

Abstract
Objective:
To define causative somatic mutations in resected brain tissue from an infant with intractable epilepsy secondary to hemispheric cortical dysplasia.
Methods:
Whole-exome sequencing was conducted on genomic DNA derived from both resected brain tissue and peripheral blood leukocytes. Comparison of the brain vs blood sequencing results was performed using bioinformatic methods designed to detect low-frequency genetic variation between tissue pairs.
Results:
Histopathology of the resected tissue showed dyslamination and dysmorphic neurons, but no balloon cells, consistent with focal cortical dysplasia type IIa. mTOR activation was observed by immunohistochemistry in the dysplasia. A missense mutation (c.4487T>G; p.W1456G) was detected in the FAT domain of MTOR in DNA from the dysplasia but not in lymphocytes. The mutation is predicted damaging (i.e., leading to mTOR activation) and was observed as a low-level mosaic with 8% of cells being heterozygous for the variant.
Conclusions:
We report the novel finding of an MTOR mutation associated with nonsyndromic cortical dysplasia. Somatic-specific mutations in MTOR and related genes should be considered in a broader spectrum of patients with hemispheric malformations and more restricted forms of cortical dysplasia.
Focal cortical dysplasia (FCD), a common cause of intractable epilepsy requiring surgery, consists of lesions varying from small bottom-of-sulcus dysplasias to hemispheric malformations with hemimegalencephaly at the severe end of the spectrum. FCD is characterized by cortical dyslamination with or without abnormal cell types, dysmorphic neurons in FCD type IIa and both dysmorphic neurons and balloon cells in FCD type IIb.1 FCD IIb shows histologic similarities to cortical tubers of tuberous sclerosis, suggesting a genetic link, but recent evidence may also suggest a link to HPV16 infection.2 The etiology of FCD IIa is unclear, although genetic causes are also suspected.3
It was hypothesized that focal cortical malformations result from somatic mutations in mTOR regulatory genes occurring in neuroglial progenitor cells.4 Subsequently, cases of hemimegalencephaly were found to be caused by somatic specific mutations in PIK3A-Akt3-mTOR signaling pathway genes, a discovery only made possible using resected brain tissue.5 Hemimegalencephaly and FCD are related lesions based on imaging and histologic overlap, evidence of mTOR dysregulation in resected tissue in both,4 and report of siblings with FCD and hemimegalencephaly.3 Further evidence of mTOR dysregulation in FCD has come from finding germline mutations in DEPDC5, coding for an mTOR regulatory protein, in rare individuals with imaging features of FCD.6 Resected tissue from these patients was not available for confirmation. Thus far, only one patient has been identified with a mutation in MTOR itself, a patient with hypomelanosis of Ito and hemispheric dysplasia.5 We hypothesized that mutations in MTOR may occur in other forms of FCD and report a patient with nonsyndromic hemispheric FCD IIa and a low-level mosaic somatic mutation in MTOR, detected only in the resected dysplastic tissue.
METHODS
Clinical details were obtained from parent interview and medical records. Brain MRI was obtained using age-specific epilepsy protocols on 1.5T and 3T scanners. Resected tissue was classified by a neuropathologist according to the proposed system of the International League Against Epilepsy Diagnostic Methods Commission.1
Genetic analysis.
Methods are briefly summarized here with details provided in appendix e-1 on the Neurology® Web site at Neurology.org. Genomic DNA isolated from resected brain tissue and peripheral blood was analyzed by whole-exome capture and massively parallel sequencing. Reads were aligned to the reference genome with Novoalign and germline variants were detected by GATK's UnifiedGenotyper and annotated by ANNOVAR. Variants were filtered in a step-wise manner against exclusion criteria including a minor allele frequency and functional impact. Inclusion criteria included predicted damaging effects on the protein using PolyPhen2 and SIFT and presence within a list of 484 candidate genes potentially associated with brain malformations (table e-1). Somatic single nucleotide variant (SNV) mutations were identified with MuTect and VarScan. Candidate variants were validated by clonal assay and Sanger sequence analysis.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Royal Children's Hospital Human Research Ethics Committee (HREC 29077E) with informed consent obtained from the patient's parent.
RESULTS
Case report.
The male patient presented at 6 weeks with multiple-daily clusters of asymmetric tonic seizures. Examination showed left hemiparesis but no dysmorphic or neurocutaneous features. He was treated unsuccessfully with anticonvulsants and at age 7 months was transferred to our hospital. EEG showed attenuation of background rhythms and excessive slow activity over the right hemisphere, frequent interictal epileptiform discharges from the right temporal and central regions, and ictal rhythms beginning over the right frontotemporal region and spreading over the right hemisphere. Brain MRI showed subtle loss of gray–white differentiation, abnormal cerebral sulcation, and white matter signal change suggestive of advanced myelination throughout the right hemisphere, particularly in the frontal and parietal regions (figure 1A). Brain PET imaging showed severe hypometabolism of the right frontal, temporal, and parietal lobes (figure 1B). Frequent life-threatening seizures with apnea continued on a daily basis. At age 10 months, he underwent a right hemispherotomy with resolution of his seizures. Histopathology showed FCD IIa (figure 2A), and phospho-S6 ribosomal immunostaining (Ser235/236, rabbit polyclonal, 1:200; Cell Signaling, Beverly, MA) was positive in cytomegalic neurons, consistent with constitutive mTOR pathway activation (figure 2B). Three years postoperatively, he had recurrence of focal dyscognitive seizures treated by removal of a residual right frontobasal connection.
Figure 1. MRI and PET imaging features.
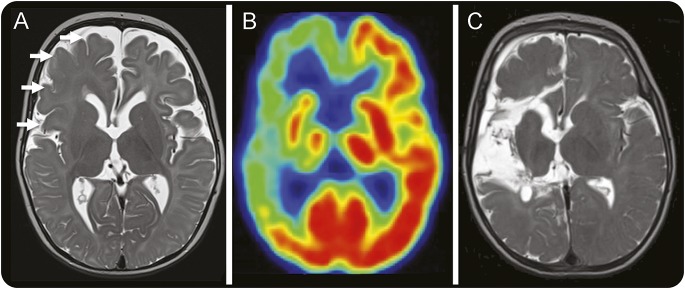
(A) Axial T2-weighted image performed at age 7 months showing blurring of the gray–white junction, simplified sulcation, and hypointense white matter signal for age maximal in the right frontal and central regions consistent with extensive focal cortical dysplasia (white arrows). (B) Interictal fluorodeoxyglucose-PET image consistent with severe hypometabolism of the majority of the right hemisphere. (C) Axial T2-weighted image performed 3 months after right hemispherotomy demonstrating the areas from which tissue was resected.
Figure 2. Histopathology, phospho-S6 immunostaining, and MTOR mutation location.

(A) Low-power and (B) high-power photomicrographs of dysplastic cortex stained with hematoxylin & eosin. (A) Cortical dyslamination and numerous dysmorphic cytomegalic neurons. (B) Dysmorphic cytomegalic neurons in greater detail with margination of Nissl substance, prominent nucleoli, and irregular cytoplasmic profiles (black arrows). No balloon cells were seen. (C) Positive phospho-S6 immunostaining (Ser235/236, rabbit polyclonal, 1:200; Cell Signaling) in dysmorphic cytomegalic neurons (black arrowheads), consistent with mTOR pathway activation (scale bar: A = 200 μm; B and C = 100 μm). (D) Schematic representation of the location of the p.W1456G variant and the MTOR domain structure. The location of the HEAT repeats, FAT domain, rapamycin-biding domain (FRB), kinase domain, and FATC motif are indicated. Protein coordinates were derived from the GeneCards human gene database (http://www.genecards.org/).
Genetic analysis.
Nine candidate heterozygous germline variants were identified in both the resected brain and lymphocyte-derived DNA (table e-2). Analysis for simple somatic variants identified 192 candidate SNVs with MuTect and 505 with VarScan using paired analysis with resected affected brain tissue and normal lymphocyte-derived DNA. VarScan also identified a further 364 INDELs. Only one SNV passed the subsequent quality filters and met the additional criterion of being a predicted damaging coding sequence variant within the candidate gene list; no INDEL met these conditions. The novel variant in MTOR (chr1:A11217312C, NM_004958.3 c4487T>G, p.W1456G) was identified in 6 reads by both MuTect and VarScan (table e-2), equivalent to a heterozygous frequency of 8.3%. We confirmed the variant in 5 of 54 subclones analyzed (approximately 9%) by clonal assay of brain-derived genomic DNA.
Of the variants observed in the germline and somatic analysis, the somatic variant in MTOR is the most compelling candidate because of accumulating knowledge regarding the role of mTOR pathway genes in cortical malformations, the previous identification of a somatic mutation of MTOR in a patient with a syndromic hemispheric dysplasia, and phospho-S6 labeling confirming mTOR signaling activation in the tissue. The p.W1456G alteration has been reported in a liver cancer cell line in the International Cancer Genome Consortium database,7 although the functional significance was not investigated. However, the amino acid is highly conserved and is localized to the FAT domain, which flanks the catalytic site and is important in regulating its activity by mediating binding to the endogenous inhibitor DEPTOR.8 The mutations p.L1460P and p.C1483F have been shown to result in reduced DEPTOR binding and upregulated MTOR activity.7 In addition, a recent in vitro study demonstrated that the analogous p.W1456R alteration significantly upregulated MTOR protein kinase activity and conferred strong tumorigenicity.9 These functional data provide further evidence supporting the likely pathogenicity of the mutation in MTOR identified in the resected dysplastic tissue.
DISCUSSION
Mutations in MTOR have been reported previously in a patient with hemispheric cortical dysplasia associated with hypomelanosis of Ito.5 As in our patient, the reported mutation was restricted to the malformation in a low-level mosaic form and not present in lymphocyte-derived DNA. The phospho-S6 staining in the resected tissue in our patient supports enhanced activation in the mTOR pathway as the likely mechanism underlying the FCD. Aberrations in the mTOR pathway are being increasingly recognized in the etiology of malformations of cortical development, not only through mutations in the tuberous sclerosis genes TSC1 and TSC2, but also by germline and postzygotic mutations in AKT3, PIK3R2, and PIK3CA,10 germline mutations in DEPDC5,6 and somatic mutations in PIK3CA, AKT3, and MTOR.5 As in our patient, identifying these mutations may require analysis of DNA from affected brain tissue using sequencing and bioinformatic techniques designed to detect low-level mosaicism. The mutations may be specific to certain clones of cells in the malformed tissue with the effects of these mutations, albeit at low level, sufficient to cause major epileptogenic malformations with potentially devastating outcomes. Assessment of this hypothesis will require mutation detection in single cells or populations of cells of the same lineage collected by cell sorting techniques, in addition to deep sequencing techniques to detect very low levels of somatic mosaicism, potentially in more accessible tissues.11 The role of MTOR and related genes in the more common and restricted forms of FCD will require further study of resected tissue using a similar brain vs blood sequencing strategy and, if confirmed, may provide opportunities for novel treatment strategies using mTOR inhibitors.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patient and his family for participating in this study and acknowledge the generous support of the Lefroy and Handbury families.
GLOSSARY
- FCD
focal cortical dysplasia
- SNV
single nucleotide variant
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R.J.L. performed patient recruitment, provided and interpreted the clinical data, formulated the study, and wrote the manuscript. T.S. performed bioinformatic analysis, interpreted the data, and revised the manuscript. A.P.L.M. performed molecular analysis and read/contributed to the manuscript. K.P. performed patient recruitment and read/contributed to the manuscript. G.G. performed patient recruitment and read/contributed to the manuscript. W.M. contributed to the design of the study, provided tissue samples, and read/contributed to the manuscript. D.M. interpreted pathologic data, provided histopathologic images, and read/contributed to the manuscript. A.S.H. contributed to the design of the study, provided clinical data and tissue samples, and read/contributed to the manuscript. M.B.D. contributed to the design of the study, provided and interpreted the clinical data, and read/contributed to the manuscript. D.J.A. contributed to the design of the study, provided and interpreted the clinical data, and read/contributed to the manuscript. P.C. contributed to the conceptualization/design of the study, performed molecular and immunohistochemical analysis, and read/contributed to the manuscript. M.B. performed bioinformatic analysis, interpreted the data, and read/contributed to the manuscript. P.J.L. formulated the study, interpreted the data, and cowrote the manuscript.
STUDY FUNDING
This work has been supported by the Victorian Government's Operational Infrastructure Support Program and Australian Government NHMRC IRIISS. Funding was provided by the National Health and Medical Research Council of Australia, the Murdoch Childrens Research Institute, and the Campbell Edwards Trust. M.B. is supported by an ARC Future Fellowship (FT100100764), A.P.L.M. is supported by an Australian Postgraduate Award, and P.J.L. is supported by an NHMRC Career Development Fellowship (APP1032364).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Blumcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, Tsai V, Parker WE, Aronica E, Baybis M, Crino PB. Detection of human papillomavirus in human focal cortical dysplasia type IIB. Ann Neurol 2012;72:881–892. [DOI] [PubMed] [Google Scholar]
- 3.Leventer RJ, Jansen FE, Mandelstam SA, et al. Is focal cortical dysplasia sporadic? Family evidence for genetic susceptibility. Epilepsia 2014;55:e22–e26. [DOI] [PubMed] [Google Scholar]
- 4.Crino PB. mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 2011;17:734–742. [DOI] [PubMed] [Google Scholar]
- 5.Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 2012;44:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 2014;75:782–787. [DOI] [PubMed] [Google Scholar]
- 7.Grabiner BC, Nardi V, Birsoy K, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov 2014;4:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009;137:873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murugan AK, Alzahrani A, Xing M. Mutations in critical domains confer the human mTOR gene strong tumorigenicity. J Biol Chem 2013;288:6511–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riviere JB, Mirzaa GM, O'Roak BJ, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet 2012;44:934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamuar SS, Lam AT, Kircher M, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med 2014;371:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.