFP-MR cells were treated for 1 h (A), 4 h (B), or 24 h (C) with vehicle (V), 10−8 m aldosterone (A), 10−6 m spironolactone (S), or finerenone (F). The upper bands, corresponding to the GFP-hMR fusion protein (150 kDa), were revealed by the 39N antibody directed toward MR and quantified with the Image Studio software. The values were normalized with those of the α-tubulin bands (55 kDa) and by the values for the vehicle conditions. The values are reported under each corresponding lanes and are the means ± S.E. of three independent experiments with 1–4 measurements. D, GFP-hMR time course degradation. Statistical significance was calculated with the Mann-Whitney nonparametric U tests with unpaired data and two-tailed calculations. * (aldosterone versus vehicle), # (aldosterone versus finerenone), and § (aldosterone versus spironolactone), p < 0.05; **, p < 0.01.
Finerenone and Spironolactone Differentially Affect Recruitment of Transcriptional Cofactors on a Mineralocorticoid Receptor Target Promoter
High throughput microscope experiments indicated that finerenone and spironolactone have different effects on MR nuclear translocation kinetics. We recently demonstrated by using ChIP assays the aldosterone-induced recruitment of MR, SRC-1, and RNA polymerase (Pol) II onto the regulatory region of the SCNN1A gene that encodes the α-subunit of the epithelial sodium channel (αENAC), a well known MR target gene. Moreover, we showed that spironolactone is able to promote MR and SRC-1 recruitment but not that of the RNA Pol II (38). We therefore compared the abilities of aldosterone, finerenone, or spironolactone to promote binding of competent transcriptional complexes onto a genomic MR target by using similar ChIP experiments. As expected, Fig. 4 shows that aldosterone induces a significant increase of MR, SRC-1, and RNA Pol II recruitment onto the SCNN1A promoter compared with the vehicle-treated condition or upon control IgG immunoprecipitation as measured by ChIP experiments. In addition, whereas aldosterone induced MR recruitment onto the SCNN1A promoter region, it did not promote a MR recruitment onto a nonpromoter region of the SCNN1A gene nor onto the 18S gene (data not shown). In addition, aldosterone was incapable of inducing MR recruitment on the 3′-untranslated region of the SCNN1A gene, whereas a dramatic Pol II recruitment was observed at this genomic region after aldosterone treatment (data not shown). These experiments indicate that aldosterone promotes MR, SRC-1, and Pol II recruitment onto the promoter region of SCNN1A gene, allowing the MR-mediated aldosterone-dependent transcriptional initiation and elongation, as demonstrated by the presence of Pol II at both the 5′ and 3′ region of the gene. Spironolactone alone also promoted a significant MR and SRC-1 recruitment onto the SCNN1A promoter but to a lesser extent than aldosterone. Spironolactone did not promote RNA Pol II recruitment, suggesting that although this compound displays an aldosterone-like behavior, it was unable to favor recruitment of RNA Pol II preventing active preinitiation complexes. In sharp contrast, finerenone alone not only impairs MR, SRC-1, and RNA Pol II recruitment but also remarkably reduces basal MR and SRC-1 binding onto the SCNN1A regulatory sequence as compared with vehicle-treated condition. In addition, finerenone also significantly inhibited MR, SRC-1, and RNA Pol II recruitment induced by aldosterone. Collectively, these results indicate that the nonsteroidal MR antagonist finerenone acts as an inverse mineralocorticoid receptor agonist.
FIGURE 4.
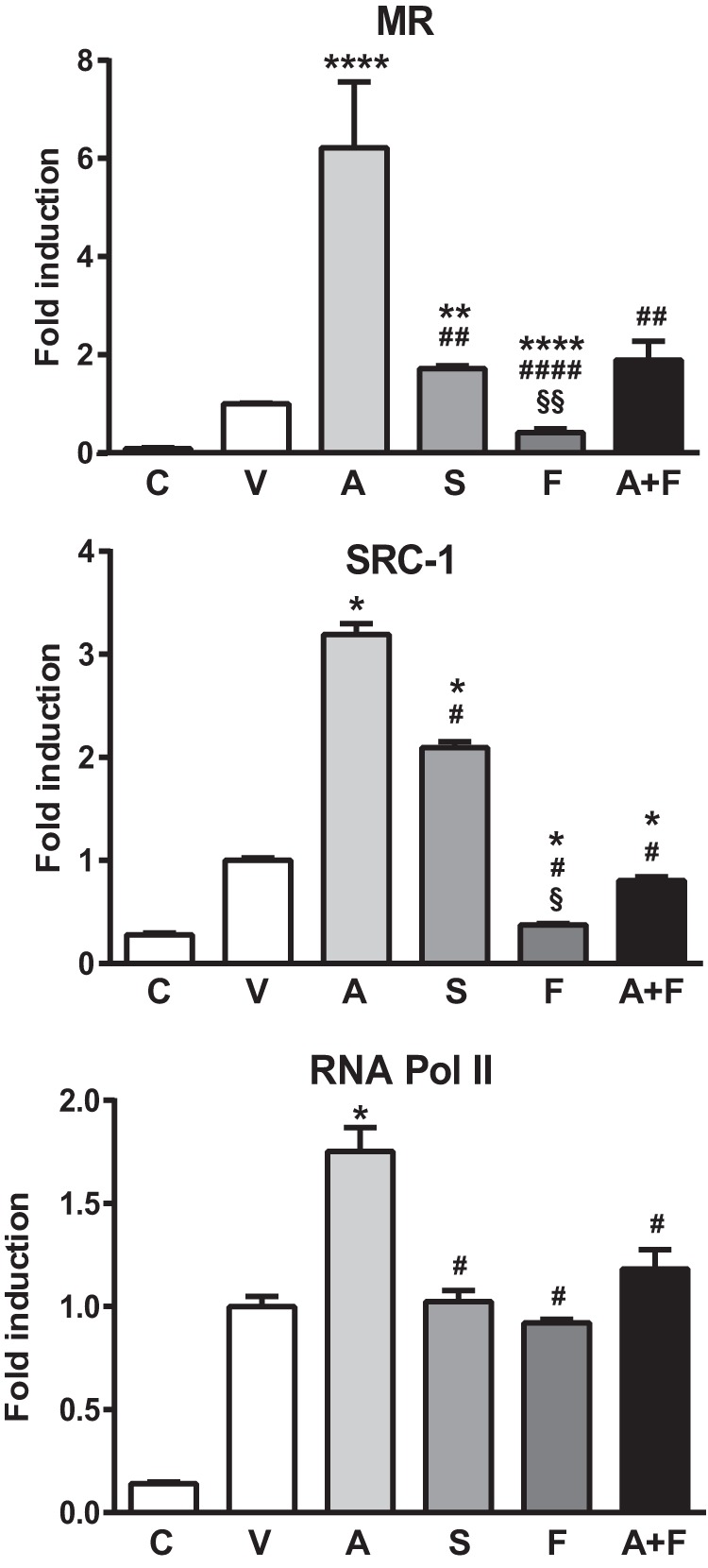
MR, SRC-1, and RNA Pol II recruitment on the promoter region of the SCNN1A gene. HK-GFP-MR cells were incubated with ethanol (V), 10−7 m aldosterone (A), 10−6 m spironolactone (S), or finerenone (F) or 10−7 m aldosterone plus 10−6 m finerenone (A+F), fixed with paraformaldehyde, lysed, and chromatin-sheared. Samples were then immunoprecipitated with the anti-MR 39N antibody or a control rabbit IgG (C) (top panel), SRC-1 (middle panel), or an antibody directed against the RNA Pol II (bottom panel) as described under “Experimental Procedures.” After elution, DNA was quantified by quantitative PCR, using primers for the genomic fragment encompassing mineralocorticoid responsive element corresponding to the MR binding site located in the promoter region of the SCNN1A gene. The results are expressed as percentages of input before immunoprecipitation. The data are means ± S.E. of three independent experiments. Statistical significance was calculated with the Mann-Whitney nonparametric U tests with unpaired data and two-tailed calculations. **** (versus vehicle), #### (versus aldosterone), and §§§§ (versus spironolactone), p < 0.0001; ***, ###, and §§§, p < 0.001; **, ##, and §§, p < 0.01; *, #, and §, p < 0.05.
Finerenone Alters the Expression of an Endogenous Mineralocorticoid Receptor Target Gene
To further evaluate finerenone's molecular mechanism of action, we investigated the ability of this molecule to inhibit expression of specific target gene. Using the murine renal cell line KC3AC1 that endogenously expresses MR (27), we evaluated the influence of aldosterone and/or finerenone on the expression of the Sgk1 gene, a well known MR target gene (39). KC3AC1 cells were treated 4 h with 10−6 m finerenone alone or in combination with 10−8 m aldosterone. Fig. 5 shows that aldosterone significantly stimulates expression of the Sgk1 gene. In contrast, finerenone alone inhibits the basal expression level of Sgk1 and is able to inhibit the aldosterone-induced expression of this gene. Under similar experimental conditions, β-actin expression is not modified, as expected.
FIGURE 5.
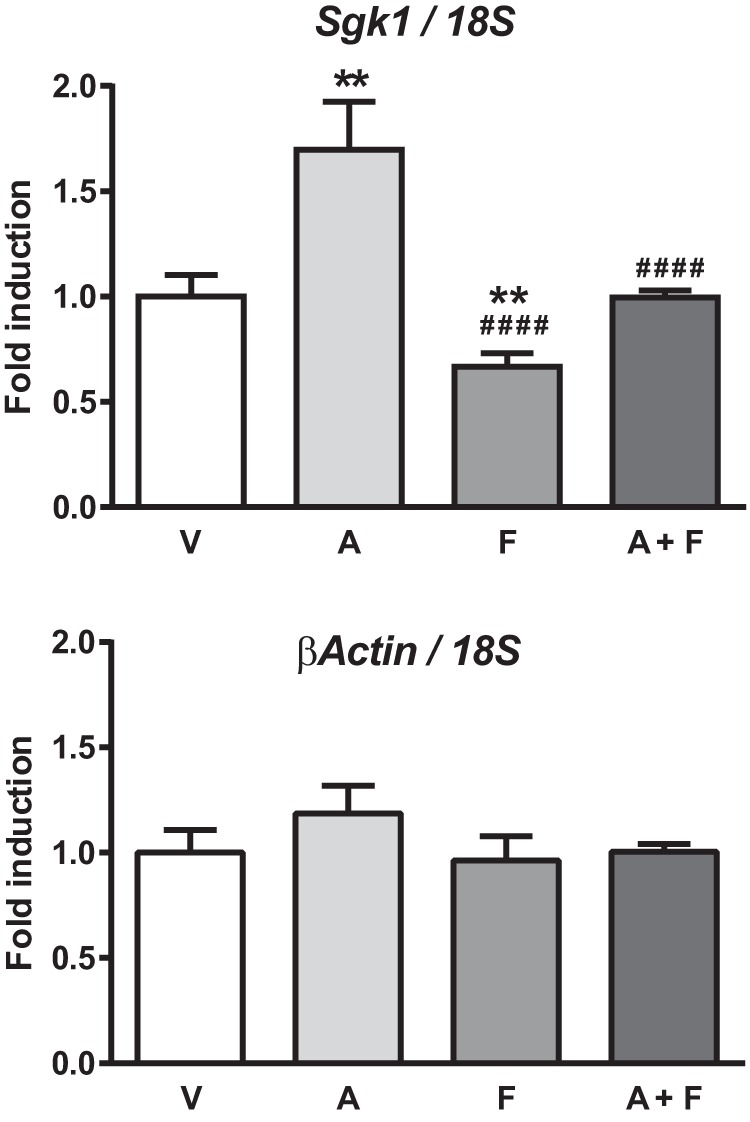
Aldosterone-induced transcriptional activation of the Sgk1 gene. KC3AC1 cells were treated for 4 h with vehicle (V), 10−8 m aldosterone (A), 10−6 m finerenone alone (F), or finerenone in the presence of 10−8 m aldosterone (A+F). Total RNAs were isolated and reverse transcribed. cDNAs for Sgk1 and β-actin were quantified by real time quantitative PCR, using a specific primer pairs. Relative gene expression values were normalized to that of 18S rRNA and expressed as fold induction compared with vehicle condition arbitrarily set at 1. The data are means ± S.E. of six independent determinations performed in duplicate. Statistical significance were calculated with Mann-Whitney nonparametric U tests with unpaired data and two-tailed calculations. **, p < 0.01 versus vehicle; ####, p < 0.0001 versus aldosterone.
Finerenone Inhibits the S810L Mutant MR
The S810L mutant MR (MRS810L) was shown to be responsible for a severe form of familial hypertension (40). Patients harboring this mutation develop early onset hypertension before the age of 15. Moreover, this arterial hypertension is exacerbated during pregnancy. Functional studies revealed that progesterone acts as a full MR agonist. This is also the case for the steroidal antagonist spironolactone and eplerenone (24, 40). We thus evaluated the agonist or antagonist properties of finerenone on the S810L mutant MR. HEK 293T cells transiently expressing the S810L mutant MR were treated with increasing concentrations of aldosterone, finerenone, or spironolactone (10−11 to 10−7 m) or with 10−9 m aldosterone plus increasing concentrations finerenone (10−9 to 10−5 m). As anticipated, aldosterone and spironolactone activate the MRS810L in a dose-dependent manner with EC50 of 4.2 ± 0.4 10−10 m and 25 ± 2 10−11 m, respectively (Fig. 6A). In contrast, finerenone alone was not only unable to activate the MRS810L but more importantly was capable of inhibiting the aldosterone-induced activity of the MRS810L activity with an IC50 value of 16.1 ± 3.1 10−6 m (Fig. 6, A and B). These results clearly demonstrate that finerenone does not display agonist activity on this mutant MR, providing additional support for distinct steroidal and nonsteroidal ligand-MR interactions and their subsequent functional consequences.
FIGURE 6.
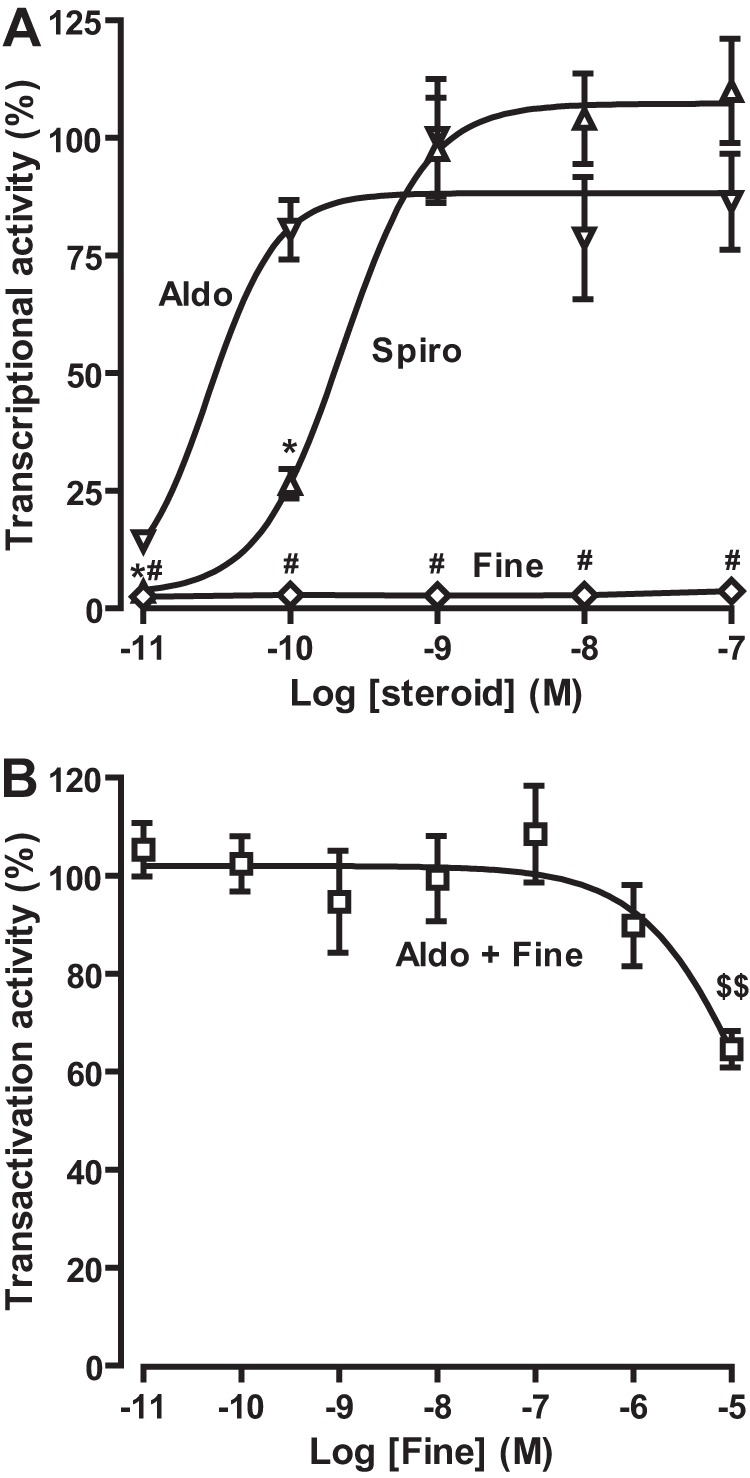
Finerenone inhibits the aldosterone-induced transactivation activity of MRS810L. HEK 293T cells transiently expressing MRS810L were incubated for 16 h with increasing concentrations aldosterone, finerenone, or spironolactone (A) or 10−9 m aldosterone in the presence of increasing concentrations of finerenone (B). The activity of MRS810L to transactivate the luciferase gene, which is under the control of a glucocorticoid response element-containing promoter, was determined by measuring the luciferase activity, which was then normalized to the β-galactosidase activity and to the value obtained with 10−9 m aldosterone. The values are means ± S.E. of at least three independent experiments performed in triplicate. GraphPad Prism software was used to fit the curves and to calculate the EC50 and IC50 values. The data are means ± S.E. of three to four independent experiments performed in sextuplet. Statistical significance were calculated with the Mann-Whitney nonparametric U tests with unpaired data and two-tailed calculations. * (spironolactone versus aldosterone) and # (finerenone versus aldosterone), p < 0.05; $$, p < 0.01 in comparison with aldosterone alone.
Finerenone Establishes Specific Contacts within Residues of the MR LBD
The crystal structures of the LBD of MR and MRS810L complexed with steroidal agonists have been recently solved, allowing their anchoring mode to be fully characterized (21, 24, 31, 41). The nonsteroidal nature of finerenone and its peculiar behavior in ChIP experiments raised the question of its mechanism of action and its binding mode within the MR binding pocket. The binding mode of dihydropyridine type MR antagonists to MR was predicted for BR-4628 first (29). This structural information was used to guide the optimization, ultimately leading to finerenone. Finerenone was docked within the x-ray structure of the wild type MR LBD (Protein Data Bank code 2ABI) (24). The binding niche of the MR LBD in its agonistic conformation appeared too small to accommodate the nonsteroidal ligand. This result suggested that DHP-type MR antagonists, including finerenone, act as bulky antagonists. Within the binding pocket, finerenone forms numerous van der Waals contacts with the neighboring amino acids (Fig. 7A) and is anchored through four hydrogen bonds. The dihydronaphtyridine NH group is hydrogen bonded to the Asn-770 carbonyl oxygen, the cyanide group is bonded to Gln-776 and Arg-817, whereas the amide NH acts as H-bond donor to Ser-810 (Fig. 7A). Superimposition of docked finerenone and the x-ray structure of spironolactone in complex with MRS810L suggests a partial overlap of the ligands (Fig. 7B). The methoxyl, cyanide, and phenyl groups of finerenone approximately occupy the same volume and space than the A and B rings of spironolactone (Fig. 7B). In contrast, the dihydronaphtyridinyl ring adopts an orthogonal orientation to the steroidal “plane” of spironolactone. With this orientation, the ethoxyl substituent points in the same direction than the spironolactone thioacethyl group, whereas the amide and the C2 methyl groups point in the direction of H12 helix and are in close contact with Ala-773.
FIGURE 7.
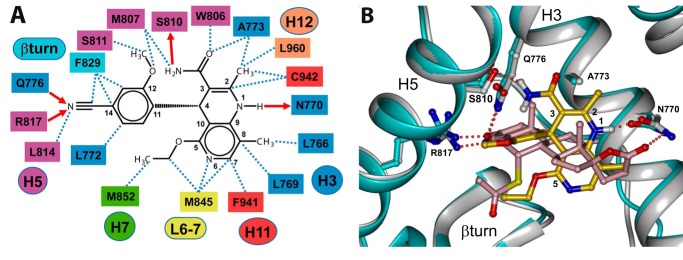
Accommodation mode of finerenone within the MR ligand-binding pocket. A, schematic representation of the contacts between finerenone and the residues lining the binding pocket of MR. The hydrogen bonds and van der Waals contacts are depicted as solid red arrows and black dashed lines, respectively. The numbers of the secondary structure elements are indicated in colored circles or ovals. The residue numbers are indicated in boxes and colored according to the secondary structure element to which they belong. B, superimposition of finerenone and spironolactone within the MR-binding pocket. Only residues that anchor the ligands are shown. Hydrogen bonds between finerenone or spironolactone, and the polar residues are depicted as dashed red lines. In the MR-spironolactone model, the protein is colored in light blue with its side chains atoms in light blue, blue, and red for the carbon, nitrogen, and oxygen atoms, respectively. Spironolactone atoms are colored in pink, red, and yellow for the carbon, oxygen, and sulfur atoms, respectively. In the MR-finerenone model, the protein is in white with its side chains atoms in white, blue, and red for the carbon, nitrogen, and oxygen atoms, respectively. Finerenone atoms are in gold, blue, and red for the carbon, nitrogen, and oxygen atoms, respectively. This figure was produced using DINO.
To validate such a docking mode, we analyzed the incidence of point mutations within the binding cavity. The residues were selected as being polar residues that anchor BR-4628 (28) and delimit the volume of the binding niche. The potency of finerenone in antagonizing the agonist-induced activity of the resulting mutant MRs was tested. In a first set of experiments, HEK 293T cells were transiently cotransfected with or without the vector allowing the expression of the wild type hMR and with a reporter vector in which the luciferase gene is under the control of a MR-sensitive promoter and treated with vehicle or 10−9 m aldosterone. A 32-fold stimulation of the luciferase activity was obtained in MR-expressing cells, whereas no significant stimulation of luciferase activity was obtained from cells devoid of transiently expressed MR (data not shown), indicating that the luciferase activity measured in transfected HEK 293T cells directly reflects the transactivation activity of the transiently expressed MR. HEK 293T cells were then transfected with the luciferase reporter vector in combination with the expression vector of the wild type or mutant MRs and treated with increasing finerenone concentrations in the presence aldosterone. The Q776A, R817A, M852A, C942A, and T945A mutations have little or no effect on the finerenone antagonistic potency, indicating that these residues form moderate interactions with the ligand (Table 1). In sharp contrast, the substitution of an alanine for Asn-770 and Ser-810 drastically reduced by ∼33- and 9-fold, respectively, the antagonistic potency of finerenone, confirming that the hydrogen bonds between finerenone and Asn-770 and Ser-810 are critical for high potency and affinity (Table 1).
It has been previously reported that two residues from the ligand-binding cavity strongly diverge between the oxo-steroid receptors. It concerns Ala-773 and Ser-810 in MR, which correspond to a glycine and a methionine at the corresponding positions in androgen receptor, glucocorticoid receptor, and progesterone receptor (20, 42). Because finerenone is highly selective for MR and establishes a peculiar interaction with Ala-773 and Ser-810 (a short van der Waals contact and a hydrogen bond, respectively), we wondered whether these residues are involved in the high MR selectivity. The A773G and S810M mutations have no effect on the aldosterone potency to activate MR as illustrated by identical ED50 values (29). The A773G mutation has also no effect on the spironolactone antagonistic potency, whereas the S810M mutation converts this ligand as a full agonist (ED50: 6 × 10−9 m) (29). In sharp contrast, both A773G and S810M mutations lead to a dramatic decrease in the antagonistic potency of finerenone (increase of the IC50 values by ∼24- and 88-fold, respectively; 1.4 ± 0.3 × 10−6 and 5.1 ± 0.8 × 10−6 m, for MRA773G and MRS810M, respectively; versus 5.8 ± 0.9 × 10−8 m for MRWT; Fig. 8). These results strongly indicated that the presence of an alanine and a serine at the 773 and 810 positions favors finerenone binding to MR, whereas a glycine and a methionine, as in the other oxo-steroid receptors, impairs finerenone binding.
FIGURE 8.
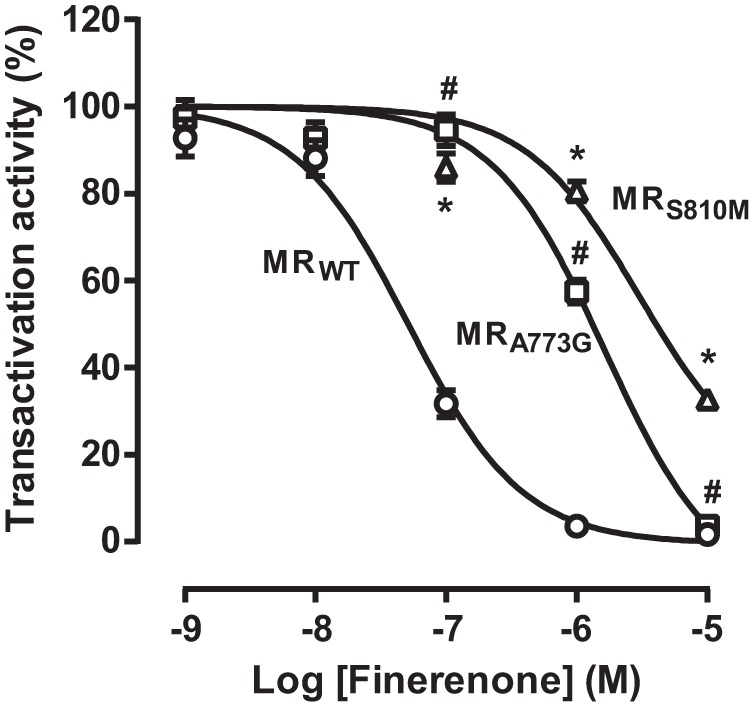
Transactivation properties of MRA773G and MRS810M in response to finerenone. HEK 293T cells transiently expressing MRA773G or MRS810M were incubated for 16 h with 10−9 m aldosterone in the presence of increasing concentrations of finerenone. The activity of MRA773G or MRS810M to transactivate the luciferase gene, which is under the control of a GRE-containing promoter, was determined by measuring the luciferase activity, which was then normalized to the β-galactosidase activity and to the value obtained with 10−9 m aldosterone alone. The values are means ± S.E. of at least three independent experiments performed in triplicate. GraphPad Prism software was used to fit the curves and to calculate the IC50 values. Statistical significance was calculated with the Mann-Whitney nonparametric U tests with unpaired data and two-tailed calculations. * (MRS810M versus MRWT) and # (MRA773G versus MRWT), p < 0.05.
Discussion
In this study, we delineated the molecular mechanisms underlying the antagonistic effects of the nonsteroidal compound, finerenone, at the biochemical and cellular levels. Binding studies revealed that this molecule has a higher antagonistic potency for MR than spironolactone. We provided evidence that finerenone is able to impair several key steps of the MR signaling pathway. In situ quantification of MR subcellular localization under hormonal treatment showed that finerenone alters nuclear translocation of the receptor more efficiently than does spironolactone. The critical phosphorylation-induced upshifting and the ligand-dependent down-regulation of MR following aldosterone treatment were both abrogated in the presence of finerenone. We also established that finerenone behaves as an inverse agonist ligand by demonstrating that RNA Pol II recruitment onto a regulatory sequence within the promoter region of the SCNN1A gene was not only impaired by finerenone in the presence of aldosterone but also markedly diminished under basal conditions, i.e. in the absence of aldosterone. Residues Asn-770 and Ser-810 within the LBD of MR were identified as critical contacts responsible for prominent potency and affinity of finerenone.
Docking experiments showed that finerenone establishes contacts with the Ala-773 and Ser-810 residues from the binding pocket, which are specific to MR as compared with the other steroid receptors. Of particular interest, these specific contacts are responsible for the high MR selectivity of this molecule because replacement of these residues by those present in androgen receptor, glucocorticoid receptor, and progesterone receptor (i.e. a glycine and a methionine) induces a dramatic decrease in finerenone antagonistic potency. Many efforts have been made over the last years to generate highly selective steroidal MR antagonists by introducing various substituents at the 7α and/or 9α-11α positions (6, 43–46). In sharp contrast, finerenone is the result of an intensive chemical optimization program on nonsteroidal dihydropyridine-based lead structures, which were identified in approximately one million screened molecules (13). The superimposition of finerenone and spironolactone indicated a global similar occupancy of the ligand-binding pocket. However, finerenone protrudes from the binding pocket, a feature unobserved with steroidal spirolactones. Moreover, in sharp contrast to spirolactone type molecules, finerenone is able to establish highly stabilizing contacts with the Ala-773 and Ser-810 residues that are MR selective, allowing this compound to display its remarkable MR selectivity. Indeed, the A773G mutation has no effect on the MR response to spironolactone, whereas it dramatically impairs that of finerenone. Moreover, the S810M mutation converts spironolactone to an agonist, whereas it almost completely abolishes MR sensitivity to finerenone. It has also to be noted that Ser-810 is a key residue to define the agonist or antagonist behavior of the ligand. The MR Ala-773 methyl side chain is surrounded by the phenyl moiety, the 2-methyl, and the carboxamide groups of finerenone. This compound interacts with Ala-773 like a socket on a joint ball. Indeed, its substitution by a leucine residue has been identified in a North American family to be responsible for an early onset form of arterial hypertension exacerbated during pregnancy (40). Progesterone, spironolactone, and eplerenone behave as full agonists at MRS810L, preventing their use as therapeutic agents for patients harboring this mutation (40). In sharp contrast, our results show that finerenone does not display any agonistic activity when acting through the MRS810L and, more interestingly, inactivates this mutant receptor. In comparison with the wild type MR, the MRS810A, MRS810M, and MRS810L mutant receptors have decreased sensitivities for finerenone by 9-, 88-, and 280-fold, respectively. This is in line with the loss of a hydrogen bond (to Ser-810), burying the polar functionality of the carboxamide side chain of finerenone in a more apolar environment and the introduction of steric hindrances (Met and Leu).
Upon aldosterone exposure, MR is shifted to the nucleus consecutively to conformational modification (47). Our results demonstrated that finerenone competes with aldosterone by inhibiting the nuclear accumulation of GFP-hMR and also impedes MR nuclear translocation slightly more efficiently than spironolactone. Compromising MR nuclear import constitutes a robust mechanism by which finerenone impairs transcriptional activation of MR target genes. Consistent with these observations, we and others have previously shown that other anti-mineralocorticoids like ZK91587, 18-vinylprogesterone, or spironolactone decrease MR nuclear localization by slowing down its nuclear transfer (16, 48). Several antagonists for steroid receptor have been shown to act through sequestration of the steroid receptor in the cytoplasmic compartment (34, 49–51). Our experiments indicated that finerenone does not permanently sequestrate MR into the cytoplasmic compartment because 24 h of antagonist exposure led to comparable MR nuclear accumulation as with aldosterone treatment.
Western blot analyses showed that aldosterone treatment, but not finerenone, rapidly induces an apparently higher molecular mass of both the endogenous and stably expressed GFP-hMR, suggesting a change in the receptor phosphorylation level (48, 52). Interestingly we also showed that this aldosterone-induced MR phosphorylation is associated with a reduction in the MR protein level, as was previously described (53). Interestingly, Tirard et al. (54) reported that a robust proteasome activity is required for optimal MR transactivation. Interestingly, finerenone likewise spironolactone totally blocked the aldosterone-induced MR phosphorylation, as well as its degradation, indicating that the antagonist compounds not only regulate receptor subcellular trafficking but also inhibit its turnover.
ChIP experiments demonstrated that MR antagonists differentially affect MR transcriptional complexes. Although spironolactone promoted MR and SRC-1 binding to the promoter of the SCNN1A gene, finerenone was unable to trigger such recruitment and moreover partly inhibited the basal MR and SRC-1 positioning onto the promoter regulatory sequence. These results indicated that spironolactone possesses at least partial aldosterone-like activity, whereas finerenone acts as an inverse agonist, indicating a distinct mechanism of action for both molecules. Three types of mechanism of antagonism have been described. The first one, known as “active,” concerns molecules that are characterized by a bulky substituent that protrudes from the binding pocket in the direction of the H12 helix, impairing it to adopt the agonist conformation. In this conformation, the receptor is able to recruit transcriptional corepressors. These molecules are also known as selective steroid receptor modulators because, depending on the cellular context and the coactivator/corepressor ratio, they may promote the transcriptional coactivator recruitment (55). The second mechanism of antagonism is called “passive.” It concerns small molecules such as spironolactone (22) or the progesterone receptor antagonist APR19 (34), which quickly dissociate from the receptor and are unable to maintain it in the agonist conformation (the H12 helix being mobile and not stabilized in the agonist conformation), impairing the recruitment of transcriptional coregulators. However, these molecules do not physically prevent the H12 helix from adopting the agonist conformation, suggesting that the equilibrium between mobile and agonist conformations could be displaced in favor of the agonistic one, allowing coactivator binding and explaining the reported partial agonistic activity of spironolactone (56). This interpretation is consistent with the spironolactone-induced recruitment of MR and SRC-1 onto the regulatory region of the SCNN1A gene without promoting that of the RNA Pol II. The third mechanism of antagonism called “bulky passive” was proposed for BR-4628, another nonsteroidal MR antagonist belonging to the same dihydropyridine-derived compound family (29). Finerenone is characterized by a bulky substituent most probably altering MR H12 helix conformation, and likewise, the BR-4628 compound rapidly dissociates from the receptor. These pharmacological properties of finerenone combined with specific molecular characteristics make this novel nonsteroidal MR ligand a full antagonistic compound with a mixed mechanism of antagonism. Finerenone is able to diminish the nuclear accumulation of MR, inhibits MR recruitment onto DNA target sequences, suppresses MR recycling, and even inhibits the S810L mutant MR. Associated with these peculiar features, finerenone has remarkable pharmacological properties.
In vivo, finerenone treatment prevented DOCA/salt-challenged rats from functional and structural heart and kidney damage at doses that did not reduce systemic blood pressure. It reduced cardiac hypertrophy, pro-B-type natriuretic peptide, and proteinuria more efficiently than eplerenone when comparing equinatriuretic doses (14). Moreover, finerenone demonstrated superior safety (i.e. less incidence of hyperkalemia and worsening of renal function) and efficacy compared with spironolactone in patients with heart failure and CKD (57). All these features could be related to its peculiar ability to inhibit MR and transcriptional coregulators recruitment onto DNA target sequences. Overall, finerenone may selectively control transcription of a subset of genes, conferring unique pharmacological indication to this molecule for clinical use. The identification of this subset of genes will allow a deeper understanding of its mechanism of action.
Author Contributions
M.-E. R.-O., M. L., and J. F. designed the study. L. A., F. L. B., K. L., S. V., M. R. F., J. A. K., A. H., and J. F. performed experiments. L. A., P. K., M. L., and J. F. wrote the paper. All authors analyzed the results, corrected, and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Lars Bärfacker for providing BAY 94–8862 (finerenone) and Dr. Peter Schmitt and Prof. Ulrich Pleiss for providing us with [3H]BAY 94–8862 ([3H]finerenone). We also thank Dr. Stuart Walsh for critical reading of the manuscript.
This work was supported by grants from INSERM, by Université Paris Sud Agence Nationale de la Recherche Grant 11-BSV1–028-01, and by Bayer Pharma AG Grants 05112/A11 and SCCV2395. P. K. and A. H. are employees of Bayer Pharma AG.
- MR
- mineralocorticoid receptor
- LBD
- ligand-binding domain
- SRC-1
- steroid receptor coactivator-1
- NI
- nuclear average intensity
- CI
- cytoplasmic average intensity
- Pol
- polymerase.
References
- 1. Viengchareun S., Le Menuet D., Martinerie L., Munier M., Pascual-Le Tallec L., Lombès M. (2007) The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl. Recept. Signal. 5, e012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Funder J. W. (2010) Aldosterone and mineralocorticoid receptors: past, present, and future. Endocrinology 151, 5098–5102 [DOI] [PubMed] [Google Scholar]
- 3. Tomaschitz A., Pilz S., Ritz E., Obermayer-Pietsch B., Pieber T. R. (2010) Aldosterone and arterial hypertension. Nat. Rev. Endocrinol. 6, 83–93 [DOI] [PubMed] [Google Scholar]
- 4. Rossi G. P., Bernini G., Desideri G., Fabris B., Ferri C., Giacchetti G., Letizia C., Maccario M., Mannelli M., Matterello M.-J., Montemurro D., Palumbo G., Rizzoni D., Rossi E., Pessina A. C., Mantero F., and PAPY Study Participants (2006) Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension 48, 232–238 [DOI] [PubMed] [Google Scholar]
- 5. Epstein M. (2006) Aldosterone blockade: an emerging strategy for abrogating progressive renal disease. Am. J. Med. 119, 912–919 [DOI] [PubMed] [Google Scholar]
- 6. Corvol P., Claire M., Rafestin-Oblin M. E., Michaud A., Roth-Meyer C., Menard J. (1977) Spirolactones: clinical and pharmacologic studies. Adv. Nephrol. Necker Hosp. 7, 199–215 [PubMed] [Google Scholar]
- 7. Pitt B., Zannad F., Remme W. J., Cody R., Castaigne A., Perez A., Palensky J., Wittes J. (1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N. Engl. J. Med. 341, 709–717 [DOI] [PubMed] [Google Scholar]
- 8. Corvol P., Michaud A., Menard J., Freifeld M., Mahoudeau J. (1975) Antiandrogenic effect of spirolactones: mechanism of action. Endocrinology 97, 52–58 [DOI] [PubMed] [Google Scholar]
- 9. Schane H. P., Potts G. O. (1978) Oral progestational activity of spironolactone. J. Clin. Endocrinol. Metab. 47, 691–694 [DOI] [PubMed] [Google Scholar]
- 10. Brass E. P. (1984) Effects of antihypertensive drugs on endocrine function. Drugs 27, 447–458 [DOI] [PubMed] [Google Scholar]
- 11. Juurlink D. N., Mamdani M. M., Lee D. S., Kopp A., Austin P. C., Laupacis A., Redelmeier D. A. (2004) Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N. Engl. J. Med. 351, 543–551 [DOI] [PubMed] [Google Scholar]
- 12. Kolkhof P., Borden S. A. (2012) Molecular pharmacology of the mineralocorticoid receptor: prospects for novel therapeutics. Mol. Cell Endocrinol. 350, 310–317 [DOI] [PubMed] [Google Scholar]
- 13. Bärfacker L., Kuhl A., Hillisch A., Grosser R., Figueroa-Pérez S., Heckroth H., Nitsche A., Ergüden J.-K., Gielen-Haertwig H., Schlemmer K.-H., Mittendorf J., Paulsen H., Platzek J., Kolkhof P. (2012) Discovery of BAY 94–8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. ChemMedChem. 7, 1385–1403 [DOI] [PubMed] [Google Scholar]
- 14. Kolkhof P., Delbeck M., Kretschmer A., Steinke W., Hartmann E., Bärfacker L., Eitner F., Albrecht-Küpper B., Schäfer S. (2014) Finerenone, a novel selective nonsteroidal mineralocorticoid receptor antagonist protects from rat cardiorenal injury. J. Cardiovasc. Pharmacol. 64, 69–78 [DOI] [PubMed] [Google Scholar]
- 15. Pascual-Le Tallec L., Lombès M. (2005) The mineralocorticoid receptor: a journey exploring its diversity and specificity of action. Mol. Endocrinol. 19, 2211–2221 [DOI] [PubMed] [Google Scholar]
- 16. Lombès M., Binart N., Delahaye F., Baulieu E. E., Rafestin-Oblin M. E. (1994) Differential intracellular localization of human mineralocorticosteroid receptor on binding of agonists and antagonists. Biochem. J. 302, 191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grossmann C., Ruhs S., Langenbruch L., Mildenberger S., Strätz N., Schumann K., Gekle M. (2012) Nuclear shuttling precedes dimerization in mineralocorticoid receptor signaling. Chem. Biol. 19, 742–751 [DOI] [PubMed] [Google Scholar]
- 18. Tallec L. P., Kirsh O., Lecomte M. C., Viengchareun S., Zennaro M. C., Dejean A., Lombès M. (2003) Protein inhibitor of activated signal transducer and activator of transcription 1 interacts with the N-terminal domain of mineralocorticoid receptor and represses its transcriptional activity: implication of small ubiquitin-related modifier 1 modification. Mol. Endocrinol. 17, 2529–2542 [DOI] [PubMed] [Google Scholar]
- 19. Coustal S., Fagart J., Davioud E., Marquet A. (1995) Synthesis of potential cytochrome P45011β-gererated intermediates. Tetrahedron 51, 3559–3570 [Google Scholar]
- 20. Auzou G., Fagart J., Souque A., Hellal-Lévy C., Wurtz J. M., Moras D., Rafestin-Oblin M. E. (2000) A single amino acid mutation of Ala-773 in the mineralocorticoid receptor confers agonist properties to 11β-substituted spirolactones. Mol. Pharmacol. 58, 684–691 [DOI] [PubMed] [Google Scholar]
- 21. Fagart J., Huyet J., Pinon G. M., Rochel M., Mayer C., Rafestin-Oblin M. E. (2005) Crystal structure of a mutant mineralocorticoid receptor responsible for hypertension. Nat. Struct. Mol. Biol. 12, 554–555 [DOI] [PubMed] [Google Scholar]
- 22. Fagart J., Wurtz J. M., Souque A., Hellal-Levy C., Moras D., Rafestin-Oblin M. E. (1998) Antagonism in the human mineralocorticoid receptor. EMBO J. 17, 3317–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fagart J., Seguin C., Pinon G. M., Rafestin-Oblin M. E. (2005) The Met852 residue is a key organizer of the ligand-binding cavity of the human mineralocorticoid receptor. Mol. Pharmacol. 67, 1714–1722 [DOI] [PubMed] [Google Scholar]
- 24. Huyet J., Pinon G. M., Fay M. R., Fagart J., Rafestin-Oblin M. E. (2007) Structural basis of spirolactone recognition by the mineralocorticoid receptor. Mol. Pharmacol. 72, 563–571 [DOI] [PubMed] [Google Scholar]
- 25. Gouilleux F., Sola B., Couette B., Richard-Foy H. (1991) Cooperation between structural elements in hormono-regulated transcription from the mouse mammary tumor virus promoter. Nucleic Acids Res. 19, 1563–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Deppe C. E., Heering P. J., Viengchareun S., Grabensee B., Farman N., Lombès M. (2002) Cyclosporine a and FK506 inhibit transcriptional activity of the human mineralocorticoid receptor: a cell-based model to investigate partial aldosterone resistance in kidney transplantation. Endocrinology 143, 1932–1941 [DOI] [PubMed] [Google Scholar]
- 27. Viengchareun S., Kamenicky P., Teixeira M., Butlen D., Meduri G., Blanchard-Gutton N., Kurschat C., Lanel A., Martinerie L., Sztal-Mazer S., Blot-Chabaud M., Ferrary E., Cherradi N., Lombès M. (2009) Osmotic stress regulates mineralocorticoid receptor expression in a novel aldosterone-sensitive cortical collecting duct cell line. Mol. Endocrinol. 23, 1948–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williams R. G., Kandasamy R., Nickischer D., Trask O. J. Jr., Laethem C., Johnston P. A., Johnston P. A. (2006) Generation and characterization of a stable MK2-EGFP cell line and subsequent development of a high-content imaging assay on the Cellomics ArrayScan platform to screen for p38 mitogen-activated protein kinase inhibitors. Methods Enzymol. 414, 364–389 [DOI] [PubMed] [Google Scholar]
- 29. Fagart J., Hillisch A., Huyet J., Bärfacker L., Fay M., Pleiss U., Pook E., Schäfer S., Rafestin-Oblin M. E., Kolkhof P. (2010) A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J. Biol. Chem. 285, 29932–29940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Claire M., Rafestin-Oblin M. E., Michaud A., Corvol P., Venot A., Roth-Meyer C., Boisvieux J. F., Mallet A. (1978) Statistical test of models and computerised parameter estimation for aldosterone binding in rat kidney. FEBS Lett. 88, 295–299 [DOI] [PubMed] [Google Scholar]
- 31. Li Y., Suino K., Daugherty J., Xu H. E. (2005) Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol. Cell 19, 367–380 [DOI] [PubMed] [Google Scholar]
- 32. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring: 1. method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 33. Jorgensen W. L., Maxwell D. S., Tirado-Rives J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 [Google Scholar]
- 34. Khan J. A., Tikad A., Fay M., Hamze A., Fagart J., Chabbert-Buffet N., Meduri G., Amazit L., Brion J. D., Alami M., Lombès M., Loosfelt H., Rafestin-Oblin M. E. (2013) A new strategy for selective targeting of progesterone receptor with passive antagonists. Mol. Endocrinol. 27, 909–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen T., Horwitz K. B., Lange C. A. (2001) Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol. Cell Biol. 21, 6122–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lonard D. M., Nawaz Z., Smith C. L., O'Malley B. W. (2000) The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 5, 939–948 [DOI] [PubMed] [Google Scholar]
- 37. Amazit L., Roseau A., Khan J. A., Chauchereau A., Tyagi R. K., Loosfelt H., Leclerc P., Lombès M., Guiochon-Mantel A. (2011) Ligand-dependent degradation of SRC-1 is pivotal for progesterone receptor transcriptional activity. Mol. Endocrinol. 25, 394–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Billan F., Khan J. A., Lamribet K., Viengchareun S., Bouligand J., Fagart J., Lombes M. (2015) Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 10.1096/fj.15-274266 [DOI] [PubMed] [Google Scholar]
- 39. Robert-Nicoud M., Flahaut M., Elalouf J. M., Nicod M., Salinas M., Bens M., Doucet A., Wincker P., Artiguenave F., Horisberger J. D., Vandewalle A., Rossier B. C., Firsov D. (2001) Transcriptome of a mouse kidney cortical collecting duct cell line: effects of aldosterone and vasopressin. Proc. Natl. Acad. Sci. U.S.A. 98, 2712–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Geller D. S., Farhi A., Pinkerton N., Fradley M., Moritz M., Spitzer A., Meinke G., Tsai F. T., Sigler P. B., Lifton R. P. (2000) Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science 289, 119–123 [DOI] [PubMed] [Google Scholar]
- 41. Bledsoe R. K., Madauss K. P., Holt J. A., Apolito C. J., Lambert M. H., Pearce K. H., Stanley T. B., Stewart E. L., Trump R. P., Willson T. M., Williams S. P. (2005) A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J. Biol. Chem. 280, 31283–31293 [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., Geller D. S. (2008) Helix 3-helix 5 interactions in steroid hormone receptor function. J. Steroid Biochem. Mol. Biol. 109, 279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossier B. C., Claire M., Rafestin-Oblin M. E., Geering K., Gäggeler H. P., Corvol P. (1983) Binding and antimineralocorticoid activities of spirolactones in toad bladder. Am. J. Physiol. 244, C24–C31 [DOI] [PubMed] [Google Scholar]
- 44. Casals-Stenzel J., Buse M., Wambach G., Losert W. (1984) The renal action of spirorenone and other 6β,7β; 15β,16β-dimethylene-17-spirolactones, a new type of steroidal aldosterone antagonists. Arzneimittelforschung 34, 241–246 [PubMed] [Google Scholar]
- 45. Chinn L. J., Salamon K. W., Desai B. N. (1981) A structure-activity relationship study of spirolactones: contribution of the cyclopropane ring to antimineralocorticoid activity. J. Med. Chem. 24, 1103–1107 [DOI] [PubMed] [Google Scholar]
- 46. de Gasparo M., Joss U., Ramjoué H. P., Whitebread S. E., Haenni H., Schenkel L., Kraehenbuehl C., Biollaz M., Grob J., Schmidlin J. (1987) Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J. Pharmacol. Exp. Ther. 240, 650–656 [PubMed] [Google Scholar]
- 47. Gekle M., Bretschneider M., Meinel S., Ruhs S., Grossmann C. (2014) Rapid mineralocorticoid receptor trafficking. Steroids 81, 103–108 [DOI] [PubMed] [Google Scholar]
- 48. Fejes-Tóth G., Pearce D., Náray-Fejes-Tóth A. (1998) Subcellular localization of mineralocorticoid receptors in living cells: effects of receptor agonists and antagonists. Proc. Natl. Acad. Sci. U.S.A. 95, 2973–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tyagi R. K., Lavrovsky Y., Ahn S. C., Song C. S., Chatterjee B., Roy A. K. (2000) Dynamics of intracellular movement and nucleocytoplasmic recycling of the ligand-activated androgen receptor in living cells. Mol. Endocrinol. 14, 1162–1174 [DOI] [PubMed] [Google Scholar]
- 50. Dauvois S., White R., Parker M. G. (1993) The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 106, 1377–1388 [DOI] [PubMed] [Google Scholar]
- 51. Devin-Leclerc J., Meng X., Delahaye F., Leclerc P., Baulieu E. E., Catelli M. G. (1998) Interaction and dissociation by ligands of estrogen receptor and Hsp90: the antiestrogen RU 58668 induces a protein synthesis-dependent clustering of the receptor in the cytoplasm. Mol. Endocrinol. 12, 842–854 [DOI] [PubMed] [Google Scholar]
- 52. Faresse N., Vitagliano J.-J., Staub O. (2012) Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J. 26, 4373–4382 [DOI] [PubMed] [Google Scholar]
- 53. Yokota K., Shibata H., Kobayashi S., Suda N., Murai A., Kurihara I., Saito I., Saruta T. (2004) Proteasome-mediated mineralocorticoid receptor degradation attenuates transcriptional response to aldosterone. Endocr. Res. 30, 611–616 [DOI] [PubMed] [Google Scholar]
- 54. Tirard M., Almeida O. F., Hutzler P., Melchior F., Michaelidis T. M. (2007) Sumoylation and proteasomal activity determine the transactivation properties of the mineralocorticoid receptor. Mol. Cell Endocrinol. 268, 20–29 [DOI] [PubMed] [Google Scholar]
- 55. Liu Z., Auboeuf D., Wong J., Chen J. D., Tsai S. Y., Tsai M.-J., O'Malley B. W. (2002) Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc. Natl. Acad. Sci. U.S.A. 99, 7940–7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cargnelli G., Trevisi L., Debetto P., Luciani S., Bova S. (2001) Effects of canrenone on aorta and right ventricle of the rat. J. Cardiovasc. Pharmacol. 37, 540–547 [DOI] [PubMed] [Google Scholar]
- 57. Pitt B., Kober L., Ponikowski P., Gheorghiade M., Filippatos G., Krum H., Nowack C., Kolkhof P., Kim S.-Y., Zannad F. (2013) Safety and tolerability of the novel nonsteroidal mineralocorticoid receptor antagonist BAY 94–8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur. Heart J. 34, 2453–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]