

Significance
Huntington’s disease (HD) is a progressive neurodegenerative disorder affecting the striatum. The striatal-enriched transcription factor COUP-TF-interacting protein 2 (Ctip2) is depleted in HD and has been identified as a putative transcription factor for the enzyme inositol polyphosphate multikinase (IPMK). IPMK displays soluble inositol phosphate kinase activity, lipid kinase activity, and several noncatalytic activities including its role as a transcriptional coactivator. We describe severe depletion in IPMK protein in HD patients and several animal and cell models of the disease. IPMK overexpression rescues the metabolic impairments in a cell model of HD. Furthermore, delivery of IPMK in a transgenic HD model improves pathological changes and motor performance. The Ctip2–IPMK–Akt signaling pathway provides a previously unidentified therapeutic target for HD.
Keywords: Huntington's disease, inositol polyphosphate multikinase, IPMK, Ctip2, Akt
Abstract
Huntington’s disease (HD) is a progressive neurodegenerative disease caused by a glutamine repeat expansion in mutant huntingtin (mHtt). Despite the known genetic cause of HD, the pathophysiology of this disease remains to be elucidated. Inositol polyphosphate multikinase (IPMK) is an enzyme that displays soluble inositol phosphate kinase activity, lipid kinase activity, and various noncatalytic interactions. We report a severe loss of IPMK in the striatum of HD patients and in several cellular and animal models of the disease. This depletion reflects mHtt-induced impairment of COUP-TF-interacting protein 2 (Ctip2), a striatal-enriched transcription factor for IPMK, as well as alterations in IPMK protein stability. IPMK overexpression reverses the metabolic activity deficit in a cell model of HD. IPMK depletion appears to mediate neural dysfunction, because intrastriatal delivery of IPMK abates the progression of motor abnormalities and rescues striatal pathology in transgenic murine models of HD.
Huntington’s disease (HD) is an autosomal dominant disorder manifesting profound neurodegeneration and dementia with motor abnormalities deriving from the expansion of glutamine repeats in mutant huntingtin (mHtt) (1). This disease mainly affects the striatum, resulting in the dysfunction and death of striatal medium spiny neurons (2). Although the genetics of HD are well delineated, the specific mechanisms whereby mHtt leads to selective neurodegeneration have been elusive. Numerous defects of neurotransmission have been reported in HD, such as glutamate-mediated excitotoxicity of neurons (3), as well as abnormalities in trophic factor signaling, including the brain-derived neurotrophic factor (BDNF) (4). Dysregulation of various transcription factors also occurs in HD (5). Krainc and colleagues noted that mHtt binds with high affinity to the transcription factor Sp1, impairing biosynthesis of diverse proteins and leading to abnormalities in dopamine receptor disposition (6).
The striatal-enriched transcription factor COUP-TF-interacting protein 2 (Ctip2) (Bcl11b) (7) similarly binds mHtt and is depleted in the striatum of HD patients (8). Thomas and colleagues subsequently performed a genome-scale Ctip2 overexpression screen identifying Ctip2 targets, which include inositol polyphosphate multikinase (IPMK) (9). IPMK physiologically generates inositol tetrakisphosphate (IP4) and inositol pentakisphosphate (IP5) (10, 11) as part of the inositol polyphosphate pathway. The family of inositol polyphosphates also includes inositol 1,4,5-trisphosphate (IP3), which releases intracellular calcium, as well as higher inositol phosphates including those containing diphosphate moieties with energetic pyrophosphate properties (12, 13). IPMK additionally possesses PI3-kinase (lipid kinase) activity and is a physiologic source of phosphatidylinositol (3,4,5)-triphosphate [PIP3 or PtdIns (3,4,5)P3] (14, 15). Furthermore, IPMK displays functions independent of its kinase activity. It binds and stabilizes the mTOR complex (16) and serves as a transcriptional coactivator for CREB-binding protein (17), p53 (18), and serum response factor (19). Despite these insights into the functions of IPMK, its physiologic and pathologic regulation have been largely unexplored.
We report that IPMK is profoundly depleted in HD, reflecting the influence of mHtt upon Ctip2, a putative striatal-enriched transcription factor for IPMK. We show that IPMK depletion mediates neural dysfunction, because virally mediated restoration of IPMK delays the progression of behavioral abnormalities and rescues striatal pathology.
Results
IPMK Protein Is Depleted in HD Striatum.
A useful model of HD is the immortalized striatal progenitor cell line with 111 glutamine repeats, STHdhQ111/Q111 (Q111), and the control cell line with seven glutamine repeats, STHdhQ7/Q7 (Q7) (20). IPMK protein is depleted by 75% in Q111 cells (Fig. 1A). This deficit appears to reflect, at least in part, defective IPMK transcription, because IPMK mRNA levels are reduced by 40% in Q111 cells (Fig. 1B). Furthermore, IPMK produces the soluble inositol phosphates IP4 and IP5, both of which are depleted in Q111 cells (Fig. S1 A and B). Levels of inositol hexakisphosphate (IP6), the catalytic product of IP5 2-kinase (IPPK or IPK1), are unaltered, likely because of elevated IPPK protein expression in Q111 cells (Fig. S1C). The R6/2 transgenic murine model of HD involves about 150 glutamine repeats (21). We examined IPMK levels in the striatum of R6/2 mice, because this portion of the brain is most prominently affected in HD (2). Striatal IPMK protein levels are reduced in R6/2 striatum (Fig. 1C) and in cortex and hippocampus but not cerebellum (Fig. S1D). The zQ175 knockin model of HD (22) displays a similar decrease in IPMK protein expression in the striatum (Fig. 1D). Most importantly, IPMK levels are diminished by 75% in the striatum of human patients with HD (Fig. 1E). Information on age, sex, and postmortem delay (PMD) of these striatal tissues is provided in Table S1.
Fig. 1.

IPMK protein and mRNA levels are decreased in HD. (A) IPMK protein levels are decreased in Q111 cells. Bars represent means ± SEM normalized to β-actin (n = 3). **P < 0.01 relative to Q7 cells. (B) IPMK mRNA levels are also reduced in Q111 cells. Bars represent means ± SEM normalized to β-actin mRNA (n = 3). **P < 0.01 relative to Q7 cells. (C) R6/2 striatal samples contain less IPMK than littermate controls. (D) IPMK levels also are decreased in zQ175 striatum. In C and D, bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to wild type. (E) IPMK protein is decreased in postmortem HD striatum. Bars represent means ± SEM normalized to GAPDH (n = 5 for control group and n = 7 for HD group). ***P < 0.001 relative to control.
Fig. S1.

Catalytic activities of IPMK. (A) Both IPMK and the p110 PI3-kinase (PI3K) phosphorylate PtdIns(4,5)P2 (PIP2) to produce PtdIns(3,4,5)P3 (PIP3). PtdIns(4,5)P2 also can be cleaved by phospholipase C (PLC), thereby generating Ins(1,4,5)P3 (IP3). IPMK additionally phosphorylates IP3 to produce inositol-1,4,5,6-tetrakisphosphate [Ins(1,4,5,6)P4] and inositol-1,3,4,5-tetrakisphosphate [Ins(1,3,4,5)P4], both known as IP4. Ins(1,3,4,5)P4 also results from the catalytic activity of Ins(1,4,5)P3-kinase (IP3K). IPMK phosphorylates IP4 to produce inositol-1,3,4,5,6-pentakisphosphate (IP5), [Ins(1,3,4,5,6)P5], which is subsequently phosphorylated by inositol 1,3,4,5,6-pentakisphosphate 2-kinase (IPPK or IPK1) to produce Ins(1,2,3,4,5,6)P6. The higher inositol phosphates, containing pyrophosphate moieties, are not shown. Circles indicate phosphate groups. (B) IP4 and IP5 levels are depleted in Q111 cells, but IP6 levels remain normal. Bars represent mean disintegrations/min (D.P.M.) ± SEM normalized to myo-inositol levels (n = 6). *P < 0.05, **P < 0.01 relative to Q7 controls. (C) IPPK levels are elevated in Q111 cells. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to Q7 control. (D) IPMK protein levels are depleted in the cortex and hippocampus of R6/2 mice but not in the cerebellum. Bars represent means ± SEM normalized to β-actin (n = 4). *P < 0.05 relative to wild-type control.
Table S1.
Patient demographics for postmortem control and HD striatal tissues
| Sample | Grade | Sex | Age in y | PMD in h |
| 1 | Control | Male | 60 | 6 |
| 2 | Control | Female | 68 | 11 |
| 3 | Control | Male | 64 | 12 |
| 4 | Control | Male | 42 | 19 |
| 5 | Control | Female | 46 | 24 |
| 6 | HD grade 4 | Male | 63 | 5 |
| 7 | HD grade 4 | Male | 63 | 15 |
| 8 | HD grade 4 | Male | 41 | 21 |
| 9 | HD grade 4 | Male | 41 | 8 |
| 10 | HD grade 3 | Female | 50 | 4 |
| 11 | HD grade 3 | Male | 43 | 23 |
| 12 | HD grade 3 | Female | 45 | 24 |
IPMK Transcription and Protein Stability Are Impaired in HD Cells.
Ctip2 recently was revealed as a putative transcription factor for IPMK (9). Accordingly, we explored its relevance to HD. We confirmed the loss of Ctip2 in R6/2 striatum (Fig. S2A), cortex, and hippocampus, but not cerebellum, which does not express Ctip2 (Fig. S2B). Depletion of Ctip2 in Q7 cells reduces IPMK levels by about 60% (Fig. 2A). Conversely, overexpression of Ctip2 reverses the loss of IPMK protein in Q111 cells, restoring them to normal values, but does not alter IPMK levels in Q7 cells (Fig. 2B). Thus, IPMK depletion in this cellular model of HD occurs in part through transcriptional regulation by Ctip2.
Fig. S2.
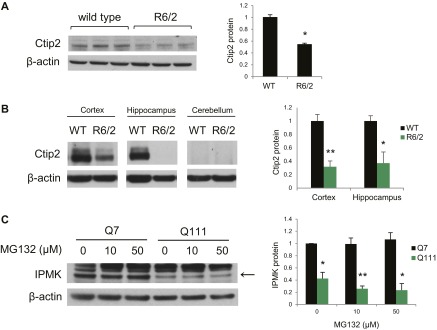
Ctip2 protein is depleted in HD. (A) Ctip2 protein levels are reduced in the R6/2 striatum relative to wild-type mice. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to wild type. (B) Ctip2 protein levels are also reduced in the cortex and hippocampus of R6/2 mice. Ctip2 is not expressed in the cerebellum. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05, **P < 0.01 relative to wild-type control. (C) Treatment of Q7 and Q111 cells with the proteasomal inhibitor MG132 did not alter IPMK protein expression. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05, **P < 0.01 relative to Q7 control.
Fig. 2.

Transcriptional regulation and protein stability of IPMK are altered in Q111 striatal cells. (A) Ctip2 knockdown resulted in decreased IPMK expression. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to scrambled siRNA control. (B) Overexpression of Ctip2 in Q7 and Q111 cells rescues IPMK protein levels. Ctip2 also increases the expression of the unknown upper band. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to Q111 empty vector control. (C) IPMK protein levels in Q7 and Q111 cells following treatment with the translational inhibitor cycloheximide (CHX). Bars represent means ± SEM normalized to β-actin (n = 4). For Q7 cells, **P < 0.01 and *P < 0.05 relative to the Q7 0-h CHX treatment control. For Q111 cells, **P < 0.01 and ****P < 0.0001 relative to the Q111 0-h CHX control. (D) Bafilomycin (Baf), an inhibitor of lysosomal degradation, restores IPMK protein levels in the Q111 cells without altering IPMK protein levels in the Q7 cells. Bars represent means ± SEM normalized to β-actin (n = 3). *P < 0.05 relative to Q111 empty vector control. (E) Coimmunoprecipitation assay of HEK293 cells transfected with plasmids expressing myc-tagged human IPMK and the N-terminal fragment of either wild-type Htt (Htt-flag) or mHtt (mHtt-flag). IPMK binds selectively to the N-terminal fragment of mHtt but not to wild-type Htt.
Because the depletion of IPMK protein in Q111 cells is substantially greater than the loss of IPMK mRNA, we investigated whether HD is associated with alterations in IPMK protein stability. We monitored the turnover of IPMK by examining its rate of depletion following inhibition of protein synthesis with cycloheximide (Fig. 2C). In Q7 cells, cycloheximide treatment requires about 8 h to secure 45% depletion. In contrast, 4 h after cycloheximide administration, IPMK protein levels in Q111 cells are reduced about 80%. The calculated half-life for IPMK turnover in Q7 cells is 8.9 h, which is reduced to 2 h in Q111 cells. Impairing the proteasomal degradation pathway using MG132 does not alter IPMK protein levels in Q7 and Q111 cells (Fig. S2C). In contrast, inhibition of the lysosomal degradation pathway using bafilomycin rescues IPMK protein levels in Q111 cells (Fig. 2D). Thus, IPMK depletion in Q111 cells is associated with decreased protein stability involving lysosomal degradation as well as diminished transcription. One potential mechanism for depleting IPMK might involve mHtt binding IPMK and increasing turnover. We examined this possibility by monitoring binding between the two proteins (Fig. 2E). IPMK binds robustly to the N-terminal fragment of mHtt (N171–82Q) but not to wild-type Htt (N171–18Q).
IPMK Expression Rescues mHtt-Induced Deficits in Mitochondrial Metabolic Activity.
We wondered whether the depletion of IPMK in HD is responsible for the molecular and behavioral abnormalities of HD. If so, restoring the depleted IPMK should be beneficial. We assessed mitochondrial metabolic activity of cells by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay (Fig. 3A). The metabolic activity of Q111 cells is only half that of Q7 cells (23). Overexpressing IPMK alleviates this abnormality (Fig. 3A). IPMK possesses both inositol phosphate kinase and PI3-kinase activities as well as displaying various noncatalytic actions (11, 14–17). To ascertain which of these activities mediates the beneficial effects of IPMK, we overexpressed IPMK K129A–S235A (IPMK-KASA), which is devoid of both inositol phosphate kinase and PI3-kinase activities. We also overexpressed the Arabidopsis thaliana ortholog, atIPK2β, which possesses inositol phosphate kinase but not PI3-kinase activity and has been shown to restore inositol phosphate production in IPMK−/− mouse embryonic fibroblasts (14). Neither IPMK-KASA nor atIPK2β rescue the depressed metabolic activity of Q111 cells (Fig. 3B). The lack of activity of IPMK-KASA indicates that the catalytic activity of IPMK is required for rescue. The inactivity of atIPK2β suggests that the PI3-kinase activity of IPMK is responsible for restoring the metabolic activity of Q111 cells.
Fig. 3.

The lipid kinase activity of IPMK rescues the mitochondrial metabolic activity deficit in Q111 striatal cells and restores Akt signaling. (A) IPMK overexpression rescues the mitochondrial metabolic activity deficit in Q111 cells as measured by the MTT assay. (B) Overexpression of the kinase-dead mutant of IPMK (IPMK-KASA) or atIPK2β does not rescue the metabolic activity deficit measured by the MTT assay. In A and B, bars represent means ± SEM (n = 4). *P < 0.05, **P < 0.01, ****P < 0.0001 relative to the Q7 empty vector control unless otherwise indicated. (C) Akt phosphorylation at the T308 site [p-Akt (T308)] is decreased in Q111 cells. (D) Akt phosphorylation at the S473 site [p-Akt (S473)] also is reduced in Q111 cells. In C and D, bars represent means ± SEM normalized to total Akt (n = 3). **P < 0.01 relative to the Q7 control. (E) IPMK overexpression rescues the loss of p-Akt (T308) in Q111 cells but not in Q7 cells. (F) IPMK overexpression similarly rescues the loss of p-Akt (S473) in Q111 cells. In E and F, bars represent means ± SEM normalized to total Akt (n = 4). *P < 0.05 relative to the Q111 empty vector control sample.
PIP3 physiologically activates Akt protein kinase. Akt signaling deficits have been described previously in HD striatum and lymphoblasts (24). We observe a 70% depletion of phospho-Akt levels in Q111 cells at the T308 and S473 sites (Fig. 3 C and D). The loss of phospho-Akt at both sites is reversed by overexpressing IPMK in Q111 cells (Fig. 3 E and F).
Adeno-Associated Virus Serotype 2–Mediated Delivery of IPMK Improves Psychomotor Performance in a Transgenic Mouse Model of HD.
Is the IPMK deficit in HD responsible for the pathological and motor abnormalities of HD? We investigated whether direct administration of IPMK-expressing adeno-associated virus serotype 2 (AAV2) in the striatum of R6/2 HD mice (Fig. 4 A and B) influences the pathology and behavioral phenotype of these animals over time (Fig. S3A). Although no effects are observed on weight and survival (Fig. S3 B and C), viral overexpression of IPMK in the striatum of R6/2 mice reduces the number of mHtt aggregates and the size of these aggregates by ∼75% and 30%, respectively, at 10 wk of age (Fig. 4 C–E). These pathological changes correspond with the delay in motor deficits observed in R6/2 animals. Repletion of IPMK restores central locomotor activity of the R6/2 mice to levels that are not significantly lower than those of wild-type mice at 6 wk (Fig. 4F). However, IPMK overexpression does not significantly improve rotarod performance (Fig. S4A), likely because of the earlier onset of this particular deficit. In a balance beam model, the time to cross is increased eightfold in R6/2 mice compared with wild-type animals (Fig. 4G). This time is reduced by half in IPMK-replenished mice. We also evaluated a composite phenotype (25) of HD abnormalities, which consist of hindlimb clasping, gait abnormalities, kyphosis, and ledge walking (Fig. 4H). The composite phenotype score is reduced almost by half with IPMK repletion. This reduction is consistent with improvement in gait, specifically stride length, in R6/2 mice receiving the IPMK-expressing virus (Fig. S4B). Furthermore, there is reduced fore footprint–hind footprint overlap in the R6/2 animals, which appears to improve with IPMK delivery. We did not observe significant differences in balance beam and composite scores relative to wild-type mice before 10 wk of age.
Fig. 4.

Virus-mediated expression of IPMK delays motor impairment of R6/2 mice. (A) Coronal section of mouse brain demonstrating intrastriatal virus expression 2 wk postinjection. (B) Striatal tissue obtained from R6/2 mice 10 wk postinjection with either GFP control AAV2 (denoted “C”) or IPMK-expressing AAV2 (denoted “I”) confirms increased IPMK expression (arrow) in wild-type and R6/2 animals receiving IPMK-expressing AAV2. The expression of the unknown upper band is also increased in tissues obtained from animals receiving the IPMK virus. (C) EM48-positive mHtt aggregates are present in R6/2 striatum and are absent in wild-type striatum. Virus-mediated overexpression of IPMK reduces the number and the size of mHtt aggregates. (Scale bar, 50 µm.) (D) Quantitation of number of mHtt aggregates per 40× field of view measuring 0.1 mm2. Bars represent the mean number of aggregates ± SEM (n = 3 animals). **P < 0.01 relative to the number of R6/2 control aggregates. (E) Quantitation of the size of mHtt aggregates based on the cross-sectional area of aggregates. Bars represent the mean size of aggregates ± SEM (n = 3 animals). *P < 0.05 relative to the size of aggregates in R6/2 control sections. (F) IPMK overexpression delays impairment of central locomotor activity. Bars represent mean beam breaks ± SEM (n = 9–12 animals per group). *P < 0.05, **P < 0.01 relative to either wild-type or R6/2 mice injected with control virus. (G) IPMK restores motor coordination and balance assessed by balance beam performance. Bars represent the mean of the total time required to cross the beam ± SEM (n = 7–10 animals per group). **P < 0.01, ****P < 0.0001 relative to either wild-type or R6/2 mice injected with control virus. (H) General phenotype based on clasping, kyphosis, gait, and ledge walking is presented as a composite score and is improved by IPMK overexpression. Bars represent means ± SEM (n = 9–10 animals per group). *P < 0.05 relative to R6/2 mice receiving control virus. (I) In normal striatal cells, Ctip2 up-regulates IPMK expression. IPMK displays several functions, including the lipid kinase activity, which enhances Akt signaling, as well as a soluble inositol phosphate kinase activity and various noncatalytic activities. In HD, Ctip2 transcriptional activity and expression is inhibited by mHtt, resulting in decreased IPMK transcription. Decreased IPMK protein stability, likely caused by the selective interaction with mHtt, further reduces IPMK protein levels, resulting in the loss of Akt phosphorylation. (J) Our current model indicates that Ctip2 up-regulates IPMK, which in turn enhances Akt phosphorylation. Rhes also has been shown to increase Akt phosphorylation in healthy cells. Additional mechanisms in HD cells (indicated in red), include Akt-mediated phosphorylates and inhibition of mHtt. Furthermore, Ctip2 and IPMK are both impaired by mHtt. The roles of Rhes in modulating Ctip2 function in healthy and HD cells and the effect of Rhes on Akt in the presence of mHtt remain to be elucidated.
Fig. S3.

Survival and weight of wild-type and R6/2 mice injected with either control or IPMK-expressing virus. (A) Intrastriatal injections were performed at age 3 wk followed by behavior testing at 6, 8, and 10 wk of age. Pathology was assessed at 10 wk. (B) R6/2 mice injected with either control or IPMK virus show a similar and gradual decrease in weight over time. IPMK overexpression did not affect wild-type mice (n = 10 for wild type with control virus, n = 9 for wild type with IPMK virus, n = 17 for R6/2 with control virus, and n = 13 for R6/2 with IPMK virus). (C) The Kaplan–Meier curve indicates that striatal overexpression of IPMK did not alter survival in wild-type or R6/2 mice (n = 10 for wild type with control virus, n = 11 for wild type with IPMK virus, n = 11 for R6/2 with control virus, n = 11 for R6/2 with IPMK virus).
Fig. S4.

Effect of virus-mediated expression of IPMK on additional motor performance. (A) IPMK overexpression does not affect rotarod performance at 6, 8, and 10 wk of age. Bars represent the mean time to fall in seconds ± SEM (n = 7–11 animals per group). *P < 0.05, **P < 0.01, ***P < 0.001 relative to wild-type mice injected with either control or IPMK virus. (B) IPMK overexpression improves R6/2 animal gait, specifically stride length at 10 wk of age. (Upper) Forepaw and hindpaw prints are shown in red and blue, respectively. (Lower) Bars represent mean stride length in centimeters ± SEM (n = 6–10 animals per group). *P < 0.05, **P < 0.01, ****P < 0.0001 relative to wild-type mice or R6/2 mice injected with control virus as indicated.
Discussion
In the present study we report a dramatic depletion of IPMK in the striatum of humans with HD as well as in Q111 HD cells and in the R6/2 and zQ175 murine models of HD. The depletion of IPMK occurs at both the transcriptional and protein stability levels and corresponds with decreased Akt signaling (Fig. 4I). IPMK depletion appears to mediate, at least in part, the pathology and motor deficits in HD, because viral expression of IPMK in the striatum of R6/2 mice, the brain region primarily affected in clinical HD, delays locomotor deficits of the animals and reduces the number and size of aggregates. Although grade III and IV HD are advanced and are characterized by severe neuronal loss in the striatum, numerous proteins are down-regulated at these stages because of mHtt-mediated transcriptional dysregulation rather than merely reflecting pathogenic tissue loss (5, 6, 8, 26–28). Additional studies during earlier stages of HD may discriminate differential sensitivities of specific neuronal populations in HD.
The striatal-enriched transcription factor Ctip2 appears to determine IPMK transcription. Its overexpression reverses the IPMK depletion in Q111 cells. Ctip2 protein itself also is selectively expressed in striatal medium spiny neurons (29), the cell type uniquely lost in HD (2). The depletion of Ctip2 in the striatum, cortex, and hippocampus of R6/2 mice is consistent with the altered expression pattern of IPMK in these mice, which reflects the HD pathology in the cortex and additional brain tissues (30). Interestingly, Ctip2 overexpression in Q7 cells does not change IPMK protein levels, suggesting a potential negative feedback effect of IPMK or other targets on Ctip2 transcriptional activity.
Altered IPMK protein stability and lysosomal degradation also contribute to the loss of IPMK protein in the cellular model of HD. Although both macroautophagy and the ubiquitin proteasome system are impaired in HD, chaperone-mediated autophagy is constitutively up-regulated in early stages of the disease, thereby increasing the turnover of both wild-type Htt and mHtt fragments (31). The selective interaction of IPMK with the N-terminal fragment of mHtt might explain the loss of IPMK through lysosomal degradation in Q111 cells but not Q7 cells.
We provide several lines of evidence that the depletion of IPMK is pathogenic in HD. Overexpressing IPMK restores to normal the depressed mitochondrial metabolic activity of Q111 cells, an action that appears to be determined by the PI3-kinase activity of IPMK. IPMK and the p110 PI3-kinase act coordinately in generating PIP3 (14), a classic stimulant of Akt (32). Phospho-Akt depletion in Q111 cells is rescued by overexpressing IPMK. Overexpression of constitutively active Akt is sufficient to rescue pathologic phenotypes, such as the formation of nuclear inclusions, in primary rat brain cultures expressing mHtt (33). Akt also regulates inclusions indirectly by phosphorylating other proteins such as the ADP ribosylation factor-interacting protein arfaptin2, which rescues mHtt-induced proteasomal impairment (34).
mHtt is phosphorylated by Akt at the S421 site, which restores fast axonal transport by altering the mHtt interaction with dynactin (35). Conversely, excitotoxic stimulation of NMDA receptors (NMDARs) reduces mHtt S421 phosphorylation (36). Because the R6/2 animals used in this study express the N-terminal fragment of mHtt rather than full-length mHtt, the observed effects of IPMK likely do not require direct phosphorylation of mHtt by Akt. Thus, through the multiple downstream effects of Akt, IPMK deficit may account for the notably pleiotropic manifestations of HD. IPMK also may have additional functions, because its lipid kinase activity is required for the selective export of mRNA (37).
Several signaling pathways implicated in HD converge on Akt. The BDNF pathway is altered in HD because of decreased transcription and release of BDNF at the corticostriatal synapses (4, 38) as well as impaired TrkB receptor signaling (39). Similarly, synaptic (but not extrasynaptic) NMDARs enhance Akt phosphorylation and neuroprotection (40).
Intrastriatal delivery of IPMK in R6/2 mice improved motor deficits in the R6/2 model of HD. IPMK overexpression had the greatest effects on balance beam performance and gait. The motor deficits assessed using these tests appear during later symptomatic stages (after age 8.5 wk) in R6/2 animals (41). Mild to no effects were observed in open field and rotarod testing, respectively, likely because the corresponding motor deficits occur at age 5 wk. Thus, earlier delivery of IPMK may have greater effects on the behavioral phenotype of these R6/2 animals. In addition to motor deficits, early clinical manifestations of HD include cognitive symptoms (42). IPMK regulates the induction of immediate early genes required for learning, memory, and behavior, because IPMK-deleted mice display aberrant spatial memory (17).
Additional clinical features of HD include peripheral organ dysfunction such as weight loss, skeletal muscle wasting, and metabolic and endocrine alterations (43). IPMK influences metabolism through its inhibition of AMP-activated kinase (AMPK) activation (44). AMPK promotes catabolic pathways while inhibiting various anabolic pathways such as cholesterol and triglyceride synthesis (45). Interestingly, a pathologic increase in AMPK phosphorylation has been demonstrated in HD patients and in the R6/2 model (46). IPMK delivery in R6/2 animals did not improve body weight and survival, probably because IPMK was expressed only in the striatum. Conceivably, widespread expression of IPMK in HD models may improve peripheral organ dysfunction.
The small G protein Ras homolog enriched in striatum (Rhes) sumoylates mHtt, resulting in cytotoxicity (47) (Fig. 4J), which is further enhanced through the interaction of Rhes and mHtt with acyl-CoA binding domain containing 3 (ACBD3) (48). An in vivo screen for SUMO1 substrates demonstrated that Ctip2 is sumoylated (49). Perhaps Rhes also modulates the Ctip2–IPMK–Akt signaling pathway. Interestingly, Rhes has been shown to recruit the regulatory subunit of PI3K to the cell membrane, subsequently enhancing Akt phosphorylation in healthy cells (Fig. 4J) (50). We also reported a marked depletion of cystathionine γ-lyase (CSE) in HD (51). CSE is a rate-limiting enzyme in the biosynthesis of cysteine and generation of the gasotransmitter hydrogen sulfide (51, 52). Treating R6/2 mice with cysteine or cysteine precursors markedly improves the motor performance of R6/2 mice, similar to the improvement that we have observed with viral delivery of IPMK.
Our findings suggest that the IPMK depletion in HD is pathogenic by dint of diminished PI3-kinase activity with less PIP3 available to activate Akt. Approaches targeting the Ctip2–IPMK pathway thus may ameliorate neuronal dysfunction in HD.
Experimental Procedures
Cell Cultures and Reagents.
The immortalized striatal cell lines STHdhQ7/Q7 (Q7) and STHdhQ111/Q111 (Q111) express endogenous wild-type Htt and mHtt with seven or 111 glutamine repeats, respectively. These cell lines were provided by M. MacDonald of the Department of Neurology, Massachusetts General Hospital, Boston. The Q7 and Q111 cells were maintained at 33 °C in DMEM supplemented with 10% (vol/vol) FBS, 2 mM l-glutamine, 400 μg/mL Geneticin, and antibiotics (penicillin and streptomycin). Experiments were performed in the absence of Geneticin.
Animals.
Animals were housed and cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (53), and animal experiments were approved by the Johns Hopkins University Animal Care and Use Committee (JHU ACUC). Animals were kept on a 12-h light/dark cycle and were provided food and water ad libitum.
Postmortem Brain Tissues.
Striatal tissues from control and HD patients were obtained from J. Troncoso and O. Pletnikov (Brain Resource Center, Johns Hopkins University).
Stereotaxic Surgery.
AAV2 containing either a GFP-only control vector or IPMK was generated by Vector BioLabs at a titer of 3.1 × 1012 genome copies (GC)/mL Three-week-old male mice were anesthetized using 300 μL Avertin (20 mg/mL solution). Virus was injected at the following coordinates: anterior (A) −0.8, lateral (L) 2, ventral (V) −3.5; A −0.8, L 2, V −3.3; A −0.8, L 2, V −3.1; and A −0.8, L 2, V −2.9 for a total of 4 μL virus in each striatum.
Statistical Analysis.
Statistical analysis was performed using Excel software (Analysis ToolPak). Student’s t test and single-factor ANOVA were performed. All error bars represent ± SEM. Significance was determined as P < 0.05.
Acknowledgments
We thank R. Barrow and J. Crawford for their assistance and G. Ho, M. Koldobskiy, S. Vandiver, P. Scherer, R. Mealer, P. Guha, C. Fu, M. Ma, B. Selvakumar, R. Tokhunts, and other members of the S.H.S. laboratory as well as R. Kuruvilla, M. Mattson, and H. Song for discussions. M. MacDonald provided the Q7 and Q111 cells, and J. Troncoso and O. Pletnikov provided the postmortem tissues. This work was funded by the US Public Health Service Grant MH-18501 (to S.H.S.), CHDI Foundation (to S.H.S.), Medical Scientist Training Grant T32 GM007309 (to I.A.), National Science Foundation Graduate Research Fellowship Award (to J.C.G.) and Thomas Shortman Training Fund Graduate Scholarship (to J.C.G.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511810112/-/DCSupplemental.
References
- 1.The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 2.Reiner A, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85(15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beal MF, et al. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321(6066):168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- 4.Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293(5529):493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 5.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19(5):233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- 6.Dunah AW, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296(5576):2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 7.Desplats PA, et al. Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J Neurochem. 2006;96(3):743–757. doi: 10.1111/j.1471-4159.2005.03588.x. [DOI] [PubMed] [Google Scholar]
- 8.Desplats PA, Lambert JR, Thomas EA. Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington’s disease. Neurobiol Dis. 2008;31(3):298–308. doi: 10.1016/j.nbd.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang B, et al. Genome-wide identification of Bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS One. 2011;6(9):e23691. doi: 10.1371/journal.pone.0023691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odom AR, Stahlberg A, Wente SR, York JD. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science. 2000;287(5460):2026–2029. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- 11.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol. 1999;9(22):1323–1326. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty A, Kim S, Snyder SH. Inositol pyrophosphates as mammalian cell signals. Sci Signal. 2011;4(188):re1. doi: 10.1126/scisignal.2001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irvine RF, Schell MJ. Back in the water: The return of the inositol phosphates. Nat Rev Mol Cell Biol. 2001;2(5):327–338. doi: 10.1038/35073015. [DOI] [PubMed] [Google Scholar]
- 14.Maag D, et al. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc Natl Acad Sci USA. 2011;108(4):1391–1396. doi: 10.1073/pnas.1017831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Resnick AC, et al. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci USA. 2005;102(36):12783–12788. doi: 10.1073/pnas.0506184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim S, et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13(2):215–221. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu R, et al. Inositol polyphosphate multikinase is a transcriptional coactivator required for immediate early gene induction. Proc Natl Acad Sci USA. 2013;110(40):16181–16186. doi: 10.1073/pnas.1315551110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu R, et al. Inositol polyphosphate multikinase is a coactivator of p53-mediated transcription and cell death. Sci Signal. 2013;6(269):ra22. doi: 10.1126/scisignal.2003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim E, et al. Inositol polyphosphate multikinase is a coactivator for serum response factor-dependent induction of immediate early genes. Proc Natl Acad Sci USA. 2013;110(49):19938–19943. doi: 10.1073/pnas.1320171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trettel F, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9(19):2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 21.Mangiarini L, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 22.Menalled LB, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One. 2012;7(12):e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gines S, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet. 2003;12(5):497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- 24.Colin E, et al. Akt is altered in an animal model of Huntington’s disease and in patients. Eur J Neurosci. 2005;21(6):1478–1488. doi: 10.1111/j.1460-9568.2005.03985.x. [DOI] [PubMed] [Google Scholar]
- 25.Guyenet SJ, et al. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp. 2010;39:1787–1789. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boutell JM, et al. Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin. Hum Mol Genet. 1999;8(9):1647–1655. doi: 10.1093/hmg/8.9.1647. [DOI] [PubMed] [Google Scholar]
- 27.Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413(6857):739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 28.Steffan JS, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97(12):6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arlotta P, Molyneaux BJ, Jabaudon D, Yoshida Y, Macklis JD. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J Neurosci. 2008;28(3):622–632. doi: 10.1523/JNEUROSCI.2986-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de la Monte SM, Vonsattel JP, Richardson EP., Jr Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J Neuropathol Exp Neurol. 1988;47(5):516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Koga H, et al. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31(50):18492–18505. doi: 10.1523/JNEUROSCI.3219-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 33.Humbert S, et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2(6):831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 34.Rangone H, et al. Phosphorylation of arfaptin 2 at Ser260 by Akt Inhibits PolyQ-huntingtin-induced toxicity by rescuing proteasome impairment. J Biol Chem. 2005;280(23):22021–22028. doi: 10.1074/jbc.M407528200. [DOI] [PubMed] [Google Scholar]
- 35.Zala D, et al. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum Mol Genet. 2008;17(24):3837–3846. doi: 10.1093/hmg/ddn281. [DOI] [PubMed] [Google Scholar]
- 36.Metzler M, et al. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J Neurosci. 2010;30(43):14318–14329. doi: 10.1523/JNEUROSCI.1589-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wickramasinghe VO, et al. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell. 2013;51(6):737–750. doi: 10.1016/j.molcel.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 38.Zuccato C, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35(1):76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 39.Plotkin JL, et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. 2014;83(1):178–188. doi: 10.1016/j.neuron.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YB, et al. Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J Neurosci. 2012;32(35):11919–11929. doi: 10.1523/JNEUROSCI.3852-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19(8):3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross CA, Tabrizi SJ. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10(1):83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 43.van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington’s disease. Lancet Neurol. 2009;8(8):765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- 44.Bang S, et al. AMP-activated protein kinase is physiologically regulated by inositol polyphosphate multikinase. Proc Natl Acad Sci USA. 2012;109(2):616–620. doi: 10.1073/pnas.1119751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase--development of the energy sensor concept. J Physiol. 2006;574(Pt 1):7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ju TC, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol. 2011;194(2):209–227. doi: 10.1083/jcb.201105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science. 2009;324(5932):1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sbodio JI, Paul BD, Machamer CE, Snyder SH. Golgi protein ACBD3 mediates neurotoxicity associated with Huntington’s disease. Cell Reports. 2013;4(5):890–897. doi: 10.1016/j.celrep.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tirard M, et al. In vivo localization and identification of SUMOylated proteins in the brain of His6-HA-SUMO1 knock-in mice. Proc Natl Acad Sci USA. 2012;109(51):21122–21127. doi: 10.1073/pnas.1215366110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bang S, Steenstra C, Kim SF. Striatum specific protein, Rhes regulates AKT pathway. Neurosci Lett. 2012;521(2):142–147. doi: 10.1016/j.neulet.2012.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paul BD, et al. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington’s disease. Nature. 2014;509(7498):96–100. doi: 10.1038/nature13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paul BD, Snyder SH. H₂S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 53. Committee on Care and Use of Laboratory Animals (1996) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23.