

Background: DnaK is a Hsp70 (heat shock protein) molecular chaperone whose function is controlled in part by the nucleotide exchange factor GrpE.
Results: We describe the structures of several complexes formed between GrpE and different DnaK variants.
Conclusion: The DnaK-GrpE complex has several conformations, an important factor in regulating DnaK function.
Significance: The information obtained explains the nucleotide exchange role of GrpE in much more detail.
Keywords: 70-Kilodalton Heat Shock Protein (Hsp70), Chaperone, Chaperone DnaK (DnaK), Electron Microscopy (EM), Protein Folding, GrpE, Nucleotide Exchange Factor
Abstract
Hsp70 chaperones comprise two domains, the nucleotide-binding domain (Hsp70NBD), responsible for structural and functional changes in the chaperone, and the substrate-binding domain (Hsp70SBD), involved in substrate interaction. Substrate binding and release in Hsp70 is controlled by the nucleotide state of DnaKNBD, with ATP inducing the open, substrate-receptive DnaKSBD conformation, whereas ADP forces its closure. DnaK cycles between the two conformations through interaction with two cofactors, the Hsp40 co-chaperones (DnaJ in Escherichia coli) induce the ADP state, and the nucleotide exchange factors (GrpE in E. coli) induce the ATP state. X-ray crystallography showed that the GrpE dimer is a nucleotide exchange factor that works by interaction of one of its monomers with DnaKNBD. DnaKSBD location in this complex is debated; there is evidence that it interacts with the GrpE N-terminal disordered region, far from DnaKNBD. Although we confirmed this interaction using biochemical and biophysical techniques, our EM-based three-dimensional reconstruction of the DnaK-GrpE complex located DnaKSBD near DnaKNBD. This apparent discrepancy between the functional and structural results is explained by our finding that the tail region of the GrpE dimer in the DnaK-GrpE complex bends and its tip contacts DnaKSBD, whereas the DnaKNBD-DnaKSBD linker contacts the GrpE helical region. We suggest that these interactions define a more complex role for GrpE in the control of DnaK function.
Introduction
In all cell types, molecular chaperones control cell proteostasis by preventing misfolding and aggregation or by directing proteins to degradation (1, 2). The Hsp70 are a ubiquitous, diverse group of chaperones involved in a large number of protein processing reactions that include folding, transport across membranes, and remodeling of specific proteins or complexes (3, 4). They share a conserved sequence that encompasses two domains (Fig. 1A), a nucleotide-binding domain (NBD)6 (Hsp70NBD; ∼45 kDa) and a substrate-binding domain (Hsp70SBD; ∼25 kDa). Hsp70SBD is composed in turn of two subdomains, a β-sandwich (SBDβ) and an α-helical domain (SBDα); the latter acts as a lid on the former and blocks release of the trapped polypeptide. Hsp70SBD is thus a tweezers that traps unfolded polypeptide when ADP is bound to Hsp70NBD, whereas protein is released in the presence of ATP (5). There is cross-talk between the two domains thanks to a flexible, highly conserved linker (6). ATP binding to Hsp70NBD induces substrate release in the Hsp70SBD, whereas substrate binding in the Hsp70SBD stimulates ATP hydrolysis in Hsp70NBD (7, 8). Hsp70 are thus ATPases controlled by two groups of cofactors. On the one hand, the J-domain proteins (i.e. Hsp40 chaperones) can interact with Hsp70 through the J-domain and stimulate their ATPase activity to induce their closed, ADP conformation (9, 10). On the other hand, the NEF induce ADP replacement with ATP, by significantly increasing the ADP dissociation rate (42).
FIGURE 1.
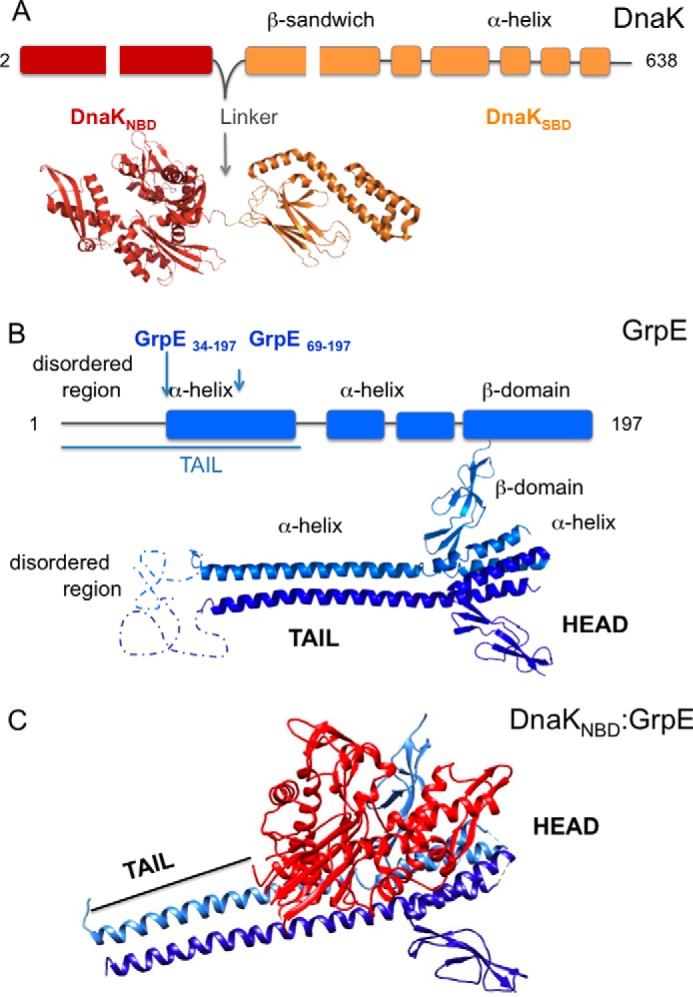
Crystal structures of DnaK, GrpE, and DnaKNBD-GrpE. A, crystal structure of DnaK (PDB code 2KHO). In both the crystal structure (bottom) and the sequence (top), the DnaKNBD and DnaKSBD domains are connected by a linker. B, crystal structure of GrpE. Top, scheme of the GrpE sequence; bottom, crystal structure of the GrpE dimer (extracted from PDB code 1DKG), showing the two structural regions (head and tail). C, crystal structure of the DnaKNBD-GrpE complex (PDB code 1DKG).
Four classes of NEF have been found to date, three in eukaryotes (BAG-1, HspBP1, and Hsp110) (11–13); the fourth (GrpE) acts as a NEF for the prokaryotic Hsp70, DnaK (14, 15). GrpE is an elongated dimer consisting of two structural regions, the tail and the head (Fig. 1B). The tail is formed by an N-terminal disordered region, and a long α-helix domain that forms a coiled-coil structure with the opposite monomer. The head is formed by a four-helix bundle with two helices from each monomer, followed by a compact β-sheet domain (15, 16). The atomic structure of a complex between the GrpE dimer and DnaKNBD was determined several years ago (17) (Fig. 1C) and showed that the GrpE dimer interacts with a single DnaKNBD molecule. The interaction takes place between one GrpE monomer, mostly through β-sheet domain residues but also with α-helix domain residues, and several regions of the DnaKNBD, especially with residues in the ATP-binding cleft. The interaction induces asymmetry in the GrpE dimer, which explains why it binds only one DnaKNBD molecule, as well as distortion in the DnaKNBD structure, which also explains the co-chaperone-induced ADP release.
Although the role of the C-terminal region of GrpE in control of DnaK activity is well defined, that of the N-terminal region (which includes a 33-residue disordered region) remains controversial. Some authors suggest a thermosensor role, to regulate DnaK folding activity in heat-shock conditions (18, 19), whereas others indicate a role in modulating dynamics of the chaperone substrate-binding domain through interaction with DnaKSBD (7). A recent crystal structure of a DnaK-GrpE complex from the archaea Geobacillus kaustophilus HTA426 (20) showed that it is composed of two DnaK molecules bound to the GrpE dimer, with one of the DnaKSBD molecules interacting with the linker of the other DnaK, thus mimicking a substrate molecule. The crystal structure nonetheless does not describe a role for the GrpE N-terminal region.
Here we applied various techniques to clarify the role of the GrpE N-terminal region in DnaK activity. We used electron microscopy (EM) and image processing for three-dimensional reconstruction of the complexes between different DnaK and GrpE variants, which showed that in the DnaK-GrpE complex, the DnaKSBD domain is located near the DnaKNBD, embracing the head of the GrpE dimer. The GrpE N-terminal region interacts with DnaKSBD due to the flexibility of the former, which is able to bend and contact the latter domain. This explains the competition between the N-terminal region residues and the substrate for binding to the peptide-binding site. Fluorescence data support this protein complex arrangement, as they show that the 67 GrpE N-terminal residues modulate accessibility of the DnaK substrate-binding site, probably by modifying DnaK conformation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
DnaK and DnaKNBD (DnaK(1–385)) were cloned, expressed, and purified as described (7). The DnaKR151A mutant was expressed and purified following the protocol developed by Taneva et al. (21). GrpE was cloned, expressed, and purified as described (22). The GrpE(34–197) mutant was generated by digestion of wtGrpE with papain (125:1 (w:w), 90 min, 25 °C), followed by chromatography purification in a HiLoad Superdex 75 16/60 column (GE Healthcare) (7). The sequence of the resulting protein was confirmed by MS and N-terminal sequencing. GrpE(69–197) was cloned, expressed, and purified by the protocol of Moro et al. (7).
Interaction in Native PAGE
To test their complex-forming ability, the DnaK and GrpE variants were mixed in a 1:2.5 DnaK:GrpE (monomer) molar ratio in 20 mm HEPES, pH 7.6, 50 mm KCl, 2.5 mm MgCl2, 0.5 mm ADP (20 min, 25 °C). Aliquots of samples were loaded on a precast Native PAGE Novex 4–16% BisTris gel (Life Technologies), run at 150 V (2 h), and stained with Coomassie Blue.
Peptide Binding Kinetics
As described above, combinations of the DnaK and GrpE variants were incubated to form stable complexes in 25 mm HEPES, pH 7.6, 50 mm KCl, 5 mm MgCl2, 1 mm ADP. Trace ATP was removed from ADP solutions as described (23). The peptides used in these experiments were fluorescent α-N, dansyl-NRLLLTG (dNR) (Neosystem), unlabeled NR, and one corresponding to the first 33 GrpE residues (GrpE(1–33); ProteoGenix). Peptides were bound by adding dNR (0.5 μm) to the DnaK-GrpE complexes; binding was measured in a FluoroMax-3 spectrofluorimeter (Jobin Ibon), with a 335-nm excitation wavelength and a 535-nm emission wavelength (excitation and emission slit widths of 4 nm). Competition of NR and GrpE(1–33) with dNR for DnaK binding was analyzed by titrating DnaK-dNR (5:1 μm) complexes with increasing peptide concentrations. In these experimental conditions, 80% dNR is bound to DnaK (Kd ∼ 1 μm).
Luciferase Refolding Experiments
Luciferase (2.5 μm) was denatured in 6 m guanidine hydrochloride, 30 mm Tris/HCl, pH 7.5, 5 mm DTT (dithiothreitol) (2 h, 25 °C). Refolding was started after 100-fold dilution in 20 mm HEPES/KOH, pH 7.4, 50 mm KCl, 5 mm MgCl2, 5 mm DTT, 2 mm ATP, 20 ng ml−1 of pyruvate kinase, 4 mm phosphoenolpyruvate, and addition of 1 μm DnaK, 1 μm DnaJ, 1 μm of the corresponding GrpE variant. Samples were incubated for various times (30 °C) and luciferase activity was measured in a Synergy HT luminometer (Biotek) using the Luciferase Assay System (Promega E1500).
Electron Microscopy and Three-dimensional Reconstruction
The complexes (DnaK-GrpE, DnaKR151A-GrpE, DnaKNBD-GrpE) were purified by gel filtration on a Superdex 200 column (GE Healthcare). Aliquots of purified complexes were applied to carbon-coated copper grids (30 s) and stained with 2% uranyl acetate. Micrographs were taken in minimal dose conditions in a JEM1200EX-II microscope (JEOL) operated at 100 kV. Micrographs of the DnaK-GrpE and DnaKNBD-GrpE complexes were recorded on Kodak SO-163 plates at 60,000 magnification and digitized on a Photoscan TD scanner (Zeiss). Images of DnaKR151A-GrpE complex were obtained in a JEOL 1230 transmission electron microscope operated at 100 kV, using a low-dose protocol and a 4k × 4k TVIPS CMOS detector under the control of EM-TOOLS software (TVIPS). Final magnification of the CMOS images was ×54,926. A total of 30,223, 15,438, and 21,910 particles of DnaK-GrpE, DnaKNBD-GrpE, and DnaKR151A-GrpE, respectively, were selected, normalized, and CTF-corrected using procedures implemented in the XMIPP package (24–26). All particles were initially classified using clustering reference-free methods in XMIPP (27). Angular refinement was performed using EMAN and XMIPP (24, 27, 28). For three-dimensional reconstructions, different starting templates were generated using the EMAN startcsym program, by common lines or using artificial noisy models and Gaussian blobs with the rough dimension of the proteins (28), subsequently refined using XMIPP. The different strategies converged to similar solutions.
For reconstruction of the two co-existing DnaK-GrpE conformations, the entire dataset was first processed assuming the complex displayed a unique conformation. The resulting structure and its projections, as well as comparison with reference-free averages, suggested that the reconstruction obtained is a mixture of two conformations. To test this hypothesis, two reconstructions were generated with two sets of reference-free averages corresponding to each of the putative conformations, using their current angular assignments. These structures were then used as templates for three-dimensional classification using maximum-likelihood methods implemented in XMIPP (29). After refinement, particles were split into groups corresponding to the two conformations, which were further refined separately. As a control, particles of each group were refined using the opposite template, and were found to converge into the structure defined by the group they belong to and not the template. Each structure and each particle subset generated compatible projections and averages, also consistent with the reference-free averages. Resolution was estimated using Fourier shell correlation at 0.5 criteria. The atomic structures were first fitted manually into the EM density maps and fine docking was performed using the automatic option in the UCSF Chimera package (30). Flexible fitting was performed using iMODfit (31). Handedness of the structures was defined as that which generated the best cross-correlation with the crystal structures.
RESULTS
DnaK-GrpE Complex Formation with the DnaK and GrpE Variants
To analyze the interaction between the chaperone DnaK and its NEF GrpE, we generated several variants of the two proteins. For the chaperone, we expressed and purified wtDnaK, a deletion mutant corresponding to the nucleotide-binding domain alone (residues 1–385; DnaKNBD), and a mutant that lacks allosteric communication between DnaKNBD and DnaKSBD (DnaKR151A; 32). For GrpE, we generated the full-length version and two deletion mutants (7); one lacked the first 33 residues corresponding to the disordered region (GrpE(34–197)), and the other also lacked part of the long α-helix (GrpE(69–197)) but maintained those residues of this region involved in DnaKNBD binding (Asn-71, Arg-73, and Arg-74) (17). The atomic structure of the DnaKNBD-GrpE complex predicts that both GrpE mutants would generate a stable complex with DnaK; this was confirmed by native electrophoresis (Fig. 2A), as it was for the allosteric mutant DnaKR151A (Fig. 2B).
FIGURE 2.
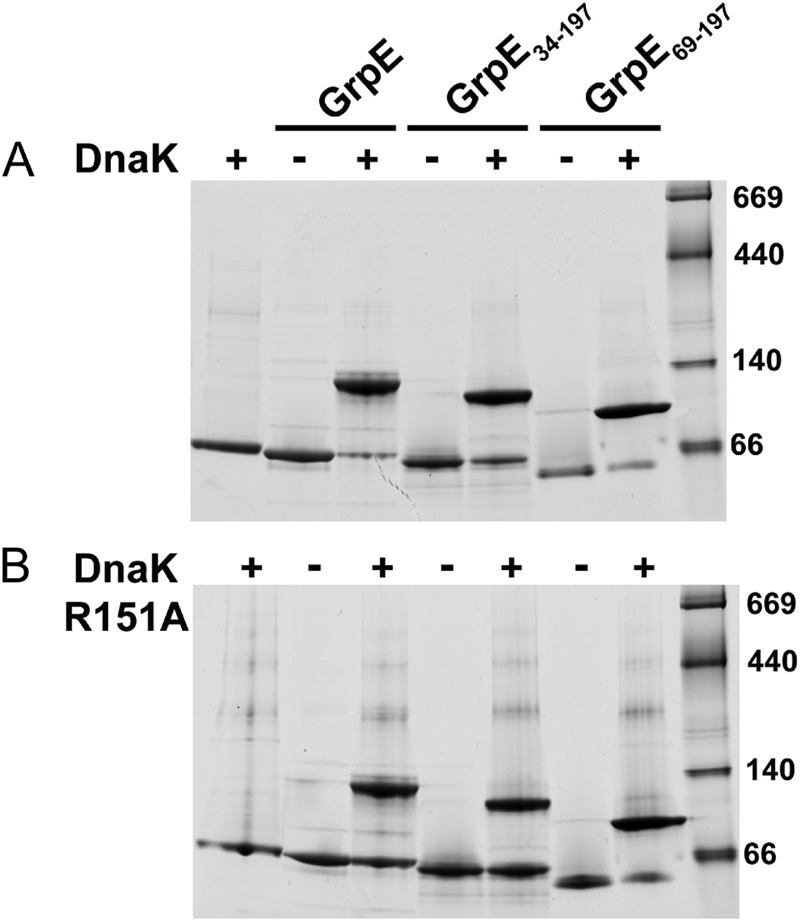
Purification of the DnaK-GrpE complexes. A, native gel showing the formation of the DnaK-GrpE complexes. DnaK is incubated alone or with GrpE, GrpE(34–197) or GrpE(69–197). B, native gel showing DnaKR151A-GrpE complex formation. DnaKR151A is incubated alone or with GrpE, GrpE(34–197), or GrpE(69–197).
DnaK-GrpE Complex Structure
There are high-resolution data for the DnaK-GrpE complex, although in one case they are derived from a complex lacking the DnaKSBD domain (17), and in the other, the DnaK-GrpE complex has a 2:2 symmetry, which has not been observed in solution (20). We thus used EM and image processing to determine the DnaK-GrpE complex structure. As this complex is small by EM standards (125 kDa), as a control we first performed a three-dimensional reconstruction of the known DnaKNBD-GrpE complex. We generated the complex and purified it by gel filtration, and negatively stained one aliquot. The sample appeared to be heterogeneous, probably due to the distinct DnaKNBD-GrpE views on the grid. The most common view showed a circular mass from which a stem protruded (see Fig. 3A). We selected and processed 15,438 particles and generated a structure (23-Å resolution; Fourier shell correlation 0.5) that can be described as composed of a head and a tail (Fig. 3B). The structure is ∼130 Å long, with a head length of ∼70 Å and a width of ∼70 × ∼90 Å. The overall complex shape is very similar to the atomic structure of the DnaKNBD-GrpE complex (Protein Data Bank 1DKG; Ref. 17), and docking of the latter into the former is good in the complex head (Fig. 3C). There are nonetheless notable differences between the tails of the two structures. The tail is longer in the complex visualized by EM, which can be explained by the first 33 residues, which were not determined in the crystal structure. The other difference is in tail shape, which is bent in the EM reconstruction and straight in the crystal structure. A flexible fitting performed on the tail structure helped to dock it within our three-dimensional reconstruction (Fig. 3D).
FIGURE 3.

Structure of the DnaKNBD-GrpE complex. A, two-dimensional reference-free averages of DnaKNBD·GrpE. The top image shows the two structural regions of the complex, the head and the tail. B, three orthogonal views of the three-dimensional reconstruction of DnaKNBD·GrpE. C, the same three views with docking of the atomic structure of the DnaKNBD-GrpE complex (PDB code 1DKG) (9) into the three-dimensional reconstruction of the same complex. D, the same docking after a flexible fitting on the same atomic structure. Bars in A and B = 50 Å.
We then determined the structure of the DnaK-GrpE complex, for which we generated and purified the complex using the same strategy as for DnaKNBD-GrpE. Aliquots of the purified complex were negatively stained and observed by EM. Heterogeneity was again indicated, which we associated to the different views of the complex. In most cases, the particles showed a circular structure, larger than that observed for DnaKNBD-GrpE, and a stem protruding from the main mass with different lengths and angles. We selected 30,223 particles and classified them two- and three-dimensionally (see “Experimental Procedures”). Our classification and processing strategy allowed us to isolate two subsets of molecule images corresponding to two distinct, albeit homogenous conformations. The two volumes generated were very similar except for tail shape, which had a different degree of bending; one was similar to that for DnaKNBD-GrpE (Fig. 4, A and C, conformation 1) and in the other, much more curved, the tip of the tail contacted the head (Fig. 4, A and C, conformation 2).
FIGURE 4.

Structure of the DnaK-GrpE and DnaKR151A-GrpE complexes. A, two-dimensional reference-free averages of the DnaK-GrpE corresponding to the two main populations obtained after three-dimensional classification. B, location of DnaKSBD in the DnaK-GrpE structure. (i) Side view of one conformation of DnaK-GrpE. (ii) The same view with the difference map between the three-dimensional reconstructions of DnaK-GrpE and DnaKNBD-GrpE (green mesh) docked into the structure. (iii) The same view with the atomic structure of DnaKSBD (PDB code 1DKG; yellow) docked in the difference map. C, three orthogonal views of the structure of the two conformations. The atomic structure of DnaKNBD-GrpE with flexible fitting as in Fig. 1C, and that of DnaKSBD as shown in B, are docked into the two three-dimensional reconstructions. Asterisks indicate the area where the DnaKNBD-DnaKSBD linker is predicted to be located. D, two-dimensional reference-free average of DnaKR151A-GrpE. In addition to the two structural regions described above (head and tail), an extra thin mass protrudes from the head (arrow). E, three orthogonal views of the structure of DnaKR151A-GrpE. F, side view of DnaKR151A-GrpE with the difference map between the three-dimensional reconstructions of DnaKR151A-GrpE and DnaKNBD-GrpE complexes (green mesh) docked into the structure. G, the same three views as in E, with the atomic structure of the DnaKNBD-GrpE complex with flexible fitting, and that of DnaKSBD as shown in B, are docked into the three-dimensional reconstruction. Bars in A, B, D, and E = 50 Å.
The main difference between the two DnaK-GrpE structures and that of DnaKNBD-GrpE was the head, which for DnaK-GrpE was larger (∼70 Å long, ∼100 Å wide) due to the presence of DnaKSBD. The difference map between DnaK-GrpE and DnaKNBD-GrpE located the DnaKSBD position more precisely (Fig. 4B, ii); we therefore docked the atomic structure of this domain (PDB code 4EZX; Ref. 33) to this position (Fig. 4B, iii). In this manner, DnaKSBD was located in the head of the DnaK-GrpE complex, in close proximity to DnaKNBD; the two domains embraced the head of the GrpE structure in an apparently symmetric manner (Fig. 4C). The complex tail is longer than that determined in the atomic structure of DnaKNBD-GrpE, confirming visualization of the first 33 GrpE residues in our EM structures. In one conformation, the tip of the tail contacted with the head of the structure, near DnaKSBD (Fig. 4C, conformation 2). The locations of DnaKNBD and DnaKSBD in the DnaK-GrpE complex leave the interdomain linker (not observed in the three-dimensional reconstruction, Fig. 4C, black asterisk), near the central region of the long GrpE α-helix (residues 65–75). This finding confirms proteolysis protection results that suggested interaction between the DnaK linker and the long GrpE α-helix N-terminal region (7).
DnaK Allosterism Influences the Structure of the DnaK-GrpE Complex
As GrpE binding appears to modify the conformational equilibrium of the chaperone, we explored the relationship between DnaK allosterism and complex structure. We generated a complex between GrpE and DnaKR151A, a mutant that harbors a single substitution resulting in defective interdomain communication. We purified the complex by gel filtration and negatively stained one aliquot. EM analysis showed different particles, the majority with two thin masses protruding from a central, round mass (Fig. 4D). We selected and processed 21,910 particles to generate a three-dimensional reconstruction of DnaKR151A-GrpE (Fig. 4E), which confirmed two long masses protruding from the central mass. The volume generated was reminiscent of that of DnaKNBD-GrpE except for an extra protruding mass, which we associated to DnaKSBD, which is not present in DnaKNBD-GrpE (Fig. 4F). Docking of the modified atomic structure of the DnaKNBD-GrpE complex used in Fig. 3D was good in this three-dimensional reconstruction, as was docking of the DnaKSBD atomic structure in the extra protruding mass (Fig. 4G). The atomic model placed the DnaKSBD far from the main structure of the complex, and strongly suggested lack of interaction between the DnaK linker and the GrpE tail. Defective interdomain communication thus maintains DnaK in an ADP-like, extended conformation when in complex with GrpE.
GrpE Modifies DnaKSBD Dynamics
EM reconstruction of the DnaK-GrpE complex showed that the ADP-bound, high substrate affinity conformation of DnaK is modified by GrpE, which displaces the DnaKSBD to a position nearer DnaKNBD and the GrpE head. To determine whether this conformational change modulates DnaKSBD dynamics, we analyzed the kinetics of substrate association to DnaK alone or with the GrpE variants (GrpE, GrpE(34–197), and GrpE(69–197)). As substrate we used dNR, a peptide modified with a dansyl fluorescent probe and high affinity for DnaK. The kinetics of peptide binding to DnaK was slow, with a Kobs value of 0.0017 s−1 (t½ 10 min) (Fig. 5, A and C), whereas association to the DnaK-GrpE complex was 3-fold faster. The lower fluorescence increase suggested that less peptide was bound to DnaK-GrpE than to free DnaK. In contrast, whereas the amount of dNR bound was similar for DnaK-GrpE(34–197) complex and isolated DnaK, the kinetics was 4-fold faster. These data suggest that the first 33 GrpE residues act as a pseudo-substrate that competes with dNR for the DnaK binding site, as reported (7, 34, 35). To further confirm this hypothesis, this GrpE fragment (GrpE(1–33)) was synthesized and its ability to interact with the DnaK binding site analyzed. Titration of DnaK-dNR complexes with GrpE(1–33) showed that within the 0–200 μm concentration range, the peptide was soluble and unable to displace DnaK-bound dNR (Fig. 5D). This finding is not surprising, because GrpE(1–33) does not have a high-affinity binding site for DnaK, as its sequence consists almost entirely of charged and polar residues, different from the hydrophobic residues normally recognized by the chaperone binding site. This apparent contradiction could be explained by our observation that DnaK-GrpE binding places the NEF N-terminal, disordered region near the DnaK substrate-binding pocket (Fig. 4, A and C), thus facilitating its interaction, as previously proposed (34). We therefore suggest that the DnaK binding site is more accessible in the presence of GrpE, due to the conformational changes induced in DnaK by GrpE binding. This last observation coincides well with the higher protease accessibility of residues in the DnaK lid subdomain when GrpE is present (7).
FIGURE 5.

Kinetics of peptide binding to different DnaK-GrpE complexes. A, binding kinetics of dNR to DnaK (in the presence of purified ADP), alone (black line) or with GrpE (green), GrpE(34–197) (blue), or GrpE(69–197) (magenta). Binding of dNR (0.5 μm) was followed by monitoring the increase in dansyl moiety fluorescence at 25 °C (535 nm). B, kinetics was determined as in A for dNR binding to DnaKR151A and to the corresponding complexes with GrpE or its mutants. C, binding constants (Kobs) for the kinetics shown in A and B. In addition, binding curves to DnaK and complexes with GrpE and GrpE(34–197) were performed at 45 °C. Curves were fitted to a single exponential equation: F = F0 + ΔF exp(−Kobs/t). D, GrpE(1–33) competition with dNR for DnaK binding. Titration of DnaK-dNR (5:1 μm) complexes with increasing GrpE(1–33) concentration (closed circles). As a control, the same titration was performed with peptide NR (open circles). E, determination of the on- and off-rate constants for dNR binding to DnaK and DnaK-GrpE(34–197). Binding curves were performed as in A at increasing DnaK concentrations alone (closed circles) or with 5 μm GrpE(34–197) (open circles). Experiments were repeated at least three times (data shown as mean ± S.E.). On- (k+1) and off-rate (k−1) constants were obtained, respectively, from the slope and y intercept of the linear plots of Kobs versus DnaK concentration. For DnaK, k+1 and k−1 were 1100 ± 100 m−1 s−1 and 0.001 ± 0.0001 s−1, respectively. For DnaK-GrpE(34–197), values of k+1 = 3800 ± 300 m−1 s−1 and k−1 = 0.002 ± 0.0001 s−1 were obtained. F, refolding of luciferase aggregates (25 nm) by 1 μm DnaK, 1 μm DnaJ, and 1 μm GrpE (green circles), GrpE(34–197) (blue), or GrpE(69–197) (magenta). Initial refolding rates (% refolding min−1) were obtained by linear regression of initial time points (gray lines).
To better understand the GrpE effect in dNR binding kinetics, on- and off-rate constants were determined in association experiments with increasing DnaK concentrations (Fig. 5E). We used the GrpE(34–197) variant to avoid competition of the first 33 GrpE residues with dNR. Plots of Kobs versus DnaK concentration were linear, and on- and off-rate constants were obtained from the slope and y intercept, respectively. The values for isolated DnaK (k+1, 1100 ± 100 m−1 s−1 and k−1, 0.001 ± 0.0001 s−1) were comparable with those reported (36), and increased 3.5- and 2-fold, respectively, for the complex with GrpE(34–197). These findings confirmed the greater DnaK binding site accessibility when the chaperone interacts with GrpE, and indicated that the nucleotide exchange factor increases affinity for the substrate only slightly (Kd = k−1/k+1); GrpE thus does not induce substrate release, as was postulated (8).
A distinct scenario is observed when DnaK is complexed with GrpE(69–197). This deletion variant constitutes the GrpE “head,” the main element responsible for interaction with DnaKNBD (17). We observed no fluorescence increase for the DnaK-GrpE(69–197) complex (Fig. 5, A and C), which suggests that DnaK has adopted a conformation unable to bind dNR stably, in accordance with the strong stimulation of peptide dissociation from DnaK(ADP) induced by this mutant (7). Similar kinetic curves were obtained when we assayed dNR binding to DnaK in the presence of ATP (not shown), which induces docking of the chaperone domains and opening of the peptide binding site (37). These data suggest that DnaKNBD interaction with the GrpE head (see Fig. 1C) promotes an open DnaKSBD conformation.
We performed similar dNR binding experiments to DnaKR151A in the presence of these GrpE variants (Fig. 5, B and C). dNR binding to DnaKR151A(ADP) was almost 2-fold slower than to wtDnaK, reflecting the allosteric defect in the mutant. The presence of GrpE or GrpE(34–197) did not modify the Kobs of dNR binding to DnaKR151A, whereas DnaK-GrpE(69–197) slightly increased this value (Fig. 5C). In accordance with the EM reconstruction of this complex, these results indicate that in the absence of interdomain communication, GrpE is unable to modify DnaKSBD dynamics.
The data indicated that interaction of the GrpE N-terminal segments with DnaK has two main effects: 1) when tethered to the rest of the NEF molecule, the 33 disordered N-terminal residues compete with the substrate for DnaK binding, and 2) interaction of the N-terminal half of the GrpE tail (residues 34–68) with the DnaK linker and/or DnaKSBD increases accessibility of the chaperone peptide binding site and stabilizes the DnaK-substrate complex. This helical GrpE segment unfolds reversibly at heat shock temperatures and modulates the functional cycle of DnaK (18). We therefore studied DnaKSBD dynamics at 45 °C, alone, or with GrpE and GrpE(34–197). As predicted, binding kinetics was greatly accelerated for isolated DnaK(ADP), with a Kobs increase from 0.0017 s−1 at 25 °C to 0.051 s−1 at 45 °C. Neither GrpE nor GrpE(34–197) modified the Kobs value in these experimental conditions, in contrast to observations at 25 °C (Fig. 5C). The results suggest that unfolding of the long N-terminal helices in heat shock conditions blocks the GrpE-induced structural rearrangement of DnaK(ADP) that takes place at permissive temperatures.
The GrpE N-terminal Domain Regulates Chaperone Activity of the DnaK System
To address whether interaction of the N-terminal GrpE regions (the first 34 or 69 residues) with DnaK influenced the chaperone activity of the DnaK system, we measured activity after refolding of chemically denatured luciferase, using GrpE, GrpE(34–197), or GrpE(69–197) as NEF (Fig. 5F). Deletion of the first 33 residues induced a 25% reduction in the initial refolding rate (from 4.9 ± 0.2 to 3.7 ± 0.4% min−1). A similar deletion variant was able to refold thermally denatured luciferase in the DnaK system as efficiently as wtGrpE (34), indicating that the chaperone activity depends on the conformational properties and stability of the unfolded and aggregated substrates. When GrpE(69–197) was used as NEF, the initial refolding rate was reduced by 65% (1.6 ± 0.2% min−1), coinciding with data reported for the shorter GrpE(89–197) construct (38). Based on the ability of the GrpE(69–197) mutant to dissociate DnaK-substrate complexes (7), premature release of unfolded luciferase might hamper correct refolding of the substrate. These results indicate functional significance for the interaction between the GrpE tail, specifically the N-terminal part of the helical region, with the chaperone.
DISCUSSION
NEF are important cofactors in the Hsp70 functional cycle; they promote ADP exchange for ATP in the Hsp70NBD, which leads to peptide release from Hsp70SBD via the linker that connects the two domains. In general, this exchange involves distortion of the V-shaped NBD structure and allows release of the tightly bound ADP (17, 39, 40). This is also the case for dimeric GrpE, which regulates DnaK by forming a stable complex with the chaperone (15). The stoichiometry of this interaction is debated, as the atomic structure of the E. coli DnaKNBD-GrpE complex shows one DnaKNBD molecule bound to a GrpE dimer (17), whereas the atomic structure of the G. kaustophilus HTA426 DnaK-GrpE complex indicates interaction of a GrpE dimer with two DnaK molecules (20). Our three-dimensional reconstructions of the various DnaK-GrpE complexes show one DnaK molecule in the complex in all cases (Figs. 3 and 4). We nonetheless cannot rule out the possibility that this stoichiometry difference arises from the nature of the organism, mesophilic in the former case and thermophilic in the latter (20).
The structural and functional information obtained here shows that the DnaK and GrpE interaction is complex and takes place in three tiers. First, DnaKNBD binds mainly to the head of the NEF (Figs. 1C, 3, and 4); second, the N-terminal region of the GrpE long α-helix (residues 34–68) interacts with the DnaK linker; and third, the first, flexible 33 residues of GrpE can act as a pseudosubstrate, binding to the DnaKSBD peptide binding pocket. Our results show that these interactions precisely control chaperone conformation and function. The three-dimensional reconstructions of the DnaK-GrpE complex in the presence of ADP clearly show DnaKSBD near the head of the GrpE structure, possibly in contact with DnaKNBD (Fig. 4); this contrasts with the loosely connected conformation adopted by both domains in the absence of NEF (6).
Both DnaK domains appear to embrace the GrpE head symmetrically, leaving the DnaK linker near GrpE residues 34–68, which might explain GrpE-mediated protection of the linker against papain digestion (7). In the absence of DnaKNBD, however, the interaction between DnaKSBD and the GrpE head is not strong enough to form a stable complex (41). DnaKNBD and DnaKSBD proximity is accompanied by an increase in accessibility of the DnaK substrate-binding site. The higher k+1 and k−1 values suggest that when in complex with GrpE, the DnaK lid opens more frequently; this facilitates substrate entry into the binding pocket and results in a high DnaK substrate affinity state. This conformation might be an intermediate between the ADP-bound conformation, in which DnaKNBD and DnaKSBD are separated by the linker and the substrate-binding site is closed, and the ATP conformation, where DnaKSBD is in contact with DnaKNBD, promoting displacement of the lid and opening of the substrate-binding site (6, 37). Our interpretation is supported by the observation that GrpE binding to DnaK in the presence of ADP induces changes in the fluorescence properties of DnaK; these changes resemble those observed after ATP binding to the chaperone alone, albeit to a lesser extent (8).
Our study clearly indicates an extreme reduction in the GrpE effect on DnaK conformation when allosteric communication between the chaperone domains is compromised. This finding is supported by the similarity of dNR binding kinetics to the allosteric mutant DnaKR151A, alone or in the presence of GrpE (Fig. 5, B and C), and by our three-dimensional reconstruction of the DnaKR151A-GrpE complex, which shows the linker and DnaKSBD located far from the long N-terminal helical region of the GrpE dimer (Fig. 4, F and G). Interaction of this region with the linker, and probably with DnaKSBD, is essential for maintaining a high substrate affinity conformation of the chaperone; this idea is supported by the inability of wtDnaK to bind stably to substrates when GrpE(69–197) is present (Fig. 5A), and by the rapid substrate dissociation promoted by this GrpE mutant (7). Results were similar when we assayed dNR binding to DnaK in the presence of ATP, or when ATP was added to preformed substrate-DnaK complexes. Our data therefore suggest that DnaK adopts an ATP-like conformation when in complex with GrpE(69–197), emphasizing the role of the 34–68 segment in maintaining substrate bound to DnaK.
The three-dimensional reconstructions of the distinct DnaK-GrpE complexes generated in this study indicate that the long α-helices of the GrpE dimer are flexible. This flexibility is found in two places. The first is the N-terminal region of the α-helix (residues 34–68), whose plasticity was observed in the crystal structures of different GrpE molecules (16, 17, 20) and is clear in the three-dimensional reconstructions of the DnaK-GrpE complexes we generated here. Bending of the N-terminal half of the helices, including residues 34–68, allows this segment to interact with the linker (Fig. 4C, conformation 2). In the absence of the linker (the case of the DnaKNBD mutant) or of interdomain allosteric communication (the DnaKR151A variant), the helical kink in this region is less pronounced, as shown by comparison of the three DnaK-GrpE complexes (Figs. 3 and 4). The second flexible region is in the N-terminal, 33-residue disordered region of GrpE.
Classification of the DnaK-GrpE particles shows two major populations that differ in the position of this disordered region, located in one of the populations in contact with the head of the DnaK-GrpE complex, specifically with DnaKSBD (Fig. 4C, conformation 2). For such an interaction to take place, it is thought that DnaKSBD must be located near the GrpE N-terminal region (7, 20), in proximity to the 33-residue flexible region. Our three-dimensional reconstructions of the DnaK-GrpE complex show a different picture. DnaKSBD is found in the head of the GrpE structure, near DnaKNBD and far from its N-terminal tail (Fig. 4C), and the GrpE tail bends at the two hinge regions. We suggest that the DnaK-GrpE complex cycles between at least two conformations (Fig. 6A), one with the tail in a straighter conformation and the other with the tip interacting with DnaKSBD. This interaction could provide the structural basis for the competition observed between substrates and GrpE residues 1–33 for the DnaK substrate-binding site (7, 22, 34).
FIGURE 6.

Proposed action mechanism for the DnaK-GrpE complex. A, movement of the GrpE tail in the DnaK-GrpE complex. The N-terminal region of the GrpE dimer is flexible and can contact the head of the DnaK-GrpE, near DnaKSBD. B, dual action of GrpE as a NEF. The model is described in the text.
Based on our results, we suggest a mechanism to explain how GrpE controls DnaK conformation (Fig. 6B). In this model, (i) the chaperone recognizes and binds unfolded substrates in the ATP (open) conformation. Thereafter, (ii) ATP hydrolysis in the DnaKNBD induces the closed conformation and substrate trapping at the chaperone substrate-binding site. Substrate release requires ADP/ATP exchange, which promotes opening of the DnaKSBD. This conformational transition is energetically unfavorable; ATP binding overcomes the energetic barrier between the two conformational states of DnaK (21). Our findings here suggest that GrpE facilitates this transition by modulating the DnaK conformational equilibrium; this stabilizes a chaperone intermediate that will promote nucleotide exchange while maintaining the substrate bound until the nucleotide exchange reaction is complete. This is achieved (iii) through strong interaction of the GrpE dimer head with DnaKNBD and the more subtle, although functionally important interaction of the N-terminal region of the GrpE α-helix (residues 34–68) with the DnaK linker (Fig. 6B, red arrow). This intermediate DnaK conformation is characterized by (a) a faster nucleotide exchange reaction due to the modifications imposed by GrpE in DnaKNBD (17), (b) high affinity for substrates, as our peptide binding kinetics demonstrate, and (c) DnaKSBD displacement close to DnaKNBD (Fig. 4C). This would facilitate (iv) interaction of the two domains and consequent opening of the substrate-binding site after ATP binding. The DnaK-GrpE interaction model proposed here lends a structural basis to the mechanism of the thermosensor activity attributed to GrpE. dNR binding experiments (Fig. 5C) suggest that segment 34–68 unfolding in heat shock conditions block DnaK conversion into the intermediate conformation, which our model predicts would slow down transition to the ATP-bound state. Finally, (v) our data also show that, due to GrpE tail flexibility, the 33 first residues of GrpE can bind to the chaperone substrate-binding site and act as a pseudo-substrate.
In summary, our biochemical, biophysical, and structural results identify a complex interaction between the chaperone DnaK and its NEF GrpE. We show that the function of GrpE is more than that of a nucleotide exchange factor, because it also modulates DnaK allosterism that could facilitate the energetically unfavorable transition to the ATP-bound open conformation.
Acknowledgment
We thank C. Mark for editorial assistance.
This work was supported in part by Spanish Ministry of Economy and Innovation Grants BFU2013-44202 (to J. M. V.), SAF2011-22988 (to O. L.), and BFU2013-47059 (to A. M.), Madrid Regional Government Grants S2013/MIT-2807 (to J. M. V.) and S2010/BMD-2316 (to O. L.), and Basque Government Grant IT709-13 (to A. M.).
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- dansyl
- 5-dimethylaminonaphthalene-1-sulfonyl
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank.
REFERENCES
- 1. Hartl F. U., Hayer-Hartl M. (2009) Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581 [DOI] [PubMed] [Google Scholar]
- 2. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 [DOI] [PubMed] [Google Scholar]
- 3. Young J. C., Barral J. M., Ulrich Hartl F. (2003) More than folding: localized functions of cytosolic chaperones. Trends Biochem. Sci. 28, 541–547 [DOI] [PubMed] [Google Scholar]
- 4. Mayer M. P., Bukau B. (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bukau B., Horwich A. L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92, 351–366 [DOI] [PubMed] [Google Scholar]
- 6. Bertelsen E. B., Chang L., Gestwicki J. E., Zuiderweg E. R. (2009) Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc. Natl. Acad. Sci. U.S.A. 106, 8471–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moro F., Taneva S. G., Velázquez-Campoy A., Muga A. (2007) GrpE N-terminal domain contribuyes to the interaction with DnaK and modulates the dynamics of the chaperone substrate binding domain. J. Mol. Biol. 374, 1054–1064 [DOI] [PubMed] [Google Scholar]
- 8. Han W., Christen P. (2003) Interdomain communication in the molecular chaperone DnaK. Biochem. J. 369, 627–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walsh P., Bursać D., Law Y. C., Cyr D., Lithgow T. (2004) The J-protein family: modulating protein assembly, disassembly and translocation. EMBO Rep. 5, 567–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kampinga H. H., Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alberti S., Esser C., Höhfeld J. (2003) BAG-1: a nucleotide exchange factor of Hsc70 with multiple cellular functions. Cell Stress Chaperones 8, 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McLellan C. A., Raynes D. A., Guerriero V. (2003) HspBP1, an Hsp70 cochaperone, has two structural domains and is capable of altering the conformation of the Hsp70 ATPase domain. J. Biol. Chem. 278, 19017–19022 [DOI] [PubMed] [Google Scholar]
- 13. Andréasson C., Fiaux J., Rampelt H., Mayer M. P., Bukau B. (2008) Hsp110 is a nucleotide-activated exchange factor for Hsp70. J. Biol. Chem. 283, 8877–8884 [DOI] [PubMed] [Google Scholar]
- 14. Groemping Y., Klostermeier D., Herrmann C., Veit T., Seidel R., Reinstein J. (2001) Regulation of ATPase and chaperone cycle of DnaK from Thermus thermophilus by the nucleotide exchange factor GrpE. J. Mol. Biol. 305, 1173–1183 [DOI] [PubMed] [Google Scholar]
- 15. Harrison C. (2003) GrpE, a nucleotide exchange factor for DnaK. Cell Stress Chaperones 8, 218–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakamura A., Takumi K., Miki K. (2010) Crystal structure of a thermophilic GrpE protein: insight into thermosensing function for the DnaK chaperone system. J. Mol. Biol. 396, 1000–1011 [DOI] [PubMed] [Google Scholar]
- 17. Harrison C. J., Hayer-Hartl M., Di Liberto M., Hartl F., Kuriyan J. (1997) Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science 276, 431–435 [DOI] [PubMed] [Google Scholar]
- 18. Grimshaw J. P., Jelesarov I., Schönfeld H. J., Christen P. (2001) Reversible thermal transition in GrpE, the nucleotide exchange factor of the DnaK heat-shock system. J. Biol. Chem. 276, 6098–6104 [DOI] [PubMed] [Google Scholar]
- 19. Muga A., Moro F. (2008) Thermal adaptation of heat shock proteins. Curr. Protein Pept. Sci. 9, 552–566 [DOI] [PubMed] [Google Scholar]
- 20. Wu C. C., Naveen V., Chien C. H., Chang Y. W., Hsiao C. D. (2012) Crystal structure of DnaK protein complexed with nucleotide exchange factor GrpE in DnaK chaperone system: insight into intermolecular communication. J. Biol. Chem. 287, 21461–21470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taneva S. G., Moro F., Velázquez-Campoy A., Muga A. (2010) Energetics of nucleotide-induced DnaK conformational states. Biochemistry 49, 1338–1345 [DOI] [PubMed] [Google Scholar]
- 22. Mehl A. F., Heskett L. D., Neal K. M. (2001) A GrpE mutant containing the NH2-terminal “tail” region is able to displace bound polypeptide substrate from DnaK. Biochem. Biophys. Res. Commun. 282, 562–569 [DOI] [PubMed] [Google Scholar]
- 23. Horst M., Oppliger W., Feifel B., Schatz G., Glick B. S. (1996) The mitochondrial protein import motor: Dissociation of mitochondrial hsp70 from its membrane anchor requires ATP binding rather than ATP hydrolysis. Protein Sci. 5, 759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marabini R., Masegosa I. M., San Martin M. C., Marco S., Fernandez J. J., de la Fraga L. G., Vaquerizo C., Carazo J. M. (1996) Xmipp: an image processing package for electron microscopy. J. Struct. Biol. 116, 237–240 [DOI] [PubMed] [Google Scholar]
- 25. Mindell J. A., Grigorieff N. (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 [DOI] [PubMed] [Google Scholar]
- 26. Scheres S. H., Núñez-Ramírez R., Sorzano C. O., Carazo J. M., Marabini R. (2008) Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 3, 977–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sorzano C. O., Bilbao-Castro J. R., Shkolnisky Y., Alcorlo M., Melero R., Caffarena-Fernández G., Li M., Xu G., Marabini R., Carazo J. M. (2010) A clustering approach to multireference alignment of single-particle projections in electron microscopy. J. Struct. Biol. 171, 197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 29. Scheres S. H., Valle M., Grob P., Nogales E., Carazo J. M. (2009) Maximum likelihood refinement of electron microscopy data with normalization errors. J. Struct. Biol. 166, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 31. Lopéz-Blanco J. R., Chacón P. (2013) iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. (2013) J. Struct. Biol. 184, 261–270 [DOI] [PubMed] [Google Scholar]
- 32. Vogel M., Bukau B., Mayer M. P. (2006) Allosteric regulation of Hsp70 chaperones by a proline switch. Mol. Cell 21, 359–367 [DOI] [PubMed] [Google Scholar]
- 33. Zahn M., Berthold N., Kieslich B., Knappe D., Hoffmann R., Sträter N. (2013) Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J. Mol. Biol. 425, 2463–2479 [DOI] [PubMed] [Google Scholar]
- 34. Brehmer D., Gässler C., Rist W., Mayer M. P., Bukau B. (2004) Influence of GrpE on DnaK-substrate interactions. J. Biol. Chem. 279, 27957–27964 [DOI] [PubMed] [Google Scholar]
- 35. Chesnokova L. S., Slepenkov S. V., Protasevich I. I., Sehorn M. G., Brouillette C. G., Witt S. N. (2003) Deletion of DnaK's lid strengthens binding to the nucleotide exchange factor. GrpE: a kinetic and thermodynamic analysis. Biochemistry 42, 9028–9040 [DOI] [PubMed] [Google Scholar]
- 36. Buczynski G., Slepenkov S. V., Sehorn M. G., Witt S. N. (2001) Characterization of a lidless form of the molecular chaperone DnaK: deletion of the lid increases peptide on- and off-rate constants. J. Biol. Chem. 276, 27231–27236 [DOI] [PubMed] [Google Scholar]
- 37. Kityk R., Kopp J., Sinning I., Mayer M. P. (2012) Structure and dynamics of the ATP-bound open conformation of Hsp70 chaperones. Mol. Cell. 48, 863–874 [DOI] [PubMed] [Google Scholar]
- 38. Gelinas A. D., Toth J., Bethoney K. A., Langsetmo K., Stafford W. F., Harrison C. J. (2003) Thermodynamic linkage in the GrpE nucleotide exchange factor, a molecular thermosensor. Biochemistry 42, 9050–9059 [DOI] [PubMed] [Google Scholar]
- 39. Sondermann H., Scheufler C., Schneider C., Hohfeld J., Hartl F. U., Moarefi I. (2001) Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science 291, 1553–1557 [DOI] [PubMed] [Google Scholar]
- 40. Shomura Y., Dragovic Z., Chang H. C., Tzvetkov N., Young J. C., Brodsky J. L., Guerriero V., Hartl F. U., Bracher A. (2005) Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol. Cell 17, 367–379 [DOI] [PubMed] [Google Scholar]
- 41. Buchberger A., Schröder H., Büttner M., Valencia A., Bukau B. (1994) A conserved loop in the ATPase domain of the DnaK chaperone is essential for stable binding of GrpE. Nat. Struct. Biol. 1, 95–101 [DOI] [PubMed] [Google Scholar]
- 42. Kabani M. (2009) Structural and functional diversity among eukaryotic Hsp70 nucleotide exchange factors. Protein Pept. Lett. 16, 623–660 [DOI] [PubMed] [Google Scholar]