

ABSTRACT
Feline infectious peritonitis and virulent, systemic calicivirus infection are caused by certain types of feline coronaviruses (FCoVs) and feline caliciviruses (FCVs), respectively, and are important infectious diseases with high fatality rates in members of the Felidae family. While FCoV and FCV belong to two distinct virus families, the Coronaviridae and the Caliciviridae, respectively, they share a dependence on viral 3C-like protease (3CLpro) for their replication. Since 3CLpro is functionally and structurally conserved among these viruses and essential for viral replication, 3CLpro is considered a potential target for the design of antiviral drugs with broad-spectrum activities against these distinct and highly important viral infections. However, small-molecule inhibitors against the 3CLpro enzymes of FCoV and FCV have not been previously identified. In this study, derivatives of peptidyl compounds targeting 3CLpro were synthesized and evaluated for their activities against FCoV and FCV. The structures of compounds that showed potent dual antiviral activities with a wide margin of safety were identified and are discussed. Furthermore, the in vivo efficacy of 3CLpro inhibitors was evaluated using a mouse model of coronavirus infection. Intraperitoneal administration of two 3CLpro inhibitors in mice infected with murine hepatitis virus A59, a hepatotropic coronavirus, resulted in significant reductions in virus titers and pathological lesions in the liver compared to the findings for the controls. These results suggest that the series of 3CLpro inhibitors described here may have the potential to be further developed as therapeutic agents against these important viruses in domestic and wild cats. This study provides important insights into the structure and function relationships of 3CLpro for the design of antiviral drugs with broader antiviral activities.
IMPORTANCE Feline infectious peritonitis virus (FIPV) is the leading cause of death in young cats, and virulent, systemic feline calicivirus (vs-FCV) causes a highly fatal disease in cats for which no preventive or therapeutic measure is available. The genomes of these distinct viruses, which belong to different virus families, encode a structurally and functionally conserved 3C-like protease (3CLpro) which is a potential target for broad-spectrum antiviral drug development. However, no studies have previously reported a structural platform for the design of antiviral drugs with activities against these viruses or on the efficacy of 3CLpro inhibitors against coronavirus infection in experimental animals. In this study, we explored the structure-activity relationships of the derivatives of 3CLpro inhibitors and identified inhibitors with potent dual activities against these viruses. In addition, the efficacy of the 3CLpro inhibitors was demonstrated in mice infected with a murine coronavirus. Overall, our study provides the first insight into a structural platform for anti-FIPV and anti-FCV drug development.
INTRODUCTION
Feline coronaviruses (FCoVs) and feline caliciviruses (FCVs) are important pathogens of cats and generally cause mild, self-limiting localized infection in the intestinal tract or oral cavity and upper respiratory tract, respectively. However, these viruses can also cause a life-threatening systemic illness with a high fatality rate in cats. FCoV associated with a fatal disease in cats, feline infectious peritonitis (FIP), causes systemic pyogranulomatous inflammation in various organs, which subsequently progresses to fluid accumulation in the abdominal cavity and death. In contrast to the more common asymptomatic or mild enteritis caused by feline enteric coronavirus, the enteric biotype of FCoV, FIP is relatively uncommon in the general cat population, but it is the leading cause of death in young cats (1–3). In addition to the two biotypes of feline enteric coronavirus and FIP coronavirus, FCoVs are also classified into two serotypes, I and II. FCoV serotype I is more prevalent than serotype II, which appears to be derived from recombination with canine coronavirus in the spike (S) protein (4–8). Both serotypes can cause enteritis or FIP in domestic and wild feline populations, including wildcats, cheetahs, mountain lions, and leopards (9–11). Virulent, systemic FCV (vs-FCV) is associated with systemic infection with a mortality rate as high as 67% (12–16). Unlike FCV associated with acute upper respiratory tract infection and oral ulceration, vs-FCV infection is characterized by an expanded tissue tropism, causing facial and limb edema, vasculitis, and dysfunctions in multiple organs (12–16). Despite the importance of these virus infections in cats, no effective preventive measure is currently available (reviewed in reference 17), and treatment options for FIP and vs-FCV infections are limited to supportive therapy, due to the lack of specific antiviral drugs. Therefore, effective therapeutic measures, such as antiviral drugs, are direly needed to combat these viral infections in cats.
FCoV is an enveloped, single-stranded positive-sense RNA virus that is a member of the Coronaviridae family. FCV is a nonenveloped, single-stranded positive-sense RNA virus that belongs to the Caliciviridae family. During replication, these viruses produce one (calicivirus) or multiple (coronavirus) viral polyproteins that are cleaved into functional structural or nonstructural virus proteins by virus genome-encoded proteases (reviewed in references 18 and 19). Viral 3C-like protease (3CLpro) is responsible for processing of the majority of cleavage sites; thus, it is essential in the replication of coronaviruses and caliciviruses. The 3CLpro enzymes encoded by the genomes of those viruses share several common characteristics, such as a typical chymotrypsin-like fold, the presence of a Cys nucleophile in the catalytic triad or dyad, and a preference for a Glu or Gln residue at the P1 position (in the nomenclature of Schechter and Berger [20]) in the substrate. Therefore, 3CLpro may serve as a potential target for the development of broad-spectrum antiviral agents for coronaviruses and caliciviruses.
We have previously synthesized peptidyl inhibitors based on the conserved key features of 3CLpro of coronaviruses and caliciviruses or the related 3C protease (3Cpro) of picornaviruses and reported on their broad-spectrum antiviral activities against multiple viruses in enzyme- or cell-based assay systems (21–23). However, those compounds showed minimal antiviral activity against FCV in cell culture, suggesting that further evaluation of structure-activity relationships around these peptidyl scaffolds is required for the development of broad-spectrum therapeutic agents for FCoV and FCV. In this study, we evaluated the anti-FCoV and anti-FCV activities of newly synthesized compounds as well as those of compounds that were previously reported by us but were not tested against FCoV and FCV and identified compounds that are effective against both FCoV and FCV in cell-based assays. The efficacies of representative dipeptidyl and tripeptidyl compounds were evaluated in mice infected with murine hepatitis virus (MHV) A59, a hepatotropic murine coronavirus, as a model for FIP. Our findings show that tripeptidyl compounds in general exhibit increased dual inhibitory activity against FCV and FCoV in cell culture and the dipeptidyl and tripeptidyl compounds significantly reduced the viral titers and histopathological changes in the liver of mice infected with MHV compared to the viral titers and histopathology of the liver of the control group. In summary, our peptidyl compounds, especially the tripeptidyl compounds, may have the potential to be developed as antiviral therapeutics targeting both FCoV and FCV.
MATERIALS AND METHODS
Compounds.
To identify potential broad-spectrum inhibitors against FCoV and FCV, the 3CLpro inhibitor libraries generated by our group were evaluated. The synthesis of dipeptidyl compounds GC373, GC376, GC543, GC546, GC551, and GC554 (22, 24, 25) and tripeptidyl compounds NPI52 (compound 2), NPI59 (compound 6), NPI64 (compound 7), and NPI71 (compound 8) (23) was described previously (22–25). Compounds NPI58, NPI65, and NPI66 were synthesized by modification of a previously reported method (23) and have not been previously reported. Compound confirmation and purity assessment were performed by nuclear magnetic resonance, mass spectrometry, and high-pressure liquid chromatography in the laboratory of D. H. Hua (Department of Chemistry, Kansas State University) or W. C. Groutas (Department of Chemistry, Wichita State University). The structures of the compounds are shown in Fig. 1A and B.
FIG 1.

Chemical structures of tripeptidyl (A) and dipeptidyl (B) compounds and the mean and standard error of the means (SEM) of the EC50 of the compounds against FCoV or FCV. Each compound was added to CRFK cells, and the cells were immediately infected with FCoV WSU-79-1146 or FCV Urbana. Cells were further incubated in the presence of each compound for up to 24 h. Virus titers were determined using the TCID50 method, and the EC50 were calculated. Compound cytotoxicity (CC50) was measured after incubating the cells with each compound for 24 h.
Cells and viruses.
Crandell-Rees feline kidney (CRFK) cells were maintained in minimum essential medium (MEM) containing 2 to 5% fetal bovine serum and the antibiotics chlortetracycline (25 μg/ml), penicillin (250 U/ml), and streptomycin (250 μg/ml). FCoV WSU-79-1146, non-vs-FCV strains Urbana, 131, and F9, and vs-FCV strains 5, Ari, Deuce, and Jengo were propagated in CRFK cells. CRFK cells and WSU-79-1146 were obtained from ATCC (Manassas, VA). The FCVs were kind gifts from J. Parker at Cornell University. WSU-79-1146 is a cell culture-adapted group II FCoV strain which is reported to cause FIP in experimentally inoculated cats (26).
Antiviral effects of compounds in cell culture.
Serial dilutions of each compound were added to confluent monolayers of CRFK cells in 24-well plates or cells were mock treated, and the cells were then immediately inoculated with virus at a multiplicity of infection (MOI) of 0.05 to 0.1. The cells were then further incubated at 37°C until an extensive cytopathic effect was observed in the mock-treated (untreated) well (up to 24 h). After freezing and thawing of the viruses in cell culture, virus titers were determined by the 50% tissue culture infective dose (TCID50) method (27). Stock solutions of test compounds (10 mM) were prepared in dimethyl sulfoxide (DMSO), and the concentration of DMSO in the cell culture did not exceed 0.5%. The 50% effective concentrations (EC50) were determined by nonlinear regression analyses of dose-response curves of virus titers against log inhibitor concentrations (variable slope) using GraphPad Prism software (GraphPad Software, San Diego, CA).
Nonspecific cytotoxic effect.
CRFK cells in 96-well plates were incubated with each compound at various concentrations up to 150 μM for 24 h. Cell cytotoxicity was measured by use of a CytoTox96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) following the manufacturer's instructions. The 50% cytotoxic concentration (CC50) of each compound was determined using GraphPad Prism software.
Western blot analysis.
CRFK cells were mock treated or treated with each compound and immediately infected with FCoV WSU-79-1146 or FCV Urbana at an MOI of 2. The cells were then further incubated at 37°C for 12 h. At 12 h postinfection, the cells were lysed with SDS-PAGE sample buffer containing 1% β-mercaptoethanol and the proteins were resolved on 10% Novex Tris-bis gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose membranes. Viral proteins were probed by using an antibody specific for FCV VP1 (28) or the FCoV nucleocapsid protein (Biocompare, Windham, NH) and then with peroxidase-conjugated, goat anti-mouse IgG or rabbit anti-goat IgG. β-Actin was used as a loading control. Following incubation with a chemiluminescent substrate (Pierce Biotechnology, Rockford, IL), the chemiluminescent signals were detected using a Fotodyne transilluminator/digital camera system (FX; Fotodyne, Hartland, WI).
Multiple-sequence alignment and three-dimensional structural models for 3CLpro.
Multiple-amino-acid-sequence alignment of the 3CLpro enzymes from FCV Urbana (GenBank accession number L40021.1), vs-FCV strains Jango (GenBank accession number DQ910793.1), Ari (GenBank accession number DQ910794.1), and Deuce (GenBank accession number DQ910789.1), FCoV strains WSU-79-1146 (GenBank accession number DQ010921.1), Black (GenBank accession number EU186072.1), and DF-2 (GenBank accession number JQ408981.1), and MHV A59 (GenBank accession number NC_001846.1) was performed using the ClustalW multiple-sequence-alignment program. FCoV strains WSU-79-1146, Black, and DF-2 are FIP-causing FCoVs. The three-dimensional structure of FCoV 3CLpro was built by use of the EasyModeller (version 4.0) program (29) and the 3CLpro structure of transmissible gastroenteritis virus (TGEV), a porcine coronavirus (Protein Data Bank accession number 2AMP), as the template. The FCV 3CLpro three-dimensional structure was built by use of the EasyModeller program and rhinovirus 3Cpro, poliovirus 3Cpro, human norovirus 3CLpro, and hepatitis A virus 3Cpro (Protein Data Bank accession numbers 1CQQ, 1L1N, 2LNC, and 1QA7, respectively) (30) as the templates. The quality of the models was assessed using the Verify 3D program (31).
Animal experiments.
The animal study was performed in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Kansas State University. BALB/c mice were purchased from Charles River Labs (Wilmington, MA). Prior to the animal experiments, the EC50 of GC376 and NPI52 against MHV A59 were determined to be 0.2 to 1 μM in CCL9.1 mouse liver cells. To confirm that MHV A59 infection induces consistent and high levels of virus replication in the livers of the infected mice, we inoculated 4- to 5-week-old female BALB/c mice intraperitoneally with MHV A59 at 7.2 × 104 or 5.2 × 105 TCID50/mouse. At 2 and 4 days postinfection (p.i.), the mice were sacrificed (4 to 6 mice/group), and the livers were collected and processed for virus titration by the TCID50 method. For the in vivo efficacy study, 4- to 5-week-old female BALB/c mice were inoculated intraperitoneally with MHV A59 at 7.2 × 104 or 5.2 × 105 TCID50/mouse. Mice were intraperitoneally given 50 μl of drug vehicle (10% ethanol, 70% polyethylene glycol 400, 20% phosphate-buffered saline), GC376 (10, 50, or 100 mg/kg of body weight/day), or NPI52 (10 or 100 mg/kg/day) divided into two doses per day. Compound administration started 4 h prior to virus infection and continued daily until the mice were euthanized. At 2 and 4 days p.i., the mice were sacrificed and the livers were collected and processed for virus titration. Virus titers were determined by the TCID50 method, and the liver virus titers were compared by two-tailed Student's t test. Fold changes in the geometric mean liver virus titers in each group were calculated by dividing the virus titers in the control group by those in the treated group.
Liver histopathology.
At 4 days p.i., the left lateral lobes were collected from NPI52-treated (10 and 100 mg/kg/day) mice, fixed with formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin for histopathological examination by a board-certified pathologist. Five views were examined per mouse liver, and a score of from 0 to 5 was assigned to each lesion contained in the view on the basis of the severity of the histopathological changes. Each score in each sample was added to give a final total score, and then the mean of the total score per sample was calculated for each group. The mean number of lesions per sample was also calculated for each group. The mean total score per sample and the mean number of lesions per sample were compared among the different experimental groups using two-tailed Student's t test.
RESULTS
Antiviral effects of dipeptidyl and tripeptidyl compounds on the replication of FCoV and FCV.
We evaluated the activities of dipeptidyl and tripeptidyl compounds with various R1 and R2 side chains against FCoV WSU-79-1146 and FCV Urbana in cell culture (Fig. 1A and B). For dipeptidyl compounds, replacing the R2 isobutyl (Leu side chain) with benzyl (Phe side chain) or cyclohexylmethyl (Cha side chain) on a representative dipeptidyl compound, GC373, increased the anti-FCV activity, while it decreased its potency toward FCoV. GC373 was previously shown to have potent anti-FCoV activity but minimal activity against FCV in cell culture (22). The tripeptidyl compound NPI52 has an additional residue of 1-napththylalanine compared to the structure of GC373 at the P3 position, and its activity against FCoV or FCV has not been previously tested (23). In this study, we found that NPI52 exhibited potent anti-FCoV and anti-FCV activity, with its EC50 being in the nanomolar range (Fig. 1A), which indicates that the presence of the additional residue at the P3 subsite dramatically increased its activity against FCV. When the R2 isobutyl was replaced with a benzyl or cyclohexylmethyl in NPI52, the benzyl substitution decreased the anti-FCoV activity more than the cyclohexylmethyl substitution, but the reduction in anti-FCV activity was similar between benzyl and cyclohexylmethyl substitutions. Replacement of the aldehyde warhead in NPI52 with ketoamides [(C=O)(C=O)NHCH(CH3)2 or (C=O)(C=O)NHC(CH3)3] greatly decreased the anti-FCV activity, but the activity of these compounds against FCoV was only moderately decreased. Similarly, replacement of aldehyde with α-hydroxy phosphonate [CH(OH)P(O)(OCH2CH3)2] greatly decreased the anti-FCV activity but had only a minor effect on anti-FCoV activity. NPI64, GC376, GC551, and GC554 are bisulfite adducts of NPI52, GC373, GC543, and GC546, respectively, and they showed antiviral activities against FCoV and FCV comparable to those of their aldehyde counterparts in cell culture. The CC50 values of all compounds ranged from 21.96 μM to greater than 150 μM in CRFK cells (Fig. 1A and B). Western blot analysis confirmed the effects of our compounds on the expression of the FCoV nucleocapsid protein or FCV VP1 (Fig. 2).
FIG 2.
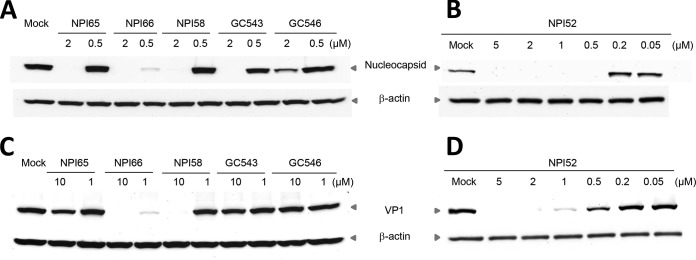
Western blot analysis of the effects of the compounds on expression of the FCoV nucleocapsid protein or FCV VP1 in CRFK cells. Cells were mock treated or treated with each compound and immediately infected with FCoV WSU-79-1146 or FCV Urbana. The cells were then further incubated for 12 h. Cell lysates were prepared and analyzed for expression of viral proteins on Western blots. β-Actin was used as a loading control.
Compound NPI52, which possesses potent antiviral activities against both FCoV WSU-79-1146 and FCV Urbana, was also tested for its activity against other non-vs-FCV and vs-FCV strains in cell culture to determine whether this compound is effective against various FCV strains. The EC50 in Table 1 show that NPI52 potently inhibited the replication of various non-vs-FCV and vs-FCV strains in cell culture.
TABLE 1.
EC50 of NPI52 against non-vs-FCV or vs-FCV strains
| Virus and strain | EC50 (μM) |
|---|---|
| vs-FCV | |
| Jengo | 0.03 ± 0.01 |
| 5 | 0.35 ± 0.27 |
| Ari | 0.10 ± 0.09 |
| Deuce | 0.22 ± 0.002 |
| non-vs-FCV | |
| 131 | 0.05 ± 0.03 |
| F9 | 0.05 ± 0.05 |
| Urbana | 0.02 ± 0.01 |
Multiple-sequence alignment and three-dimensional structural models for 3CLpro.
The amino acid sequences of the 3CLpro enzymes have high sequence homology of >95% within strains of FCV or FCoV (Fig. 3A and C). However, there are substantial differences in the 3CLpro sequences (19.72% homology) between FCV and FCoV strains. Although the sequence homology between FCV 3CLpro and FCoV 3CLpro is low, the catalytic residues are well conserved (Fig. 3A to D). MHV A59 3CLpro shares an amino acid sequence homology of 47.35% with FCoV 3CLpro, and the locations of the catalytic residues (red arrowheads in Fig. 3C) correspond well to those of FCoV strains. The residues in the catalytic dyad or triad are shown in the blue boxes in Fig. 3B and D.
FIG 3.

Multiple-sequence alignments of 3CLpro enzymes from FCV (A) and FCoV and MHV A59 (C) and ribbon presentations of three-dimensional structural models for FCV 3CLpro (B) and FCoV 3CLpro (D). (A and C) The catalytic residues E60, C122, and H39 of FCV 3CLpro (A) and H41 and C144 of FCoV 3CLpro and MHV A59 3CLpro are indicated by red arrowheads (C). (B and D) The structure model of FCoV 3CLpro was built by use of the EasyModeller (version 4.0) program (29) and the 3CLpro structure of TGEV (Protein Data Bank accession number 2AMP) as the template. The structural model of FCV 3CLpro was built by use of the EasyModeller program and the 3Cpro enzymes of rhinovirus, poliovirus, and hepatitis A virus and the 3CLpro of human norovirus (Protein Data Bank accession numbers 1CQQ, 1L1N, 1QA7, and 2LNC, respectively) (30) as the templates. The amino acids in the catalytic triad (E60, C122, and H39 for FCV 3CLpro) (B) and dyad (H41 and C144 for FCoV 3CLpro) (C) are shown in the blue boxes.
In vivo efficacy of compounds in coronavirus-infected mice.
Intraperitoneal inoculation of MHV A59 at 7.2 × 104 or 5.2 × 105 TCID50 per mouse led to high levels of virus replication in the liver, and the levels of virus replication were not significantly different between the two virus inocula, as determined by two-tailed Student's t test (P < 0.05) (data not shown). In the in vivo efficacy study, NPI52 and GC376 were tested in mice infected with MHV A59. In two separate experiments where mice were treated with GC376 or mock treated, the liver virus titers in mice treated with GC376 at 50 or 100 mg/kg were significantly lower than those in the no-treatment control mice at 4 days p.i. but not at 2 days p.i. (P < 0.01) (Fig. 4A and B). The fold reductions of the geometric mean virus titers in mice that received GC376 at 50 or 100 mg/kg at 4 days p.i. were 9.86 and 21.99, respectively, compared to the titers in the control mice (Fig. 4A and B, bar graphs). In contrast, GC376 at 10 mg/kg did not consistently lead to a significant reduction in virus titers at the two time points.
FIG 4.

Effects of 3CLpro inhibitor treatment on MHV A59 titers. Four- to 5-week-old BALB/c mice were intraperitoneally inoculated with MHV A59 at 5.2 × 105 (A) or 7.2 × 104 (B to D) TCID50/mouse and treated with drug vehicle, GC376 (10, 50, or 100 mg/kg/day), or NPI52 (10 or 100 mg/kg/day) divided into two doses per day starting at 4 h prior to virus infection. Scatter plots show the mean ± standard error of the mean virus titers in the livers of mice receiving mock treatment (drug vehicle) or treatment with GC376 (A and B) or NPI52 (C and D) at 2 or 4 days after virus infection (dpi, days postinfection). Virus titers are expressed as log10 TCID50 per gram of liver tissue. Bar graphs show the fold reduction of the geometric mean virus titers in the treatment groups compared to the titers in the control group. Asterisks indicate significant differences between the control and the treated groups (*, P < 0.05; **, P < 0.01).
In two independent NPI52 treatment experiments, treatment with NPI52 at 100 mg/kg resulted in a significant reduction in liver virus titers at 4 days p.i. (fold reductions, 19.63 to 40.27) and at 2 days p.i. (fold reductions, 3.46 to 12.3) compared to the titers in the controls (Fig. 4C and D). However, NPI52 at 10 mg/kg failed to significantly reduce the virus titers compared to those in the controls at 2 days p.i. or 4 days p.i. (Fig. 4C and D), although a significant reduction in viral titer was observed at 4 days p.i. in one of the experiments (Fig. 4C and D). The mock-infected mice did not show any signs of illness during the duration of the experiments, and no gross pathological lesions were observed on necropsy.
Histopathology of liver.
Panels 0 through 5 in Fig. 5A show microscopic lesions for increasing histopathology severity scores of from 0 through 5, respectively. On the basis of the lesion scoring, the group treated with NPI52 at 100 mg/kg had a significantly lower mean number of lesions and a significantly lower mean total histopathology scores per mouse liver than the controls (Fig. 5B and D). There was no statistically significant difference in the mean total histopathology scores and the mean number of lesions between the control group and the group treated with NPI52 at 10 mg/kg. However, the lesions in all drug-treated groups were scored 3 or lower, which is in contrast to the presence of lesions scored 4 or 5 in the no-treatment control group (Fig. 5C). Of note, the liver section from a mouse in the group treated with NPI52 at 10 mg/kg did not contain any histopathology lesion, and the data for that liver section were excluded from the statistical analysis for Fig. 5B to D. Examination of the liver samples from mock-infected mice revealed no significant microscopic lesions associated with compound toxicity.
FIG 5.

Histopathology changes in the livers of mice treated with NPI52. (A) Panels 0 through 5 show microscopic lesions for increasing histopathology severity scores of 0 through 5, respectively. Score 0, minimal change; scores 1 and 2, multifocal areas of necrosis; and scores 3 to 5, coalescing areas of necrosis. (B) A box-and-whisker plot showing the mean total liver histopathology score for each group. (C) A table showing the frequency of histopathology scores in four liver samples per group (for the group treated with NPI52 at 10 mg/kg) or five liver samples per group (for the control group and the group treated with NPI52 at 100 mg/kg). (D) A box-and-whisker plot showing the mean number of lesions per mouse liver for each group. Asterisks indicate statistically significant differences between control mice and mice treated with NPI52 at 100 mg/kg (P < 0.05). The whiskers represent the 5% and 95% confidence intervals, and the boxes represent the 25% and 75% confidence intervals. The lines in the middle of the boxes represent the medians for the data.
DISCUSSION
Although infections with FCoV or FCV are generally asymptomatic or cause mild localized symptoms in cats, they can also cause systemic diseases with high fatality rates among cats. These viruses are distinct in their genome organization, virus properties, and pathogenesis, but during replication they share a dependency on viral proteases for the production of functional structural or nonstructural virus proteins from a viral polyprotein(s). The amino acid sequence homology between FCV3CLpro and FCoV 3CLpro is less than 20%; however, they have similar active-site configurations (Fig. 3A to D) (22, 30, 32). Based on the highly conserved key site of 3CLpro expressed by coronaviruses and caliciviruses, we have previously synthesized peptidyl compounds and identified compounds that exhibit broad-spectrum antiviral efficacy against viruses in the Coronaviridae and Caliciviridae families and also against viruses in the Picornaviridae family that encode closely related 3Cpro enzymes (22). The dipeptidyl compounds that were previously evaluated for broad-spectrum antiviral effects consist of a warhead, a Gln surrogate structure in a position that corresponds to the P1 position, Leu in the P2 position, and a cap structure. These compounds have EC50 against many members of the caliciviruses, picornaviruses, and coronaviruses, including FCoV, in the nanomolar or low micromolar range (22). These findings demonstrate that 3CLpro could serve as a target for the development of broad-spectrum antiviral agents for viruses whose genomes encode 3CLpro or 3Cpro. However, these compounds were only minimally effective against FCV in cell culture (EC50, >30 μM) (22), and their low level of activity was speculated to be due to space constraints in the S2 pocket in the 3CLpro of FCV.
In the present study, we evaluated newly synthesized and previously reported dipeptidyl and tripeptidyl compounds that were not previously tested against FCoV and FCV in cell culture. Our findings show that the presence of an additional residue in NPI52 remarkably enhances anti-FCV activity, while the potency against FCoV compared to that of dipeptidyl compounds (including GC373) is maintained, suggesting that tripeptidyl compounds may provide a more suitable platform for dual-spectrum antiviral drug design for FCoV and FCV. Our limited structure-activity relationship study revealed that replacing the Leu side chain at the P2 site or the warhead on dipeptidyl or tripeptidyl compounds changed the antiviral activity against FCoV and FCV to various degrees. The effects of different warheads on the tripeptidyl compound were more profound on the antiviral activity against FCV than that against FCoV, which may suggest that the interaction of the warhead and the nucleophile Cys in the active site of FCV 3CLpro may require a fit more stringent than that in the active site of FCoV 3CLpro. Further investigation, such as crystallographic studies with inhibitor-FCoV 3CLpro or inhibitor-FCV 3CLpro complexes, may illuminate the structural basis of our findings. Among our tested compounds, NPI64, GC376, GC551, and GC554 are bisulfite adducts of NPI52, GC373, GC543, and GC546, respectively, and in cell culture they showed antiviral activities against FCoV and FCV comparable to those of their aldehyde counterparts. Bisulfite adduct compound GC376 was previously reported to be dissociated into the corresponding aldehyde (GC373) and bisulfite ion when incubated with 3CLpro, with the resulting aldehyde subsequently forming a covalent adduct with the active-site Cys of 3CLpro in X-ray crystallographic studies (22). We also observed the facile transformation of GC376 and NPI64 to their respective aldehyde forms in the blood of rats and cats in our preliminary animal studies (data not shown). These observations suggest that the bisulfite adduct compounds may act as prodrugs with the active aldehyde metabolites in cell culture and animals.
We also determined the antiviral effects of NPI52 on the replication of various vs-FCV strains as well as non-vs-FCV strains in cell culture to determine the sensitivity of various strains of FCV to the compound. The results showed that the potency of NPI52 against four vs-FCV strains was generally lower than that against non-vs-FCV strains, but it still remained high, with EC50 being in the nanomolar range. The higher EC50 of NPI52 against vs-FCV may be attributed to faster multicycle growth kinetics of vs-FCV strains leading to yields of virus progeny higher than those for non-vs-FCV strains (33). These results indicate that our compounds may be of potential therapeutic value for the treatment of highly fatal vs-FCV infection and non-vs-FCV infection, which are important causes of respiratory diseases and oral ulceration in cats. The compounds have minimal or low cytotoxicity in CRFK cells; the CC50 values of the dipeptidyl compounds were greater than 150 μM, with the in vitro therapeutic indexes (TIs) being 349 to higher than 7,500. Tripeptidyl compounds also have good TIs, but they are lower than those of dipeptidyl compound: compounds with EC50 of <1 μM against FCoV or FCV had TIs that ranged from 31.8 to 3,514 (Fig. 1A). The in vitro TIs are expressed as the ratio of the CC50 to the EC50, and these results indicate that our compounds have relatively high in vitro safety margins and can be suitable candidates for in vivo studies.
A number of classes of inhibitors of coronavirus 3CLpro have been identified in cell culture systems or in enzyme assays since the severe acute respiratory syndrome (SARS) coronavirus outbreaks in 2003 (34–41). However, few studies have reported on the efficacy of coronavirus 3CLpro inhibitors in experimental animals. Therefore, we evaluated a dipeptidyl compound (GC376) and a tripeptidyl compound (NPI52) in mice infected with a murine coronavirus, MHV. MHV causes systemic diseases, including hepatitis and a variety of immunological dysfunctions, in mice. Specifically, MHV A59 inoculation of mice by the peritoneal route causes severe liver disease and multiorgan infections (42, 43). This animal model was used as a surrogate for FIP, since feline coronavirus naturally infects only members of the family Felidae. In our study, the antiviral effects of GC376 and NPI52 in reducing liver viral titers compared to those in the no-treatment control group were dose dependent, and a statistically significant reduction in the viral load in the liver was consistently observed at 4 days p.i. with higher doses of GC376 or NPI52 (Fig. 4A to D). It is also important to note that GC376 and NPI52 have much weaker activity against MHV A59 than FCoV in cell culture (EC50 are at least 10-fold-higher). Nonetheless, the compounds showed marked antiviral activity against MHV (providing a reduction in the virus load of up to 40-fold) without causing toxicity in mice.
Histopathology examination of liver samples from mice treated with NPI52 or mock treated demonstrated that NPI52 at 100 mg/kg significantly reduced the mean total scores and the mean number of lesions in the livers of mice treated with NPI52 compared to the findings for mice in the no-treatment control group. There was no statistically significant difference in the mean total histopathology scores or the mean number of lesions between the no-treatment control group and the group treated with NPI52 at 10 mg/kg. However, none of the mice in the groups treated with NPI52 had a lesion score of 4 or greater, indicating that NPI52 treatment at both doses inhibited the expansion of the lesions in the liver, since lesions develop as small foci and adjacent foci coalesce to form larger lesions. These in vivo results demonstrate that inhibition of coronavirus 3CLpro is a valid therapeutic approach to suppress coronavirus replication and virus-induced pathology.
In summary, we synthesized and tested derivatives of peptidyl compounds that target 3CLpro and identified compounds with dual antiviral activity against FCoV and FCV in cell culture. Their efficacy in a mouse model of coronavirus infection and wide safety margin in cell culture suggest that these compounds may be suitable for further investigation as broad-spectrum antiviral drugs targeting the 3CLpro enzymes of FCoV and FCV.
ACKNOWLEDGMENTS
This work was supported by Winn Feline Foundation grant W13-020, Morris Animal Foundation grant D14FE-012, and NIH grants U01AI081891 and R01AI109039.
We thank David George and Takashi Taguchi for technical assistance. We also thank Tracy Miesner and the Comparative Medicine Group for their support for animal research.
REFERENCES
- 1.Rohrbach BW, Legendre AM, Baldwin CA, Lein DH, Reed WM, Wilson RB. 2001. Epidemiology of feline infectious peritonitis among cats examined at veterinary medical teaching hospitals. J Am Vet Med Assoc 218:1111–1115. doi: 10.2460/javma.2001.218.1111. [DOI] [PubMed] [Google Scholar]
- 2.Foley JE, Poland A, Carlson J, Pedersen NC. 1997. Risk factors for feline infectious peritonitis among cats in multiple-cat environments with endemic feline enteric coronavirus. J Am Vet Med Assoc 210:1313–1318. [PubMed] [Google Scholar]
- 3.Pedersen NC. 2009. A review of feline infectious peritonitis virus infection: 1963–2008. J Feline Med Surg 11:225–258. doi: 10.1016/j.jfms.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benetka V, Kubber-Heiss A, Kolodziejek J, Nowotny N, Hofmann-Parisot M, Mostl K. 2004. Prevalence of feline coronavirus types I and II in cats with histopathologically verified feline infectious peritonitis. Vet Microbiol 99:31–42. doi: 10.1016/j.vetmic.2003.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pedersen NC, Black JW, Boyle JF, Evermann JF, McKeirnan AJ, Ott RL. 1984. Pathogenic differences between various feline coronavirus isolates. Adv Exp Med Biol 173:365–380. doi: 10.1007/978-1-4615-9373-7_36. [DOI] [PubMed] [Google Scholar]
- 6.Kummrow M, Meli ML, Haessig M, Goenczi E, Poland A, Pedersen NC, Hofmann-Lehmann R, Lutz H. 2005. Feline coronavirus serotypes 1 and 2: seroprevalence and association with disease in Switzerland. Clin Diagn Lab Immunol 12:1209–1215. doi: 10.1128/CDLI.12.10.1209-1215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motokawa K, Hohdatsu T, Hashimoto H, Koyama H. 1996. Comparison of the amino acid sequence and phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline, canine and porcine coronaviruses. Microbiol Immunol 40:425–433. doi: 10.1111/j.1348-0421.1996.tb01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrewegh AA, Smeenk I, Horzinek MC, Rottier PJ, de Groot RJ. 1998. Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J Virol 72:4508–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stephenson N, Swift P, Moeller RB, Worth SJ, Foley J. 2013. Feline infectious peritonitis in a mountain lion (Puma concolor), California, USA. J Wildl Dis 49:408–412. doi: 10.7589/2012-08-210. [DOI] [PubMed] [Google Scholar]
- 10.Heeney JL, Evermann JF, McKeirnan AJ, Marker-Kraus L, Roelke ME, Bush M, Wildt DE, Meltzer DG, Colly L, Lukas J, Manton VJ, Caro T, O'Brien SJ. 1990. Prevalence and implications of feline coronavirus infections of captive and free-ranging cheetahs (Acinonyx jubatus). J Virol 64:1964–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennedy M, Citino S, McNabb AH, Moffatt AS, Gertz K, Kania S. 2002. Detection of feline coronavirus in captive Felidae in the USA. J Vet Diagn Invest 14:520–522. doi: 10.1177/104063870201400615. [DOI] [PubMed] [Google Scholar]
- 12.Hurley KE, Pesavento PA, Pedersen NC, Poland AM, Wilson E, Foley JE. 2004. An outbreak of virulent systemic feline calicivirus disease. J Am Vet Med Assoc 224:241–249. doi: 10.2460/javma.2004.224.241. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen NC, Elliott JB, Glasgow A, Poland A, Keel K. 2000. An isolated epizootic of hemorrhagic-like fever in cats caused by a novel and highly virulent strain of feline calicivirus. Vet Microbiol 73:281–300. doi: 10.1016/S0378-1135(00)00183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foley J, Hurley K, Pesavento PA, Poland A, Pedersen NC. 2006. Virulent systemic feline calicivirus infection: local cytokine modulation and contribution of viral mutants. J Feline Med Surg 8:55–61. doi: 10.1016/j.jfms.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulz BS, Hartmann K, Unterer S, Eichhorn W, Majzoub M, Homeier-Bachmann T, Truyen U, Ellenberger C, Huebner J. 2011. Two outbreaks of virulent systemic feline calicivirus infection in cats in Germany. Berl Munch Tierarztl Wochenschr 124:186–193. [PubMed] [Google Scholar]
- 16.Reynolds BS, Poulet H, Pingret JL, Jas D, Brunet S, Lemeter C, Etievant M, Boucraut-Baralon C. 2009. A nosocomial outbreak of feline calicivirus associated virulent systemic disease in France. J Feline Med Surg 11:633–644. doi: 10.1016/j.jfms.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedersen NC. 2014. An update on feline infectious peritonitis: diagnostics and therapeutics. Vet J 201:133–141. doi: 10.1016/j.tvjl.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziebuhr J, Snijder EJ, Gorbalenya AE. 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81:853–879. [DOI] [PubMed] [Google Scholar]
- 19.Green KY. 2007. Caliciviruses: the noroviruses, p 582–608 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 20.Schechter I, Berger A. 1967. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27:157–162. doi: 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- 21.Kim Y, Mandadapu SR, Groutas WC, Chang K-O. 2013. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antiviral Res 97:161. doi: 10.1016/j.antiviral.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y, Lovell S, Tiew KC, Mandadapu SR, Alliston KR, Battaile KP, Groutas WC, Chang KO. 2012. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J Virol 86:11754–11762. doi: 10.1128/JVI.01348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prior AM, Kim Y, Weerasekara S, Moroze M, Alliston KR, Uy RA, Groutas WC, Chang KO, Hua DH. 2013. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg Med Chem Lett 23:6317–6320. doi: 10.1016/j.bmcl.2013.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tiew KC, He G, Aravapalli S, Mandadapu SR, Gunnam MR, Alliston KR, Lushington GH, Kim Y, Chang KO, Groutas WC. 2011. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg Med Chem Lett 21:5315–5319. doi: 10.1016/j.bmcl.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandadapu SR, Gunnam MR, Tiew KC, Uy RA, Prior AM, Alliston KR, Hua DH, Kim Y, Chang KO, Groutas WC. 2013. Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg Med Chem Lett 23:62–65. doi: 10.1016/j.bmcl.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedersen NC, Evermann JF, McKeirnan AJ, Ott RL. 1984. Pathogenicity studies of feline coronavirus isolates 79-1146 and 79-1683. Am J Vet Res 45:2580–2585. [PubMed] [Google Scholar]
- 27.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497. [Google Scholar]
- 28.Sosnovtsev S, Green KY. 1995. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VpG for infectivity. Virology 210:383–390. doi: 10.1006/viro.1995.1354. [DOI] [PubMed] [Google Scholar]
- 29.Kuntal BK, Aparoy P, Reddanna P. 2010. EasyModeller: a graphical interface to MODELLER. BMC Res Notes 3:226. doi: 10.1186/1756-0500-3-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oka T, Yamamoto M, Yokoyama M, Ogawa S, Hansman GS, Katayama K, Miyashita K, Takagi H, Tohya Y, Sato H, Takeda N. 2007. Highly conserved configuration of catalytic amino acid residues among calicivirus-encoded proteases. J Virol 81:6798–6806. doi: 10.1128/JVI.02840-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowie JU, Luthy R, Eisenberg D. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 32.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. 2003. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 33.Ossiboff RJ, Sheh A, Shotton J, Pesavento PA, Parker JS. 2007. Feline caliciviruses (FCVs) isolated from cats with virulent systemic disease possess in vitro phenotypes distinct from those of other FCV isolates. J Gen Virol 88:506–517. doi: 10.1099/vir.0.82488-0. [DOI] [PubMed] [Google Scholar]
- 34.Ramajayam R, Tan KP, Liu HG, Liang PH. 2010. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg Med Chem 18:7849–7854. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bacha U, Barrila J, Gabelli SB, Kiso Y, Mario Amzel L, Freire E. 2008. Development of broad-spectrum halomethyl ketone inhibitors against coronavirus main protease 3CL(pro). Chem Biol Drug Des 72:34–49. doi: 10.1111/j.1747-0285.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang PH. 2006. Characterization and inhibition of SARS-coronavirus main protease. Curr Top Med Chem 6:361–376. doi: 10.2174/156802606776287090. [DOI] [PubMed] [Google Scholar]
- 37.Kuo CJ, Liu HG, Lo YK, Seong CM, Lee KI, Jung YS, Liang PH. 2009. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett 583:549–555. doi: 10.1016/j.febslet.2008.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Xie W, Xue X, Yang K, Ma J, Liang W, Zhao Q, Zhou Z, Pei D, Ziebuhr J, Hilgenfeld R, Yuen KY, Wong L, Gao G, Chen S, Chen Z, Ma D, Bartlam M, Rao Z. 2005. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto N, Yang R, Yoshinaka Y, Amari S, Nakano T, Cinatl J, Rabenau H, Doerr HW, Hunsmann G, Otaka A, Tamamura H, Fujii N, Yamamoto N. 2004. HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus. Biochem Biophys Res Commun 318:719–725. doi: 10.1016/j.bbrc.2004.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh AK, Gong G, Grum-Tokars V, Mulhearn DC, Baker SC, Coughlin M, Prabhakar BS, Sleeman K, Johnson ME, Mesecar AD. 2008. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett 18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agnihothram S, Yount BL Jr, Donaldson EF, Huynh J, Menachery VD, Gralinski LE, Graham RL, Becker MM, Tomar S, Scobey TD, Osswald HL, Whitmore A, Gopal R, Ghosh AK, Mesecar A, Zambon M, Heise M, Denison MR, Baric RS. 2014. A mouse model for Betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. mBio 5(2):e00047-14. doi: 10.1128/mBio.00047-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hingley ST, Leparc-Goffart I, Weiss SR. 1998. The mouse hepatitis virus A59 spike protein is not cleaved in primary hepatocyte and glial cell cultures. Adv Exp Med Biol 440:529–535. [DOI] [PubMed] [Google Scholar]
- 43.Kim KD, Zhao J, Auh S, Yang X, Du P, Tang H, Fu YX. 2007. Adaptive immune cells temper initial innate responses. Nat Med 13:1248–1252. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]