

ABSTRACT
O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) is an essential cellular enzyme that posttranslationally modifies nuclear and cytoplasmic proteins via O-linked addition of a single N-acetylglucosamine (GlcNAc) moiety. Among the many targets of OGT is host cell factor 1 (HCF-1), a transcriptional regulator that is required for transactivation of the immediate-early genes of herpes simplex virus (HSV). HCF-1 is synthesized as a large precursor that is proteolytically cleaved by OGT, which may regulate its biological function. In this study, we tested whether inhibition of the enzymatic activity of OGT with a small molecule inhibitor, OSMI-1, affects initiation of HSV immediate-early gene expression and viral replication. We found that inhibiting OGT's enzymatic activity significantly decreased HSV replication. The major effect of the inhibitor occurred late in the viral replication cycle, when it reduced the levels of late proteins and inhibited capsid formation. However, depleting OGT levels with small interfering RNA (siRNA) reduced the expression of HSV immediate-early genes, in addition to reducing viral yields. In this study, we identified OGT as a novel cellular factor involved in HSV replication. Our results obtained using a small molecule inhibitor and siRNA depletion suggest that OGT's glycosylation and scaffolding functions play distinct roles in the replication cycle of HSV.
IMPORTANCE Antiviral agents can target viral or host gene products essential for viral replication. O-GlcNAc transferase (OGT) is an important cellular enzyme that catalyzes the posttranslational addition of GlcNAc sugar residues to hundreds of nuclear and cytoplasmic proteins, and this modification regulates their activity and function. Some of the known OGT targets are cellular proteins that are critical for the expression of herpes simplex virus (HSV) genes, suggesting a role for OGT in the replication cycle of HSV. In this study, we found that OGT is required for efficient expression of viral genes and for assembly of new virions. Thus, we identify OGT as a novel host factor involved in the replication of HSV and a potential target for antiviral therapy.
INTRODUCTION
Herpes simplex virus (HSV) causes a variety of infections that can lead to considerable morbidity and mortality in immunocompromised patients and neonates (1). Lytic replication of HSV occurs in a highly regulated fashion with sequential expression of immediate-early (IE), early (E), and late (L) viral genes (1). IE gene transcription occurs in the absence of de novo viral protein synthesis and requires the viral VP16 tegument protein, which, in infected cells, forms a complex with two host transcription factors: octamer-binding protein 1 (Oct-1) and the transcriptional coregulator host cell factor 1 (HCF-1) (2–5). The VP16–Oct-1–HCF-1 multiprotein complex binds to conserved core elements on IE gene promoters and recruits cellular transcription and chromatin-modifying factors to initiate high levels of viral gene expression (6–8). HCF-1 is essential for VP16-mediated transactivation of IE viral promoters, and its depletion abrogates initiation of gene expression (9). In uninfected cells, HCF-1 is synthesized as a large precursor protein that is posttranslationally modified and cleaved at six central amino acid repeats (pro-repeats) into N- and C-terminal subunits that remain stably associated (10, 11). The proteolytic cleavage of the HCF-1 precursor is carried out by the cellular enzyme O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) (12–14), an essential glycosyltransferase that catalyzes posttranslational addition of a single N-acetylglucosamine (GlcNAc) sugar to Ser and Thr residues of hundreds of nuclear and cytoplasmic proteins via an O-glycosidic linkage (15). In a recently defined mechanism, the glycosyltransferase active site of OGT binds and cleaves the C-terminal portion of the HCF-1 pro-repeat sequence while catalyzing O-GlcNAcylation of several residues within the HCF-1 N terminus (14). Cleavage of HCF-1 is required for cell cycle progression (16), and it can also alter the interaction of HCF-1 with transcriptional coactivators and repressors (17). Although one study shows that proteolysis of HCF-1 may affect the expression of HSV IE genes (13), the importance of HCF-1 cleavage for its ability to bind VP16 and transactivate viral gene expression is unclear.
In addition to HCF-1, OGT O-GlcNAc glycosylates many cellular proteins involved in regulation of gene expression, including RNA polymerase II and its associated transcription factors, transcriptional coactivators and repressors, histones, and chromatin remodeling factors, as well as components of the translational machinery. O-GlcNAcylation plays a pivotal role in regulating the cellular localization, stability, interactions, and activity of the modified proteins (15, 18). Because HSV relies on the cellular transcriptional machinery for expression of its genes, we determined if OGT plays a role during HSV infection. To address this question, we utilized a recently developed small molecule inhibitor of OGT, OSMI-1, which was shown to specifically inhibit the catalytic activity of OGT, decrease global O-GlcNAcylation, and inhibit O-GlcNAc modification of known OGT substrates (19). We observed that blocking OGT's catalytic activity significantly decreases replication of HSV-1, HSV-2, and cytomegalovirus. The major effect of the inhibitor on HSV was exerted late in the viral replication cycle. In contrast, we demonstrate that depletion of OGT by small interfering RNA (siRNA) decreases viral IE gene expression. Our results thus far identify OGT as a novel factor involved in multiple steps of the replication cycle of HSV. In total, our results suggest that OGT may have distinct scaffolding and glycosylation functions in HSV replication.
MATERIALS AND METHODS
Cell culture and viruses.
Human foreskin fibroblasts (HFFs; ATCC catalog number CRL-1634) and Vero, HEp-2, HeLa, and HEK-293 cells were obtained from the ATCC (Manassas, VA). HFFs were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS). Vero and HEp-2 cells were cultured in DMEM with 5% FBS and 5% heat-inactivated bovine calf serum (BCS). HSV-1 KOS and HSV-2 186 strains were propagated in Vero cells, and both viruses were titrated on Vero cells. K26GFP recombinant virus (20) was a gift from Prashant Desai (Johns Hopkins School of Medicine). The human cytomegalovirus (HCMV) laboratory strain AD169 was kindly provided by Donald Coen (Harvard Medical School).
Viral infections and inhibitor treatment.
For infections, viruses were diluted in phosphate-buffered saline (PBS) containing 1% BCS and 0.1% glucose and incubated with cells for 1 h on a 37°C shaker. After viral adsorption, the cells were washed with PBS and overlaid with DMEM supplemented with 1% BCS and containing dimethyl sulfoxide (DMSO) (0.5%) or OSMI-1 at the desired concentrations. Progeny virus titers were determined by a plaque assay as described previously (21). To assess HCMV replication in the presence of OSMI-1, HFFs were infected with HCMV at the desired multiplicities of infection (MOIs). After incubation for 1 h at 37°C, the viral inoculum was removed and replaced with DMEM (10% FBS) containing DMSO or OSMI-1. The cells were incubated for a total of 96 h, and the inhibitor was replenished at 48 h postinfection (hpi). To determine viral titers, cells and supernatants were collected in an equal volume of sterile milk and stored at −80°C. Serial dilutions of each sample were titrated on HFF monolayers and overlaid with methylcellulose and DMEM at 1:1 ratio. Viral plaques were counted at 14 days postinfection. The OGT inhibitor OMSI-1 and the control compound PG34 were identified and characterized in a recent publication (19).
siRNA transfections.
HFFs were transfected with an siRNA pool targeting OGT (Santa Cruz) or with a nontarget control siRNA pool (Dharmacon) using DarmaFECT 2 transfection reagent (Dharmacon) at a final siRNA concentration of 50 nM according to the manufacturer's instructions. At 72 h after transfection, the knockdown efficiency was determined by quantitative reverse transcription-PCR (qRT-PCR) and Western blotting, and the cells were used for HSV-1 infections.
Isolation of nuclear, supernatant, and cell-associated DNA.
Nuclei were isolated from HSV-1-infected, siRNA-transfected HFFs using the NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Scientific). The nuclei were resuspended in PBS, and DNA was isolated using the DNA blood and tissue kit (Qiagen) according to the manufacturer's instructions. The purity of the nuclear and cytosolic fractions was assessed by an immunoblot detecting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or histone H3. Relative gene copy numbers were determined by qRT-PCR.
qRT-PCR analysis.
To measure transcript levels, total RNA was harvested from cells using TRIzol reagent (Invitrogen). Samples were DNase treated with the DNA-free kit (Ambion), and 1 μg of RNA was reverse transcribed using the high-capacity cDNA reverse transcription kit (Applied Biosystems). Gene copy numbers and transcripts were quantified using the Fast SYBR green PCR master mix (Applied Biosystems) on an ABI 7900 detection system (ABI). Relative copy numbers of genes and transcripts were determined using the standard curve method. Transcript levels were normalized to 18S rRNA. Viral and cellular DNA copy numbers were normalized to levels of a GAPDH pseudogene (22).
SDS-PAGE and immunoblotting.
HFFs were infected with HSV-1 KOS at the desired MOI and treated with OSMI-1 or a DMSO vehicle control. Cell lysates were collected at the times postinfection indicated below and processed as previously described (23). The antibodies used were mouse anti-O-GlcNAc (RL2; Santa Cruz Biotechnology; 1:1,000), mouse anti-ICP27 (ab31631; Abcam; 1:1,000), rabbit anti-gC (R46; 1:1,000), rabbit anti-actin (ab8227; Abcam; 1:2,000), rabbit anti-ICP8 (3-83; 1:2,000), mouse anti-gD (ab6507; Abcam; 1:10,000), mouse anti-GAPDH (ab9484; Abcam; 1:4,000), and rabbit anti-histone H3 (ab1791; Abcam, 1:3,000). Rabbit and mouse horseradish peroxidase (HRP)-conjugated antibodies (Santa Cruz Biotechnology) were used for secondary detection at 1:5,000.
Immunofluorescence.
HFFs grown on glass coverslips were mock or HSV-1 infected and overlaid with medium containing DMSO or 50 μM OSMI-1-containing medium. At the desired times postinfection, cells were fixed with 2% formaldehyde and processed as described previously (24). The coverslips were incubated with mouse anti-VP5 antibody (HA018; EastCoast Bio; 1:500) followed by a secondary anti-mouse Alexa Fluor 488 antibody (1:1,000; Jackson ImmunoResearch). Cells were imaged using a Nikon TE2000 w/C1 point scanning confocal microscope at a ×60 magnification.
Electron microscopy.
HFFs were grown to confluent monolayers in wells of a 12-well plate, infected with HSV-1 KOS at an MOI of 0.1, and maintained in the presence of DMSO or 50 μM OSMI-1. At 18 hpi, the infected cells were fixed with 2.5% paraformaldehyde, 5% glutaraldehyde, and 0.1 M cacodylate buffer (pH 7.2) for 1 h at room temperature. Fixed cells were embedded in resin and sectioned for imaging. For imaging of supernatant-purified virions, the samples were diluted in PBS and adsorbed onto a hydrophilic carbon-coated grid, followed by a negative staining with 0.75% uranyl acetate. Sections were examined by Tecnai G2 Spirit Bio Twin electron microscope.
RESULTS
Inhibition of OGT activity in HFFs.
The small molecule inhibitor of OGT, OSMI-1 (Fig. 1A), was previously developed and optimized from a high-throughput screen hit (19). The compound was evaluated for OGT inhibition in an in vitro assay and in several mammalian cell lines (19). In this study, we first evaluated whether OSMI-1 can inhibit OGT activity in human foreskin fibroblasts (HFFs) by examining the change in global O-GlcNAc modification in the presence of the compound. Treatment of HFFs with increasing concentrations of OSMI-1 for 24 h led to a dose-dependent decrease in global O-GlcNAc levels, as measured by immunoblotting (Fig. 1B), without significant toxic effects on the cells (Fig. 1C).
FIG 1.
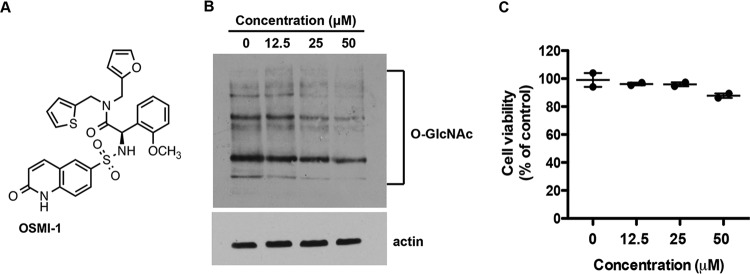
OSMI-1 inhibits OGT activity in HFFs. (A) Chemical structure of OSMI-1. (B) Effect of OSMI-1 on O-GlcNAcylation in HFF cells. HFFs were incubated with increasing concentrations of OSMI-1 for 24 h. Cell lysates were analyzed by immunoblotting using O-GlcNAc-specific RL2 antibody. (C) HFFs were treated with increasing concentrations of OSMI-1. After 24 h, cell viability was measured with CellTiter-Glo Luminescent Cell Viability assay (Promega) and expressed as a percentage of DMSO-treated control cells.
Effect of OGT inhibition on HSV-1 yields.
To test whether OGT inhibition affects HSV-1 replication, we infected HFFs with HSV-1 strain KOS at a multiplicity of infection (MOI) of 0.1 PFU per cell, and immediately following viral adsorption, we treated the cells with either a vehicle control (DMSO) or increasing concentrations of OSMI-1. At 48 hpi, we determined the production of progeny virus by a plaque assay on Vero cells. OGT inhibition by OSMI-1 reduced viral yields over 1,000-fold at the highest concentration of inhibitor (50 μM) (Fig. 2A). We further tested whether the OSMI-1 effect was MOI dependent by infecting HFFs with HSV-1 KOS virus at low or high MOIs and treating them with OSMI-1 at a 50 μM concentration, which previously resulted in the highest yield reduction. We observed a decrease in viral yield in OSMI-1-treated HFFs at both low (950-fold) and high (890-fold) MOIs (Fig. 2B), arguing that OSMI-1 efficiently inhibits a single cycle of replication as well as multiple cycles. OGT inhibition also caused a dose-dependent reduction in HSV-1 yields in HeLa (Fig. 2C), HEp-2 carcinoma cells (Fig. 2D), and HEK-293 cells (Fig. 2E), demonstrating that the effect of the inhibitor was independent of the cell type. A cell viability assay after OSMI-1 treatment showed that the inhibitor did not significantly affect cell viability, as 91% (HeLa), 89% (HEp-2), and 92% (HEK-293) of the cells remained viable after 24 h of treatment (results not shown). We found the concentration of OMSI-1 that resulted in a 50% decrease in HSV-1 yield (EC50) in HFFs to be 6.34 μM (Fig. 2F).
FIG 2.

Effect of OSMI-1 on HSV-1 replication. (A) HFFs were infected with HSV-1 at an MOI of 0.1 and incubated with media containing increasing concentrations of OSMI-1 or vehicle (DMSO). At 48 hpi, viral yields were determined by plaque assay on Vero cells. (B) HFFs infected at MOIs of 0.1 and 5 and treated with 50 μM OSMI-1 or DMSO control for 48 and 24 h, respectively. Bars represent mean values of viral yields ± SEMs (n = 3). Statistical analysis was done using a multiple measurements one-way analysis of variance (ANOVA) followed by Bonferroni posttest. **, P < 0.001. (C to E) HeLa (C), HEp-2 (D), and HEK-293 (E) cells were infected with HSV-1 at an MOI of 5 and treated with increasing concentrations of OSMI-1 for 24 h. Viral titers were determined by plaque assays on Vero cells. (F) EC50 for OSMI-1 was determined in HFFs infected with KOS at an MOI of 0.1 and treated for 24 h. Values are expressed as percent yield relative to cells treated with DMSO vehicle control. EC50 was calculated using nonlinear regression curve fitting with a variable slope (GraphPad Prism 5 software). (G) HFFs were infected with HSV-1 at an MOI of 5 and treated with 50 μM PG34, OSMI-1, or DMSO for 24 h, and viral yields were measured by a plaque assay.
To determine whether the antiviral effect of OSMI-1 was due to OGT inhibition and not due to off-target effects, we tested a structural analog of OSMI-1 in an HSV-1 inhibition assay. The analog PG-34 is structurally similar to OSMI-1, but it contains a phenylalanine in place of the 2-methoxyphenylglycine of OSMI-1 (19). PG34 has similar cell permeability but demonstrates poor in vitro inhibitory activity against OGT and does not reduce global O-GlcNAcylation in cells (19). HSV-1 viral yields remained relatively unaffected by treatment with a 50 μM concentration of the control compound (Fig. 2G). These results were consistent with the interpretation that OSMI-1's effects on viral yields are due to inhibition of OGT.
Effect of OGT inhibition on replication of HSV-2 wild-type and acyclovir-resistant viruses.
To determine if OSMI-1 is effective at inhibition of HSV-2, we infected HFFs with HSV-2 186 virus at 0.1 or 5 PFU/cell and treated the cells with 50 μM OSMI-1 for the duration of the infection. At 24 hpi, the amount of infectious virus was determined by plaque assay. OSMI-1 decreased viral titers by 1,700-fold at the low MOI, whereas there was a lower (60-fold) yield reduction at the high MOI (Fig. 3A). We also tested an HSV-2 mutant lacking thymidine kinase (TK−) that is resistant to acyclovir (25). We infected HFFs with wild-type (WT) HSV-2 186 or the TK− mutant virus at 0.1 PFU/cell and measured viral yields. The TK− virus was resistant to acyclovir treatment, while OSMI-1 reduced replication of both WT HSV-2 and the TK− mutant (Fig. 3B). In total, the results argued that OGT is necessary for efficient replication of HSV.
FIG 3.
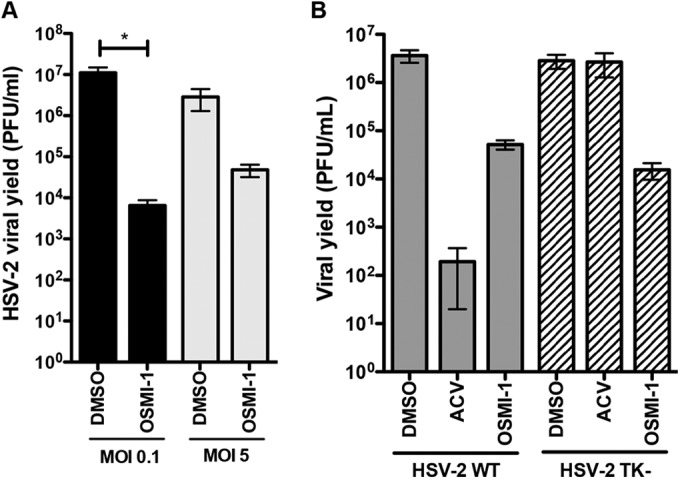
Effect of OSMI-1 on HSV-2 replication. (A) HFFs were infected with WT HSV-2 at MOIs of 0.1 and 5 and treated with 50 μM OSMI-1 or DMSO. Viral yields were determined at 24 hpi (MOI of 5) and 48 hpi (MOI of 0.1) by plaque assays on Vero cells. Means ± SEMs (n = 3) were analyzed by one-way ANOVA with repeated measures followed by Bonferroni posttest. *, P < 0.05. (B) HFFs were infected with WT HSV-2 or 186 TK− virus at an MOI of 0.1, followed by addition of 50 μM OSMI-1, 100 μM acyclovir (ACV), or DMSO control. Virus yield was determined at 24 hpi by titration on Vero cells. Bars represent mean values ± SEMs from three independent experiments.
Effect of OGT inhibition on replication of HCMV.
In addition, we tested the effect of OSMI-1 on replication of human cytomegalovirus (HCMV), another human herpesvirus. We infected HFFs with the HCMV lab-adapted AD169 virus strain at MOIs of 0.1 and 1, and we treated infected cells with OSMI-1 or DMSO. Viral yields measured at 96 h after infection showed that the inhibitor reduced HCMV yields in a dose-dependent manner at both low (Fig. 4A) and high (Fig. 4B) MOIs. Treatment with OSMI-1 did not significantly affect cell viability, as over 89% of the cells remained viable during the duration of the treatment (results not shown). These results argue that OGT is involved in the replication cycle of other herpesviruses.
FIG 4.
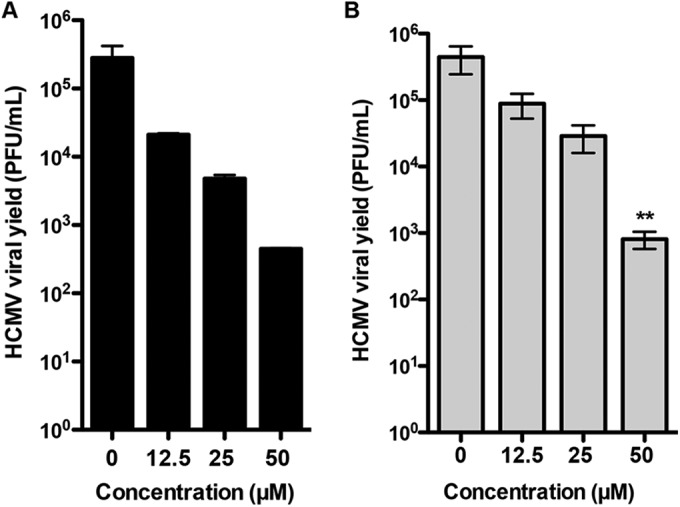
Effect of OSMI-1 on HCMV replication. HFFs were infected with HCMV AD169 at MOIs of 0.1 (A) and 1 (B) and treated with 50 μM OSMI-1 or DMSO. Viral yields were determined at 96 hpi by plaque assays on HFFs. Means ± SEMs (n = 2) of viral yields were analyzed by one-way ANOVA with repeated measures followed by Bonferroni posttest. *, P < 0.05.
Effect of OGT inhibition on HSV-1 gene expression.
To determine the stage in the HSV-1 replication cycle that is targeted by the compound, we measured the levels of IE, E, and L transcripts in the presence of OSMI-1. We infected HFFs with HSV-1 at an MOI of 5 and measured IE ICP27, E ICP8, and L gC gene transcripts by qRT-PCR at 3, 6, and 9 hpi. Levels of transcripts for all gene classes were not affected by OSMI-1 over the course of 9 h, as shown by equal levels of IE ICP27 (Fig. 5A), E ICP8 (Fig. 5B), and L gC (Fig. 5C) transcripts. Treatment of the cells with OSMI-1 for 16 h prior to HSV-1 infection also had no effect on the expression of viral genes (results not shown). Similarly, the levels of the IE ICP27 and E ICP8 proteins did not decrease throughout the course of infection (Fig. 5D), and a slight increase in protein levels was even observed at 6 and 9 hpi. However, the levels of the L gC protein were markedly reduced in the presence of the inhibitor. To confirm that this decrease was not specific for gC, we measured the expression of two other late proteins, glycoprotein D (gD) and the major capsid protein VP5, and we saw a similar decrease in both proteins at 6 and 9 hpi (Fig. 5E). The decreases in gD and VP5 levels were not observed at the mRNA level (results not shown), arguing that the inhibitor either interferes with the synthesis of late viral proteins or increases their turnover. To confirm that OSMI-1 acts at late times of the viral replication cycle, we performed a time-of-addition assay (Fig. 5F). We added OSMI-1 to HSV-1-infected HFFs at different times after infection and analyzed the viral yields at 24 hpi. We observed that antiviral activity of OSMI-1 was preserved when OSMI-1 was added to infected cells up to 4 h after infection but declined thereafter. Together, these results argued that OSMI-1 acts late in the viral cycle.
FIG 5.

Effect of OSMI-1 on HSV-1 gene expression. HFFs were infected with HSV-1 at MOI of 5 or mock infected and treated with a vehicle control or 50 μM OSMI-1. Total RNA was harvested at 3, 6, and 9 h postinfection, and the levels of transcripts from IE ICP27 (A), E ICP8 (B), and L gC (C) genes were measured by qRT-PCR. The mRNA levels were normalized to cellular 18S rRNA. The results are expressed as means ± SEMs from three independent experiments. (D) HFFs were infected with HSV-1 at an MOI of 5 and treated with 50 μM OSMI-1 or DMSO. Protein lysates were collected at 3, 6, and 9 hpi and analyzed for expression of ICP27, ICP8, and gC or GAPDH as a loading control by immunoblotting. (E) HFFs were treated as described for panel B and blotted for expression of gD, VP5, and GAPDH to serve as a loading control. (F) Time-of-addition assay. HFFs were infected with HSV-1 at an MOI of 5 and treated with DMSO or 50 μM OSMI-1, which was added at different times after infection, as indicated. HSV-1 replication was measured after 24 h by plaque assay. Results represent means ± SEMs from two independent experiments.
Effect of OGT inhibition on viral capsid formation.
Late viral genes encode structural proteins that are mainly involved in the formation of progeny virions (1). Of those, VP5 is the major constituent of the viral capsid and, together with VP26, VP19C, and VP23, forms the immature capsid shell (26). Because we saw lower VP5 protein levels in the presence of the inhibitor, we speculated that capsid assembly and subsequent virion maturation would be negatively affected by OSMI-1. We first examined the effect of OSMI-1 on capsid assembly by evaluating the localization of VP5 and the minor capsid protein VP26 by immunofluorescence. We infected HFFs with the HSV-1 K26GFP recombinant virus that expresses the VP26 small capsid protein fused to green fluorescent protein (GFP), followed by treatment with DMSO control or 50 μM OSMI-1. At 8 hpi, we examined the localization of GFP-VP26 and VP5 by immunofluorescence. Control-treated cells showed GFP-VP26 and VP5 colocalization in a punctate pattern within the cell nuclei (Fig. 6A). In the presence of the inhibitor, we observed fewer and less distinct GFP puncta, and while VP5 appeared to be concentrated in the nucleus, it did not form distinct foci that colocalized with GFP-VP26. Moreover, in many infected cells, we found more cytoplasmic VP5 staining. To confirm that capsid assembly was impaired in OSMI-1-treated cells, we examined infected cells by transmission electron microscopy (TEM). The vehicle control-treated cell nuclei contained a mixed population of capsids in different stages of assembly: empty A capsids, scaffold-containing B capsids, and DNA-filled, mature C capsids (Fig. 6B, panel a). In contrast, the nuclei of OSMI-1-treated cells (Fig. 6B, panel b) contained fewer capsids than those of the control-treated cells. Moreover, particles budding out of inhibitor-treated infected cells lacked the characteristic virion shape (Fig. 6B, panels c and d). EM images of virions isolated from the medium of OSMI-1-treated cells revealed an abnormal shape compared to control cells (Fig. 6, panel e). To quantify the effect of OSMI-1 on capsid formation, we determined the number and the types of viral capsids in the DMSO- and OMSI-1-treated cells (Fig. 7C). In the presence of the inhibitor, we observed an approximately 10-fold decrease in the number of A, B, and C capsids. Therefore, we concluded that inhibition of OGT by OMSI-1 disrupts the assembly of viral capsids and the production of mature viral particles.
FIG 6.

Effect of OGT inhibition on capsid assembly. (A) Fibroblasts were infected with K26GFP recombinant virus at an MOI of 10 and treated with 50 μM OSMI-1 or DMSO. Cells were fixed at 8 hpi, stained with an antibody against VP5, and imaged with a Nikon TE2000 w/C1 point scanning confocal microscope (60× objective). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). (B) Electron microscopy of viral capsids and virions in OMSI-1-treated cells. (a to d) HFF cells were infected with HSV-1 at an MOI of 5 and treated with 50 μM OSMI-1 (b and d) or a vehicle control (a and c) for 16 h. Infected cells were fixed and processed for EM. White arrows point to C capsids, black arrows point to A capsids, and arrowheads designate B capsids. N, nucleus; C, cytoplasm. (e and f) KOS-infected HFFs were treated with a vehicle (e) or OMSI-1 (f) for 24 h. Extracellular virus released in the supernatant was isolated by centrifugation and resuspended in PBS. Virions were stained by uranyl acetate and imaged by EM. (C) Capsids were quantified by counting the number and type of capsids in 15 random microscopic fields. Counts are generated from two independent experiments.
FIG 7.

Effect of OGT depletion on HSV replication. (A) HFFs were transfected with a nontargeting or OGT-specific siRNA pools. At 72 hpi, the levels of OGT were measured by qRT-PCR and Western blotting. (B) siControl- or siOGT-transfected cells were infected with HSV-1 at an MOI of 0.1. At 24 hpi, viral yields were determined by plaque assay on Vero cells. The graph presents means (±SEMs) of viral yields from 4 independent experiments. *, P < 0.05. (C) Cell viability at 96 h after siRNA treatment was measured with CellTiter-Glo Luminescent Cell Viability assay (Promega) and expressed as percentage of siControl-treated cells. (D) siRNA-treated cells were infected with HSV-1 at an MOI of 5, and total protein was harvested at 6 hpi. Levels of viral IE (ICP27), E (ICP8), and L (gC) proteins were measured by Western blotting. Cellular actin was used as a loading control. (D) siRNA-treated cells were infected with HSV-1 at an MOI of 5, and total RNA was isolated at 4 hpi. Relative transcript levels of viral IE (ICP27), E (ICP8), and L (gC) genes were measured by qRT-PCR and normalized to cellular 18S rRNA. Values represent means from 3 independent experiments (±SEMs). (E) siControl- or siOGT-treated HFFs were infected with HSV-1 at an MOI of 5, and at 2 hpi, cells were fractionated into cytoplasmic and nuclear extracts. DNA isolated from nuclear fractions and total cellular DNA were analyzed for ICP27 levels by qRT-PCR. Viral gene copy numbers were normalized to cellular GAPDH pseudogene levels. Values represent means from three independent experiments (±SEMs).
Depletion of OGT reduces viral replication and gene expression.
To validate the OSMI-1 phenotype using an siRNA approach, we transfected HFFs with a pool of siRNAs targeting OGT (siOGT) or with a control nonsilencing RNA pool (siControl). At 72 h after siRNA transfection, the levels of OGT mRNA and protein in the siOGT-transfected samples were notably reduced (Fig. 7A). To assess the effect of OGT knockdown on viral yields, we treated cells with siRNAs, infected them with HSV-1 at an MOI of 0.1, and measured viral yields by a plaque assay at 24 hpi. We observed a 10-fold decrease in the production of infectious virus in the OGT knockdown cells (Fig. 7B) that was not due to nonspecific effects such as cell toxicity due to depletion of OGT (Fig. 7C). When we assessed viral mRNA, we observed a 10-fold downregulation in IE (ICP4, ICP27), early (ICP8), and late (gC) transcripts (Fig. 7D); therefore, the reduction of viral yield correlated with the reduction of viral transcripts. Similarly, viral IE (ICP4), E (ICP8), and L (gC) proteins were decreased in the OGT-depleted cells (Fig. 7E). This phenotype was specific for viral genes because OGT depletion appeared to have no effect on the levels of cellular TATA binding protein (TBP) or 18S rRNA transcripts (results not shown). O-GlcNAc modification is the most common posttranslational modification of nuclear pore complex (NPC) proteins (27). Depletion of OGT has been shown to reduce the expression of several members of the NPC (27), and inhibition of O-GlcNAc modification can inhibit protein transport into the nucleus (28). Because the NPC facilitates attachment of viral capsids and delivery of viral DNA into the nucleus (29), we tested whether the OGT-mediated decrease in viral gene expression was due to fewer viral genomes entering the nuclei of OGT-depleted cells. We measured the amount of viral DNA in the nuclei of OGT-depleted and control cells by qRT-PCR and observed equal numbers of viral genomes (Fig. 7F). The absence of GAPDH protein in the Western blot analysis confirmed the purity of the nuclear fractions (results not shown). Thus, the decrease in viral yield and viral gene expression upon OGT knockdown, together with the OSMI-1 phenotype, confirmed that OGT plays a role in the replication cycle of HSV.
DISCUSSION
Posttranslational addition of an O-GlcNAc sugar moiety to nuclear and cytoplasmic proteins by the conserved cellular enzyme OGT plays a major role in intracellular signaling by altering the activity, stability, localization, and interactions of a myriad of cellular proteins (15). O-GlcNAcylation by OGT is one of the most abundant posttranslational modifications in mammalian cells, with more than 30% of the human proteome consisting of O-GlcNAcylated proteins (30). This modification is essential for development and proper cell function, and deletion of the OGT gene in vertebrates leads to developmental defects and embryonic lethality (31, 32). OGT is highly conserved in higher eukaryotes and is abundantly expressed in all tissues in the body (33). In addition to the many O-GlcNAcylated cellular proteins, OGT-modified residues have been found on proteins from several human viruses. This is not surprising, as many viruses commonly exploit cellular posttranslational modifications like phosphorylation and ubiquitination to alter cellular pathways or to modify the activity of viral proteins. Little is known, however, about the role of O-GlcNAcylation in viral infection, and the biological function of this modification for viruses is unclear. Reports have shown that O-GlcNAcylation inhibits replication of Kaposi's sarcoma-associated herpesvirus (KSHV) (34) and human immunodeficiency virus type 1 (HIV-1) (35), suggesting that O-GlcNAcylation can negatively regulate viral replication. In this study, we demonstrated that inhibition of OGT's glycosyltransferase activity with a small molecule inhibitor notably decreased HSV replication.
Small molecules that regulate the activity of proteins are valuable tools to study the function of proteins. The inhibitor that we used was characterized in a recent study (19). It selectively inhibits >80% of OGT's activity in vitro and has high cell permeability, allowing us to efficiently probe the function of OGT in the context of HSV infection. Treatment with OSMI-1 significantly reduced replication of HSV-1 in HFFs and HeLa, HEK-293, and HEp-2 cells, without affecting cell viability. The compound also reduced replication of HSV-2, the acyclovir-resistant TK− HSV-2 strain, and HCMV. However, we did not see inhibition of HSV-1 replication when we treated cells with an inactive compound that is structurally analogous to OSMI-1, demonstrating that our phenotype is correlated to OGT inhibition.
To gain insight into the mechanism of action of OSMI-1, we examined its effect on HSV gene expression. Transactivation of HSV IE genes upon infection is dependent on association of the VP16–Oct-1 complex with the major transcriptional coactivator HCF-1 (2, 10). HCF-1 is cleaved in a unique site-specific maturation process in which several amino acid repeats within its proteolytic processing domain (PPD) are bound and cleaved by OGT, generating several cleavage products (12). Inhibiting OGT's catalytic activity has been shown to block HCF-1 cleavage, leading to accumulation of full-length unprocessed protein (13). How HCF-1 proteolysis affects HSV gene transactivation is unclear. Daou et al. reported that depletion of OGT in HFFs results in an increase of HSV IE gene expression (13). The authors propose that full-length, uncleaved HCF-1 is a better substrate for VP16, and cleavage could disrupt interaction of HCF-1 with the transcriptional coactivator/corepressor four-and-a-half LIM domain-2 (FHL2), which, in turn, downregulates transactivation of IE genes. However, the change in IE gene expression observed upon OGT depletion was rather modest (∼1.5- to 2.5-fold). When we inhibited OGT with OSMI-1, we saw a reduction in the amount of the HCF-1 cleavage product, confirming inhibition of OGT's catalytic activity (results not shown). In contrast to the aforementioned study, we observed no change in the IE or E gene expression and protein levels upon OGT inhibition. The differences in the results may be due to the fact that Daou et al. (13) used low-MOI infections (0.1 PFU/cell), while we used a higher MOI (5 PFU/cell). However, the inhibitor notably reduced the levels of viral late proteins, while their mRNA levels remained unchanged. Thus, inhibition of OGT may either interfere with the synthesis of the late viral proteins or affect their stability and degradation. The relationship between O-GlcNAcylation and protein synthesis and degradation is complex, in that O-GlcNAcylation has been shown to be important for protein synthesis, as many ribosome and translation-associated proteins are O-GlcNAcylated (36). Therefore, it is possible that the inhibitor directly affects the synthesis of late viral proteins by inhibiting cellular or viral factors needed for their translation. O-GlcNAc modification has also been implicated in regulating protein turnover, by possibly protecting the glycosylated proteins from ubiquitination and proteasomal degradation (37, 38). In addition, HSV glycoproteins have been shown to be O-glycosylated (39). Therefore, it is possible that OGT inhibition by OSMI-1 leads to the loss of O-GlcNAc residues from late viral glycoproteins, which could enhance their susceptibility to proteasomal degradation. We will further dissect if the decrease in late viral proteins by OSMI-1 is due to a block in translation or enhanced degradation.
Many of the HSV late viral genes encode structural proteins that form the viral capsid. The capsid shell is comprised of four proteins, the major capsid protein VP5 and three less abundant proteins: VP19C, VP23, and VP26 (26). Because we saw a decrease in the levels of VP5, we hypothesized that capsid formation and maturation would be affected by OGT inhibition. Indeed, immunofluorescence analysis showed decreased nuclear staining for VP26 and VP5 in the presence of the OGT inhibitor. In support of these results, EM analysis of infected HFFs showed fewer capsids in the nuclei of inhibitor-treated cells, further demonstrating that formation of capsids is reduced upon OGT inhibition. Prediction software shows that these proteins possess multiple O-GlcNAcylation sites.
Although the mechanism of capsid inhibition by OSMI-1 is unclear at this stage, our results argue that OGT's glycosylation activity is needed for late stages of viral maturation. Consistent with a previous study (40), we observed that global O-GlcNAc levels increase at 12 hpi of HSV-1 (results not shown). Although the O-GlcNAcylated proteins could represent both viral and cellular proteins, this observation further suggests that O-GlcNAcylation of viral and/or host proteins is important during HSV infection.
Role of OGT as a scaffold.
The catalytic site of OGT resides in its C terminus, while the N terminus contains multiple tetratricopeptide (TRP) repeats, which form an elongated, flexible scaffold that provides a large electrostatic surface for multiple protein-protein interactions (41). The TRP scaffold allows for protein binding in many different binding orientations and modes, thus explaining the absence of a conserved sequence for OGT binding (42). One study shows that a catalytically inactive mutant OGT appears to be unaffected in substrate binding (43), arguing that OGT can serve as a scaffolding platform for the assembly of multiprotein complexes independent of its role as a glycosyltransferase. Moreover, OGT remains associated with its substrates even after glycosylation (41), further suggesting that the function of OGT is not solely modification of proteins. Thus, OGT whose catalytic function is inhibited by OSMI-1 can still retain its ability to interact with proteins and mediate formation of protein complexes. Depletion of OGT with an siRNA pool resulted in a 10-fold reduction in viral yields, which is a more modest phenotype than the 1,000-fold reduction we observed with the inhibitor. Even though the control compound PG34 confirmed OSMI-1's specificity, the reduced effect of OGT knockdown compared to OSMI-1 treatment may be due to off-target effects by the inhibitor. This smaller magnitude of inhibition may result from incomplete depletion of OGT by the siRNA, as we see that 15 to 20% of OGT transcripts remain after knockdown, which could provide enough enzymatic activity for viral replication. When we examined viral gene expression, we noted that unlike OSMI-1, which did not affect the levels of viral transcripts, OGT knockdown decreased expression of viral IE, E, and L genes by ∼10-fold. This difference in phenotypes between the OGT inhibitor and the knockdown most likely reflects the multiple functions of OGT in the cell. We speculate that OGT may serve as a scaffold to facilitate the binding of HCF-1 and other transcription factors to the IE multiprotein complex, and its depletion disrupts this interaction and negatively affects viral gene expression. Moreover, unpublished results from our lab show that OGT is a part of the HCF-1–VP16–OCT transactivation complex (44).
The role of OGT in replication of other viruses.
Several studies implicate O-GlcNAcylation as an important modification that can regulate the replication cycle of viruses. O-GlcNAc modification has been found on many viral proteins, including HCMV UL32 tegument protein (45), adenovirus fiber protein (46), baculovirus tegument protein gp41 (47), rotavirus NS26 protein (48), and several KSHV proteins involved in DNA replication (34, 49). A few studies suggest that high O-GlcNAc levels have an inhibitory effect on viral replication. Increased O-GlcNAcylation of the cellular protein Sp1, which is a critical transcription factor for HIV-1 gene expression, was shown to inhibit transcription from the HIV-1 long terminal repeat (LTR) promoter (35). Also, increasing O-GlcNAcylation by overexpression of OGT reduced KSHV replication (49). In another study, increased O-GlcNAcylation of the KSHV major replication and transcriptional activator (RTA) reduced its ability to transactivate viral genes (34). Consistent with this study, we observed that OGT inhibition did not prevent reactivation of latent KSHV, as measured by the levels of lytic gene expression (results not shown). Unlike the negative effect of O-GlcNAc modification for KSHV and HIV-1, we demonstrate that OGT is needed for the efficient replication of HSV and HCMV, as the OGT inhibitor decreased replication over 1,000-fold.
The results from this study demonstrate that OGT is involved in multiple steps of the HSV replication cycle. The OGT depletion results argue that OGT is needed for efficient formation of the viral IE transactivation complex, while the enzymatic activity of OGT, as demonstrated by the OSMI-1 studies, may be required late in infection, for translation of late viral genes and capsid assembly. In summary, the results described here implicate OGT as a novel factor involved in HSV-1 replication, and further studies are needed to define its precise role in the viral cycle.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI 063106 awarded to D.M.K. and NIH grant GM094263 awarded to S.W.
We thank Patrick T. Waters for assistance in the preparation of the manuscript.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Gerster T, Roeder RG. 1988. A herpesvirus trans-activating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc Natl Acad Sci U S A 85:6347–6351. doi: 10.1073/pnas.85.17.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kristie TM, LeBowitz JH, Sharp PA. 1989. The octamer-binding proteins form multi-protein–DNA complexes with the HSV alpha TIF regulatory protein. EMBO J 8:4229–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Hare P, Goding CR, Haigh A. 1988. Direct combinatorial interaction between a herpes simplex virus regulatory protein and a cellular octamer-binding factor mediates specific induction of virus immediate-early gene expression. EMBO J 7:4231–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson AC, Cleary MA, Lai JS, LaMarco K, Peterson MG, Herr W. 1993. Combinatorial control of transcription: the herpes simplex virus VP16-induced complex. Cold Spring Harb Symp Quant Biol 58:167–178. doi: 10.1101/SQB.1993.058.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Kristie TM, Roizman B. 1987. Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1 alpha genes. Proc Natl Acad Sci U S A 84:71–75. doi: 10.1073/pnas.84.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKnight JL, Pellett PE, Jenkins FJ, Roizman B. 1987. Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate alpha-trans-inducing factor-dependent activation of alpha genes. J Virol 61:992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKnight JLC, Kristie TM, Roizman B. 1987. Binding of the virion protein mediating alpha gene induction in herpes simplex virus 1 infected cells to its cis site requires cellular proteins. Proc Natl Acad Sci U S A 84:7061–7065. doi: 10.1073/pnas.84.20.7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narayanan A, Nogueira ML, Ruyechan WT, Kristie TM. 2005. Combinatorial transcription of herpes simplex virus and varicella zoster virus immediate early genes is strictly determined by the cellular coactivator HCF-1. J Biol Chem 280:1369–1375. doi: 10.1074/jbc.M410178200. [DOI] [PubMed] [Google Scholar]
- 10.Wilson AC, LaMarco K, Peterson MG, Herr W. 1993. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell 74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 11.Kristie TM, Pomerantz JL, Twomey TC, Parent SA, Sharp PA. 1995. The cellular C1 factor of the herpes simplex virus enhancer complex is a family of polypeptides. J Biol Chem 270:4387–4394. doi: 10.1074/jbc.270.9.4387. [DOI] [PubMed] [Google Scholar]
- 12.Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, Herr W. 2011. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 13.Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, Affar el B. 2011. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A 108:2747–2752. doi: 10.1073/pnas.1013822108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S. 2013. HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 342:1235–1239. doi: 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart GW, Housley MP, Slawson C. 2007. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 16.Julien E, Herr W. 2003. Proteolytic processing is necessary to separate and ensure proper cell growth and cytokinesis functions of HCF-1. EMBO J 22:2360–2369. doi: 10.1093/emboj/cdg242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vogel JL, Kristie TM. 2006. Site-specific proteolysis of the transcriptional coactivator HCF-1 can regulate its interaction with protein cofactors. Proc Natl Acad Sci U S A 103:6817–6822. doi: 10.1073/pnas.0602109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozcan S, Andrali SS, Cantrell JE. 2010. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta 1799:353–364. doi: 10.1016/j.bbagrm.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S. 9 March 2015. A small molecule that inhibits OGT activity in cells. ACS Chem Biol doi: 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desai P, DeLuca NA, Person S. 1998. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 247:115–124. doi: 10.1006/viro.1998.9230. [DOI] [PubMed] [Google Scholar]
- 21.Bryant KF, Yan Z, Dreyfus DH, Knipe DM. 2012. Identification of a divalent metal cation binding site in herpes simplex virus 1 (HSV-1) ICP8 required for HSV replication. J Virol 86:6825–6834. doi: 10.1128/JVI.00374-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol 82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang L, Godinez WJ, Kim IH, Tektonidis M, de Lanerolle P, Eils R, Rohr K, Knipe DM. 2011. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc Natl Acad Sci U S A 108:E136–E144. doi: 10.1073/pnas.1103411108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC. 1993. Structure of the herpes simplex virus capsid. Molecular composition of the pentons and the triplexes. J Mol Biol 232:499–511. [DOI] [PubMed] [Google Scholar]
- 27.Mizuguchi-Hata C, Ogawa Y, Oka M, Yoneda Y. 2013. Quantitative regulation of nuclear pore complex proteins by O-GlcNAcylation. Biochim Biophys Acta 1833:2682–2689. doi: 10.1016/j.bbamcr.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Lefebvre T, Ferreira S, Dupont-Wallois L, Bussiere T, Dupire MJ, Delacourte A, Michalski JC, Caillet-Boudin ML. 2003. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—a role in nuclear localization. Biochim Biophys Acta 1619:167–176. doi: 10.1016/S0304-4165(02)00477-4. [DOI] [PubMed] [Google Scholar]
- 29.Malik P, Tabarraei A, Kehlenbach RH, Korfali N, Iwasawa R, Graham SV, Schirmer EC. 2012. Herpes simplex virus ICP27 protein directly interacts with the nuclear pore complex through Nup62, inhibiting host nucleocytoplasmic transport pathways. J Biol Chem 287:12277–12292. doi: 10.1074/jbc.M111.331777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta R, Brunak S. 2002. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput 2002:310–322. [PubMed] [Google Scholar]
- 31.Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. 2000. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci U S A 97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webster DM, Teo CF, Sun Y, Wloga D, Gay S, Klonowski KD, Wells L, Dougan ST. 2009. O-GlcNAc modifications regulate cell survival and epiboly during zebrafish development. BMC Dev Biol 9:28. doi: 10.1186/1471-213X-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lubas WA, Frank DW, Krause M, Hanover JA. 1997. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J Biol Chem 272:9316–9324. doi: 10.1074/jbc.272.14.9316. [DOI] [PubMed] [Google Scholar]
- 34.Ko YC, Tsai WH, Wang PW, Wu IL, Lin SY, Chen YL, Chen JY, Lin SF. 2012. Suppressive regulation of KSHV RTA with O-GlcNAcylation. J Biomed Sci 19:12. doi: 10.1186/1423-0127-19-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jochmann R, Thurau M, Jung S, Hofmann C, Naschberger E, Kremmer E, Harrer T, Miller M, Schaft N, Sturzl M. 2009. O-linked N-acetylglucosaminylation of Sp1 inhibits the human immunodeficiency virus type 1 promoter. J Virol 83:3704–3718. doi: 10.1128/JVI.01384-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeidan Q, Wang Z, De Maio A, Hart GW. 2010. O-GlcNAc cycling enzymes associate with the translational machinery and modify core ribosomal proteins. Mol Biol Cell 21:1922–1936. doi: 10.1091/mbc.E09-11-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han I, Kudlow JE. 1997. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol Cell Biol 17:2550–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. 2003. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115:715–725. doi: 10.1016/S0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- 39.Johnson DC, Spear PG. 1983. O-linked oligosaccharides are acquired by herpes simplex virus glycoproteins in the Golgi apparatus. Cell 32:987–997. doi: 10.1016/0092-8674(83)90083-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofemeister H, O'Hare P. 2008. Nuclear pore composition and gating in herpes simplex virus-infected cells. J Virol 82:8392–8399. doi: 10.1128/JVI.00951-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iyer SP, Hart GW. 2003. Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J Biol Chem 278:24608–24616. doi: 10.1074/jbc.M300036200. [DOI] [PubMed] [Google Scholar]
- 42.Zeytuni N, Zarivach R. 2012. Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20:397–405. doi: 10.1016/j.str.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Schimpl M, Zheng X, Borodkin VS, Blair DE, Ferenbach AT, Schuttelkopf AW, Navratilova I, Aristotelous T, Albarbarawi O, Robinson DA, Macnaughtan MA, van Aalten DM. 2012. O-GlcNAc transferase invokes nucleotide sugar pyrophosphate participation in catalysis. Nat Chem Biol 8:969–974. doi: 10.1038/nchembio.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh HS, Knipe DM. Proteomic analysis of the herpes simplex virus 1 virion protein 16 transactivator protein in infected cells. Proteomics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greis KD, Gibson W, Hart GW. 1994. Site-specific glycosylation of the human cytomegalovirus tegument basic phosphoprotein (UL32) at serine 921 and serine 952. J Virol 68:8339–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mullis KG, Haltiwanger RS, Hart GW, Marchase RB, Engler JA. 1990. Relative accessibility of N-acetylglucosamine in trimers of the adenovirus types 2 and 5 fiber proteins. J Virol 64:5317–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Novelli A, Boulanger PA. 1991. Deletion analysis of functional domains in baculovirus-expressed adenovirus type 2 fiber. Virology 185:365–376. doi: 10.1016/0042-6822(91)90784-9. [DOI] [PubMed] [Google Scholar]
- 48.González SA, Burrone OR. 1991. Rotavirus NS26 is modified by addition of single O-linked residues of N-acetylglucosamine. Virology 182:8–16. doi: 10.1016/0042-6822(91)90642-O. [DOI] [PubMed] [Google Scholar]
- 49.Jochmann R, Pfannstiel J, Chudasama P, Kuhn E, Konrad A, Sturzl M. 2013. O-GlcNAc transferase inhibits KSHV propagation and modifies replication relevant viral proteins as detected by systematic O-GlcNAcylation analysis. Glycobiology 23:1114–1130. doi: 10.1093/glycob/cwt028. [DOI] [PubMed] [Google Scholar]