

Abstract
Aims:
To study the histological features on muscle biopsy and correlate them with clinical features, other laboratory data in adult patients to make a diagnosis of dermatomyositis (DM), applying the European Neuromuscular center (ENMC) criteria.
Materials and Methods:
Adult patients who fulfilled clinical, laboratory, and muscle biopsy findings according to ENMC criteria for DM during the period 2010–2013 were included in the study. Cryostat sections of muscle biopsy were reviewed with emphasis on Perifascicular atrophy (PFA), perivascular/endomysial inflammation. Muscular dystrophies and metabolic myopathies were excluded by appropriate immunohistochemistry and special stains.
Results:
The diagnosis of adult DM was made in 45 patients out of 170 clinically suspected idiopathic inflammatory myopathies. These included 33 definite, 4 probable, 7 possible sine dermatitis, and 1 amyopathic DM. All patients with definite DM had typical rash and proximal muscle weakness and muscle biopsy showed PFA with or without inflammation. Thirteen patients had quadriparesis, neck muscle weakness, dysphagia/dysphonia at presentation. Patients with probable DM had rash and showed perivascular/endomysial inflammation with no PFA. Possible DM sine dermatitis showed PFA with perivascular/endomysial infiltrates. One patient of amyopathic DM had typical heliotrope rash and characteristic skin biopsy.
Conclusions:
Histological features are important for the diagnosis of DM. Relying on PFA for diagnosis of definite DM underestimates the true frequency of DM.
Keywords: Dermatomyositis, perifascicular atrophy, perivascular inflammation
Introduction
The idiopathic inflammatory myopathies (IIM) are heterogenous group of acquired muscle diseases that include polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM).[1,2] With the recent advances in the understanding of immunopathogenesis, new classification criteria were designed which distinguish nonspecific myositis and immune mediated necrotizing myopathy as additional subtypes.[3] Apart from IBM, all the other four subtypes may occur as isolated conditions or in association with a connective disease (CTD) or with cancer. These criteria make histopathological examination of muscle, a mandatory requirement for diagnosis and differential diagnosis.[3] The diagnosis is made on the basis of clinical symptoms, serum creatine kinase (CK), electromyogram (EMG), and muscle biopsy findings. Histopathological features characterize each entity and are helpful, especially in the absence of skin rash or autoantibodies.[4]
DM is a unique disease in the category of IIM with characteristic skin involvement. It is also characterised by its association with interstitial lung disease (ILD), CTD, or malignancy(5). We aim to apply the European Neuromuscular center (ENMC) criteria to diagnosis adult DM based on clinical, laboratory, and muscle biopsy features.
Materials and Methods
All the records of patients referred for a muscle biopsy between 2010 and 2013 with a clinical diagnosis of IIM from specialist neurologists and rheumatologists were reviewed. Only adult patients who fulfilled the clinical, laboratory, and muscle biopsy findings according to the ENMC criteria for DM were included in the study. The demographic data, type of onset (aute/insidious), and clinical features with particular attention to rash and proximal muscle weakness were noted. Serum CK and reports of EMG, wherever available were retrieved from medical records. Magnetic resonance imaging (MRI) and myositis specific antibodies (MSA) were not done as a part of routine workup due to financial constraints.
The muscle biopsies were done by open method from the vastus lateralis muscle in all patients. The cryostat sections of the muscle biopsy stained with hematoxylin and eosin (H&E), Masson-trichrome (MT), modified Gomori trichrome (MGT), Adenosine triphosphatase (pre-incubated at pH 9.4, 4.6 and 4.3), Nicotinamide adenine dehydrogenase reductase (NADH-TR), succinate dehydrogenase (SDH) were reviewed. Stains for cytochrome C oxidase (COX), COX-SDH, neutral lipid (oil red O), and glycogen (periodic acid Schiff- PAS) were done wherever necessary to rule out metabolic myopathies. Immunohistochemistry (IHC) with antibodies against dystrophin, sarcoglycans (alpha, beta, gamma, and delta), and dysferlin were done wherever necessary to rule out muscular dystrophy. The histological features reviewed were:
Presence/absence of PFA.
Presence of inflammation.
Distribution of inflammatory cells (perivascular/perimysial/endomysial).
Invasion of non-necrotic fibers by lymphocytes.
Type of inflammatory cells- lymphocytes, mononuclear cells, plasma cells, eosinophils, and granulomas.
Presence/absence of necrotic/degenerating/regenerating fibers/infarcts.
Fibrosis.
Endothelial swelling, thickening of blood vessels.
Rimmed vacuoles/red ragged fibers/sarcoplasmic vacuoles.
Others including type 1 predominance, type 2 atrophy, type grouping.
The diagnosis was made definite, probable, amyopathic, and possible sine dermatitis according to ENMC criteria. Immunohistochemistry or immunoflourescence (IF) studies were not done to characterize the inflammatory cells or deposition of membranolytic attack complex (MAC) or major histocompatability complex (MHC). Electron microscopic (EM) studies were also not done.
Results
There were 170 patients who had muscle biopsies in the study period with a clinical diagnosis/suspicion of inflammatory myopathy. The diagnosis of adult DM was made in 45 patients which included 33 definite, 4 probable, 7 possible sine dermatitis, and 1 amyopathic DM. The patients with definite DM had association with CTD in 2 and association with malignancy in one. Hypothyroidism was noted in two patients. The clinical and laboratory features are summarized in Tables 1 and 2.
Table 1.
Demographic, clinical, laboratory features of adult dermatomyositis (n = 45)
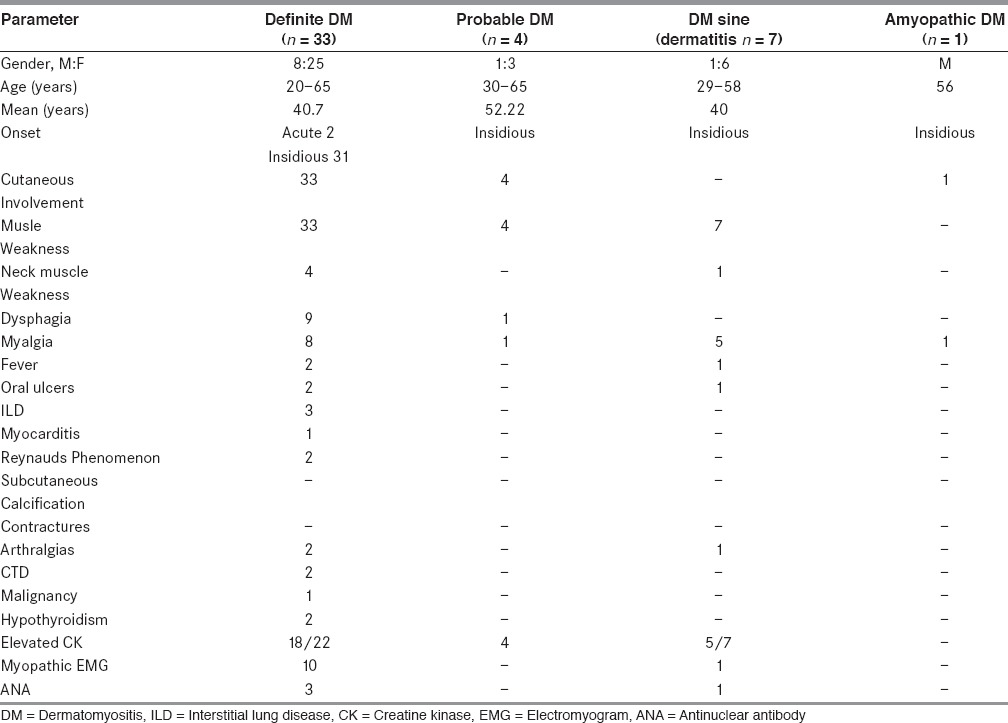
Table 2.
Muscle biopsy features of adult dermatomyositis (n = 45)
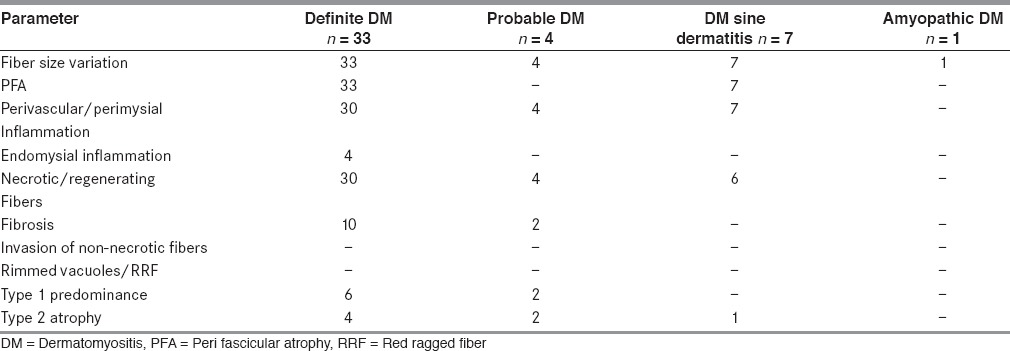
Muscle biopsy showed PFA in all biopsies. The involvement was patchy and it was highlighted on ATPase [Figures 1a and b]. There were micro infarcts in two biopsies. PFA was associated with inflammation in 30 biopsies and PFA alone without inflammation in three biopsies. The inflammation was perivascular and/or perimysial in 30 biopsies [Figures 2a and b]. Four biopsies showed endomysial inflammation in addition to perivascular and perimysial inflammation. However, there was no invasion of non-necrotic fibers by inflammatory cells. The inflammatory cells included lymphocytes, momonuclear cells, and few plasma cells. Necrotic, degenerating, and regenerating fibers were seen in majority of the biopsies predominantly in perifascicular distribution. Sarcoplasmic vacuoles were seen in degenerating fibers in four biopsies. However, the vacuoles were negative for neutral lipid and PAS positive material. There were no rimmed vacuoles or ragged red fibers. The vessel walls were thickened and hyalinised with endothelial swelling. Interstitial fibrosis was seen in 10. Type 1 predominance was seen in six and type 2 atrophy in four biopsies.
Figure 1.

(a) Photomicrograph showing perifascicular atrophy (PFA) (H and E ×40) (b) PFA (ATPase pH 9.4 ×40)
Figure 2.
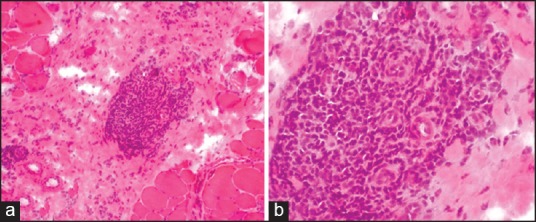
(a) Photomicrograph showing perivascular/perimysial inflammation (H and E ×40), (b) Perivascular/perimysial inflammation (H and E ×100)
Probable DM (n = 4): Muscle biopsy showed perivascular/perimysial lymphomononuclear infiltrate in all biopsies. There was no PFA. Few necrotic and regenerating fibers were seen. Type 1 predominance and type 2 atrophy were seen in two biopsies each [Figure 3a].
Figure 3.
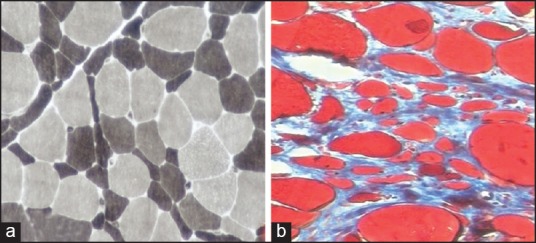
(a) Type 2 atrophy (ATPase pH 9.4 X100) (b) Fibrosis (Masson's Trichrome x100)
Possible DM sine dermatitis (n = 7): Muscle biopsy showed PFA and perivascular/perimysial inflammation in all biopsies. Necrotic and regenerating fibers were seen in six biopsies. One biopsy showed type 2 atrophy.
Amyopathic DM (n = 1): This was a 56-year-old male who presented with typical heliotrope rash on face and neck. He also had myalgias but no muscle weakness. Serum CK and EMG were within normal limits. Skin biopsy showed vacuolar type of interface dermatitis with folliculitis. Muscle biopsy showed few scattered atrophic fibers.
Discussion
The original classification of IIM by Bohan and Peter is widely used.[6] However, with advances in the understanding of pathogenesis of IIM, these criteria were found to have low specificity and failed to distinguish sporadic IBM and non-inflammatory myopathies like muscular dystrophies and other myopathies with cellular infiltrates.[7] Hence, the classification proposed by Bohan and Peter was challenged. New classification criteria on behalf of the muscle study group (MSG) for adult IIM (except IBM) were suggested to ensure homogenous groups for clinical trials. These revised diagnostic criteria were approved by MSG and 119th European Neuromuscular Center international workshop held in Naarden, Netherlands.[3] This classification requires histopathological features on muscle biopsy in all cases. However, these criteria need validation. We applied these criteria to diagnose adult DM in the present study.
DM is a chronic inflammatory disorder of the skin and muscles. Bendewald et al. (2010) in a 32-year retrospective study showed that the incidence of DM including all subtypes (age and sex adjusted) was 9.63 per 1,000,000 people.[8]
The onset of DM is insidious that usually develops over a period of weeks to months but rarely acutely.[1] Only two patients in our series presented with less than 2 weeks onset of disease and in all others, the duration of disease varied from 1-6 months.
DM is identified by a characteristic rash which accompanies, preceeds (more commonly), or occurs after the onset of muscle weakness.[2,5] The skin manifestations include a heliotrope rash on the upper eyelids with or without periorbital edema. Thirty-eight of our patients had characteristic heliotrope rash and/or Gottron's papules with erythema and photosensitivity.
Patients with DM present with varying degrees of muscle weakness from mild to severe leading to quadriparesis.[2] Distal muscle weakness, quadriparesis, and neck muscle weakness occur late in the course of the disease.[5] Forty-four of our patients had proximal muscle weakness at presentation. Proximal more than distal in 6, quadriparesis in 3, Dysphagia and dysphonia in 10, neck muscle weakness in 7 indicating delayed presentation and advanced disease at initial presentation.
The diagnosis of DM sine dermatitis is made on the basis of the characteristic immunopathological muscle biopsy findings of DM but in the absence of a rash. The skin rash is reported to be transient or poorly recognized due to dark skin in this group of patients.[2,3] Characteristic muscle biopsy findings in the absence of skin rash helped for making the diagnosis in seven patients.
The term amyopathic DM is applied to patients with classic cutaneous manifestations for more than 6 months without clinical, laboratory or other evidence of muscle disease.[9] The term clinically amyopathic DM includes both amyopathic and hypomyopathic DM. Gerami et al. (2006) reported that clinically amyopathic patients developed muscle weakness at 15 months to 6 years after skin disease and hence recommended follow-up of these patients.[9] Otero et al. (1992) reported that muscle biopsy from such cases shows perivascular and perimysial inflammation.[10] Dalakas et al. (2003) opined that amyopathic and myopathic DM are part of the range of DM affecting skin and muscle to a varying degree.[2] The ENMC criteria of define rash typical of DM with no objective muscle weakness, normal CK and EMG with characteristic skin biopsy, and no diagnostic features on muscle biopsy.[3] The present study was based on muscle biopsy findings, hence there was only one patient with amyopathic DM. Skin biopsy was diagnostic and muscle biopsy showed occasional atrophic fibers.
One patient in the present series had associated malignancy, which was carcinoma breast. Association with malignancy is reported with a frequency from 9-42%.[11] The risk of malignancy was highest at the time of or within one year of diagnosis of myositis.[5] The patient with carcinoma breast in our series developed myositis during treatment for the malignancy within first year. The prevalence of malignancy in a biopsy proven series from our institute was reported to be 7% and the most common malignancy was carcinoma breast.[12]
The other manifestations of DM include ILD in about 35-40% of patients and this association is reported to be associated with rapidly progressive subgroups.[13] ILD was seen in only three patients of definite DM in our series. Cardiac involvement is usually subclinical in DM[5] and it was seen in only one patient in our series. The other manifestations in our series included fever (2), oral ulcers (1), polyarthralgia (1), and Raynaud's phenomenon (2). These symptoms are reported in patients of DM associated with CTD. The most common CTD associated with DM is systemic sclerosis (SS).[2] In our series, two patients of DM had associated SS.
Serum CK is often increased in IIM and is a useful clinical parameter to muscle disease.[14] The ENMC criteria did not define normal values or activity as the values depend on the technique used, gender and ethnic groups.[3] Serum CK values were available in 30 patients and they were normal in 2 definite DM and elevated in the other 28 patients (200–20000 IU/L).
The ENMC criteria include EMG, MRI, and MSA in the other laboratory criteria.[3] The EMG studies are useful early in disease and show abnormal findings in 70% patients.[5] They may be non-specific and seen in other muscle diseases. The ENMC criteria did not establish the reliability of EMG as the interpretation depends on the skills of the electromyographer and the number of muscles tested. EMG reports were available in 12 patients and it was myopathic in all patients tested. MRI and MSA are useful for evaluation of myositis; however, they were not done as a routine in our patients due to financial constraints and availability.
Histopathologic features on muscle biopsy are the most important criteria for establishing the diagnosis as histology characterizes each subtype of IIM.[3] A highly characteristic histologic feature of DM is PFA. The muscle fibers undergo phagocytosis and necrosis resulting in microinfarcts involving a portion/periphery of the fascicle.[2] The causes of PFA and capillary pathology in DM are not completely understood. It was proposed that PFA is caused by ischemia due to the loss of endomysial capillary bed and it affects the distal most part of the fascicle, which is a water shed region.[2] Infarcts are also reported in DM as seen in two of our biopsies. All biopsies of definite DM and DM sine dermatitis showed PFA in our study. PFA is diagnostic of DM even in the absence of inflammation as seen in three of our definite DM biopsies. However, PFA is a late finding and is found in only 50% of adult cases when biopsied early in the course of illness.[15] However, the ENMC criteria indicate PFA as the diagnostic feature on muscle biopsy for definite diagnosis of DM.[3] As 40/45 of our biopsies showed PFA when the diagnosis was made, it indicates either late presentation before diagnosis or under diagnosis of DM till PFA became evident.
DM is a humorally mediated autoimmune disorder in which activation of complement leads to the formation of MAC resulting in destruction of capillaries in muscle tissue and other tissues.[2] The microangiopathy in DM leads to the characteristic features of infarction and PFA.[16,17] The earliest histologic abnormality in DM is the deposition of MAC on small blood vessels.[18,19] MAC precedes inflammation and other structural abnormalities in the muscle on light microscopy and is considered specific for DM.[18,19] Immunohistochemical studies to demonstrate MAC deposition, MHC-1 expression or type of the inflammatory infiltrate and EM studies are not done as routine diagnostic tests in the present study, resulting in late or under diagnosis in our series.
Five of the patients in this series received treatment before muscle biopsy was done including 4 cases of definite DM and one case of probable DM. Type 1 predominance and Type 2 atrophy were seen as additional features on muscle biopsy in these patients.
The other histological features include perivascular and perimysial inflammatory cell infiltrates. The inflammatory cells are found around blood vessels, in the septae between muscle fascicles, and in the fibroadipose tissue around the muscle.[2,20] Perivascular/perimysial infiltrates were seen in all biopsies except in three biopsies of definite DM, and one amyopathic DM biopsies in our series. The inflammatory cells in DM are reported to be predominantly B cells with few CD4+ T cells, and plasmacytoid dendritic cells.[2,21] However, immunological studies were not done in our study.
The distribution of inflammatory infiltrates in DM is predominantly perivascular. The number of inflammatory cells vary from few to large infiltrates as seen in our biopsies and all biopsies except amyopathic DM in our series showed inflammatory infiltrates. However, the perivascular changes may be seen in patients without skin rash as seen in our patients of DM sine dermatitis. Biopsies with less well defined pattern of infiltrates or combined endomysial and perivascular infiltrates are reported.[3,22,23] These observations point out that there is an overlap between clinical phenotype, histopathology and immune types and suggest that the pathogenesis is determined by more than one factor.[24]
All subtypes of IIMs may not exhibit pathophysiology relevant histopathologic features or histopathologic features may be scarce, nonspecific and overlapping. Many patients do not fit into any subcategory.[25] Hence, there is still a need for review and revision of diagnostic criteria of IIM and in the 193rd ENMC international workshop on pathology diagnosis of IIM, it was suggested that analysis of individual muscle biopsy abnormalities like cellular infiltrates, vascular changes, and muscle fiber abnormalities should be given importance rather than pattern recognition.[23] Pestronk suggested an alternate classification of IIMs based on myopathologic features.[26] This myopathologic classification is reported to provide useful diagnostic specificity. We have not tested or used this classification in the present study.
The ENMC criteria help in differentiating IIMs from muscular dystrophies and metabolic myopathies as validated in our series.[3] However, much stress on pathologic criteria may result in delayed diagnosis as in our series.[23] Hoojendijk et al. strongly stressed the need for validating the criteria proposed by them.[3] We tested the criteria for routine diagnostic purposes, and conclude that using light microscopic muscle biopsy findings alone may underestimate the frequency of DM. Clinical features along with muscle biopsy findings are important for making correct diagnosis. Incorporation of immunopathological studies in routine diagnosis will help in making early diagnosis.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
References
- 1.Dalakas MC. Polymyositis, dermatomyositis and Inclusion-body myositis. N Engl J Med. 1991;325:1487–98. doi: 10.1056/NEJM199111213252107. [DOI] [PubMed] [Google Scholar]
- 2.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971–82. doi: 10.1016/S0140-6736(03)14368-1. [DOI] [PubMed] [Google Scholar]
- 3.Hoogendijk JE, Amato AA, Lecky BR, Choy FH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion-body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:337–45. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Lazarou IN, Guerne PA. Classification, diagnosis and management of idiopathic inflammatory myopathies. J Rheumatol. 2013;40:550–64. doi: 10.3899/jrheum.120682. [DOI] [PubMed] [Google Scholar]
- 5.Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375–81. doi: 10.4103/0019-5154.100486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292:344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 7.Confalonieri P, Oliva L, Andretta F, Lorenzoni R, Dassi P, Mariani E, et al. Muscle inflammation and MHC class I up-regulation in muscular dystrophy with lack of dysferlin: An immunopathological study. J Neuroimmunol. 2003;142:130–6. doi: 10.1016/s0165-5728(03)00255-8. [DOI] [PubMed] [Google Scholar]
- 8.Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: A population based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146:26–30. doi: 10.1001/archdermatol.2009.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerami P, Schope JM, Mc Donald L, Walling HW, Sontheimer RD. A systematic review of clinically amyopathic dermatomyositis (dermatomyositis sine myositis): A missing link within the spectrum of idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54:597–61. doi: 10.1016/j.jaad.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 10.Otero C, Illa I, Dalakas MC. Is there dermatomyositis (DM) without myositis? Neurology. 1992;42:388. [Google Scholar]
- 11.Zahr ZA, Baer AN. Malignancy in myositis. Curr Rheumatol Rep. 2011;13:208–15. doi: 10.1007/s11926-011-0169-7. [DOI] [PubMed] [Google Scholar]
- 12.Kannan MA, Sundaram C, Uppin M, Mridula R, Jabeen SA, Borgohain R. Incidence of malignancies in biopsy-proven inflammatory myopathy. Neurol India. 2013;61:152–5. doi: 10.4103/0028-3886.111121. [DOI] [PubMed] [Google Scholar]
- 13.Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies. What progress has been made in the past 35 years? Chest. 2010;138:1464–74. doi: 10.1378/chest.10-0180. [DOI] [PubMed] [Google Scholar]
- 14.Hilton-Jones D. Observation on the classification of the inflammatory myopathies. Presse Med. 2011;40(4 Pt 2):e199–208. doi: 10.1016/j.lpm.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 15.Hak AE, de Paepe B, de Bleecker JL, Tak PP, de Visser M. Dermatomyositis and polymyositis: New treatment targets on the horizon. Neth J Med. 2011;69:410–21. [PubMed] [Google Scholar]
- 16.Cappelletti C, Morandi L, Mora M, Salerno F, Confalonieri P, Mantegazza R, et al. Idiopathic inflammatory myopathies: A review of immunopathological features and current models of pathogenesis. InTechOpen. 2011:1–24. [Google Scholar]
- 17.Rayavarapu S, Coley W, Kinder TB, Nagaraju K. Idiopathic inflammatory myopathies: Pathogenic mechanisms of muscle weakness. Skelet Muscle. 2013;3:13. doi: 10.1186/2044-5040-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emslie-Smith AM, Engel AG. Microvascular changes in early and advanced dermatomyositis: A quantitative study. Ann Neurol. 1990;27:343–56. doi: 10.1002/ana.410270402. [DOI] [PubMed] [Google Scholar]
- 19.Kissel JT, Mendell JR, Rammohan KW, Mendell JR. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol. 1991;48:26–30. doi: 10.1001/archneur.1991.00530130034016. [DOI] [PubMed] [Google Scholar]
- 20.Dalakas MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:779–98. doi: 10.1016/s0889-857x(02)00030-3. [DOI] [PubMed] [Google Scholar]
- 21.Mammen AL. Dermatomyositis and polymyositis. Clinical presentation, autoantibodies and pathogenesis. Ann >N Y Acad Sci. 2010;1184:134–53. doi: 10.1111/j.1749-6632.2009.05119.x. [DOI] [PubMed] [Google Scholar]
- 22.Fasth AE, Dastmalchi M, Rahbar A, Salomonsson S, Pandya JM, Lindroos E, et al. T cell infiltrates in the muscle of patients with dermatomyositis and polymyositis are dominated by CD 28(null) T cells. J Immunol. 2009;183:4792–9. doi: 10.4049/jimmunol.0803688. [DOI] [PubMed] [Google Scholar]
- 23.De Bleecker JL, Lundberg IE, de Visser M. ENMC Myositis Muscle biopsy study group, 193rd ENMC International workshop. Pathology diagnosis of idiopathic inflammatory myopathies 30 November-2 December 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:945–51. doi: 10.1016/j.nmd.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Nagaraju K, Casciola-Rosen I, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis, potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–35. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 25.Gherardi RK. Pthogenic aspects of dermatomyositis, polymyositis and overlap myositis. Presse Med. 2011;40(4 Pt 2):e209–18. doi: 10.1016/j.lpm.2010.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Pestronk A. Acquired immune and inflammatory myopathies: Pathologic classification. Curr Opin Rheumatol. 2011;23:595–604. doi: 10.1097/BOR.0b013e32834bab42. [DOI] [PubMed] [Google Scholar]