

Significance
How ATP hydrolysis is coupled to promoter DNA unwinding and open complex formation at RNA polymerase II (Poll II) promoters is a longstanding question. Of the multisubunit RNA polymerases, only Pol II requires ATP for DNA unwinding. Here we show that the general transcription factor TFIIH subunit Ssl2 is a double-stranded DNA translocase. These and other data suggest that Ssl2 promotes DNA opening by tracking along the nontemplate promoter strand, rotating and inserting DNA into the Pol II active site cleft, leading to DNA unwinding. Our accompanying biochemical studies explain why the open complex is unstable and how TFIIH can promote Pol II escape from the promoter. Our findings also have important implications for the mechanism of TFIIH-mediated DNA repair.
Keywords: transcription initiation, DNA unwinding, DNA helicase
Abstract
Formation of the RNA polymerase II (Pol II) open complex (OC) requires DNA unwinding mediated by the transcription factor TFIIH helicase-related subunit XPB/Ssl2. Because XPB/Ssl2 binds DNA downstream from the location of DNA unwinding, it cannot function using a conventional helicase mechanism. Here we show that yeast TFIIH contains an Ssl2-dependent double-stranded DNA translocase activity. Ssl2 tracks along one DNA strand in the 5′ → 3′ direction, implying it uses the nontemplate promoter strand to reel downstream DNA into the Pol II cleft, creating torsional strain and leading to DNA unwinding. Analysis of the Ssl2 and DNA-dependent ATPase activity of TFIIH suggests that Ssl2 has a processivity of approximately one DNA turn, consistent with the length of DNA unwound during transcription initiation. Our results can explain why maintaining the OC requires continuous ATP hydrolysis and the function of TFIIH in promoter escape. Our results also suggest that XPB/Ssl2 uses this translocase mechanism during DNA repair rather than physically wedging open damaged DNA.
For all multisubunit RNA polymerases (Pols), a universal step in the transcription initiation pathway is formation of the open complex (OC) (1, 2). The OC forms when Pol and its associated transcription machinery bind to promoter DNA, generating a series of conformational changes in both DNA and protein, including the unwinding of ∼11 bp of DNA upstream from the transcription start site (TSS). This open state is stabilized by interactions of Pol with the unwound strands of promoter DNA and by the binding of downstream double-stranded promoter DNA to the Pol Cleft/Jaw domains (3–5). All tested multisubunit Pols spontaneously form OCs, except for RNA Pol II, where ATP and the general transcription factor TFIIH are required for DNA unwinding (6–8). For Pol II, this unwound state is unstable, decaying with a half-life of 30–60 s (8, 9).
The general transcription factor TFIIH contains two subunits with DNA-dependent ATPase activity, XPD/Rad3 and XPB/Ssl2 (human/yeast proteins), which are members of the SF2 helicase-translocase family (10). The XPB/Ssl2 ATPase is required for DNA unwinding in the OC, whereas the XPD/Rad3 ATPase activity does not function in transcription (11–14). In DNA strand displacement assays, the two isolated human subunits have DNA helicase activity of opposite polarity with XPD having 5′ → 3′ activity and XPB having 3′ → 5′ activity. XPD is at least eightfold more active than XPB in strand displacement (15). In contrast to the activity of purified XPB, the most purified preparations of native or recombinant human TFIIH have only the XPD 5′ → 3′ helicase activity, with no detectable 3′ → 5′ helicase function (14, 15). In addition to its role in transcription initiation, TFIIH can assist in Pol II promoter escape in an XPB-dependent mechanism (16, 17), and both the XPB and XPD subunits of TFIIH play an essential role in general and transcription-coupled nucleotide excision repair (NER) (18, 19).
Mapping the location of XPB/Ssl2 in RNA Pol II preinitiation complexes (PICs) revealed that this factor binds promoter DNA downstream from the site of DNA unwinding in the OC (20–24). Therefore, XPB/Ssl2 cannot function as a conventional helicase to promote OC formation (20). Three models have been proposed to explain the role of XPB/Ssl2 in transcription. First, it was postulated that XPB acts as a molecular wrench, binding to its site on downstream DNA and using its ATPase to rotate upstream DNA within the Pol II cleft (20). Because upstream DNA is constrained by TBP, TFIIB, and other factors, DNA rotation could lead to DNA opening. Second, it was proposed that the XPB ATPase activity promoted DNA opening via a conformational change in PIC components, leading to a structural rearrangement of both protein and DNA, analogous to the ATP-dependent mechanism of OC formation in the bacterial σ54 system (25). Third, comparing structure models of the PIC and OC suggested that ∼15 bp of downstream promoter DNA inserts into the Pol II cleft upon OC formation (22). Based on this and other data, it was proposed that XPB/Ssl2 functions as a double-stranded DNA (dsDNA) translocase (22, 23, 26). According to this model, XPB/Ssl2 attempts to track away from the PIC, but, because it is bound to the PIC via TFIIH, it instead feeds and rotates dsDNA into the Pol II cleft, leading to DNA opening. However, a key feature of this latter model, whether XPB/Ssl2 has dsDNA translocase function, had not been tested.
Here we show that yeast TFIIH has Ssl2-dependent dsDNA translocase activity and that it primarily tracks along one DNA strand in the 5′ → 3′ direction. The kinetic properties of the enzyme suggest a rate-limiting step between DNA binding and translocation and a processivity on the order of a turn of DNA, consistent with the amount of DNA thought to be unwound in the OC. Our findings have important implications for the mechanism of OC formation, the relative instability of the OC, and the role of XPB/Ssl2 in NER.
Results
TFIIH Has Ssl2-Dependent dsDNA Translocase Activity.
We first measured DNA helicase activity of purified yeast TFIIH using a substrate consisting of a 100-base oligonucleotide with a labeled 25-base oligonucleotide annealed at either the 3′ or 5′ end (Fig. 1A). Consistent with the function of human TFIIH, we found that yeast TFIIH contains 5′ → 3′ helicase but not 3′ → 5′ helicase function (Fig. 1B). The 5′ → 3′ helicase activity requires both hydrolysable ATP and the Rad3 ATPase, because a TFIIH derivative containing a mutation in the Rad3 Walker B motif (Rad3 E236Q) that is predicted to be defective in ATP hydrolysis (27) is defective in helicase function. In contrast, both WT and Rad3 mutant TFIIH preparations are equally active in supporting transcription with purified factors and Pol II from the yeast HIS4 promoter (Fig. S1).
Fig. 1.

ATP-dependent helicase and translocase activities of TFIIH. (A) Helicase and triple helix substrates. Helicase substrates are designed to distinguish 5′ → 3′ and 3′ → 5′ enzyme polarity. The short displaced oligonucleotide is fluorescently labeled for visualization. The triplex substrate is formed via Hoogsteen base pairs between the 22 nucleotide (nt) 32P-labeled triplex forming oligo (TFO) and the purine rich strand of the PCR generated duplex. (B) Helicase assay testing ATP-dependence and polarity of WT TFIIH or TFIIH containing the ATPase mutant Rad3-E236Q. (C) Triplex disruption assay testing ATPase requirements for triplex displacement with either WT TFIIH or TFIIH containing the ATPase defective mutants Rad3-E236Q or Ssl2-E489Q.
As an initial test for dsDNA translocase function, we assayed whether TFIIH can displace a 22 nucleotide DNA triplex, a well-established assay for dsDNA translocase function (28–30). The DNA triplex was formed by annealing a polypyrimidine triplex-forming oligonucleotide (TFO) to a 142-bp dsDNA containing a complementary polypurine sequence at one end (Fig. 1A). The triplex is disrupted by heating, but is stable at 26 °C with or without the addition of ATP (Fig. 1C, lanes 1–3). Incubation of WT TFIIH, ATP, and the triplex substrate results in near complete dissociation of the TFO (Fig. 1C, lanes 4–6). The triplex displacement activity of TFIIH is unaffected by the Rad3 E236Q ATPase mutation but is abolished by the equivalent Walker B ATPase mutation in Ssl2 (E489Q; Fig. 1C, lanes 7–11). To assay whether TFIIH can simply bind and displace the triplex without translocation, we used a 22 nucleotide triplex substrate that did not contain additional dsDNA (Fig. 2A). The short triplex substrate is less stable than the longer triplex, likely due to the lack of stabilizing dsDNA. Nevertheless, as predicted for a dsDNA translocase, TFIIH cannot displace the TFO from this shorter substrate lacking dsDNA (Fig. 2B). The TFIIH triplex displacement activity was not dependent on a free DNA end, because TFIIH can also displace the TFO annealed to a 3.2-kb double-stranded plasmid, although with much slower kinetics (Fig. 2C). Both ATP and dATP function to promote OC formation (7) and, as expected, both nucleotides can promote triplex displacement (compare Fig. 1C and Fig. 2C, Right). Our combined results are consistent with the prediction that TFIIH contains an Ssl2-dependent dsDNA translocase activity.
Fig. 2.

Template requirements for triplex disruption by TFIIH. (A) Triple helix substrates used to monitor TFIIH translocation. TFO22 was generated by annealing the 22-nt TFO to the 22-bp triplex target sequence. This smaller triplex was somewhat less stable than the 142-bp triplex DNA, likely due to no additional dsDNA. To make the 142-bp and circular triplex templates, the TFO was annealed to a PCR-generated 142-bp duplex or a 3.2-kb plasmid containing the triplex target sequence. (B) Triplex disruption assay using the TFO22 triple helix template and the TFIIH derivative Rad3 E236Q. (C) Time course of triplex disruption comparing the circular 3.2-kb plasmid triplex and the linear 142-bp triplex templates. Intact triplex was measured at each time point and quantified by comparison with a standard curve. The percent disruption of each triplex is indicated. ATP and dATP both function in the TFO displacement assay (compare with Fig. 1C) and in OC formation (7).
Ssl2 Tracks in the 5′ → 3′ Direction Along the DNA Duplex.
dsDNA translocases are thought to track along one strand of the DNA backbone (31). If true for Ssl2, discerning the polarity of translocation will reveal which promoter strand is used by Ssl2 during DNA unwinding. As an initial test of polarity, we assayed triplex displacement using substrates where the TFO was annealed to either the top or bottom strand of the 142-bp duplex DNA (Fig. 3A). As shown above, TFIIH can readily displace the TFO from the bottom strand triplex substrate (Fig. 3B, lanes 4–6). In contrast, the top strand triplex substrate is almost completely resistant to TFO displacement (Fig. 3B, lanes 1–3). One model consistent with this result is that Ssl2 tracks along one strand of duplex DNA in the 5′ → 3′ direction and that this tracking is blocked by the annealed TFO. As a further test of this model, we introduced biotin as a tracking barrier on the triplex substrates (Fig. 3A). These substrates contain a single-strand DNA nick with biotin attached to the 5′ end of one strand of duplex DNA. We found that the top-strand biotin was a much stronger block to TFO displacement, with only one third of the TFO displaced after 6 h (Fig. 3 C and D). Although the bottom-strand biotin modestly inhibited TFO displacement, the TFO was >80% displaced after 6 h, similar to the nonbiotinylated template. As a further test of whether the integrity of the top strand is the most critical for TFO displacement, we generated triplex substrates with 5-bp single-stranded gaps on either the top or bottom duplex strand, directly adjacent to the triplex (Fig. S2). Consistent with the above results, we found that TFO displacement was most inhibited by the gap on the top strand. Under conditions of the assay, 80–90% of the TFO was displaced from the nongapped template, 45% displaced from the template with the gap on the bottom strand, and only 20–25% displaced from the template with the gap on the top strand. Our combined results strongly suggest that Ssl2 primarily tracks along one strand of duplex DNA in the 5′ → 3′ direction.
Fig. 3.

Polarity of Ssl2 catalyzed TFIIH translocation. (A) Triple helix substrates designed to test the polarity of Ssl2 translocation. Top and bottom strand triplexes are annealed as shown using PCR-generated duplex DNA and the TFO. PAGE-purified oligonucleotides were annealed with the TFO to generate the biotin-containing triplexes where a 5′-biotin labeled oligonucleotide is positioned immediately upstream of the triplex on the top or bottom duplex strand. (B) Triplex disruption assay comparing templates with the TFO annealed to either the top or bottom strand of the duplex. Either WT or Rad3 E236Q TFIIH was used as indicated. (C) Time course of triplex disruption from biotin templates by TFIIH (Rad3-E236Q). Reactions were incubated for the indicated times with 1 mM dATP at 26 °C and quantified for intact triplex remaining. (D) Quantitation of results in C.
Blocking Ssl2 Translocation Inhibits Transcription Initiation.
The above results predict that blocking Ssl2 translocation should inhibit transcription initiation. Mapping the location of Ssl2/XPB-DNA binding in PICs has suggested Ssl2 binds 30–36 (yeast) and 40–50 bp (human) downstream from TATA (20–23). To test the role of Ssl2 translocase function in transcription initiation, we created three promoter derivatives with Cy3 dye inserted in the phosphodiester backbone of the nontemplate DNA strand 37, 41, or 46 bp downstream from the HIS4 TATA (Fig. 4A). Cy3 positioned in the DNA backbone is a strong inhibitor of Ssl2 translocation in the TFO displacement assay (Fig. S3).
Fig. 4.

In vitro transcription from promoters with DNA backbone blocks on the nontemplate DNA strand. (A) HIS4 promoter derivatives with Cy3 DNA backbone insertions. DNAs were constructed from synthetic oligonucleotides and contained Cy3 positioned 37, 41, or 46 bp downstream from HIS4 TATA. Transcription was assayed by primer extension using the lacI oligonucleotide as shown. (B) In vitro transcription using the reconstituted yeast Pol II system and the promoters in A. Lanes 1–4 contain the indicated amounts of a transcription reaction used to generate a standard for quantitation of transcription signals relative to the unmodified template. Lanes 5–7 are transcription reactions using the indicated Cy3 templates. Percent transcription relative to the unblocked template is indicated. No transcription is observed when TFIIH is omitted (lanes 8–11). (C) HIS4 promoter derivatives identical to those in A except that they contain the 12 nucleotide single-stranded DNA bubble as shown. This bubble allows transcription initiation by Pol II in the absence of other general factors (33). (D) In vitro transcription using purified yeast Pol II on the bubble templates and assayed by primer extension. Lanes 1–4 contain the indicated amounts of a transcription reaction using the non-Cy3 bubble template. Lanes 5–7 are transcription reactions using the Cy3-modified bubble templates. Lanes 8–9 are mock transcription reactions lacking Pol II and assayed by primer extension. The products marked by * are due to primer extension of the DNA template, which is blocked by Cy3. These blocked products are not visible in B, lanes 9–11, as they are significantly longer than the RNA products initiated at the HIS4 TSS.
The modified and unmodified DNA templates were tested for in vitro transcription activity using the reconstituted system (32), and transcription from these templates was completely TFIIH dependent (Fig. 4B, compare lanes 4–7 and 8–11). We found that Cy3 positioned on the nontemplate strand 41 and 46 bp from TATA inhibited transcription approximately fivefold (Fig. 4B, lanes 4, 6, and 7), consistent with the dsDNA translocase model. Cy3 positioned 37 bp downstream from TATA inhibited transcription approximately twofold (Fig. 4B, lanes 4–5). We speculate that transcription escaping this more upstream Cy3 insertion may be due to inherent flexibility in the Ssl2–DNA interaction, allowing Ssl2 to escape the Cy3 block by binding DNA just downstream of Cy3 37 in a position nearly equivalent to the observed XPB-human promoter interaction (23).
To confirm that the observed transcription inhibition by Cy3 could not be explained by an inhibition of transcription elongation, we created HIS4 promoter derivatives containing a 12 nucleotide single-strand bubble beginning 21 bp downstream from TATA (Fig. 4C). Pol II initiating from this bubble in the absence of any general factors transcribed past the Cy3 block with 47–77% efficiency compared with a bubble template lacking Cy3 (Fig. 4D, lanes 4–7). Taken together, our results show that the integrity of the nontemplate strand is important for transcription initiation and is consistent with the dsDNA translocase model for open complex formation.
Ssl2 Translocates with Low Processivity.
To further characterize the Ssl2 motor within the TFIIH complex, we next investigated stimulation of the ATPase by nucleic acid. Measurements of how the steady-state rate of ATP hydrolysis depends on DNA template length and concentration can be used to assay for translocation and to evaluate different models of translocation (33–35). For these measurements, we used the TFIIH preparation containing Rad3 E236Q so that Ssl2 was the only functional ATPase. TFIIH and DNA were preincubated for 40 min, and then ATP was added, and phosphate release was quantitated at different times (1–20 min). The steady-state rate of ATP hydrolysis was determined by a linear fit of these data (Fig. 5A). ATP hydrolysis in the absence of nucleic acid was undetectable, and dsDNA consistently stimulated ATPase activity four- to fivefold higher than that of single-stranded DNA (Table 1), consistent with the enzyme being a dsDNA translocase.
Fig. 5.

Dependence of the ATP hydrolysis rate on DNA length. In all reactions, TFIIH (Rad3 E236Q) was preincubated with DNA for 40 min before ATP addition. (A) The phosphate released (Pi) as a function of time for a range of DNA lengths shows a linear dependence that can be fit to give a steady-state rate of ATP hydrolysis. (B) The steady-state rate of ATPase hydrolysis as a function of DNA length and DNA concentration. Concentration is plotted as micromolar-base pairs (µMbp), and the data are fit to Michaelis–Menten curves, allowing for the extraction of KM and Vmax parameters. The Vmax of the plasmid data and its associated error are shown by two dashed horizontal lines. (C) KM as a function of DNA length. (D) Vmax as a function of DNA length. Fit shown using analytical expression for the dependence of Vmax on DNA length for the model described in the text with a step size of 1 bp and a processivity of 10 bp (SI Materials and Methods).
Table 1.
Vmax of the Ssl2 ATPase with single- or double-stranded DNA
| DNA | Length [bp (ds) or nt (ss)] | Vmax (ATP/Ssl2 s) |
| ds | 30 | 5.19 |
| ds | 40 | 6.41 |
| ds | 60 | 8.82 |
| ds | 80 | 9.49 |
| ss | 30 | 1.03 |
| ss | 40 | 1.20 |
| ss | 60 | 2.46 |
| ss | 80 | 2.58 |
Reactions with single-stranded DNA were carried out as described in Materials and Methods and contained 12.5 μM nt of each template. The average Vmax values from two independent experiments are given. For comparison, the predicted Vmax from an analogous ds DNA template is shown (data from Fig. 4D).
The steady-state rate of ATP hydrolysis was measured as a function of DNA concentration and DNA template length over a range of DNA lengths from 30 to 80 bp and on a circular plasmid representing an infinitely long template (Fig. 5 A and B). As the dependence of rate on DNA concentration follows a Michaelis–Menten curve, we were able to determine both a Michaelis constant (KM) and a maximal velocity (Vmax) for each length of DNA (Fig. 5B). Although KM does not show a length dependence (Fig. 5C), Vmax is clearly sensitive to DNA length (Fig. 5D). The dependence of Vmax on template length is a clear indication that Ssl2 is a dsDNA translocase as nontranslocating enzymes do not show this behavior (33–35) (Fig. S4A).
Models of translocation can be used to fit the dependence of Vmax on DNA length to extract quantitative values for parameters such as occluded site size, kinetic step size, and motor processivity, i.e., the probability that the motor takes a step forward instead of dissociating (Materials and Methods) (33–35). In the simplest model for translocation, the motor binds DNA at any site and steps forward coupled to ATP hydrolysis. At each site, the motor may either hydrolyze ATP and take a step forward or dissociate (Fig. S4B). At the end of the template, the motor either dissociates or translocates off the end of the DNA. This model predicts that KM will vary with the length of the DNA template, whereas Vmax will not (Fig. S4B). This behavior is inconsistent with our observed results (Fig. 5 C and D).
However, in contrast to the simple translocation model above, models that incorporate a slow kinetic step between unbound and the active DNA-bound motor states result in a length-dependent Vmax and a length-independent KM (33–35). Two limiting cases of this behavior are (i) a slow step after binding and before translocation or (ii) a slow step before dissociation from the end of the DNA (Fig. S4C). These two cases predict different pre–steady-state behavior in the ATPase rate (34). A slow step at the end of the DNA predicts a burst of rapid ATP hydrolysis on ATP addition compared with the steady-state rate. This burst phase arises because initial rounds of hydrolysis do not encounter the rate-limiting step that takes place at the DNA end. In contrast, a slow step only between DNA binding and translocation predicts a lag in ATP hydrolysis rate. This lag phase arises because the rate-limiting step occurs before translocation and before ATP hydrolysis occurs. Our data are consistent with the latter case, as there is a lag in ATP hydrolysis after adding ATP to TFIIH prebound to DNA (Fig. 5A). The observed lag suggests that Ssl2, in complex with TFIIH, first binds DNA in an inactive conformation and then, after ATP addition, isomerizes to the active state before translocation.
To extract quantitative measures of translocase parameters, we analyzed the DNA length dependence of the Ssl2 ATPase Vmax with an expression derived from the isomerization model (SI Materials and Methods) (34). Assuming a translocation step size of 1 bp and a contact size on the DNA of 16 bp (Table S1 and Materials and Methods), we determined a processivity of Ssl2 in the context of TFIIH of 0.90 with a fit error of 0.01 corresponding to an average of 10.0 ± 0.9 bp translocated before dissociation from the DNA. Because we have no independent measure of step size, we note that a larger step size would lead to a lower processivity. For example, analyzing the same data with a step size of 2 bp would correspond to 5.3 ± 0.3 bp translocated before dissociation from the DNA.
Discussion
Our results on the translocase activity of Ssl2 in the context of the TFIIH complex are completely consistent with the translocase model of OC formation and explain how TFIIH might generate torsional stress in promoter DNA, leading to unwinding. Our finding that Ssl2 tracks in the 5′ → 3′ direction implies that Ssl2 uses the nontemplate strand of promoter DNA as it attempts to track away from the PIC but instead reels downstream dsDNA toward the PIC, creating open DNA via torsional or mechanical stress that is then fed into the Pol II cleft (Fig. 6). Interestingly, this is the opposite direction to the characterized helicase activity of the Ssl2 subunit alone, suggesting that the enzyme does not use the same motor mechanism for helicase and dsDNA translocase activities. Our conclusion that the processivity of Ssl2 translocation is similar to the amount of DNA unwound in the OC can explain why the Pol II OC is unstable. We speculate that DNA opening requires continuous ATP hydrolysis because Ssl2 likely dissociates from the DNA after translocating a short distance (while remaining in complex with TFIIH and the PIC), resetting the initiation complex to its closed state. In contrast to the situation with other prokaryotic and eukaryotic RNA Pols, it seems likely that the contacts between Pol II and unwound DNA are not strong enough to stabilize the fully unwound state in the absence of continual Ssl2 translocation activity.
Fig. 6.
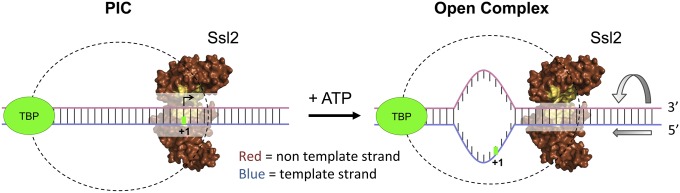
dsDNA translocase model for open complex formation. The Ssl2 subunit of TFIIH tracks in the 5′ → 3′ direction on the nontemplate promoter DNA strand (red). Because TFIIH movement is constrained due to interaction with other PIC components, translocation results in insertion and rotation of promoter DNA into the Pol II cleft, leading to DNA unwinding (right arrows indicate rotation and direction of dsDNA movement). The short persistence length of Ssl2/TFIIH predicts that the OC state is unstable, in agreement with experimental observations (9, 10).
This mechanism predicts multiple cycles of DNA opening and closing during the initial stages of transcription. It was found that the Pol II OC is stabilized by a four-nucleotide RNA (8), so this may be the minimum length of transcript to prevent reversion to the PIC dsDNA state. However, translocase function may be necessary to assist in DNA opening until a 7- to 8-base RNA:DNA hybrid is formed. This model can explain the observation that TFIIH stimulates promoter escape from templates with a short stretch of premelted DNA from −9 to −1 with respect to the TSS (17).
Finally, our results have important implications for the action of XPB during NER. In the general genome repair pathway, TFIIH is recruited to DNA lesions bound by factor XPC, where it opens an asymmetric ∼27-bp DNA bubble surrounding the lesion (18, 19). Both XPB and XPD are required for this DNA unwinding, although XPB does not seem to promote unwinding via a conventional helicase mechanism. Mutations that abolish the XPB ATPase abolish DNA unwinding activity during NER, but two XPB mutations with reduced 3′ → 5′ helicase function were reportedly active for NER (13). Based on these results, it was proposed that XPB functions indirectly in DNA opening by using its ATPase function to promote a conformational change in the XPC–DNA–TFIIH complex, physically wedging open 5 bp of the DNA duplex, and positioning the XPD helicase to open the DNA surrounding the DNA lesion (10, 18, 19). Given our results, it seems more likely that XPB opens 5 bp of DNA using a dsDNA translocase mechanism similar to that in OC formation. By generating torsional strain in rotationally fixed damaged DNA, the XPB subunit of TFIIH can lead to initial unwinding, generating a substrate for XPD to generate the fully opened 27-bp asymmetric bubble surrounding DNA lesions.
Materials and Methods
DNA Helicase Assay.
TFIIH helicase activity was monitored using a fluorescent dye-labeled oligonucleotide (IR700-TTCACCAGTGAGACGGGCAACAGCC) annealed to PAGE-purified 100-base oligonucleotides, with the resulting templates having 5′ or 3′ overhangs of 75 bases (Fig. 1). The assay was performed as previously described (36), with the following modifications: 10-μL reactions contained 10 mM Hepes (pH 7.6), 100 mM potassium glutamate, 10 mM magnesium acetate, 3.5% (vol/vol) glycerol, 1 mM DTT, 1 μg BSA, 60 fmol holo-TFIIH, and 30 fmol template DNA. ATP or ATPγS was added to 1 mM, and reactions were incubated 1–2 h at 26 °C. Controls received TE buffer (10 mM Tris, pH 7.5, 1 mM EDTA) in lieu of ATP or were heated to 95 °C for 30 s. Reactions were quenched by the addition of 10 μL 33% glycerol, 40 mM EDTA, and 0.5% SDS and analyzed by PAGE using 10% acrylamide gels in 0.5× TBE buffer (134 mM Tris, 44 mM boric acid, 2.5 mM EDTA, pH 8.8) plus 0.1% SDS. Following electrophoresis, gels were visualized using an Odyssey IR scanner (LI-COR).
In Vitro Transcription Assay.
In vitro transcription using recombinant and purified factors was performed similarly to previously described assays (32). See SI Materials and Methods for additional information.
TFIIH Purification.
WT TFIIH was purified from strain SHY869 (RAD3-(HA)1-TAP tag, tfb6Δ) as previously described (32) except that, following the ultracentrifugation step, potassium acetate was added to the extract to a final concentration of 0.6 M before binding to IgG-Sepharose (GE Healthcare). TFIIH with ATPase-defective Rad3 was purified by the same method from strain SHY887 (leu2Δ rad3Δ::KanMX, tfb6Δ::HPH) carrying plasmid pJF82 [ars cen LEU2 RAD3 (E236Q)-(HA)1-TAP tag]. Because the Ssl2 E489Q mutation is lethal, this TFIIH derivative was purified from a WT strain containing the Tap-tagged Ssl2 mutation on a plasmid. Strain SHY861 (leu2Δ SSL2) carrying plasmid pJF62 [ars cen LEU2 ssl2 (E489Q)-(FLAG)1-TAP tag] was grown in glucose complete media lacking leucine, and TFIIH was purified by the method described above.
Triplex Disruption Assay.
Triplex DNA template formation and disruption reactions were performed as previously described (28), with the following modifications: templates were assembled from duplex DNA containing a triplex target sequence (AAGAAAAGAAAGAAGAAAGAAA) and a fluorescent or 32P-labeled TFO (TTCTTTTCTTTCTTCTTTCTTT). The DNA and TFO were combined at 1 μM final concentration in 25 mM Mes (pH 5.5) and 10 mM MgCl2 and heated to 57 °C for 15 min and then cooled at 1 °C/min over 35 min to allow annealing. Triplex DNA was stored at -20 °C and diluted in 50 mM Tris⋅HCl, pH 8.0, 10 mM MgCl2, and 1 mM DTT before the assay. Ten-microliter reactions contained 10 mM Hepes (pH 7.6), 100 mM potassium glutamate, 10 mM magnesium acetate, 3.5% glycerol, 1 mM DTT, 1 μg BSA, 0.01% Nonidet P-40, 15 fmol holo-TFIIH, and 30 fmol triplex DNA. ATP or dATP was added to 1 mM, and reactions were incubated at 26 °C for the indicated times before stopping with 2.5 μL 5× GSMB (15% glucose, 3% SDS, 250 mM Mops, pH 5.5, and 0.04% bromophenol blue). The reactions were analyzed by PAGE using 6% acrylamide gels with buffer: 40 mM Tris-acetate (pH 5.5), 5 mM sodium acetate, and 1 mM magnesium chloride. Gels were visualized using either an Odyssey IR scanner (LI-COR) or dried and visualized by PhosphorImager (Molecular Dynamics).
ATPase Assay.
DNA-dependent ATPase activity of TFIIH (Rad3-E236Q) was measured using a colorimetric assay kit (Innova; 601-0120). Forty-microliter reactions contained 10 mM Hepes (pH 7.6), 100 mM potassium glutamate, 10 mM magnesium acetate, 3.5% glycerol, 1 mM DTT, 4 μg BSA, 40 fmol holo-TFIIH, and 1.25 nM to 1.5 μM template DNA. After 40 min at room temperature, purified ATP was added to 0.5 mM, and reactions were incubated 1–20 min at 26 °C. Reactions were stopped by the addition of 10 μL gold mix and, after 4 min, 4 μL stabilizer 2. After 30 min at room temperature, absorbance was measured at 635 nm and plotted against DNA concentration. A standard curve was established for every experiment using the kit-included phosphate standard and used to determine TFIIH catalyzed ATP hydrolysis. Templates from 30 to 80 bp were tested at nine concentrations in triplicate, spanning a 1,200-fold range of DNA concentration. See SI Materials and Methods for information on extracting translocation kinetic parameters from the ATPase data.
Supplementary Material
Acknowledgments
We thank members of the S.H. laboratory and Ted Young for comments and suggestions during the course of this work and S. Grünberg for Fig. 6 and comments on the manuscript. This work was supported by National Institute of General Medical Sciences Grant 2RO1GM053451 (to S.H.) and National Science Foundation-Molecular and Cellular Biosciences Grant 1243918 (to E.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417709112/-/DCSupplemental.
References
- 1.Saecker RM, Record MT, Jr, Dehaseth PL. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J Mol Biol. 2011;412(5):754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grünberg S, Hahn S. Structural insights into transcription initiation by RNA polymerase II. Trends Biochem Sci. 2013;38(12):603–611. doi: 10.1016/j.tibs.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung ACM, Sainsbury S, Cramer P. Structural basis of initial RNA polymerase II transcription. EMBO J. 2011;30(23):4755–4763. doi: 10.1038/emboj.2011.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sainsbury S, Niesser J, Cramer P. Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature. 2013;493(7432):437–440. doi: 10.1038/nature11715. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, et al. Structural basis of transcription initiation. Science. 2012;338(6110):1076–1080. doi: 10.1126/science.1227786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunick D, Zandomeni R, Ackerman S, Weinmann R. Mechanism of RNA polymerase II—specific initiation of transcription in vitro: ATP requirement and uncapped runoff transcripts. Cell. 1982;29(3):877–886. doi: 10.1016/0092-8674(82)90449-4. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Carey M, Gralla JD. Polymerase II promoter activation: Closed complex formation and ATP-driven start site opening. Science. 1992;255(5043):450–453. doi: 10.1126/science.1310361. [DOI] [PubMed] [Google Scholar]
- 8.Holstege FC, Fiedler U, Timmers HT. Three transitions in the RNA polymerase II transcription complex during initiation. EMBO J. 1997;16(24):7468–7480. doi: 10.1093/emboj/16.24.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dvir A, et al. A role for ATP and TFIIH in activation of the RNA polymerase II preinitiation complex prior to transcription initiation. J Biol Chem. 1996;271(13):7245–7248. doi: 10.1074/jbc.271.13.7245. [DOI] [PubMed] [Google Scholar]
- 10.Compe E, Egly J-M. TFIIH: When transcription met DNA repair. Nat Rev Mol Cell Biol. 2012;13(6):343–354. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- 11.Feaver WJ, et al. Dual roles of a multiprotein complex from S. cerevisiae in transcription and DNA repair. Cell. 1993;75(7):1379–1387. doi: 10.1016/0092-8674(93)90624-y. [DOI] [PubMed] [Google Scholar]
- 12.Guzder SN, et al. DNA repair gene RAD3 of S. cerevisiae is essential for transcription by RNA polymerase II. Nature. 1994;367(6458):91–94. doi: 10.1038/367091a0. [DOI] [PubMed] [Google Scholar]
- 13.Oksenych V, Bernardes de Jesus B, Zhovmer A, Egly J-M, Coin F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009;28(19):2971–2980. doi: 10.1038/emboj.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tirode F, Busso D, Coin F, Egly JM. Reconstitution of the transcription factor TFIIH: Assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol Cell. 1999;3(1):87–95. doi: 10.1016/s1097-2765(00)80177-x. [DOI] [PubMed] [Google Scholar]
- 15.Coin F, et al. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Genet. 1998;20(2):184–188. doi: 10.1038/2491. [DOI] [PubMed] [Google Scholar]
- 16.Dvir A, Conaway RC, Conaway JW. A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc Natl Acad Sci USA. 1997;94(17):9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moreland RJ, et al. A role for the TFIIH XPB DNA helicase in promoter escape by RNA polymerase II. J Biol Chem. 1999;274(32):22127–22130. doi: 10.1074/jbc.274.32.22127. [DOI] [PubMed] [Google Scholar]
- 18.Egly J-M, Coin F. A history of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10(7):714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 19.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10(7):697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim TK, Ebright RH, Reinberg D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science. 2000;288(5470):1418–1422. doi: 10.1126/science.288.5470.1418. [DOI] [PubMed] [Google Scholar]
- 21.Miller G, Hahn S. A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat Struct Mol Biol. 2006;13(7):603–610. doi: 10.1038/nsmb1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grünberg S, Warfield L, Hahn S. Architecture of the RNA polymerase II preinitiation complex and mechanism of ATP-dependent promoter opening. Nat Struct Mol Biol. 2012;19(8):788–796. doi: 10.1038/nsmb.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495(7442):481–486. doi: 10.1038/nature11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murakami K, et al. Architecture of an RNA polymerase II transcription pre-initiation complex. Science. 2013;342(6159):1238724. doi: 10.1126/science.1238724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin YC, Choi WS, Gralla JD. TFIIH XPB mutants suggest a unified bacterial-like mechanism for promoter opening but not escape. Nat Struct Mol Biol. 2005;12(7):603–607. doi: 10.1038/nsmb949. [DOI] [PubMed] [Google Scholar]
- 26.Goel S, Krishnamurthy S, Hampsey M. Mechanism of start site selection by RNA polymerase II: Interplay between TFIIB and Ssl2/XPB helicase subunit of TFIIH. J Biol Chem. 2012;287(1):557–567. doi: 10.1074/jbc.M111.281576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lammens A, Schele A, Hopfner K-P. Structural biochemistry of ATP-driven dimerization and DNA-stimulated activation of SMC ATPases. Curr Biol. 2004;14(19):1778–1782. doi: 10.1016/j.cub.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 28.Firman K, Szczelkun MD. Measuring motion on DNA by the type I restriction endonuclease EcoR124I using triplex displacement. EMBO J. 2000;19(9):2094–2102. doi: 10.1093/emboj/19.9.2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha A, Wittmeyer J, Cairns BR. Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev. 2002;16(16):2120–2134. doi: 10.1101/gad.995002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitehouse I, Stockdale C, Flaus A, Szczelkun MD, Owen-Hughes T. Evidence for DNA translocation by the ISWI chromatin-remodeling enzyme. Mol Cell Biol. 2003;23(6):1935–1945. doi: 10.1128/MCB.23.6.1935-1945.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: Mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9(5):391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 32.Fishburn J, Hahn S. Architecture of the yeast RNA polymerase II open complex and regulation of activity by TFIIF. Mol Cell Biol. 2012;32(1):12–25. doi: 10.1128/MCB.06242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young MC, Kuhl SB, von Hippel PH. Kinetic theory of ATP-driven translocases on one-dimensional polymer lattices. J Mol Biol. 1994;235(5):1436–1446. doi: 10.1006/jmbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 34.Fischer CJ, Saha A, Cairns BR. Kinetic model for the ATP-dependent translocation of Saccharomyces cerevisiae RSC along double-stranded DNA. Biochemistry. 2007;46(43):12416–12426. doi: 10.1021/bi700930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khaki AR, et al. The macroscopic rate of nucleic acid translocation by hepatitis C virus helicase NS3h is dependent on both sugar and base moieties. J Mol Biol. 2010;400(3):354–378. doi: 10.1016/j.jmb.2010.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung P, Prakash L, Matson SW, Prakash S. RAD3 protein of Saccharomyces cerevisiae is a DNA helicase. Proc Natl Acad Sci USA. 1987;84(24):8951–8955. doi: 10.1073/pnas.84.24.8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.