

Abstract
Corneal allograft rejection has been described as a Th1-mediated process involving IFN-γ production. However, recent evidence has also implicated IL-17 as being involved during acute corneal allograft responses. Our data supports those that maintain that IL-17 is involved in early acute corneal allograft acceptance. However, we decided to extend these studies to include a later phase of rejection in which there is a peak of IL-17 production that is >15 fold higher than seen during acute rejection that occurs >45 days post-engraftment at the onset of late-term rejection. We demonstrate that neutralizing IL-17A at this time significantly reduced corneal graft rejection. Surprisingly, when corneal grafts that are undergoing this later phase of rejection are treated with anti-IL17A there is a reversal of both opacity and neovascularization. When compared to the early phase of rejection, the cellular infiltrate is significantly less with a greatly reduced presence of Gr-1+ neutrophils with a relative increase in CD4+ T cells and macrophages. We went on to identify that the cells expressing IL-17 were CD4+ IL-17+ T cells and somewhat surprisingly, IL-17+ F4/80+ macrophages within the rejecting corneal allografts. Taken together, these findings describe a distinct late phase of corneal allograft rejection which is likely mediated by Th17 cells and that therapeutic neutralization of IL-17A reverses this rejection. This further suggests that IL-17 might serve as an excellent therapeutic target to reduce this form of corneal allograft rejection.
Introduction
Due to advances in surgical management, critical care, administration of traditional and novel immunosuppressants over the last two decades, the incidence of acute corneal allograft rejection has been significantly reduced. Consequently, late-term corneal graft rejection, which tends to have an increased incidence of chronic graft rejection and a higher rate of resistance to conventional anti- rejection therapy, is the main problem in clinical penetrating keratoplasty. This has fueled the desire to understand the mechanisms responsible for late-term corneal allograft rejection and develop clinically relevant protocols to better control them. Studies have shown that late-term corneal graft rejection consists of a series of reversible rejections and ends up with an irreversible rejection (1). During the first 5 postoperative years, irreversible rejection accounts for 28% to 35% of all corneal graft loss, the number rises to 30% to 56% when performing on “high risk” corneal recipients (1–3). Thus, it is important to understand the mechanisms underlying late-term rejection to prevent long-term graft loss and improve the overall results of cornea transplantation. A major complication in studying late-term corneal allograft rejection is the lack of a robust animal model. Such a model would enable investigators to better characterize late-term rejection and determine if there exist significant differences from those mechanisms that are responsible for acute allorejection. This manuscript will describe a murine model of spontaneous late-term cornea allorejection model that we believe approximates what occurs during clinical cornea transplant rejection in the aspects of severity and chronicity (tempo) as well as lower response rate to conventional anti-rejection therapy (4,5). We further believe that this model would provide a tool that will provide a better means of understanding the immunopathogenesis of the late-term corneal graft rejection.
In addition, there has been a growing understanding that IL-17 is an important cytokine not only for autoimmunity (7–9) but also for resistance to microbial infection (10,11) and in some forms of transplantation rejection (12,13). This proinflammatory cytokine has been shown to not only stimulate inflammation but is also pro-angiogenic, both during tumor development (14) and during herpes infection of the cornea (15). Since we observed that there was a peak in production of IL-17A shortly prior to late-term corneal allorejection, we evaluated its role during this type of rejection. We present data indicating that IL-17A is a key factor mediating late-term corneal allograft rejection and that targeting this cytokine might hold significant therapeutic efficacy.
Materials and Methods
Animals
Investigations with mice conformed to the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. Male six week old C57BL/6 (B6) and BALB/c mice were purchased from Jackson Lab and Taconic respectively. These mice were then used for corneal transplantation between 9 to 12 weeks of age with BALB/c mice serving as recipients and B6 mice as cornea donors. The mice were housed under specific pathogen-free conditions and treated in accordance of NIH and IACUC guidelines. All procedures and protocols were approved by the Animal Research Committee of Saint Louis University.
Orthotopic corneal transplantation and grafts opacity, neovascularization evaluation
Each mouse was placed under general anesthesia by IP injections of ketamine (86.98 mg/kg) and xylazine (13.04 mg/kg). C57BL/6 corneas were grafted onto the right eye of BALB/c recipients as described previously (4). Briefly, the center of the donor cornea was marked with a 2mm trephine, excised and placed onto the 1.5 mm cornea bed of recipient marked and excised in the center of the cornea, then was secured with eight to ten interrupted 11-0 nylon sutures. Antibiotic ointment was applied to the finished graft. All grafted eyes were examined after 48 hours, recipient mice with signs of surgical complications such as hyphema, cataract, shallow or flat anterior chamber, or opacity score of 4, were considered technical failures and excluded from the study. The sutures were removed on day 7 postoperatively. Clinical examination was performed 3 times a week. The grafts were scored for opacity and neovascularization as previously described (4). At each time point, grafts were graded for opacity using a 0–5 scale and for neovascularization using a 0–8 scale. Those corneal grafts with persistent opacity score >2 within 2–5 weeks postoperative, were considered to be undergoing acute rejection. Mice whose corneas were clear through 5– 6 weeks postoperative but develop opacity at times >45 days were defined as undergoing late-term rejection.
Bio-Plex cytokine analysis
Cornea allografts were removed from recipient mice at various time points following transplantation. These include time points shortly following corneal engraftment (3,5 and 7 days postoperative), during subclinical rejection and time points when clinical rejection is apparent. Both the acute and late-term phase of the response was evaluated in this fashion. Samples were generated by removing individual graft buttons which were mechanically disrupted in 100 μl of cell lysis buffer provided by Bio- Rad (Bio-Rad, Hercules, CA), and stored at −80°C until processing. Samples were then thawed and sonicated on ice twice for 30 s and centrifuged for 4 min at 515 × g at 4°C to remove debris. Supernatant was then transferred to a new tube and protein analysis was performed with Bradford assay to ensure similar amounts of protein to be used for analysis. The BioPlex assay (Bio-Rad) was performed as described in the kit protocol. Briefly, protein from individual allografts were added to each well of a Th1 and Th17 multiplex mouse cytokine BioPlex plate. Cytokine concentrations were determined by comparison to a standard curve provided by Bio-Rad, and the results are reported as pg/ml protein.
Flow cytometric characterization of IL-17A producing leukocytes in corneas, spleens, and lymph nodes of recipient mice
Corneas, spleens and lymph nodes were harvested from recipient mice at the same time points described in the preceding section and single cell suspensions were isolated. The single cell suspensions from allografts were prepared as previously described (16). Briefly, they were digested with 84 U of collagenase type 1 (Sigma-Aldrich, St. Louis, MO) for 1 hour at 37°C, then triturated and filtered through a 40 μ cell strainer (BD Labware, Bedford, MA). Once isolated, these cell samples were resuspended in RPMI 1640 media supplemented with 10% FBS, antibiotics and L-glutamine and stimulated with 50ng/ml PMA and 500ng/ml ionomycin for 6 hours, the last 2 hours with the addition of Golgiplug (BD Bioscience, San Jose, CA). The cells were then stained for surface markers with PerCP-conjugated anti-CD45 (clone 30-F11), Alexa Fluor700-Gr-1 (clone RB6-8C5), and APC-conjugated anti-F4/80 (clone BM8) (BioLegend, San Diego, CA), FITC-conjugated anti-CD4 (clone RM4-5), PE-conjugated anti-CD8a (clone 53-6.7) (BD Bioscience) and eFlour 450-conjugated CD11b (clone M1/70) (eBioscience, San Diego, CA). Separate samples were also stained with APC labelled mouse CD1d Tetramer (CD1d Alpha Gal Cer) (Proimmune, Inc. Sarasota, FL) to detect NKT cells. These cells were then permeabilized using a Cytofix/Cytoperm kit (BD Bioscience), and stained for IL-17A (clone eGio17B7) and IFNγ (clone XMG1.2) both purchased from eBioscience (San Diego, CA). Samples were assayed with a BD FACSCalibur flow cytometer, and the data were acquired using FlowJo software (TreeStar, Ashland, OR). The strategy for analysis was to initially gate on live cells and then the CD45+ cells. These cells were further gated on either CD4 or F4/80 expressing cells and then analyzed for either IL-17A or IFNγ expression. The F4/80+ cells expressing IL-17A were further analyzed for CD4, CD8, and CD11b expression.
H&E staining and Immunohistochemistry
Normal and grafted corneas were removed at various time points corresponding to acute clinical rejection, subclinical late-term rejection and clinical late-term rejection. These corneas were snap- frozen in OCT with liquid nitrogen and stored at −80°C until sectioned. To evaluate inflammation, sections were stained by hematoxylin and eosin by the Saint Louis University Pathology core facility. For immunohistochemistry, sections (10 μm) were fixed in fresh 4% paraformaldehyde (Electron Microscopy Science, Hatfield, PA) for 10 minutes, then washed and permeabilized with ice - cold acetone for 2 minutes at −20°C, and then treated with 2.5% Triton X-100 15 minutes. nonspecific binding was blocked with non-serum blocking buffer (Dako, Carpinteria, CA) for 10 minutes. Sections then were labeled with rat anti-mouse CD4 antibody (1:200, 2.5 μg/ml; clone GK1.5, eBioscience, San Diego, CA.), goat anti-mouse anti-Ly-6G and Ly-6C (Gr-1, clone RB6-8C5) antibody (1:200, Abcam, Cambridge, MA), CD-11b (1:1000.5mg/ml; clone M1/70, BD Bioscience, San Diego, CA) CD-1d (1: 200,0.5mg/ml; clone 1B1, Abcam, Cambridge, MA), F4/80 (1:100, BioLegend, San Diego, CA) and rabbit polyclonal anti-mouse IL-17 antibody (Abcam, Cambridge, MA) overnight at 4 °C. After washing, samples were incubated with Alexa Fluor 488 conjugated donkey anti-rabbit antibody (1:1000; Invitrogen, Carlsbad, CA.) and NorthernLights 557 conjugated anti-rat monoclonal antibody (1:1000, 1mg/ml; R&D systems) for 2 hour at room temperature. All specimens were mounted with DAPI (Electron Microscopy Sciences). Photomicrographs were obtained using a 5000B Leica microscope (Leica, Wetzlar, Germany) equipped with a charge-coupled device camera (Retiga 200R) interfaced with QCapture Pro software (both from Q Imaging, Surrey, Canada). Images were globally adjusted for contrast and brightness.
In vivo treatment with neutralizing anti-IL-17A
Monoclonal rat anti-mouse IL-17A (JL7.1D10; kindly provided by Edward Bowman at Merck Research Labs, Palo Alto, CA) was used at a dose of 30 mg/kg and was administrated to recipient mice by subcutaneous injection at the nape of the neck by using a volume of 100μl. Administration frequency varied depending on the specific experiment. The rat IgG1 isotype control antibody was also provided by Merck Research Labs, Palo Alto, CA and was administrated at the same time and dose as was JL7.1D10.
Statistics
SigmaPlot 11.0 (Systat Software Inc., San Jose, CA) and SlideWrite (Statcon Software, Rancho Santa Fe, CA) were used to analyze and present data. The Rank Sum test was used to compare corneal opacity and neovascularization scores. All other data was compared with an unpaired 2-tailed Student t-test and confirmed by parametric and F tests. Significant differences were considered when comparisons had a P value less than 0.05.
Results
Neutralization of IL-17A during acute corneal allograft rejection reduced corneal graft acceptance
Reports evaluating the role of IL-17 during acute murine corneal allograft rejection have yielded inconsistent results. These results have ranged from reports suggesting that IL-17 is involved in rejection (17,18), that it has no role (19) and most recently that it is involved in generating Treg cells that promote acceptance (20,21). We decided to address whether IL-17 was associated with acute rejection in mice. When corneas from BALB/c mice that displayed signs of rejection were evaluated for cytokine production, we did not observe a significant increase in IL-17 production during acute rejection (Fig. 1A). In contrast, our data did demonstrate increased production of both IL-6 and IFN-γ (Fig. 1B, 1C), which is consistent with previous reports suggesting their involvement in acute rejection (21). Thus our data does not indicate that there is a significant amount of IL-17 produced during the first 30 days post-engraftment, when approximately 50% of the cornea allografts are rejected. However, when we neutralized IL-17A with an anti-IL-17A antibody our results were very similar to that reported by Cunnusamy, et al. (20,21), namely that such treatment significantly increased corneal allograft rejection (Fig. 2). Therefore we support the notion that IL-17 production during the early stages of corneal allograft rejection might be involved in promoting acceptance of such grafts.
Figure 1.
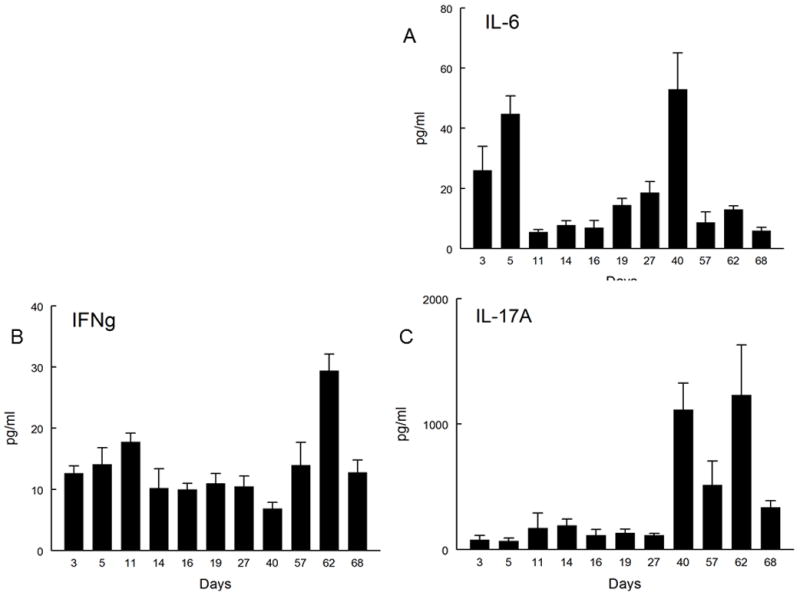
Multiplex cytokine analysis of engrafted corneas. The corneas of BALB/c mice engrafted with B6 allogeneic corneas were removed at various time points including time points shortly following corneal engraftment when only a few corneas show any evidence of inflammation (3, 5 and 7 days post-engraftment), during subclinical rejection and time points when clinical rejection is apparent and corneal lysates were made. These corneal lysates were analyzed with a multiplex cytokine array. Average cytokine concentrations determined from at least four mice/time point are reported as pg/ml (y axis). The cytokines measured were IL-17 (A), IL-6 (B) and IFN- γ (C) and values indicate mean ± SEM of IL-17, IL-6 and IFN- γ.
Figure 2.
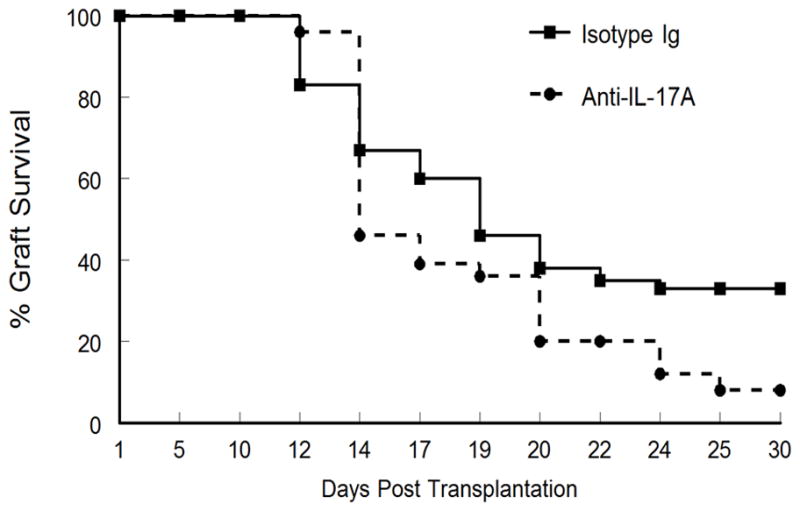
Neutralization of IL-17A during acute corneal allograft decreases allograft survival. BALB/c mice were engrafted with B6 corneas and then injected S.C. with either control Ig (n=12) or anti-IL-17A (n=12) and observed for 30 days. Mice receiving anti-IL-17A displayed reduced corneal allograft survival (P<0.01).
Late-term rejection is preceded by an increase in the amount of IL-17 found in the cornea
The aforementioned results do not explain why the production of both IL-6 and IFN-γ display a second peak at Day 40 and post-Day 50 respectively (Fig. 1A, 1B). Furthermore, these results also do not explain why production of IL-17A shows a dramatic increase (15–30 fold) from Day 40 to Day 70 than is seen during acute corneal allograft rejection (Fig. 1C). This secondary production of these cytokines was coincident with our studies demonstrating that over 80% of corneal allografts that had survived the acute rejection period were being rejected at some time post 45 days after engraftment. Note that the gross appearance of this late-term rejection (Fig. 3C) is very similar to that seen in acute rejection (Fig. 3A) and is clearly differentiated from an accepted corneal allograft (Fig. 3B). Thus increases in IL-17 production peaked during subclinical late-term rejection and gradually dropped until the allograft was fully rejected. It should also be noted that the second peak of IL-6 (Fig. 1A) could be interpreted as a factor leading to IL-17A production since IL-6 is a critical factor in the differentiation of Th17 cells (23,24).
Figure 3.
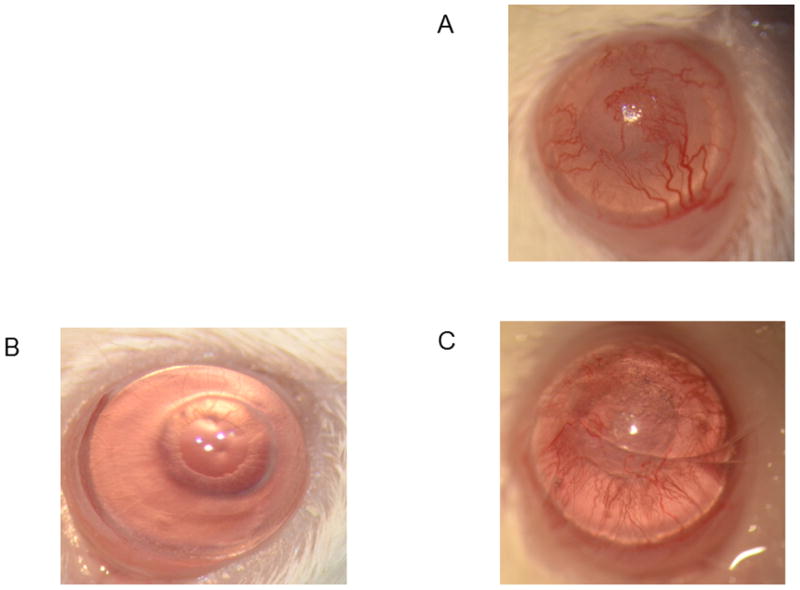
Late-term corneal allograft rejection appears to be very similar to that seen in acutely rejected corneal allografts. Eyes of mice were photographed using a vertical slit-lamp during acute allografts rejection (A), allograft acceptance (B), or late-term allograft rejection (C).
Prophylactic neutralization of IL-17 significantly increased the survival of corneal allografts
The profound increase in IL-17 found as corneal grafts undergo late-term rejection suggests that this cytokine might be involved in mediating graft rejection. To that end, recent studies have shown that IL-17 not only mediates certain forms of autoimmunity and anti-microbial responses (7–11, 25), but has also been implicated in alloimmune responses leading to graft rejection (12). Due to these reports and our observation of increased IL-17 production during late-term corneal allograft rejection, we decided to target IL-17A with a neutralizing antibody. We therefore treated mice bearing clear corneas with neutralizing anti-mouse IL-17A mAb or control isotype IgG beginning at Day 40 post-engraftment. Mice were administered antibody twice weekly for two weeks. Data indicated that treatment of mice with anti-IL-17A antibody prevented rejection during the time of treatment and reduced the incidence of rejection for up to 4 weeks after treatment was discontinued (Fig. 4). These results suggest that late-term corneal allograft rejection might even be prevented if treatment were extended.
Figure 4.
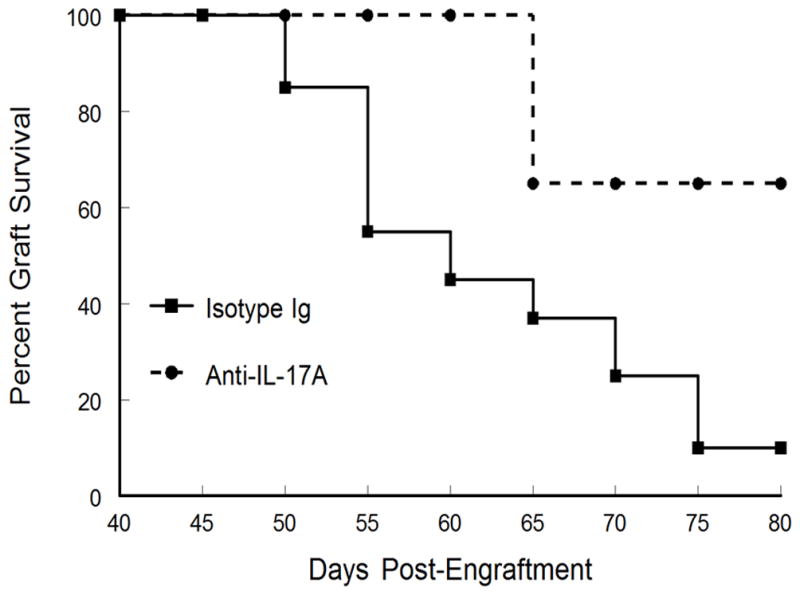
Prophylactic neutralization of IL-17A beginning at Day 40 significantly increased corneal allograft survival. Mice bearing clear corneas at Day 40 were treated with anti-mouse IL-17A mAb (n=6) or control isotype IgG (n=10) at a dose of 30mg/kg, twice weekly for two weeks by SC injection. Corneal allograft survival for mice treated with anti-IL-17A during late-term rejection were enhanced compared to mice treated with control antibody (p<0.05).
Therapeutic neutralization of IL-17 reverses late-term corneal allorejection
Once we established that targeting IL-17 prior to corneal pathology was effective, it raised the question as to whether similar neutralization of IL-17A, when used to treat corneas that displayed signs of rejection, might prevent further destruction of the allograft. Consequently, we treated mice displaying both opacity and neovascularization scores indicative of a corneal graft undergoing rejection with anti-IL-17A twice weekly for 3 weeks. To our surprise, such treatment of mice with corneas undergoing rejection, displayed not only a lack of further rejection, but that signs of rejection were reversed leading to both significantly reduced opacity and reduced blood vessels (Fig. 5A and Fig. 5B). This is also demonstrated in Figure 5C, where corneas treated with anti-IL-17A become clear and blood vessels show no sign of blood in them. Much like results seen when anti-IL-17A treatment is discontinued prior to signs of rejection (See dashed line in Fig. 4 where the appearance of new rejection is noted at day 65, which is 10 days after anti-IL-17 treatment was discontinued.), these therapeutically treated corneas once again show signs of rejection two to three weeks after treatment was discontinued (Fig. 5A). These corneas once again show increasing opacity and the blood vessels once again have blood in them. These data indicate that treatment only targets the cytokine and not the cell that produces IL-17A, which remain intact and functional at least following three weeks of anti-IL-17A treatment.
Figure 5.

Therapeutic neutralization of IL-17A during late-term rejection for mice demonstrating rejection of donor corneas reversed both opacity (A) and neovascularization (B). Representative mice displaying the corneas appearance at the start of treatment and then after treatment were photographed using a vertical slit-lamp (C). Mice were treated with anti-IL-17A antibody or control antibody by twice weekly injections for three weeks beginning at the point when grafted corneas were displaying signs of late-term rejection (first photo of C). Treatment was initiated between day 45 and day 60 post-grafting depending on when the corneal allograft showed signs of rejection. The subsequent data is expressed as a consequence of when treatment began. Results are expressed as mean opacity (A) or mean neovascularization (B) ± SEM. The opacity was compared using ANOVA analysis and asterisk indicates significant differences (P< 0.01–0.001). Neovascularization was analyzed by unpaired 2-tailed Student t test indicating significant differences between the two groups (p<0.05).
Corneal increases in IL-17 were not accompanied by increases in IL-17 in lymph nodes or spleen
In an effort to determine the source of this IL-17 production, we initially evaluated spleens and lymph nodes for the presence of IL-17 at the same time as IL-17 was seen to increase in cornea. However, the levels of IL-17 found in these organs were not statistically different from controls (data not shown). When we evaluated whether there was an increase in splenic or lymph node cells that produced IL-17, there was a slight increase (approximately 2 fold) in both IL-17+ and IFN-γ+ cells but these data were not statistically significant. Thus these studies clearly indicated that at no time, were there significant increases in total number of IL-17+ cells in spleen or lymph nodes for mice undergoing late-term rejection. Thus the preponderance of these data indicates that enhanced IL-17 production was only found locally within corneal allografts that were undergoing rejection and not in any of the secondary lymphoid organs.
The phenotype of inflammatory cells during late-term rejection is not the same as during acute rejection
While late-term corneal allograft rejection appears similar to acute, we know that the cytokine factors involved in rejection are not similar (see Figs. 1, 4, 5). Thus to characterize the inflammatory cells within allografts undergoing late-term rejection we initially did an H&E stain of section from acute and late-term rejected corneas as well as accepted and ungrafted corneas. As shown in Figure 6, there is a significant difference between acute and late-term rejected corneas with the acutely rejected corneas displaying a much greater cellular infiltrate and generally more edema than did corneas rejecting after 45 days. We next performed a series of flow cytometric studies to characterize these cells. As is seen in Figure 7, the inflammatory infiltrate during acute corneal allograft rejection is overwhelmingly neutrophilic, with almost 60% of the cells GR-1+. In contrast, the percentage of neutrophils during late-term rejection was only about 20%. Consequently, the percentages of the other subsets of cells during late-term rejection was greater than seen during acute rejection, though there was no preponderance of any one particular subset. We also compared intracellular levels of IL-17A and IFNγ and much like the data shown in Figure 1, there were significantly more IL-17A expressing cells in late rejection than seen during acute rejection while the number of cells making IFNγ were very similar between these two phases of rejection (Fig. 8). Thus it is clear that the inflammatory cell infiltrate during late-term rejection is not the same as during acute rejection. Nor was the magnitude of the infiltrate as large during late-term rejection as during acute rejection (data not shown).
Figure 6.
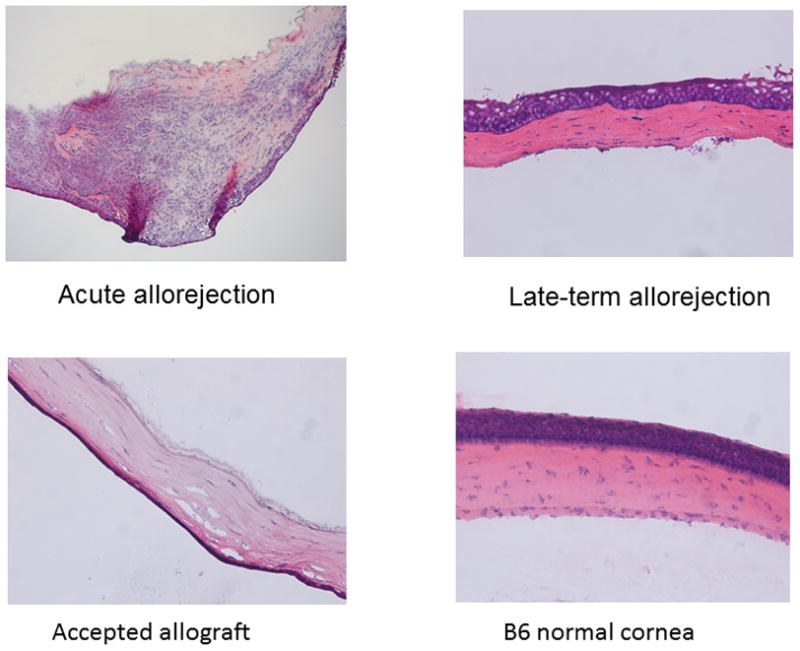
Histological sections indicate that acute corneal allograft rejection has significantly greater numbers of inflammatory cells than corneas from mice undergoing late-term corneal allograft rejection. Hematoxylin and eosin stain of corneal sections are from acute rejection (day 17 post-engraftment), late-term rejection (day 65 post-engraftment), accepted corneal allograft (day post-engraftment), and an ungrafted B6 cornea (B). (original magnification × 20).
Figure 7.
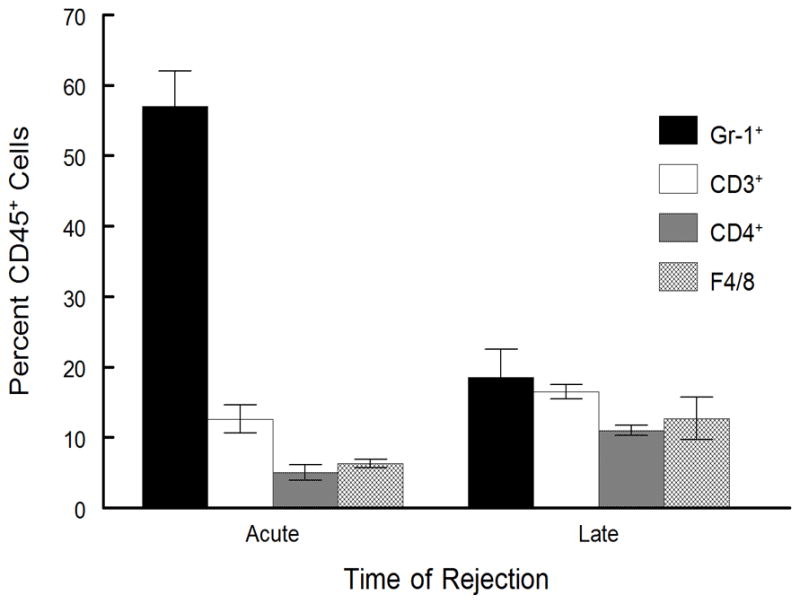
Inflammatory infiltrate from acute and late-term rejection are not equivalent. Rejected corneas were removed at days 18 (acute rejection, n=4) or day 65 (late-term rejection, n=6) and disaggregated into single-cell suspensions and stained with anti-CD45. The CD45+ cells were gated and further analyzed for staining with antibodies directed against CD3, CD4, Gr-1, CD11b, and F4/80 surface markers. Cells were analyzed by flow cytometry. Late-term rejected corneas had a much smaller infiltrate of Gr-1+ neutrophils than did acutely rejected corneas (p< 0.01).
Figure 8.
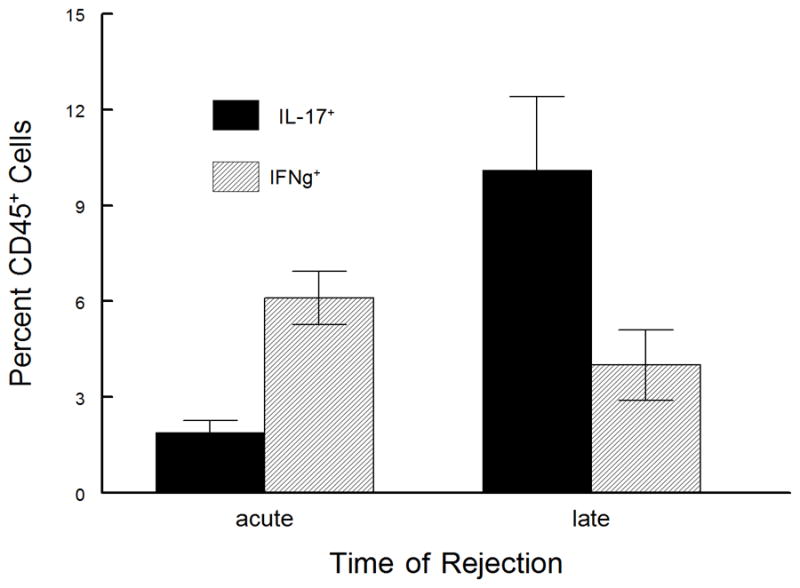
Inflammatory infiltrate from acute and late-term rejection do not express equivalent numbers of IL-17 expressing cells. Rejected corneas were removed at days 18 (acute rejection, n=5) or days 55 – 65 (late-term rejection, n=5) and disaggregated into single-cell suspensions and stained with anti-CD45. The CD45+ cells were gated and further analyzed for staining with antibodies directed against IL-17A and IFNγ. Cells were analyzed by flow cytometry. Late-term rejected corneas contained a much greater number of IL-17A expressing cells than did acutely rejected corneas (p< 0.05).
CD4+ T cells and F4/80+ macrophages express IL-17 during late-term allograft rejection
In order to identify which cells were expressing IL-17A we performed immunocytochemistry on sections from corneas undergoing late-term allorejection. From flow cytometry we determined that the primary cells found in late-term rejecting corneas were neutrophils, CD4+ T cells, and macrophages. It has been documented that CD4+IL-17+, Th17 cells can mediate solid organ allograft rejection in heart and lung transplantation (26, 27), even in the absence of IFN-γ or T-bet expression (26). When corneal sections were stained with markers for CD4+ T cells and costained for IL-17A, we observed several cells in one field with high magnification (100X) that co-stained for these markers indicating the presence of Th17 cells in these corneas (Fig. 9). Likewise, when corneal sections were costained for F4/80 and IL-17A several cells in one field (40X) costained for these markers as well indicating that macrophages were another source of IL-17 in late-term rejecting corneas (Fig. 10). However, while co-staining for Gr-1 and CD11b confirmed the presence of neutrophils in these rejecting corneas, they did not display unmistakable co-localization of Gr-1 and IL-17A (data not shown). In order to confirm and extend these results, we next isolated cells from grafted corneas that were undergoing rejection. Our strategy for analyzing these cells was to perform flow cytometry and gate on live CD45+ cells. These CD45+ cells were further analyzed by evaluating those cells that expressed CD4, CD8, Gr-1, and F4/80 to determine if they co-expressed intracellular IL-17 and/or IFNγ. Figure 11B is a representative plot of CD45+ cells that express IL-17A. When these IL-17A expressing cells were further evaluated for CD4 and F4/80 expression, we confirmed our observations with immunohistochemistry by demonstrating that both CD4+ T cells and F4/80+ macrophages expressed IL-17A intracellularly (Fig. 11C, D). The relative numbers of CD4+IL-17A+ T was always greater than the number of F4/80+IL-17A+ cells with a ratio that tended to be around 4 to 1 (Fig. 11B, C). In addition, while there were CD4+IFNγ+ cells, they were significantly less than those CD4+ cells expressing IL-17A and there were no F4/80+ cells found to have intracellular IFNγ (Fig. 11C). We also monitored for any CD4+ cells that expressed both IL-17A and IFNγ and as Figure 11A demonstrates, there were no such cells found, demonstrating that the CD4+ cells were either of a Th17 or a Th1 phenotype. Furthermore, there were no CD1d+ cells indicating that NKT cells are not found in late-term rejecting corneas.
Figure 9.
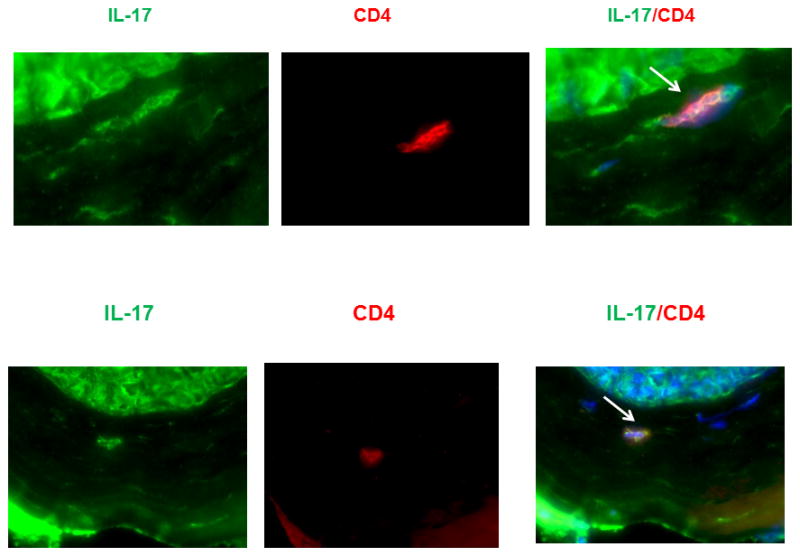
IL-17A is produced by CD4+ T cells. Sections were stained with CD4, Gr-1 or CD11b and each were then stained for IL-17A. DAPI was used to counterstain the nuclei. Results are only shown for CD4 (100X) as Gr-1 and CD11b did not demonstrate convincing co-localization of IL-17A.
Figure 10.
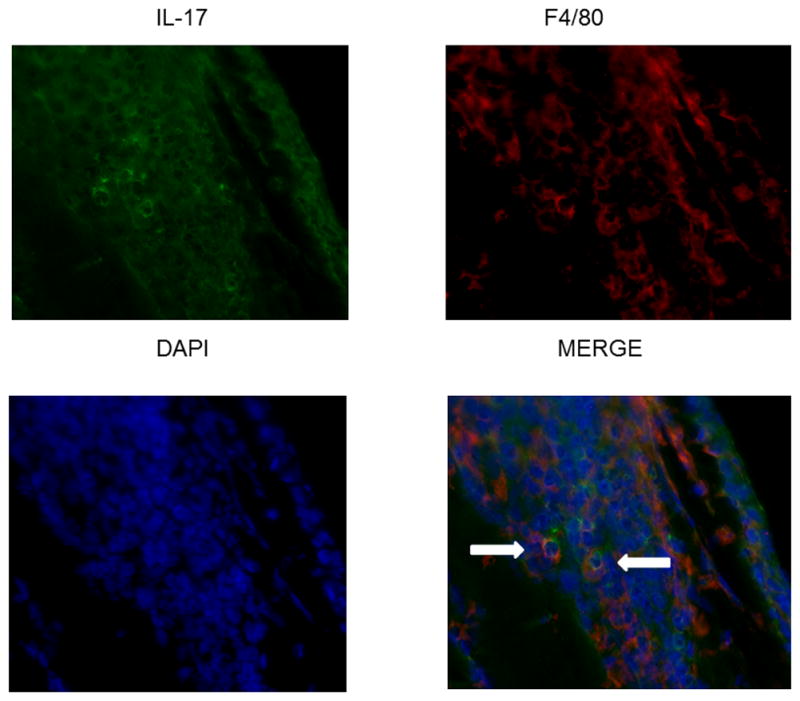
IL-17A is produced by F4/80+ macrophages. Sections were stained with F4/80 and then stained for IL-17A. DAPI was used to counterstain the nuclei. Results are only shown for F4/80 (40X).
Figure 11.

Flow cytometric analysis confirms that both CD4+ and F4/80+ cells produce IL-17A and that no cells express both at the same time. A. Cells isolated from the indicated corneas were stained with CD45, then CD4 and permeabilized and further stained for IL-17A and IFN-γ. Figure displays a series of representative dot plots where the CD45+CD4+F4/80− cells were analyzed for IL-17A and IFN-γ expression. Figure displays a representative dot-plot of cells gated first for CD45+ cells and then those gated for CD4+ cells and assayed for IL-17A and IFN-γ expression. B. Representative plot of corneal inflammatory cells that were analyzed for CD45 and IL-17A expression to visualize the CD45+ cells that express IL-17A. C. The IL-17A+ cells shown in part B were further analyzed for CD4 and F4/80 expression. This demonstrates that there are both CD4+ T cells that express IL-17A and F4/80+ cells that express IL-17A. D. Graphic representation of cells isolated from corneas undergoing late-term rejection that were stained with surface markers for CD4, F4/80 and CD1d tetramer and then permeabilized and stained with either IL-17A or IFN-γ. Data show that no CD4+ cells were producing both IL-17A and IFN-γ, they also demonstrate significantly more CD4+IL-17A+ cells than CD4+IFNγ+ cells (P<0.01). No F4/80+IFNγ+ or CD1d tetramer+ cells were detected.
Discussion
It is widely believed that corneal allograft rejection is predominantly mediated by CD4+ T cells expressing the Th1 phenotype. However, therapies like CsA and FK506 directed at these cells and their products have shown limited success in preventing clinical graft rejection (6). This is likely due to the fact that most clinical graft rejection occurs as the result of late-term graft rejection and not acute graft rejection. At present there is a disturbing lack of experimental models to study late-term or chronic corneal allograft rejection. In this report we describe a murine model for late-term rejection that approximates some of the features of clinical late-term corneal allograft rejection. In this model we define acute corneal allograft rejection as that which occurs within 5 weeks of engraftment (accounts for 50–70% of allorejection) and late-term rejection that which begins at least 45 days post-surgery. Our data shows that 90% of late-term rejection occurs from 45 days to sometime short after 80 days post-engraftment. Our data also demonstrates that late-term rejection has several distinct features not shared with acute allograft rejection. Namely, the magnitude of the inflammatory infiltrate is much less than is seen during acute rejection. The phenotypic makeup of the infiltrate is also different during late-term rejection as there is not the preponderance of neutrophils seen during acute rejection. Finally, the cytokine response is different both qualitatively and quantitatively. We believe these factors indicate that this murine model for late-term corneal allorejection displays some of the histological hallmarks of clinic late-term (or chronic) allorejection including interstitial fibrosis, mononuclear cell infiltration, neovascularization, and tissue destruction (5,26,28).
Results demonstrating that production of IL-17A during acute rejection appears to be associated with corneal graft survival, which is consistent with Cunnusamy, et al. (20,21). These data are not, however, consistent with the idea that IL-17A production during acute rejection is important to rejection (17,18). The underlying difference between these different results is not known since the same strain combination was used for all of these studies. That said, while we and others typically see 50 to 70% rejection during the acute rejection period (20,21), the reports demonstrating IL-17A as destructive during acute rejection report 100% rejection (17,18). We do not know why they see universal rejection but it could be due to different suppliers of animals.
Using the late term model of corneal allograft rejection we investigated the role that IL-17 and/or IFN-γ plays. Unlike what is seen when these cytokines are measured in corneas undergoing acute rejection where IFN-γ is associated with rejecting grafts and IL-17 is not, we observe that peak levels of corneal IL-17A and not IFN-γ are most associated with late-term rejection. This observation suggested that enhanced production of IL-17A is likely important for late-term corneal allograft rejection. In addition, we also observed a biphasic peak in IL-6 levels. The first appeared shortly following corneal engraftment and was not followed by a significant peak in IL-17A production. The second peak occurred at around Day 40 post-engraftment and was followed shortly thereafter by the peak production of IL-17A. IL-6 which is known to have pro-inflammatory activity (24,29), is also important in the development of IL-17A producing cells, particularly CD4+ Th17 cells (30). Thus it seemed likely that IL-6 might be involved in the transition to an environment where IL-17A could directly affect the viability of corneal allografts. In this scenario IL-17A would, at least in part, be responsible for mediating late-term rejection. This is supported by the fact that recent publications have indicated that IL-17A is involved in angiogenesis following transplantation, likely due its ability to induce multiple proinflammatory cytokines from a wide range of cell types including those of haemopoietic, endothelial, epithelial and mesenchymal lineages (24,27,32,33). Thus it was reasonable to think that IL-17A might mediate corneal allograft angiogenesis based on data that the peak of IL-17A expression was coincident with onset of allograft infiltration and angiogenesis.
When we tested this hypothesis by targeting IL-17A with a neutralizing antibody at a time shortly after the second peak in IL-6 production corneal allografts displayed significantly greater survival than those treated with isotype control antibody. These findings raised the possibility that late-term rejection, unlike acute rejection which is mediated by Th1 cells, is primarily the result of a response mediated by the production of IL-17A. Interestingly, previous studies have shown that the Th17/IL-17 pathway is involved in mediating chronic inflammatory diseases, such as rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis, psoriasis, and experiment models of uveitis (34,35). The role that these cells play in chronic allorejection of solid organs is the subject of several recent studies (26,27,36,37). These studies have demonstrated that Th17 cells play an important role in responses to kidney grafts (26), lung grafts (27), graft vs. host disease (38), and also minor histocompatibility antigen disparate allograft rejection (39). In fact, one such study determined that the magnitude of Th17 infiltrating cells was correlative to the kinetics of chronic rejection seen in 11 samples of human renal grafts, and further point out that Th17 cells could hasten graft destruction by promoting lymph angiogenesis (26). In addition to lymph angiogenesis, it has been reported that IL-17A regulates corneal vacular endothelial growth factor (VEGF-A) expression after infection with herpes simplex. That targeting IL-17A results in significantly reduced production of VEGF, and reduced neovascularization (15). Consequently, we support the notion that IL-17 is likely promoting neovascularization during late term rejection by inducing the production of VEGF-A.
We next addressed the question as to whether targeting IL-17A therapeutically would also be beneficial. Consequently, we treated mice with neutralizing antibody at a point when the corneas were displaying early and moderate rejection as determined by grafts opacity and neovascularization scores. To our surprise, not only did the corneas not progress to complete rejection of their corneal allografts, but the opacity and neovascularization for anti-IL17A treated mice almost disappeared. We had anticipated that such treatment might stabilize the cornea such that it did not become more inflamed, but were surprised by the observation that these corneas improved and did not demonstrate signs of rejection as long as the antibody treatment was maintained.
It is interesting to note that short-term treatment of mice therapeutically with anti-IL-17A does not result in permanent acceptance of corneal allografts. When mice are treated therapeutically for 3 weeks and treatment is discontinued, opacity and neovascularization began again by two weeks and the allograft went on to full rejection (data not shown). This suggests that cells producing IL-17A within the graft likely persist for at least 3 weeks and that treatment with anti-IL-17A is targeting only the effector function of this cytokine and is not affecting the cells producing it. We do not know if longer administration of anti-IL-17A would lead to more permanent acceptance of these corneal allografts.
We do believe that the main source of relevant IL-17A is found locally within the graft as neither spleen nor draining lymph nodes demonstrated any increases in IL-17A protein levels during late-term allorejection, nor was there an increase in cells producing IL-17A in these lymphoid organs (data not shown). These data do suggest that direct treatment of corneal allografts when they begin to display late term rejection with a combination of anti-IL-17A to neutralize this cytokine along with local targeting of the cells producing IL-17A might result in corneal grafts that survive late-term allo-rejection.
To address the cellular source of IL-17A, immunohistochemical and flow cytometric analysis was performed on allografts during the early stages of late-term rejection. The inflammatory infiltrate at that time consisted of mostly neutrophils, CD4+ T cells and F4/80+ macrophages. Data indicates that both CD4+ T cells and F4/80+ macrophages express IL-17A thus establishing that they are among the cells producing IL-17A during late term rejection. Of these two cell types, data indicates that CD4+IL17A+ outnumber those F4/80+IL-17A+ cells and are likely the more important subset. Future studies involving in vitro stimulation of these cells with B6 alloantigen will identify to what degree they express allo-specificity.
Due to preliminary data suggesting that Gr-1+ cells in the spleen expressed IL-17A, we had hypothesized that neutrophils could also be a source of IL-17A in the cornea which would be similar to what was reported in mouse kidney ischemia- reperfusion (IR) injury model, where neutrophils are the major source of IL-17A, and play a critical role in the pathogenesis after IR (40). However, we have not been able to definitively detect any cells producing IL-17A that also express neutrophilic surface markers in the corneas of mice undergoing late-term rejection. That said, the fact that some infiltrating macrophages produce IL-17A is a novel observation for ocular immune responses. Interestingly these F4/80+IL-17A+ are uniformly CD11b+ and in most cases also express CD4 (data not shown and see Fig. 11C as a representative of these CD4 expressing F4/80+IL-17A+ cells). To date very few reports have reported that macrophages can produce IL-17A (41).
Thus our data indicates that CD4+ T cells and macrophages produce IL-17A, but neutrophils likely do not. However, both immunocytochemistry and flow cytometry have indicated the presence of other CD45+ cells that produce IL-17A that are not T cells or macrophages. At first we thought that this might be NK T cells, which have been shown to be capable of producing IL-17A (42–47). However, we could not find any cells that bind the NK T cell CD1d tetramer by either immunocytochemistry or flow cytometry, suggesting that they too are not responsible for making IL-17A in late-term allorejection. This is not entirely surprising as NK T cells have been associated with survival during acute corneal allograft responses (48,49). The identity of these unidentified IL-17A producing cells will be the subject of future studies.
In summary, we are the first to establish a murine late term corneal allorejection model that approximates some features of what occurs during late term or chronic penetrating cornea transplant rejection in clinic. Using this model we demonstrate that late corneal allograft rejection, unlike acute corneal allograft rejection that is predominantly mediated by allo-specific CD4+ T cells expressing a Th1 phenotype, displays high level of IL-17, and expresses Th17 phenotype. The cellular sources producing IL-17A consist of primarily CD4+ T cells and, to a lesser extent F4/80+ macrophages, and possibly an as of yet undetermined cell. Neutralization of IL-17A was shown to be effective in preventing late cornea allograft rejection and more surprised to reverse such rejection response in mice model. However, acceptance of these corneas required continuous treatment with anti-IL-17A as withdrawal of treatment was followed by a resumption of neovascularization and rejection. Taken together, we believe this is the first report indicating that IL-17A is associated with late-term corneal allograft rejection and that this cytokine might prove to be a potential therapeutic target for treatment of clinical late term cornea transplant rejection.
Acknowledgments
This work was supported in part by NIH research grants RO1 EY12707 to P. M. Stuart and unrestricted funds from the Department of Ophthalmology, Saint Louis University.
We wish to acknowledge the technical assistance of Dr. Jiehong Pan for her assistance with immuno-fluorescent microscopy and Dr. Tracy Jo Pasieka and Chloe Potter for their technical assistance. We also wish to acknowledge Dr. John Forrester for critical reading of this manuscript.
Footnotes
The authors have no conflict of interest with this work.
References
- 1.Ing JJ, Ing HH, Nelson LR, Hodge DO, Bourne WM. Ten-year postoperative results of penetrating keratoplasty. Ophthalmol. 1998;105:1855–1865. doi: 10.1016/S0161-6420(98)91030-2. [DOI] [PubMed] [Google Scholar]
- 2.Yamagami S, Suzuki Y, Tsuru T. Risk factors for graft failure in penetrating keratoplasty. Acta Ophthalmol Scand. 1996;74:584–588. doi: 10.1111/j.1600-0420.1996.tb00740.x. [DOI] [PubMed] [Google Scholar]
- 3.Inoue K, Amano S, Oshika T, Tsuru T. Risk factors for corneal graft failure and rejection in penetrating keratoplasty. Acta Ophthalmol Scand. 2001;79:251–255. doi: 10.1034/j.1600-0420.2001.790308.x. [DOI] [PubMed] [Google Scholar]
- 4.Stuart PM, Yin XT, Plambeck S, Pan F, Ferguson TA. The role of Fas ligand as an effector molecule in corneal graft rejection. Eur J Immunol. 2005;35:2591–2597. doi: 10.1002/eji.200425934. [DOI] [PubMed] [Google Scholar]
- 5.Gong HQ, Gao H, Xie LX, Shi WY. Ultrastructure changes in chronic corneal allograft dysfunction after penetrating keratoplasty. Zhonghua Yanke Zazhi. 2007;43:307–312. [PubMed] [Google Scholar]
- 6.Shimazaki J, Den S, Omoto M, Satake Y, Shimmura S, Tsubota K. Prospective, randomized study of the efficacy of systemic cyclosporine in high-risk corneal transplantation. Am J Ophthalmol. 2011;152:33–39. doi: 10.1016/j.ajo.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 7.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 8.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ekinci NS, Alpsoy E, Karakas AA, Yilmaz SB, Yegin O. IL-17A has an important role in the acute attacks of Behçet’s disease. J Invest Dermatol. 2010;130:2136–2138. doi: 10.1038/jid.2010.114. [DOI] [PubMed] [Google Scholar]
- 10.Sieve AN, Meeks KD, Bodhankar S, Lee S, Kolls J, Simecka JW, Berg RE. A novel IL-17-dependent mechanism of cross protection: respiratory infection with mycoplasma protects against a secondary listeria infection. Eur J Immunol. 2009;39:426–438. doi: 10.1002/eji.200838726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nanjappa SG, Heninger E, Wüthrich M, Gasper DJ, Klein BS. Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLos Pathog. 2012;8(7):e1002771. doi: 10.1371/journal.ppat.1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, Sayegh MH, Ansari MJ. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205:3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itoh S, Nakae S, Axtell RC, Velotta JB, Kimura N, Kajiwara N, Iwakura Y, Saito H, Adachi H, Steinman L, Robbins RC, Fischbein MP. IL-17 contributes to the development of chronic rejection in a murine heart transplant model. J Clin Immunol. 2010;30:235–240. doi: 10.1007/s10875-009-9366-9. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Duan Y, Cheng X, Chen X, Xie W, Long H, Lin Z, Zhu B. IL-17 is associated with poor prognosis and promotes angiogenesis via stimulating VEGF production of cancer cells in colorectal carcinoma. Biochem Biophys Res Commun. 2011;407:348–354. doi: 10.1016/j.bbrc.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 15.Suryawanshi A, Veiga-Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J Immunol. 2012;188:3434–3446. doi: 10.4049/jimmunol.1102602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris JE, Zobell S, Yin XT, Zakeri H, Summers BC, Leib DA, Stuart PM. Mice with mutations in Fas and Fas Ligand demonstrate increased herpetic stromal keratitis following corneal infection with HSV-1. J Immunol. 2012;188:793–799. doi: 10.4049/jimmunol.1102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen H, Wang W, Xie H, Xu X, Wu J, Jiang Z, Zhang M, Zhou L, Zheng S. A pathogenic role of IL- 17 at the early stage of corneal allograft rejection. Transplant Immunol. 2009;21:155–161. doi: 10.1016/j.trim.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Chen XD, Zhao SY, Tang XL, Ge HY, Liu P. Neutralization of mouse interleukin-17 bioactivity inhibits corneal allograft rejection. Mol Vision. 2011;17:2148–2156. [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada J, Hamuro J, Fukushima A, Ohteki T, Terai K, Iwakura Y, Yagita H, Kinoshita S. MHC-matched corneal allograft rejection in an IFN-gamma/IL-17-independent manner in C57BL/6 mice. Invest Ophthalmol Vis Sci. 2009;50:2139–46. doi: 10.1167/iovs.08-2993. [DOI] [PubMed] [Google Scholar]
- 20.Cunnusamy K, Chen PW, Niederkorn JY. IL-17 promotes immune privilege of corneal allografts. J Immunol. 2010;185:4651–4658. doi: 10.4049/jimmunol.1001576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunnusamy K, Chen PW, Niederkorn JY. IL-17A-dependent CD4+CD25+ regulatory T cells promote immune privilege of corneal allografts. J Immunol. 2011;186:6737–6745. doi: 10.4049/jimmunol.1100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King WJ, Comer RM, Hudde T, Larkin DF, George AJ. Cytokine and chemokine expression kinetics after corneal transplantation. Transplantation. 2000;70:1225–1233. doi: 10.1097/00007890-200010270-00017. [DOI] [PubMed] [Google Scholar]
- 23.Park H, Zhao XL, Yang XXO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Qiang T, Chen D. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimoto M, Nakano M, Terabe F, Kawahata H, Ohkawara T, Han Y, Ripley B, Serada S, Nishikawa T, Kimura A, Nomura S, Kishimoto T, Naka T. The influence of Excessive IL-6 production in vivo on the development and function of Foxp3+ regulatory T cells. J Immunol. 2011;186:32–40. doi: 10.4049/jimmunol.0903314. [DOI] [PubMed] [Google Scholar]
- 25.Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked Cytokines associated with SjÖgren’s syndrome immunopathogenesis. Am J Pathol. 2009;175:1167–1177. doi: 10.2353/ajpath.2009.090319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, Caligiuri G, Graff-Dubois S, Morelon E, Thaunat O. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. 2010;184(9):5344–51. doi: 10.4049/jimmunol.0902999. [DOI] [PubMed] [Google Scholar]
- 27.Bobadilla JL, Love RB, Jankowska-Gan E, Xu Q, Haynes LD, Braun RK, Hayney MS, Munoz del Rio A, Meyer K, Greenspan DS, Torrealba J, Burlingham WJ, Wilkes DS, et al. Th-17, monokines, collagen type v, and primary graft dysfunction in lung transplantation. Am J Respir Crit Care Med. 2008;177:660–668. doi: 10.1164/rccm.200612-1901OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JC, Christie JD. Primary graft dysfunction. Proc Am Thorac Soc. 2009;6:39–46. doi: 10.1513/pats.200808-082GO. [DOI] [PubMed] [Google Scholar]
- 29.Fenton RR, Molesworth-Kenyon S, Oakes JE, Lausch RN. Linkage of IL-6 with neutrophil chemoattractant expression in virus-induced ocular inflammation. Invest Ophthalmol Vis Sci. 2002;43:737–743. [PubMed] [Google Scholar]
- 30.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, Takahashi T, Ripley B, Kimura A, Kishimoto T, Naka T. IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2008;105:9041–9046. doi: 10.1073/pnas.0802218105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cupedo T. An unexpected role for IL-17 in lymphoid organogenesis. Nat Immunol. 2011;12:590–592. doi: 10.1038/ni.2058. [DOI] [PubMed] [Google Scholar]
- 32.Pickens SR, Volin MV, Mandelin AM, 2nd, Kolls JK, Pope RM, Shahrara S. IL-17 contributes to angiogenesis in rheumatoid arthritis. J Immunol. 2010;184:3233–3241. doi: 10.4049/jimmunol.0903271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Numasaki M, Fukushi J, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood. 2003;101:2620–2627. doi: 10.1182/blood-2002-05-1461. [DOI] [PubMed] [Google Scholar]
- 34.Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M. Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev. 2008;226:87–102. doi: 10.1111/j.1600-065X.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- 35.Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–221. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakagiri T, Inoue M, Morii E, Minami M, Sawabata N, Utsumi T, Kadota Y, Ideguchi K, Tokunaga T, Okumura M. Local IL-17 production and a decrease in peripheral blood regulatory T cells in an animal model of bronchiolitis obliterans. Transplantation. 2010;89:1312–1319. doi: 10.1097/TP.0b013e3181d8ea16. [DOI] [PubMed] [Google Scholar]
- 37.Vanaudenaerde BM, De Vleeschauwer SI, Vos R, Meyts I, Bullens DM, Reynders V, Wuyts WA, Van Raemdonck DE, Dupont LJ, Verleden GM. The role of the IL23/IL17 axis in bronchiolitis obliterans syndrome after lung transplantation. Am J Transplant. 2008;8:1911–20. doi: 10.1111/j.1600-6143.2008.02321.x. [DOI] [PubMed] [Google Scholar]
- 38.Zhao XY, Xu LL, Lu SY, Huang XJ. IL-17-producing T cells contribute to acute graft-versus-host disease in patients undergoing unmanipulated blood and marrow transplantation. Eur J Immunol. 2011;41:514–526. doi: 10.1002/eji.201040793. [DOI] [PubMed] [Google Scholar]
- 39.Vokaer B, Van Rompaey N, Lemaître PH, Lhommé F, Kubjak C, Benghiat F, Iwakura Y, Petein M, Field KA, Goldman M, Le Moine A, Charbonnier LM. Critical role of regulatory T cells in Th17-mediated minor antigen-disparate rejection. J Immunol. 2010;185:3417–3425. doi: 10.4049/jimmunol.0903961. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Huang LP, Vergis AL, Ye H, Bajwa A, Narayan V, Strieter RM, Rosin DL, Okusa MD. IL-17 produced by neutrophils regulates IFN-γ mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120:331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vykhovanets EV, Maclennan GT, Vykhovanets OV, Gupta S. IL-17 expression by macrophages is associated with proliferative inflammatory atrophy lesions in prostate cancer patients. Int J Clin Exp Pathol. 2011;4:552–565. [PMC free article] [PubMed] [Google Scholar]
- 42.Liu XC, Zhai A, Li JQ, Qi HZ. Interliukin-23 promotes natural killer T-cell production of IL-17 during rat liver transplantation. Transplant Proc. 2011;43:1962–1966. doi: 10.1016/j.transproceed.2011.01.175. [DOI] [PubMed] [Google Scholar]
- 43.Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, Berzins SP, Smyth MJ, Godfrey DI. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4− Nk1.1−. Proc Natl Acad Sci USA. 2008;105:11287–11292. doi: 10.1073/pnas.0801631105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, Zhu M, Iwakura Y, Savage PB, DeKruyff RH, Shore SA, Umetsu DT. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385–393. doi: 10.1084/jem.20071507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KA, Kang MH, Lee YS, Kim YJ, Kim DH, Ko HJ, Kang CY. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell Immunol. 2008;251:50–55. doi: 10.1016/j.cellimm.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshiga YD, Goto D, Segawa S, Ohnishi Y, Matsumoto I, Ito S, Tsutsumi A, Taniguchi M, Sumida T. Invariant NKT cells produce IL-17 through IL-23-dependent and -independent pathways with potential modulation of Th17 response in collagen-induced arthritis. Int J Mol Med. 2008;22:369–374. [PubMed] [Google Scholar]
- 48.Sonoda KH, Taniguchi M, Stein-Streilein J. Long-term survival of corneal allografts is dependent on intact CD1d-reactive NKT cells. J Immunol. 2002;168:2028–2034. doi: 10.4049/jimmunol.168.4.2028. [DOI] [PubMed] [Google Scholar]
- 49.Watte CM, Nakamura T, Lau CH, Ortaldo JR, Stein-Streilein J. Ly49 C/I-dependent NKT cell-derived IL-10 is required for corneal graft survival and peripheral tolerance. J Leukoc Biol. 2008;83:928–935. doi: 10.1189/jlb.0807579. [DOI] [PubMed] [Google Scholar]