

Abstract
The high prevalence of Herpesviruses in the population and the maintenance of lifelong latent reservoirs are challenges to the control of herpetic diseases, despite the availability of antiviral pharmaceuticals that target viral DNA replication. In addition to oral and genital lesions, herpes simplex virus infections and recurrent reactivations from the latent pool can result in severe pathology including neonatal infection and mortality, blindness due to ocular keratitis, and viral-induced complications in immunosuppressed individuals. Herpesviruses, like their cellular hosts, are subject to the regulatory impacts of chromatin and chromatin modulation machinery that promotes or suppresses gene expression. The initiation of herpes simplex virus infection and reactivation from latency is dependent on a transcriptional coactivator complex that contains two required histone demethylases, LSD1 and JMJD2s. Inhibition of either of these enzymes results in heterochromatic suppression of the viral genome and a block to infection and reactivation in vitro. Here, the concept of epigenetic suppression of viral infection is demonstrated in three animal models of herpes simplex virus infection and disease. Inhibition of LSD1 via treatment of animals with the monoamine oxidase inhibitor tranylcypromine results in suppression of viral lytic infection, subclinical shedding, and reactivation from latency in vivo. Phenotypic suppression is correlated with enhanced epigenetic suppression of the viral genome and suggests that, even during latency, the chromatin state of the virus is dynamic. Given the expanding development of epipharmaceuticals, this approach has substantial potential for anti-herpetic treatments with distinct advantages over the present pharmaceutical options.
Introduction
The worldwide prevalence of herpes simplex virus (HSV) in the human population is estimated to be 50-90% with 70-80% serologically positive by adolescence and near 100% positive for viral DNA in autopsied trigeminal ganglia of adults >60 yrs. of age (1-3). Following the initial primary infection, herpes simplex virus establishes life-long latent infections in sensory neurons. Periodic reactivation from the latent viral reservoirs results in episodes of lytic infection, recurrent disease, or asymptomatic shedding of the virus (4).
In addition to oral and genital lesions, HSV ocular infections (primary and reactivations) remain the leading cause of virus-mediated blindness in the western world with >300,000 cases of ocular infection diagnosed yearly (1, 3, 5). Neonatal infections also represent a critical clinical issue and can result in disseminated infection with mortality, ocular disease, and continued neurological or developmental issues (3). In addition to disease directly related to HSV infection and recurrence, the virus is also a co-pathogen and has been linked to increased rates of HIV transmission (6-9). Importantly, the most prevalent means of HSV transmission is via asymptomatic viral shedding.
Current control of viral primary infection and reactivation from latency is via anti-herpetic pharmaceuticals that target viral replication components to inhibit viral DNA synthesis during the late stage of the infectious cycle. While an important advance in treatment of HSV disease, these compounds do not completely control reactivation, viral subclinical shedding, or the associated inflammation that contributes to pathologies such as ocular scarification. Additionally, as these inhibitors target virus-encoded replication components, resistant viral mutants have evolved that are particularly evident in immunosuppressed patients and those with recurrent herpetic keratitis (10). Thus, there is a need for new therapeutic approaches to control HSV infection, reactivation, and transmission.
Epigenetics represents an evolving frontier for the development of therapeutics for the control of cancers (11-17) and other diseases (18-20). Advances in the identification and small molecule targeting of epigenetic components are rapidly driving emerging epi-pharmaceuticals (21-25). However, recent recognition of the role of chromatin and chromatin modulation components in control of HSV lytic infection, latency, and reactivation suggests epi-pharmaceuticals may also be used in approaches to anti-herpes therapies.
Like the cellular genome, many viruses are subject to epigenetic regulation. Herpesviral genomes are encapsidated without histones but rapidly acquire chromatin structures upon infection (26-29). For HSV, this is a dynamic process in which repressive heterochromatin accumulates on the viral genome in the absence of critical viral and host factors (Fig. 1) (30-34). This is countered by specific chromatin modulation components, including the transcriptional coactivator HCF-1 in concert with the LSD1 (KDM1A) and JMJD2 (KDM4 family) histone demethylases that reduce the levels of heterochromatic marks associated with the viral chromatin and promote the expression of the first wave of viral genes (32, 33, 35, 36).
Fig. 1.

Multiple chromatin modulation enzymes drive the initial chromatin state of the HSV genome. Model illustrating the initial heterochromatic chromatin state of viral genome during infection or latency and the coactivator complex required to modulate this to a euchromatic chromatin structure for productive infection or reactivation from latency. TCP inhibits the required activity of the histone demethylase LSD1.
Similarly, the recurrent cycle of viral latency-reactivation in sensory neurons is modulated by chromatin and chromatin regulatory factors (26, 27, 37). During latency, nucleosomes bearing repressive histone H3-lysine 9 and lysine 27-methylation marks are associated with the silent viral lytic genes (26, 38-41). Reactivation from latency is correlated with the transition to euchromatic marks (37, 42, 43) due, at least in part, to the HCF-1/LSD1/JMJD2 modulation complex (32, 35, 44). In cell culture models of HSV infection, inhibition of the activities of either LSD1 or the JMJD2s results in a block to viral gene expression and the progression of infection as well as a block to viral reactivation from latency in a mouse ganglia explant system. Thus, epigenetic therapies could provide approaches to suppression of the initiation of HSV infection and reactivation that are distinct from the present pharmaceuticals by promoting and maintaining epigenetic suppression of the virus.
Here, this concept is demonstrated in three animal model systems where pharmaceutical (TCP, tranylcypromine) inhibition of the required histone demethylase LSD1 resulted in suppression of primary infection, a block to subclinical shedding, and reduction in recurrent lesions. Importantly, treated animals exhibited enhanced epigenetic suppression of the virus that correlated with the phenotypic inhibition of disease. The results represent a comprehensive study demonstrating the principle and the potential of epigenetic approaches to anti-HSV therapy.
Results
Three animal model systems of HSV infection were utilized to investigate the impacts of inhibition of the histone demethylase LSD1. Each animal model has distinct advantages in assessing the complex lifecycle of the virus. The mouse model is appropriate for the quantification of mortality rates and viral loads in tissues during ocular or intranasal infections. The rabbit eye model permits clinical assessment of herpetic keratitis and, unlike the mouse model, these animals exhibit viral shedding and spontaneous viral reactivation from latency. Finally, the guinea pig genital model of HSV-2 infection is highly utilized to monitor clinical lesion recurrence. Together, these animal models comprehensively represent the spectrum of HSV mediated disease.
Suppression of primary infection in the mouse model system
To initially assess the impact of LSD1 inhibition on HSV primary infection, mice were infected with HSV-1 via the ocular route and treated topically or intraperitoneally (IP) with the LSD1 inhibitor TCP (45), control ACV (Acyclovir, viral DNA replication inhibitor), or Vehicle. As shown in Fig. 2A, mice topically treated with TCP exhibited enhanced survival compared to Vehicle-treated animals (Vehicle 37.5%, TCP, 75%; ACV, 85%). In a parallel manner, mice treated IP with TCP had reduced viral loads in sensory ganglia (Fig. 2B).
Fig. 2.
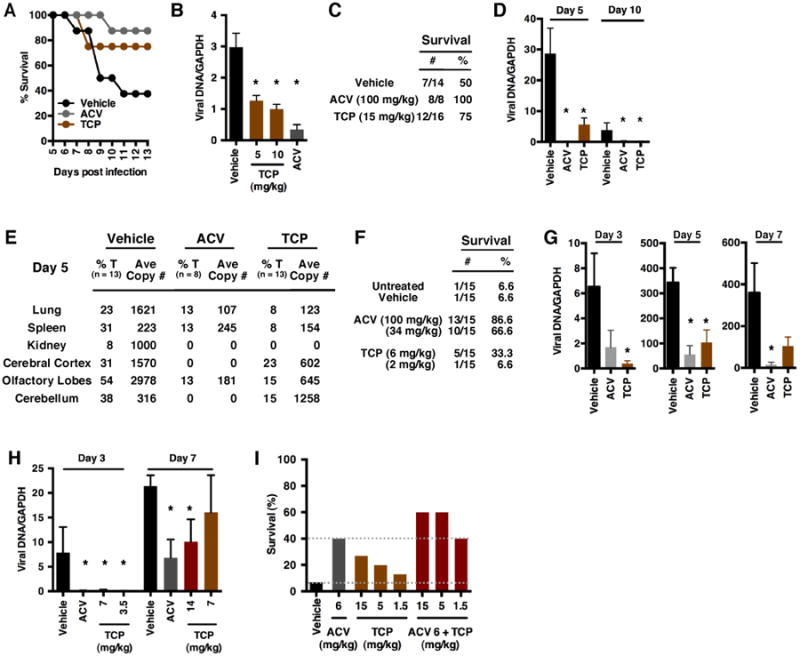
Inhibition of LSD1 reduces primary acute infection in the mouse model system. (A-B) Ocular infection with 1×105 pfu HSV-1(F)/eye. (A) Survival of mice treated topically with Vehicle, ACV (5 mM) or TCP (5 mM). (B) Viral loads in ganglia at 10 dpi of mice treated IP with Vehicle, ACV (100 mg/kg) or TCP (5 or 10 mg/kg). (C-E) Intranasal infection with 1.1×104 pfu (one LD50) HSV-2(MS). Mice were treated orally with Vehicle, ACV (100 mg/kg) or TCP (15 mg/kg). (C) Survival chart. (D) Viral loads in ganglia at 5 and 10 dpi. (E) Viral loads in select organs at 5 dpi. %T, % of total mice positive. (F-H) Intranasal infection with 1.1×105 pfu (one LD90) HSV-2(MS). (F) Survival of mice treated orally with Vehicle, ACV, or TCP at the indicated levels. (G) Viral loads in ganglia at the indicated dpi in mice treated orally with Vehicle, ACV (100 mg/kg), or TCP (6 mg/kg). (H) Viral loads in ganglia of mice implanted with Vehicle or TCP time-release pellets (ACV, oral, 100 mg/kg). (I) Survival of mice infected intranasally with 1.1×105 pfu (one LD90) HSV-2(MS) and treated orally with Vehicle, ACV, TCP, or the combination of ACV and TCP. Data are means +/- s.e.m. *Statistically significant.
As a model of neonatal infection, mice infected intranasally with an LD50 of HSV-2 and treated orally with TCP showed increased survival (Fig. 2C) and reduced viral loads in sensory ganglia (Fig. 2D) and select tissues (Fig. 2E, Fig. S1). Similar results were obtained when mice were infected at a stringent LD90 and treated orally with TCP (Fig. 2F-G) or via insertion of time-release TCP drug pellets (Fig. 2H).
Finally, because TCP blocks viral gene expression and the initiation of infection while ACV functions at the later stage of viral DNA replication, the impact of combined therapy was investigated. As shown in Fig. 2I, combined TCP and ACV treatment increased survival of animals infected with HSV-2 intranasally (LD90) over that achieved by treatment with either compound individually. Collectively, the results indicate that inhibition of the histone demethylase LSD1 suppresses HSV in vivo, even at high levels of infection, and that approaches to combinational therapy approaches can improve the impacts of the present therapeutics.
Suppression of infection in the rabbit eye model of herpetic keratitis
Primary infection of the rabbit eye provides a model of herpetic keratitis in which defined parameters of infection and clinical pathology can be assessed. Therefore, rabbits were treated orally with TCP, Vehicle, or control Valtrex (VCV) and subsequently infected with HSV-1 via the ocular route. Viral yields and parameters of ocular disease (Fig. S2) were assessed from 7-14 days post infection (dpi) (Fig. 3). Treatment with TCP and control VCV both reduced the levels of infectious virus detected in daily eye swabs relative to the Vehicle control (Fig. 3A). Furthermore, in assessment of 4 clinical parameters of ocular disease (Fig. 3B-E) in addition to slit lamp examination (Fig. 3F), TCP treatment clearly improved the clinical status of treated animals (Cumulative Scores, Fig. 3G).
Fig. 3.
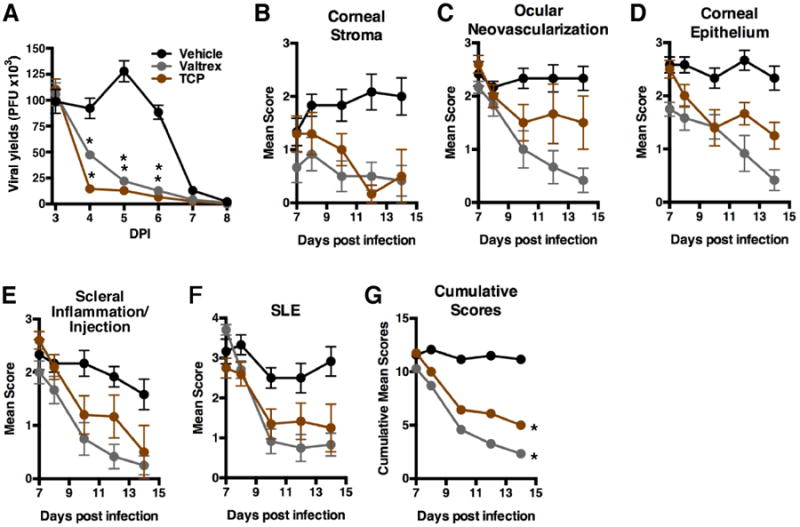
Inhibition of LSD1 reduces viral yields and ocular disease during acute infection of rabbit corneas. (A-G) Ocular infection with 2.5×105 pfu HSV-1(17sn+). Rabbits were treated orally with Vehicle, Valtrex (VCV, 30 mg/kg), or TCP (12 mg/kg) from -2 to 14 dpi. (A) Levels of infectious virus detected in eye swabs collected 3-8 dpi. (B-G) Parameters of ocular disease were assessed at 7-14 dpi. (B-F) Mean values of scores for Corneal Stroma, Ocular Neovascularization, Corneal Epithelium, Scleral Inflammation/Injection, and Slit-Lamp Examinations (SLE). (G) Cumulative scores of (B-F). Data are means +/- s.e.m. *Statistically significant.
Suppression of viral reactivation and shedding
In contrast to the mouse model, rabbits readily exhibit spontaneous viral reactivation and subclinical viral shedding in vivo. Thus, the rabbit ophthalmic model has been used extensively to investigate the biology of HSV recurrent disease. Previously, inhibitors of LSD1 or JMJD2 protein activities suppressed HSV reactivation in a mouse latently infected ganglia ex vivo explant system (32, 35). To investigate the impacts of TCP-mediated inhibition of LSD1 on viral reactivation in vivo, rabbits were infected with HSV-1 via the ocular route and maintained for 28 days to allow resolution of the primary infection and the establishment of viral latency. Subsequently animals were treated orally with TCP, Vehicle, or VCV. Eye swabs were taken daily to assess the level of viral reactivation and shedding by detection of infectious HSV-1 and viral DNA. As shown in Fig. 4A-B, 24.7% of swabs from the vehicle treated group were positive for infectious virus (Fig. 4A) while 36.6 % were positive for viral DNA (Fig. 4B). Strikingly, while VCV reduced these levels to 7.1% and 17.1%, respectively, TCP treatment abrogated the detection of infectious virus and reduced the number of swabs with detectable viral DNA to 3.3%. Additionally, those swabs positive for viral DNA in TCP treated animals had significantly reduced copy number relative to both Vehicle and VCV treated animals.
Fig. 4.
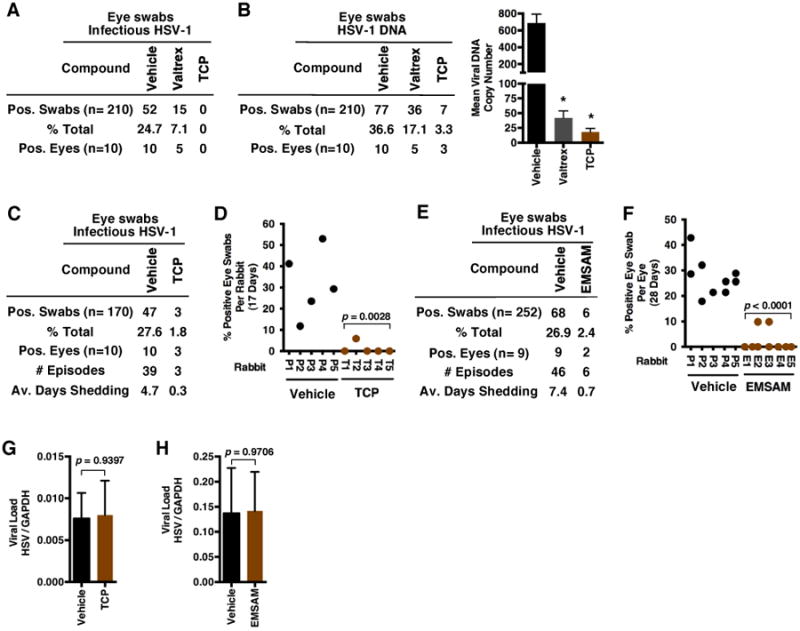
Inhibition of LSD1 blocks viral reactivation and shedding in the rabbit eye model system. (A-J) Ocular infection with 3×105 HSV-1(17syn+). (A-B) Eye swabs, collected at 33-53 dpi, were assayed for the presence of infectious HSV-1 and viral DNA in animals treated orally with Vehicle, Valtrex (250 mg/kg) or TCP (12 mg/kg). (C-D) Latently infected rabbits were implanted with placebo (Vehicle) or TCP (9.5 mg/kg) time-release pellets at 48 dpi. Eye swabs collected at 52-68 dpi were assayed for the presence of infectious HSV-1 and represented as positive per group (C) and per rabbit (D). (E-F) Latently infected rabbits were treated with Placebo or Selegiline (EMSAM) time-release topical patch (5.5 mg/kg) at 17-47 dpi. Eye swabs collected at 20-47 dpi were assayed for infectious HSV-1 and represented as positive per treatment group (E) and per eye (F). (G) Viral loads in ganglia of rabbits treated with Vehicle and TCP time-release pellets at 68 dpi. (H) Viral loads in ganglia of rabbits treated with Vehicle or EMSAM transdermal patches at 47 dpi. *Statistically significant.
Similar results showing significant reductions in the levels of viral reactivation were obtained from animals treated by insertion of time-release TCP drug pellets (Fig. 4C-D) or via transdermal delivery of a second LSD1 inhibitor (Selegiline/EMSAM patch) (Fig. 4E-F). In each case, the inhibitor treated group had significantly reduced numbers of HSV-1 positive eye swabs, reduced number of recurrences (episodes), and reduced times of shedding (Fig. 4E-F). No differences were seen in the viral loads in trigeminal ganglia of animals from the treated groups relative to the control groups (Fig. 4G-H), indicating that TCP-mediated reductions in viral shedding and reactivation were not due to inherent variance in the ganglia loads.
Enhanced epigenetic suppression of the latent HSV-1 genome
As inhibition of LSD1 results in enhanced epigenetic suppression of HSV-1 in vitro (32, 44), chromatin immunoprecipitation (ChIP) assays were performed on trigeminal ganglia removed from Vehicle, TCP (time-release drug pellet), and EMSAM treated rabbits at the conclusion of the treatment periods. As shown, viral genomes in TCP (Fig. 5A) and EMSAM (Fig. 5B) treated animals exhibited increased levels of histones (H3) and highly enriched levels of the repressive histone H3-lysine 9 methylation (H3K9-me) associated with the viral Immediate Early gene regions. In contrast, no significant changes in the levels of H3 or H3K9-me were seen on the control cellular GAPDH gene. The results illustrate that inhibition of LSD1 results in enhanced epigenetic suppression of the HSV-1 genome that translates into reduced viral shedding and reactivation in vivo.
Fig. 5.
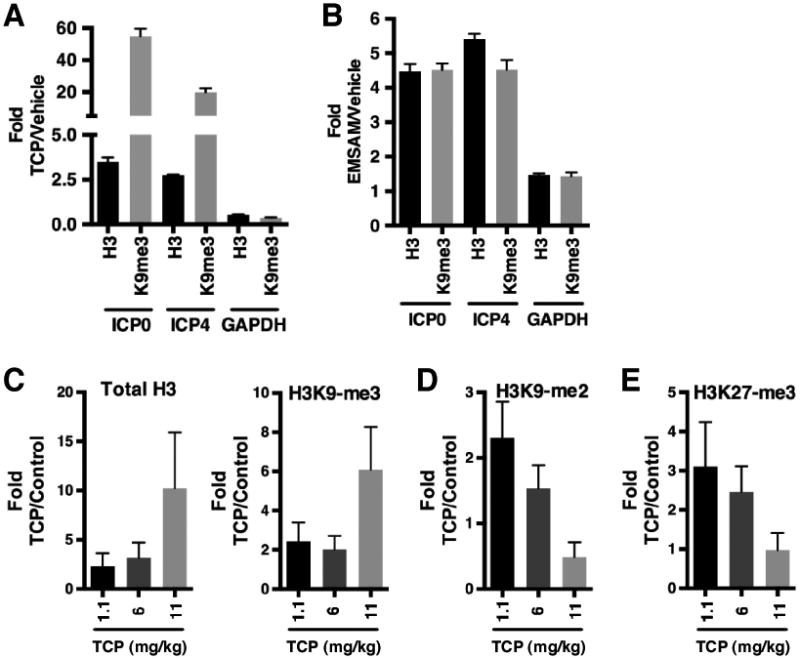
Inhibition of LSD1 results in enhanced epigenetic suppression of HSV-1. (A-B) ChIP assays showing increased levels of histone H3 and H3K9-methylation at viral genomes in ganglia of TCP-time release (A) and transdermal EMSAM treated rabbits (B). (C-E) ChIP assays showing (C) increased levels of histone H3 and H3K9-me3 and (D-E) decreased levels of H3K9-me2 and H3K27-me3 on the viral genome of latently infected mice treated with time-release TCP pellets. Data are means +/- s.e.m.
To determine if similar epigenetic suppression would be evident in ganglia of TCP treated mice, latently infected animals were implanted with time-release TCP drug pellets delivering different doses of compound. At 21 days post implantation, trigeminal ganglia were removed and ChIP assays were performed. As shown (Fig. 5C), TCP treated mice exhibited increased levels of histones and H3K9-me3 that correlated with the level of TCP dosage, further indicating that TCP treatment results in enhanced epigenetic suppression of latent HSV-1 in vivo. Concomitant with these increases, TCP treatment also resulted in reduced levels of H3K9-me2 (Fig. 5D), likely due to the conversion of the di-methyl mark to the H3K9-me3 state. Strikingly, the “facultative heterochromatin” mark H3K27-me3 was also significantly reduced in TCP treated mice (Fig. 5E), suggesting that treatment resulted in a shift in the chromatin dynamics of the latent viral pool.
Suppression of HSV-2 viral shedding and lesion recurrence in the guinea pig vaginal model
The guinea pig has been utilized extensively as a model of recurrent HSV genital lesions and viral shedding. Animals are infected intravaginally with HSV-2 and recurrent lesions are scored after recovery from primary disease according to defined criteria (Fig. S3A). Here, infected guinea pigs were treated orally with TCP, Vehicle, or ACV control from 15-35 days post infection. Recurrent lesions were scored by severity during the treatment period and extending for an additional 16 days. As shown in Fig. 6A-C, TCP reduced the cumulative HSV disease and recurrent lesion scores of infected animals in a manner comparable to that of the control ACV treated group. More strikingly, vaginal swabs assessed for HSV-2 DNA demonstrated a clear reduction in the number of positive swabs and the level of viral genomes in the TCP treated animals relative to either Vehicle or ACV (Fig. 6D-E). This difference was not due to variance in the viral loads in the dorsal root ganglia (DRG) or spinal cords (Fig. 6F).
Fig. 6.
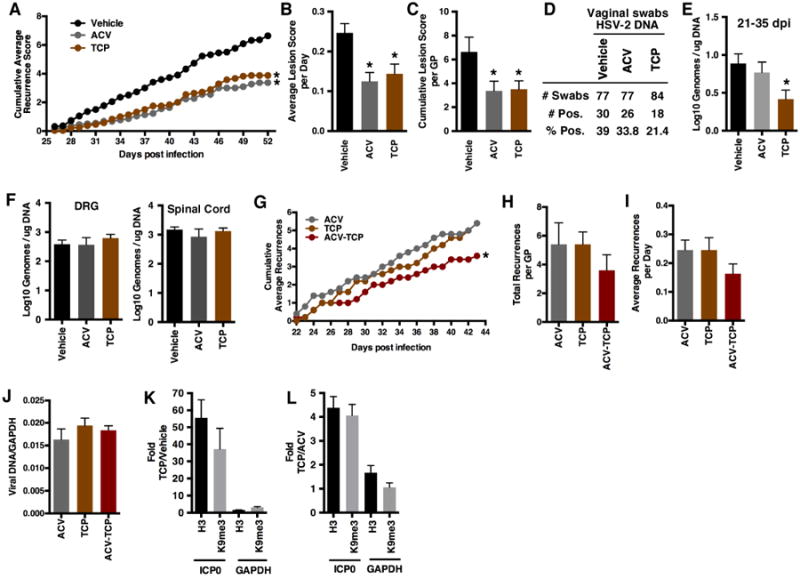
Inhibition of LSD1 reduces recurrent disease in the guinea pig model. (A-F) Vaginal infection with 1×106 pfu HSV-2(MS). Guinea pigs were treated orally with Vehicle, ACV (5 mg/ml ad libitum in water), or TCP (15 mg/kg/day) from 15-35 dpi. (A-C) Lesions were scored (26-52 dpi) and shown as (A) cumulative average disease and recurrent lesion scores per day as defined in S3A; (B) average lesion scores per day; and (C) cumulative lesion scores per guinea pig. (D-E) HSV DNA detected in vaginal swabs taken from 21-35 dpi. (F) Viral loads in DRGs and spinal cords at 52 dpi. (G-J) Vaginal infection with 2×105 pfu HSV-2(MS). Guinea pigs were treated IP with ACV (6 mg/kg/day), TCP (5 mg/kg/day), or the combination for 22-43 dpi. (G-I) Lesion recurrences were scored as defined in S3B and are shown as (G) cumulative average recurrences; (H) total recurrences per guinea pig; and (I) average recurrences per group per day. (J) Viral loads in DRGs at 43 dpi. (K-L) ChIP assays showing increased levels of histone H3 and H3K9-methylation at viral promoters in dorsal root ganglia of guinea pigs (K) treated orally with Vehicle or TCP (30 mg/kg/day) and (L) treated IP with ACV (6 mg/kg/day) or TCP (5 mg/kg/day). Data are means +/- s.e.m. *Statistically significant.
As demonstrated in the mouse model system (Fig. 2I), the combination of TCP and ACV treatment increased the survival rate to a greater extent than either compound alone. In a similar manner and based upon the previous experiment (Fig. 6A), combined ACV and TCP treatment in the guinea pig vaginal model further reduced the cumulative HSV-2 recurrences as compared to either compound alone (Fig. 6G-I, S3B). No differences in the viral loads between the groups were detected in the dorsal root ganglia (Fig. 6J).
Consistent with the impacts of TCP on the chromatin status of viral genomes in both the mouse and rabbit trigeminal ganglia, treatment of infected guinea pigs with TCP (30 mg/kg/day) resulted in enhanced histone levels and repressive heterochromatic marks associated with the viral genomes in the dorsal root ganglia (Fig. 6K). In a second study, qualitatively comparable results were also seen in animals treated with a lower (5 mg/kg/day) TCP dose (Fig. 6L) when compared to the control ACV.
Discussion
Despite the availability and widespread use of pharmaceutical inhibitors for the treatment of herpesvirus infections, herpes simplex virus remains a clinically important pathogen. A high percentage of the human population is infected and carries the virus in a latent state in sensory neurons, while asymptomatic shedding of the virus maintains the transmission rate. Importantly, neonatal HSV infections can result in mortality or continued developmental-neurological issues while HSV ocular infections and recurrences are the leading virus-mediated cause of blindness or the requirement for corneal transplants. In addition, along with other herpesviruses, HSV is a complicating factor in immunosuppressed individuals and is a cofactor in HIV transmission. Presently, the primary anti-herpetic pharmaceuticals target virus-encoded DNA replication machinery during a late stage in the infection cycle. However, these compounds do not efficiently suppress viral shedding, subclinical reactivation, and lytic recurrence. Additionally, while the incidence of resistant virus populations is low, the frequency is increased in immunosuppressed patients and those with herpetic keratitis. Importantly, recent studies indicate that prophylactic ACV treatment does not reduce the HSV-mediated enhancement of HIV transmission (46, 47). Thus, it is clear that new approaches to address deficiencies in the prevention of transmission and treatment of recurrence are needed.
Together, the recognition that viruses such as HSV, are subject to epigenetic control (27) and the emerging development of epigenetic pharmaceuticals open new avenues for developing antivirals. Inhibitors of chromatin modification enzymes required for the initiation of viral infection can prevent viral gene expression, reduce the potential for inflammation, and reduce the evolution of drug-resistant viral strains. Here, epigenetic suppression of HSV infection, shedding, reactivation, and clinical recurrence was demonstrated in the primary animal models of HSV disease using pharmaceutical inhibition of the histone demethylase LSD1.
LSD1 is a component of the regulatory paradigm that mediates the initiation of herpes simplex virus infection by modulating the epigenetic state of the virus to allow the first wave of viral gene expression. The enzyme is specifically recruited to viral gene promoters upon infection, and the catalytic demethylation activity is required to drive the chromatin-mediated repressed state of the genome to an active euchromatic state. TCP and other MAOIs inhibit the activity of LSD1 and these compounds block infection in vitro and viral reactivation in the latently infected mouse ganglia explant model.
Mechanistically, inhibition of LSD1 results in accumulation of repressive methylation on histones associated with the viral genome and a block to viral gene expression. Notably, a specific LSD1 inhibitor (OG-L002) that does not inhibit the cellular MAOA or MAOB was also effective in reducing HSV lytic infection and reactivation (44). In contrast, a compound that targets the cellular MAOA and MAOB but does not inhibit LSD1 had no effect on viral infection. Thus, LSD1 represents a potentially important therapeutic target for anti-herpetic therapeutics.
In this study TCP treatment of HSV infected mice resulted in reduced pathology as observed by reduced mortality, reduced accumulation of genomes in sensory ganglia, and reduced viral loads in tissues. Importantly, reduction in viral pathology was evident in animals infected with high doses, LD50 or LD90 doses, and using a variety of inhibitor treatments (oral, time-release, topical, intraperitoneally)#. Similarly, in the rabbit eye model of herpetic keratitis, treatment of primary infection reduced the critical clinical indicators of ocular disease.
While suppression of HSV primary infection in these experiments was effective, TCP treatment resulted in a dramatic reduction in viral shedding and reactivation or recurrences. This was clearly evident in the rabbit model of spontaneous reactivation and shedding. In this model, TCP was significantly more effective than VCV treatment.
Finally, in the guinea pig vaginal model of HSV-2 recurrent disease, inhibition of LSD1 resulted in suppression of recurrent lesions as effectively as the control ACV and recurrent shedding more effectively than ACV. Together these studies demonstrate suppression of HSV lytic infection, shedding, and reactivation that correlated with enhanced epigenetic suppression of the viral genome.
Importantly, TCP inhibition of LSD1 targets the initiation stage of viral lytic infection and reactivation. This is distinct from the predominant anti-herpetic pharmaceuticals, nucleoside analogues that target the later stage of viral DNA replication. Consistent with these two distinct mechanisms, combined treatment (TCP and ACV) resulted in enhanced suppression over the use of either compound alone. The results are more striking as the combination treatment used lower concentrations of the two compounds relative to the individual compound treatments. The results suggest that combination therapies targeting distinct stages of the HSV infectious cycle may be clinically advantageous with respect to the present antiviral therapies and may also reduce the propensity for the generation of ACV resistant viruses in the population.
The increased epigenetic silencing of the HSV genome, in sensory ganglia where the viral genome is latent, is consistent with the antiviral impacts of the LSD1 inhibitors. Strikingly, this argues that the epigenetic status of latent viral genomes is dynamic, with histone marks being continuously modulated. Interestingly, while the levels of repressive H3K9-me3 increased upon TCP treatment, the levels of the “facultative heterochromatin mark” H3K27-me3 decreased, suggesting a shift in the heterochromatin status of the population of viral genomes. This dynamic nature of HSV chromatin is consistent with recent studies of HIV where latent infection involves epigenetic silencing (48), and reactivation is a stochastic process (49, 50), involving “bursts” of reactivation which may proceed forward to a full reactivation (51).
Interestingly, the sensitivity of the state of the viral genome to LSD1 inhibition, as compared to the host GAPDH gene, may reflect the critical nature of this enzyme for HSV gene expression relative to other LSD1 targets. LSD1 demethylates both histone H3K4me1/2 and H3K9me1/2. However, the target selectivity and specificity is determined by cofactors. Additionally, multiple enzymes modulate H3K9me levels in the cell and thus, LSD1 inhibition would not be expected to have global impacts. The specific impacts on HSV gene expression may be conceptually similar to the regulation of cellular super-enhancers where it has been demonstrated that specific loci are surprisingly hypersensitive to a critical regulatory factor (52-54).
Investigation of the role of chromatin and specific chromatin modulation components in the control of viral infection, states of latency, and reactivation offers targets for antiviral therapies. In addition to HSV, human cytomegalovirus and adenovirus have also been shown to rely on the HCF-1/LSD1/JMJD2 transcriptional coactivator complex (35, 44). Therefore, compounds targeting these components may have activity against other viral pathogens in addition to HSV.
Epigenetic pharmaceuticals represent potential antivirals to control viral infection and reactivation of latent genomes. Inhibitors of LSD1 (OG-L002) as well as inhibitors of the JMJD2 demethylases (ML324) have also been shown to suppress HSV infection and reactivation in vitro (35, 44). Thus, there is potential to target multiple components to increase efficacy.
In the in vivo studies presented here, the MAOIs Tranylcypromine and Selegiline were chosen to investigate the impacts of LSD1 inhibition on HSV infection, shedding, and reactivation in vivo based upon multiple parameters. These compounds are very well characterized clinical pharmaceuticals and are available in multiple formats (oral compound, time release subcutaneous drug administration pellets, and time release transdermal patches). The characteristic TCP clinical doses range from 40-60 mg/day to 170 mg/day for refractive depression in patients. While the dose range used in these studies exceeds the levels used for the treatment of depression, this was anticipated as the IC50 for LSD1 is higher than for the target MAOA and MAOB. In the studies presented here, no toxicity was exhibited in the three animal model systems even at the highest dosage and lower doses also reduced viral infection and reactivation. Furthermore, advances in MAOI therapy, such as the transdermal or topical applications demonstrated here, have reduced the characteristic side effects of these compounds. However, it remains important to consider the potential issues that arise from off-target impacts of these compounds or of LSD1 inhibition in long-term treatment. Importantly, specific LSD1 inhibitors are in development for the treatment of various cancers. At this time, these inhibitors are less well characterized than the MAOIs but could represent antivirals that would have advantages over the non-selective MAOI compounds.
Overall, the described studies represent a comprehensive analysis of epigenetic inhibition of viral infection and reactivation from latent pools. This is in contrast to approaches used to attempt to “purge” latent HIV reservoirs by epigenetically inducing viral reactivation (55-57). Thus, epigenetic suppression can represent an approach to antivirals with extensive potential derived from studies of viral epigenetics and the emergence of the field of epipharmaceuticals.
Materials and Methods
Study Design
Three animal model systems of herpetic disease were used to study the potential for epigenetic suppression of HSV infection, subclinical shedding, and spontaneous recurrence using an inhibitor of the histone demethylase LSD1. Experiments were designed to utilize the most appropriate animal model for each aspect of HSV infection (primary infection, mouse model; spontaneous reactivation, rabbit eye model; clinical recurrence, guinea pig vaginal model).
Sample sizes for all experiments were based upon standards ascribed to the National Institute of Allergy and Infectious Diseases Anti-Viral Program and the specific expertise of each contributing research group. For rabbit and guinea pig experiments, infected animals were randomized to study groups based upon initial acute disease. Following infection in the rabbit eye model, slit-lamp examination was used to determine the extent of corneal damage and those eyes with extensive damage were discarded from the study. Animal groups were not blinded.
Animals and Infections
Mice
Ocular infections
6-7 week old Balb/c mice were infected with 1×105 pfu HSV-1(F) per eye following corneal scarification and treated topically or via IP injection as described in the appropriate figure legend and Supplementary Methods.
Intranasal infections
4 week old Balb/c mice were infected intranasally with 1.1×104 pfu (one LD50) or 1.1×105 pfu (one LD90) HSV-2(MS) and treated orally or via insertion of time-release drug pellets as described in the appropriate figure legend and Supplementary Methods.
Rabbits
Latency
New Zealand white (NZW) rabbits (McNeil Rabbitry; 2.0-2.5 kg) were infected with 2.5-3×105 pfu HSV-1 (17 Syn+ or McKrae) per eye following corneal scarification. Infection was verified by slit-lamp examination at 3 days post infection (dpi). Latently infected animals (>17 dpi) were assessed and those with normal corneas and ocular tissues were utilized. Rabbits were treated orally, via IP injection, via insertion of time-release drug pellets, or via topical drug patches as described in the appropriate figure legend and Supplementary Materials.
Acute Infection
NZW rabbits were infected as detailed above and treated as described in the appropriate figure legend. Ocular disease was assessed according to clinical parameters detailed in Supplementary Figure S2.
Eye Swabs
Rabbit eye swabs were analyzed for infectious HSV-1 or viral DNA as described (58).
Guinea Pigs
TCP Efficacy
Female Hartley guinea pigs (15 per group, 250-350 g, Charles River) were inoculated intravaginally with 1×106 pfu HSV-2(MS) by rupturing the vaginal closure membrane with a moistened calcium alginate tipped swab and instilling 0.1 ml of a virus suspension into the vaginal vault (59). Vaginal swabs were obtained at 2 dpi to assess infection. Following recovery from primary infection (15 dpi), animals were randomized based on acute disease. Animals were treated orally (15-35 dpi) with Vehicle, TCP 15 mg/kg/day (7.5 mg/kg twice daily), or ACV (5 mg/ml ad libitum in drinking water) and monitored for recurrent herpetic lesions (15-56 dpi) and recurrent viral shedding (21-35 dpi). Scoring criteria are defined in Supplementary Figure S3A. Dorsal root ganglia and spinal cords were collected from each animal at 57 dpi and processed for viral DNA loads. In a second study, animals were treated orally (15-35 dpi) with Vehicle, TCP (30 mg/kg/day), or ACV (5 mg/ml in drinking water). At 35 dpi, dorsal root ganglia were collected for ChIP analyses.
Viral Shedding
Vaginal viral swabs were collected 3 times per week (21-35 dpi). HSV-2 DNA levels were determined by qPCR as described (60).
TCP-ACV combination
Female Hartley guinea pigs (Charles River) were inoculated intravaginally with 2 × 105 PFU HSV-2(MS). At 22 dpi, animals were randomized based on acute disease. Animals were treated IP (22-43 dpi) with TCP (5 mg/kg/day), ACV (6 mg/kg/day) or TCP and ACV (5 mg/kg/day and 6 mg/kg/day, respectively). Recurrent lesions, defined as new lesions on the external genitalia, were scored (22-43 dpi). Scoring criteria are defined in Supplementary Figure S3B. Dorsal root ganglia were collected from each animal immediately after sacrifice and processed for viral DNA loads and ChIP assays.
Animal Care
Mice and guinea pigs were housed in American Association for Accreditation of Laboratory Animal Care-approved facilities. Rabbit experimental procedures were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) resolution for the use of animals in ophthalmic and vision research. All animal care and procedures were done according to Institutional Animal Care & Use Committee approved animal protocols and in accordance with the NIH Animal Care and Use Guidelines. All animals were monitored by experienced animal care staff and by the responsible research scientists. No indication of MAOI-mediated toxicity (i.e. weight loss, persistent hyper-excitability or behavioral changes, consumption of food and water, morbidity, or mortality) was evident in these experiments.
Compounds
The following compounds were used in these studies: TCP, Trans-2-phenylcyclopropylamine hydrochloride; SEL, Selegiline, R-(-)-Deprenyl hydrochloride; ACV, Acyclovir, Acycloguanosine (Sigma-Aldrich); VCV-Valtrex, Valacyclovir (GlaxoSmithKline Pharmaceuticals); TCP and Placebo time-release pellets (Innovative Research of America); EMSAM, Selegiline transdermal patches (Somerset Pharmaceuticals, Inc.).
Viral loads
Total genomic DNAs from mouse, rabbit, and guinea pig tissues were isolated and analyzed according to standard procedures as described (32, 60-62). Levels of viral DNA were determined by qPCR and are represented per ug of total DNA or as the ratio to the levels of the cellular GAPDH gene. The sequences of the relevant primer sets are listed in Supplementary Materials.
ChIP assays
Chromatin immunoprecipitation assays were done from latently infected mouse, rabbit and guinea pig sensory ganglia essentially as described (39, 42) with minor modifications detailed in Supplementary Methods.
Statistical analyses
Data are expressed as means +/- s.e.m. Statistical comparisons were made using Prism (V6.0) and included: Unpaired two-tailed t tests with a statistical significance of <0.05; ANOVA with post hoc Dunnett's multiple comparison test with a statistical significance of <0.025; Holm Sidak Multiple t tests, and Linear Regression Slope analyses. Additional details are given in the appropriate figure legends and in Supplementary Materials.
Supplementary Material
Fig. S1. Viral loads in select organs of mice treated with ACV or TCP at 10 dpi.
Fig. S2. Scoring criteria for ocular disease in the rabbit eye model.
Fig. S3. Scoring criteria for HSV disease in the guinea pig model.
Table S1. Experimental conditions.
Table S2. Statistical analyses.
Table S3. Antibodies and primers.
Numerical values of experimental data: Supplementary file
Acknowledgments
We thank J. Arbuckle & T. Pierson for editorial comments; J. Skinner (Laboratory of Immunogenetics, NIAID) for statistical analyses; NIAID Bld33 Animal Facility staff (NIH), L. L. Birke & M. Johnson (LSUHSC Animal Care), D. Pullum & H. Shen (University of Cincinnati), R. Crosby II & M. Simpson (LSUHSC); D.J. Collins & T.L. Rice (University of Alabama Birmingham) for technical assistance; and J.Arbuckle. (NIAID, NIH) for assistance to T.M.K. with mouse studies.
Funding: These studies were supported by the Laboratory of Viral Diseases, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, U.S. National Institutes of Health (T.M.K.); Contract HHSN272201000027I, Task Order HHSN2720001 (D.Q.); National Eye Institute R01-EY006311 (J.M.H.), Research to Prevent Blindness (RIB) Senior Investigator Award (J.M.H.), RIB unrestricted grant to the LSU Eye Center (J.M.H.), LSUHSC unrestricted grant to J.M.H.; U.S. Food and Drug Administration Intramural Funding (P.R.K.); Contract HHSN27220100008I (D.I.B. & R.D.C.); and NIH P01-AI098681 (D.M.K.).
Footnotes
Portions of this work, including murine studies, were presented at the 2012 & 2013 International Herpesvirus Workshops. While this manuscript was in preparation, a second study supported the use of TCP to suppress HSV infection in the mouse model system. H. W. Yao, P. H. Lin, F. H. Shen, G. C. Perng, Y. Y. Tung, S. M. Hsu, S. H. Chen, Tranylcypromine reduces herpes simplex virus 1 infection in mice. Antimicrobial agents and chemotherapy 58, 2807-2815 (2014).
Author contributions: Mouse studies: D.Q., J.L.V. & T.M.K. designed the studies and analyzed the experimental results. D.Q. conducted the intranasal infection therapeutic studies; T.M.K. & J.L.V. performed the mouse ocular infection studies; J.L.V. performed viral loads; D.M.K. & T.M.K. designed the mouse ChIP experiments and analyzed the experimental results; P.R. & J.S.L. performed mouse ganglia ChIP assays; T.M.K. performed mouse infections, drug implants, and ganglia preparation. Rabbit studies: J.M.H., C.C., T.P.F., & T.M.K. designed the studies and analyzed the experimental results. J.M.H., C.C., & T.P.F. conducted the studies. J.L.V. performed viral yields and rabbit ganglia ChIP assays. Guinea pig therapeutic studies: D.I.B., R.D.C., & T.M.K. designed and analyzed the therapeutic studies. D.I.B. & R.D.C. were responsible for study oversight and analyses. F.J.B. conducted the treatment and monitored the guinea pigs. J.L.V. performed guinea pig ChIP assays. Guinea pig combination treatment study: M.B-M., P.R.K., & T.M.K. designed the study and analyzed the experimental results. M.B-K. & P.R.K. conducted the study. J.L.V. performed guinea pig ChIP assays. T.M.K. assembled all data and wrote the manuscript. All authors commented on the manuscript prior to publication.
Competing interests: The NIH has the following patent applications: (i) Methods of preventing or treating viral infection or reactivation from latency in a host using inhibitors of the LSD1 protein (T.M.K. J.L.V., and Y.L.; U.S. patent application no. 61/083,304; International patent application no. PCT/US2009/051557); (ii) Method of preventing or treating viral infection via inhibition of the JMJD2 proteins (T.M.K., Y.L., and J.L.V.; U.S. patent application no. 61/366,563). All other authors declare no competing interests.
References
- 1.Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Survey of ophthalmology. 2012;57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hill JM, Ball MJ, Neumann DM, Azcuy AM, Bhattacharjee PS, Bouhanik S, Clement C, Lukiw WJ, Foster TP, Kumar M, Kaufman HE, Thompson HW. The high prevalence of herpes simplex virus type 1 DNA in human trigeminal ganglia is not a function of age or gender. J Virol. 2008;82:8230–8234. doi: 10.1128/JVI.00686-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitley R, Kimberlin DW, Prober CG. In: Human Herpesviruses Biology, Therapy, and Immunoprophylaxis. Arvin A, Whitley R, editors. chap. 32. Cambridge University Press; Cambridge: 2007. pp. 589–601. [PubMed] [Google Scholar]
- 4.Roizman B, Knipe DM, Whitley RJ. In: Fields Virology. 6th. Knipe DM, Howley PM, editors. Lippincott Williams & Wilkins; Philadelphia: 2013. [Google Scholar]
- 5.Kaye S, Choudhary A. Herpes simplex keratitis. Progress in retinal and eye research. 2006;25:355–380. doi: 10.1016/j.preteyeres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Duffus WA, Mermin J, Bunnell R, Byers RH, Odongo G, Ekwaru P, Downing R. Chronic herpes simplex virus type-2 infection and HIV viral load. International journal of STD & AIDS. 2005;16:733–735. doi: 10.1258/095646205774763298. [DOI] [PubMed] [Google Scholar]
- 7.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. Aids. 2006;20:73–83. doi: 10.1097/01.aids.0000198081.09337.a7. [DOI] [PubMed] [Google Scholar]
- 8.Wald A, Link K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis. 2002;185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- 9.Zhu J, Hladik F, Woodward A, Klock A, Peng T, Johnston C, Remington M, Magaret A, Koelle DM, Wald A, Corey L. Persistence of HIV-1 receptor-positive cells after HSV-2 reactivation is a potential mechanism for increased HIV-1 acquisition. Nat Med. 2009;15:886–892. doi: 10.1038/nm.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Velzen M, van de Vijver DA, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J Infect Dis. 2013;208:1359–1365. doi: 10.1093/infdis/jit350. [DOI] [PubMed] [Google Scholar]
- 11.Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, Rotolo A, Tough DF, Smithers NN, Bassil AK, Chapman TD, Harker NR, Barbash O, Tummino P, Al-Mahdi N, Haynes AC, Cutler L, Le B, Rahemtulla A, Roberts I, Kleijnen M, Witherington JJ, Parr NJ, Prinjha RK, Karadimitris A. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123:697–705. doi: 10.1182/blood-2013-01-478420. [DOI] [PubMed] [Google Scholar]
- 12.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, Gillespie SM, Fernandez D, Ku M, Wang H, Piccioni F, Silver SJ, Jain M, Pearson D, Kluk MJ, Ott CJ, Shultz LD, Brehm MA, Greiner DL, Gutierrez A, Stegmaier K, Kung AL, Root DE, Bradner JE, Aster JC, Kelliher MA, Bernstein BE. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46:364–370. doi: 10.1038/ng.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 16.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein HU, Popescu AC, Burnett A, Mills K, Casero RA, Jr, Marton L, Woster P, Minden MD, Dugas M, Wang JC, Dick JE, Muller-Tidow C, Petrie K, Zelent A. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyspianska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, Robson S, Rioja I, Li J, Wiese M, Cannizzaro E, Dawson MA, Huntly B, Prinjha RK, Green AR, Gottgens B, Kouzarides T. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia. 2014;28:88–97. doi: 10.1038/leu.2013.234. [DOI] [PubMed] [Google Scholar]
- 18.Kim DW, Park JW, Willingham MC, Cheng SY. A histone deacetylase inhibitor improves hypothyroidism caused by a TRalpha1 mutant. Hum Mol Genet. 2014;23:2651–2664. doi: 10.1093/hmg/ddt660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 2009;8:1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 21.Best JD, Carey N. Epigenetic opportunities and challenges in cancer. Drug Discov Today. 2010;15:65–70. doi: 10.1016/j.drudis.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery. Current opinion in chemical biology. 2010;14:505–510. doi: 10.1016/j.cbpa.2010.06.174. [DOI] [PubMed] [Google Scholar]
- 23.Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- 24.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 25.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with ‘epigenetic’ drugs: an update. Mol Oncol. 2012;6:657–682. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knipe DM, Cliffe A. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol. 2008;6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 27.Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels M, Parks RJ, Kristie TM. Snapshots: chromatin control of viral infection. Virology. 2013;435:141–156. doi: 10.1016/j.virol.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kristie TM, Liang Y, Vogel JL. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim Biophys Acta. 2010;1799:257–265. doi: 10.1016/j.bbagrm.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacasse JJ, Schang LM. Herpes simplex virus 1 DNA is in unstable nucleosomes throughout the lytic infection cycle, and the instability of the nucleosomes is independent of DNA replication. J Virol. 2012;86:11287–11300. doi: 10.1128/JVI.01468-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arbuckle JH, Kristie TM. Epigenetic repression of herpes simplex virus infection by the nucleosome remodeler CHD3. MBio. 2014;5:e01027–01013. doi: 10.1128/mBio.01027-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu H, Roizman B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. Journal of virology. 2009;83:4376–4385. doi: 10.1128/JVI.02515-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narayanan A, Ruyechan WT, Kristie TM. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc Natl Acad Sci U S A. 2007;104:10835–10840. doi: 10.1073/pnas.0704351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva L, Cliffe A, Chang L, Knipe DM. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008;4:e1000071. doi: 10.1371/journal.ppat.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Science translational medicine. 2013;5:167ra165. doi: 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogel JL, Kristie TM. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses. 2013;5:1272–1291. doi: 10.3390/v5051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloom DC, Giordani NV, Kwiatkowski DL. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta. 2010;1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cliffe AR, Coen DM, Knipe DM. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio. 2013;4 doi: 10.1128/mBio.00590-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cliffe AR, Garber DA, Knipe DM. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol. 2009;83:8182–8190. doi: 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwiatkowski DL, Thompson HW, Bloom DC. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. Journal of virology. 2009;83:8173–8181. doi: 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A. 2005;102:16055–16059. doi: 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Creech CC, Neumann DM. Changes to euchromatin on LAT and ICP4 following reactivation are more prevalent in an efficiently reactivating strain of HSV-1. PloS one. 2010;5:e15416. doi: 10.1371/journal.pone.0015416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J Virol. 2007;81:13248–13253. doi: 10.1128/JVI.01569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. MBio. 2013;4:e00558–00512. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt DM, McCafferty DG. trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- 46.Celum C, Wald A, Hughes J, Sanchez J, Reid S, Delany-Moretlwe S, Cowan F, Casapia M, Ortiz A, Fuchs J, Buchbinder S, Koblin B, Zwerski S, Rose S, Wang J, Corey L, Team HP. Effect of aciclovir on HIV-1 acquisition in herpes simplex virus 2 seropositive women and men who have sex with men: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:2109–2119. doi: 10.1016/S0140-6736(08)60920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson-Jones D, Weiss HA, Rusizoka M, Changalucha J, Baisley K, Mugeye K, Tanton C, Ross D, Everett D, Clayton T, Balira R, Knight L, Hambleton I, Le Goff J, Belec L, Hayes R H. S. V. t. team, Steering, C. Data Monitoring. Effect of herpes simplex suppression on incidence of HIV among women in Tanzania. The New England journal of medicine. 2008;358:1560–1571. doi: 10.1056/NEJMoa0800260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill AL, Rosenbloom DI, Fu F, Nowak MA, Siliciano RF. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weinberger AD, Weinberger LS. Stochastic fate selection in HIV-infected patients. Cell. 2013;155:497–499. doi: 10.1016/j.cell.2013.09.039. [DOI] [PubMed] [Google Scholar]
- 51.Dar RD, Hosmane NN, Arkin MR, Siliciano RF, Weinberger LS. Screening for noise in gene expression identifies drug synergies. Science. 2014;344:1392–1396. doi: 10.1126/science.1250220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA, Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ay E, Banati F, Mezei M, Bakos A, Niller HH, Buzas K, Minarovits J. Epigenetics of HIV infection: promising research areas and implications for therapy. AIDS reviews. 2013;15:181–188. [PubMed] [Google Scholar]
- 56.Choudhary SK, Margolis DM. Curing HIV: Pharmacologic approaches to target HIV-1 latency. Annual review of pharmacology and toxicology. 2011;51:397–418. doi: 10.1146/annurev-pharmtox-010510-100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirakawa K, Chavez L, Hakre S, Calvanese V, Verdin E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013;21:277–285. doi: 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hill JM, Nolan NM, McFerrin HE, Clement C, Foster TP, Halford WP, Kousoulas KG, Lukiw WJ, Thompson HW, Stern EM, Bhattacharjee PS. HSV-1 latent rabbits shed viral DNA into their saliva. Virol J. 2012;9:221. doi: 10.1186/1743-422X-9-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanberry LR, Bernstein DI, Burke RL, Pachl C, Myers MG. Vaccination with recombinant herpes simplex virus glycoproteins: protection against initial and recurrent genital herpes. J Infect Dis. 1987;155:914–920. doi: 10.1093/infdis/155.5.914. [DOI] [PubMed] [Google Scholar]
- 60.Skoberne M, Cardin R, Lee A, Kazimirova A, Zielinski V, Garvie D, Lundberg A, Larson S, Bravo FJ, Bernstein DI, Flechtner JB, Long D. An adjuvanted herpes simplex virus 2 subunit vaccine elicits a T cell response in mice and is an effective therapeutic vaccine in Guinea pigs. J Virol. 2013;87:3930–3942. doi: 10.1128/JVI.02745-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernstein DI, Cardin RD, Bravo FJ, Earwood J, Clark JR, Li Y, Mishra P, Li C, Nayak BP, Miller AT, Wu TY, Cooke MP, Valiante NM. Topical SMIP-7.7, a toll-like receptor 7 agonist, protects against genital herpes simplex virus type-2 disease in the guinea pig model of genital herpes. Antivir Chem Chemother. 2014;23:189–196. doi: 10.3851/IMP2499. [DOI] [PubMed] [Google Scholar]
- 62.Prichard MN, Kern ER, Hartline CB, Lanier ER, Quenelle DC. CMX001 potentiates the efficacy of acyclovir in herpes simplex virus infections. Antimicrobial agents and chemotherapy. 2011;55:4728–4734. doi: 10.1128/AAC.00545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Viral loads in select organs of mice treated with ACV or TCP at 10 dpi.
Fig. S2. Scoring criteria for ocular disease in the rabbit eye model.
Fig. S3. Scoring criteria for HSV disease in the guinea pig model.
Table S1. Experimental conditions.
Table S2. Statistical analyses.
Table S3. Antibodies and primers.
Numerical values of experimental data: Supplementary file