

Abstract
The ligation of Toll-like receptors (TLRs) leads to rapid activation of dendritic cells (DCs). However, the metabolic requirements that support this process remain poorly defined. We found that DC glycolytic flux increased within minutes of exposure to TLR agonists and that this served an essential role in supporting the de novo synthesis of fatty acids for the expansion of the endoplasmic reticulum and Golgi required for the production and secretion of proteins that are integral to DC activation. Signaling via the kinases TBK1, IKKε and Akt was essential for the TLR-induced increase in glycolysis by promoting the association of the glycolytic enzyme HK-II with mitochondria. In summary, we identified the rapid induction of glycolysis as an integral component of TLR signaling that is essential for the anabolic demands of the activation and function of DCs.
Dendritic cells (DCs) are highly specialized antigen-presenting cells of myeloid origin with a pivotal role in the initiation and maintenance of adaptive immune responses1. Under noninflammatory conditions, DCs in peripheral tissues exist in a resting immature state, but they express a range of germline-encoded pattern-recognition receptors, including Toll-like receptors (TLRs), that allow them to recognize and rapidly respond to microbial products or inflammatory stimuli. After encountering such danger signals, DCs become activated, a process that involves enhanced capturing and processing of antigens for the stable presentation of antigen-derived peptides in the context of major histocompatibility complex (MHC) class I and class II and induction of the expression of genes encoding chemokine receptors, cytokines and costimulatory molecules. Collectively, these changes enable DCs to promote local inflammation and traffic to T cell zones of secondary lymphoid organs, where they prime T cell responses2.
There is a growing appreciation that changes in the activation of cells of the immune system are coupled to profound changes in cellular metabolism and that cellular fate and function are metabolically regulated3. Studies have begun to characterize the metabolic programs required for the activation and function of DCs. After exposure to TLR agonists, DCs differentiated from bone marrow in the presence of the growth factor GM-CSF (GM-DCs), a model for inflammatory monocyte-derived DCs, undergo a metabolic transition characterized by a robust increase in glycolysis4. Moreover, inhibition of glycolysis substantially limits the activation and lifespan of DCs after stimulation via TLRs4,5. The commitment of GM-DCs to glycolysis after activation is a direct effect of the TLR-stimulated expression of inducible nitric oxide synthase (iNOS), which produces the toxic gas nitric oxide (NO) from arginine at a high rate6. NO inhibits mitochondrial electron transport and therefore blocks oxygen consumption and coupled ATP production7. It is clear that the prolonged commitment to glycolysis in activated DCs occurs only in DC subsets that express iNOS and that it is a direct consequence of the inhibition of oxidative phosphorylation by NO and serves the vital survival function of providing ATP in the absence of mitochondrial generation of ATP6.
While the glycolysis-biased metabolism of GM-DCs at 24 h after activation represents a response to endogenous NO production, we hypothesized that at much earlier times after stimulation with TLR agonists, metabolic reprogramming must occur to meet the bioenergetic and anabolic needs of TLR-driven DC activation itself. Here we found that the glycolytic rate increased within minutes after DCs were stimulated with TLR agonists. This process, which was iNOS independent and was controlled directly by activation of the rate-limiting glycolytic enzyme HK-II by the kinases TBK1, IKKε and Akt, was essential for support of the de novo synthesis of fatty acids that is critical for DC activation.
RESULTS
TLR agonists induce a rapid increase in glycolysis by DCs
We analyzed GM-DCs for real-time changes in the rate of extracellular acidification (ECAR), as a measure of lactate production (a surrogate for the glycolytic rate), and the mitochondrial rate of oxygen consumption (OCR), directly following stimulation with lipopolysaccharide (LPS). While the OCR remained stable after stimulation, there was a rapid increase in the ECAR (Fig. 1a); this was independent of iNOS (Supplementary Fig. 1). However, consistent with published work6, the long-term commitment to glycolysis was dependent on NO, since in the presence of a general inhibitor of NOS, the TLR-induced increase in the glycolytic rate returned toward baseline by 9 h after stimulation, whereas in the absence of the inhibitor, the ECAR remained elevated beyond that time (Supplementary Fig. 1). We confirmed the rapid induction of glycolysis by LPS by measuring increases in extracellular lactate concentrations (Fig. 1b) and glucose consumption (Fig. 1c). Those results were further supported by analysis of 1,2-13C-glucose tracing by gas chromatography–mass spectrometry, with which we observed more rapid incorporation of glucose-derived carbon into pyruvate and lactate after stimulation with LPS for 1 h than in unstimulated (control) conditions (Fig. 1d). Finally, global profiling of metabolites in GM-DCs that had been stimulated for 1 and 3 h with LPS revealed selective LPS-induced changes in the abundance of metabolic intermediates of glycolysis and associated pathways, such as the pentose phosphate pathway (PPP) and tricarboxylic acid (TCA) cycle (Fig. 1e and Supplementary Table 1). Other TLR agonists induced a similar response (Fig. 1f). In in vitro cultures of conventional SIRPα+ and CD24+ DCs differentiated from bone marrow in the presence of the growth factor Flt3L, the ECAR was also rapidly increased after stimulation of TLRs (Fig. 1g). Thus, rapidly increased glycolysis was a cardinal feature of the TLR-induced DC activation.
Figure 1.

TLR ligation induces a rapid increase in glycolytic metabolism in DCs. (a) Real-time changes in the ECAR and OCR of GM-DCs left untreated (−) or treated with LPS (vertical dotted line indicates initiation of treatment throughout). (b,c) Lactate production (b) and glucose consumption (c) in GM-DCs left unstimulated (−) or stimulated for 6 h with LPS. (d) 13C-labeled metabolites in GM-DCs incubated for 1 h with medium (−) or LPS in the presence of 1,2-13C-glucose, detected by NMR spectrometry and presented relative to the total pool of that metabolite (m+1, m+2, m+3 (horizontal axes), mass of metabolite (m) + mass units (1, 2, 3) derived from incorporated glucose-derived 13C). (e) Total metabolite profiling of GM-DCs stimulated for 1 and 3 h with LPS, assessed by pathway-enrichment analysis, based on changes in the relative abundance of metabolites; key lists the five pathways with the most significant change (in ranked order, i–v). (f,g) Real-time changes in the ECAR and OCR of GM-DCs (f) and mouse DCs generated by stimulation with Flt3L and cultured in vitro (g), treated with various TLR agonists (TLR-L (key); f) or with poly(I:C) or LPS (g). *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test). Data are from one experiment representative of over ten experiments (a) or three experiments (f,g; mean ± s.e.m. of triplicates) or represent three independent experiments (b–d; mean and s.e.m.) or are representative of five independent experiments (e).
DC activation depends on an early increase in glycolysis
We sought to determine whether the early increase in glycolysis was important for DC activation. We stimulated GM-DCs with LPS in the presence of 2-deoxyglucose (2-DG), which inhibits hexokinase activity8; 2-DG effectively blocked glycolysis, as shown by the lack of an increase in the ECAR after stimulation with LPS (Fig. 2a). The LPS-induced surface expression of the costimulatory receptor CD40 and costimulatory molecule CD86 and MHC class I and class II and production of interleukin 6 (IL-6), IL-12p70 and tumor-necrosis factor (TNF) at 6 h were substantially impaired by 2-DG (Fig. 2b,c and Supplementary Fig. 2a). Notably, that regulation seemed to be entirely at the translational level, as mRNA encoding each of those proteins was induced equivalently in GM-DCs stimulated with LPS in the presence and absence of 2-DG (Fig. 2d). The production of effector cytokines in T cells is controlled post-transcriptionaIly by aerobic glycolysis through a mechanism that involves the binding of GAPDH to AU-rich elements in the 3′ untranslated region of mRNA encoding interferon-γ (IFN-γ), which results in impaired translation of IFN-γ 9. Given the presence of AU-rich elements in the 3′ untranslated region of Il6, Il12b and Tnf mRNA10, we assessed whether glycolysis controls cytokine expression in DCs in a similar manner. However, the binding of Il6, Il12b and Tnf transcripts to GAPDH was very low in DCs after stimulation with LPS and did not increase much in the presence of 2-DG (Supplementary Fig. 2b), which suggested that the observed post-transcriptionaI regulation of cytokine expression was controlled by other processes. iNOS is not yet expressed at that time point after LPS stimulation, and mitochondria are active6, and consistent with the presence of still-functional mitochondria, cell viability was unaffected by the presence of 2-DG (Supplementary Fig. 2c). We observed a similar impairment in the TLR-induced expression of surface markers in 2-DG-treated DCs generated from bone marrow in the presence of Flt3L (Supplementary Fig. 2d). Furthermore, blocking glycolysis in iNOS-deficient (Nos2−/−) GM-DCs suppressed the LPS-induced expression of the chemokine CCL19 receptor CCR7 (Fig. 2e) and consequently impaired migration toward CCL19 (Fig. 2f). In addition, inhibition of glycolysis compromised the ability of Nos2−/− GM-DCs to induce T cell proliferation (Fig. 2g) and IFN-γ secretion (Fig. 2h), although notably, under these conditions, the frequency of T cells that produced IL-17A and expressed the transcription factor Foxp3 increased (Fig. 2h). Thus, an increase in aerobic glycolysis early after stimulation with TLR agonists was required for the activation and function of DCs.
Figure 2.

The glycolytic burst is required for the activation and function of DCs. (a) Real-time changes in the ECAR and OCR (as in Fig. 1a) of GM-DCs left untreated (−) or treated with LPS with or without 2-DG (key). (b,c) Expression of markers CD86 and CD40 or of MHC class I and II (b) or cytokines IL-6, IL-12 and TNF (c) in GM-DCs either left untreated (−) or given no pretreatment (LPS) or pretreated with 2-DG (2-DG + LPS) and then stimulated for 6 h with LPS, analyzed by flow cytometry with surface (b) or intracellular (c) staining. Numbers in top right corners (b) indicate percent CD86+CD40+ cells (top row) or MHCI+MHCII+ cells (bottom row). Numbers above bracketed lines (c) indicate percent cells with cytokine expression; gray filled curves and black lines (c) indicate unstimulated cells and stimulated cells, respectively. (d) Expression of mRNA (vertical axes) in GM-DCs left unstimulated or stimulated for 3 h with LPS with or without 2-DG (key), normalized to that of mRNA encoding β-actin and presented relative to that of unstimulated cells, set as 1 (100). (e) CCR7 expression by Nos2−/− GM-DCs left unstimulated or stimulated for 16 h with LPS with or without 2-DG (key); numbers above lines indicate percent cells with CCR7 expression (right of vertical line). Isotype, isotype-matched control antibody. (f) Migration of Nos2−/− GM-DCs, stimulated for 16 h with LPS with or without 2-DG (horizontal axes), toward CCL19 or no cytokine (−) in a Transwell chamber, assessed after 2 h of migration and presented relative to total input, set as 100%. *P < 0.05 (Student’s t-test). (g) Dilution of the cytosolic dye CFSE in CFSE-labeled OT-I or OT-II T cells (which have transgenic expression of an OVA-specific T cell antigen receptor) cultured for 3 d together with Nos2−/− GM-DCs (that had been stimulated for 6 h with LPS and OVA with or without 2-DG; key), at a DC/T cell ratio of 1:50 (left) or at various DC/T cell ratios (horizontal axes; right). (h) Expression of IFN-γ, IL-4, IL-17A and Foxp3 in OT-II T cells cultured for 6 d together with GM-DCs treated as in g (DC/T cell, 1:5). Numbers adjacent to outlined areas indicate percent cells with cytokine expression. Data are from one experiment representative of three (a,e–h) or five (b,c) experiments (mean and s.e.m. of triplicates (a,f) or duplicates (g)) or are from three independent experiments (d; mean and s.e.m.).
Glycolysis feeds the TCA cycle to support DC activation
TLR signaling drives the bioenergetically demanding expression of a large set of genes11. Since we observed that ATP concentrations in GM-DCs dropped by ~40% within 2 h after stimulation with LPS (Supplementary Fig. 3a), we first determined whether that increase in glycolysis was needed to provide extra ATP for DC activation. For cells that depend on glycolysis as their chief source of ATP, the recycling of NADH generated during glycolysis back to NAD+ by the conversion of pyruvate into lactate catalyzed by the enzyme lactate dehydrogenase A (LDHA) is essential for maintenance of a high glycolytic flux. However, we found that although inhibition of LDHA by oxamate blocked the early increase in the ECAR (Supplementary Fig. 3b), it had no effect on DC activation (Supplementary Fig. 3c,d), which challenged the proposal of an essential role for glycolytic ATP in DC activation.
On the basis of those findings, we tested the alternative hypothesis that glucose was serving as a key source of carbon for other anabolic programs that underpinned the DC activation. Since metabolite profiling revealed that DC activation was associated with changes in pyruvate metabolism and the TCA cycle (Fig. 1e), we hypothesized that the increase in glycolysis may have served to generate more pyruvate to feed the TCA cycle. In this case, an increase in glycolysis would be expected to be associated with coincident changes in mitochondrial activity. We found that the mitochondrial membrane potential and spare respiratory capacity, a measure of mitochondrial fitness12,13, increased rapidly after stimulation with LPS (Fig. 3a,b), indicative of increased mitochondrial activity. Consistent with those findings, tracing of 1,2-13C-glucose by gas chromatography–mass spectrometry revealed more rapid incorporation of glucose-derived carbon into TCA cycle intermediates in cells stimulated for 1 h with LPS than in control cells left unstimulated for that same period of time (Fig. 3c). In addition, the LPS-induced increase in the spare respiratory capacity was prevented by 2-DG (Fig. 3d), which indicated that the increase in mitochondrial fitness was mediated by that enhanced flux of glycolytic carbon into the TCA cycle. To address whether flux of pyruvate into the TCA cycle is important for DC activation, we suppressed expression of the mitochondrial pyruvate carrier MPC-1 (ref. 14) (Fig. 3e). GM-DCs transduced with short hairpin RNA (shRNA) specific for Mpc1 (which encodes MPC-1) were less activated after stimulation for 6 h with LPS than were GM-DCs transduced with control shRNA (Fig. 3f,g); pharmacological inhibition of MPC-1 with α-cyanocinnimate resulted in a phenocopy of those results (Supplementary Fig. 4a,b). In contrast, when we inhibited fatty acid oxidation or glutaminolysis, two other chief potential sources of carbon for the TCA cycle, we noted no major effects on DC activation (Supplementary Fig. 4a,b). The proposal of the importance of glycolysis for fueling the TCA cycle for DC activation was further supported by data from experiments in which we manipulated the flux of pyruvate into lactate or acetyl-CoA through the use of retroviral transduction to promote or inhibit the expression of LDHA and the pyruvate dehydrogenase kinase PDK1. We found that preventing the flux of pyruvate (and therefore glucose-derived carbon) into the TCA cycle by overexpressing LDHA and PDK1 inhibited activation, whereas reducing expression of those enzymes led to more activation (Supplementary Fig. 4c).
Figure 3.

TLR-induced activation of DCs depends on the flux of glucose-derived carbon into the TCA cycle. (a) Change in mitochondrial membrane potential (ΔΨm) of GM-DCs left unstimulated or stimulated for various times (horizontal axis) with LPS, then stained with the mitochondria-selective dye DiOC6; results are presented relative to the potential at time 0, set as 1. (b) Real-time changes in the OCR of GM-DCs stimulated for 1 h with control medium (−) or LPS, assessed during subsequent sequential treatment with oligomycin (Oligo; inhibitor of ATP synthase), FCCP (ionophore) and rotenone plus antimycin A (Ant-Rot; inhibitors of the electron-transport chain). SRC, spare respiratory capacity (double-headed arrow). (c) 1,2-13C-glucose tracing (left and right; as in Fig. 1d) in GM-DCs incubated for 1 h with medium (−) or LPS; middle, TCA cycle. (d) Spare respiratory capacity (as in b) of GM-DCs either left untreated (−) or given no pretreatment (LPS) or pretreated with 2-DG (2-DG + LPS) and then stimulated with LPS. *P < 0.001 (Student’s t-test). (e) Expression of Mpc1 mRNA in GM-DCs transduced with retroviral vector expressing control shRNA (Ctrl-hp) or Mpc1-specific shRNA (Mpc1-hp), normalized to that of mRNA encoding β-actin and presented relative to that of cells transduced with control shRNA, set as 1. (f,g) Expression of CD86 and CD40 (f) or IL-6, IL-12 and TNF (g) in GM-DCs transduced as in e and then left untreated (−) or treated with LPS. Numbers in top right corners (f) indicate percent CD86+CD40+ cells; numbers above bracketed lines (g) indicate percent cells with cytokine expression (as in Fig. 2b,c). Data are from one experiment representative of three (a,b,f,g) or two (d) experiments (mean and s.e.m. of duplicates (a) or triplicates (b,d)) or are from three (c) or two (e) independent experiments (mean and s.e.m.).
DC activation depends on glycolysis-fueled synthesis of fatty acids
Inhibition of the mitochondrial production of ATP by oligomycin had a minimal effect on the early activation of GM-DCs by LPS (Supplementary Fig. 5a,b). That result indicated that the increase in the flux of glucose-derived carbon into the TCA cycle in response to stimulation via TLRs was not needed to fuel oxidative phosphorylation to generate ATP for DC activation. We therefore reasoned that the increase in glycolysis served to replenish intermediates of the TCA cycle that were being extracted for other biosynthetic processes important for DC activation. Consistent with that, we found that intracellular concentrations of citrate and isocitrate, but not of subsequent intermediates in the TCA cycle, dropped after stimulation with LPS (Fig. 4a). Moreover, the percentage of glucose-derived carbon was significantly lower in intermediates of the TCA cycle downstream of citrate than in citrate itself (Fig. 3c), which indicated ‘preferential’ removal of citrate from the TCA cycle during DC activation. Citrate is one of the main sources of carbon for the de novo synthesis of fatty acids14, which requires the export of citrate from the mitochondria into the cytosol via the citrate shuttle Slc25a1 (ref. 15). Citrate is subsequently converted to acetyl-CoA, followed by its conversion by acetyl-CoA carboxylase into malonyl-CoA, which is then used for the elongation of fatty acid chains by fatty acid synthase. Our finding of lower citrate concentrations in activated DCs led us to hypothesize that the rapid increase in glycolytic flux after stimulation via TLRs was needed to support the de novo synthesis of fatty acids. Consistent with that, stimulation with LPS resulted in the accumulation of lipids in Nos2−/− DCs (Fig. 4b) that was diminished when we inhibited either glycolysis or the de novo synthesis of fatty acids by treatment with 2-DG or the fatty-acid-synthase inhibitor C75, respectively (Fig. 4c). Moreover, stimulation of GM-DCs with LPS resulted in increased incorporation of glucose-derived carbon into lipids that was prevented by C75 (Fig. 4d). We addressed the importance of the synthesis of fatty acids from citrate for DC activation by measuring the effect of LPS on GM-DCs in which expression of Slc25a1 had been inhibited by retrovirally introduced shRNA (Fig. 4e) or on GM-DCs were incubated with the acetyl-CoA carboxylase inhibitor TOFA or with C75. We found that each of those three treatments impaired the LPS-induced activation of GM-DCs (Fig. 4f–h), including secretion of the inflammatory lipid mediator prostaglandin E2 (Supplementary Fig. 5c).
Figure 4.

TLR-induced activation of DCs requires de novo synthesis of fatty acids supported by glycolysis. (a) Abundance of metabolites in DCs stimulated for 1 h with LPS, relative to that in untreated control cells, set as 1 (dotted line). (b) Confocal microscopy (left) of Nos2−/− GM-DCs left unstimulated (−) or stimulated for 24 h with LPS, then stained with the DNA-intercalating dye DAPI and the fluorescent dye BODIPY. Scale bars, 10 μm. Right, quantification of BODIPY staining, assessed by flow cytometry and presented as the geometric mean fluorescence intensity (geoMFI). (c) Uptake of BODIPY by Nos2−/− GM-DCs left unstimulated or stimulated for 24 h with LPS with or without 2-DG or C75 (key). (d) Incorporation of 14C into the lipid fraction of GM-DCs left unstimulated or stimulated for 6 h with LPS with or without C75, in the presence of U-14C-glucose (glc). (e,f) Expression of Slc25a1 mRNA (e) and of the cytokines IL-6, IL-12 and TNF (f) in GM-DCs transduced with control shRNA (Ctrl-hp) or Slc25a1-specific shRNA (Slc25a1-hp); mRNA results (e) are as above (Fig. 3e), and numbers above bracketed lines (f) indicate percent cells with cytokine expression. (g,h) Expression of CD86 and CD40 (g) or of IL-6, IL-12 and TNF (h) in GM-DCs left untreated (far left, g) or given no pretreatment (LPS) or pretreated with TOFA (TOFA + LPS) or C75 (C75 + LPS) and then stimulated with LPS (numbers in plots, as in Fig. 3f,g). (i) Expression of mRNA (vertical axes) in GM-DCs left unstimulated (−) or stimulated for 3 h with LPS with or without C75 (presented as in Fig. 2d). (j,k) Quantification of ER and Golgi (j) and electron microscopy (k) of GM-DCs treated as in i. Scale bars (k), 500 nm. *P < 0.05, **P < 0.01 and ***P < 0.001 (Student’s t-test). Data are from three experiments (a), five (b) or three (e,i) independent experiments (mean and s.e.m.), or one experiment representative of three (c,f), two (d,j,k) or four (g,h) experiments (mean and s.e.m. of triplicates (c,d) or 20 cells (j)).
Next we addressed the role of the de novo synthesis of fatty acids in TLR-driven activation of DCs. De novo–synthesized fatty acids may act as ligands for transcription factors, such as peroxisome proliferator-activated receptors, that could regulate TLR-induced gene expression16,17. However, rosiglitazone, a synthetic ligand for peroxisome proliferator-activated receptor-γ, did not abrogate the defect in LPS-driven activation when fatty acid synthesis was inhibited (Supplementary Fig. 5d). Consistent with that finding, inhibition of fatty acid synthesis had no effect on the abundance of mRNA encoding cytokines or costimulatory molecules (Fig. 4i), which suggested post-transcriptional regulation of LPS-induced gene expression by fatty acid synthesis. Those observations led us to hypothesize that de novo lipogenesis may be important for the synthesis of additional membranes to expand the endoplasmic reticulum (ER) and Golgi to support the increase in the synthesis, transport and secretion of proteins in response to stimulation via TLRs. We addressed this possibility by using transmission electron microscopy to visualize ER and Golgi. We found that both compartments were expanded considerably in GM-DCs at 3 h after stimulation with LPS and that this process was significantly attenuated by the inhibition of fatty acid synthesis (Fig. 4j,k). These data suggested that TLR-driven glycolysis in DCs, through the generation of citrate, supported the de novo synthesis of fatty acids to promote the expansion of ER and Golgi networks required for DC activation.
Glycolysis-driven PPP is required for DC activation
The PPP, an offshoot of the glycolytic pathway, has a crucial role in the generation of NADPH, which is critical for the synthesis of fatty acids as well as for the maintenance of cellular redox balance by controlling the concentration of reduced glutathione (GSH) and the production of ribose-5-phosphate used for nucleotide synthesis. Metabolite analysis indicated considerable changes in the PPP after DC activation (Fig. 1e). Specifically, consistent with a high demand for products of the PPP, we observed that intracellular concentrations of NADPH (Fig. 5a) and ribose-5-phosphate (Fig. 5b) dropped rapidly in DCs after stimulation with LPS. Indeed when we suppressed expression of glucose-6-phosphate dehydrogenase, the first enzyme in the PPP (Fig. 5c), LPS-driven expression of proinflammatory cytokines (Fig. 5d) and lipid accumulation (Fig. 5e) were diminished. Those results suggested that the flux of glucose into the PPP had a crucial role in DC activation by supporting fatty acid synthesis through the generation of NADPH. Together these data indicated that the increased flux of glucose-derived carbon into the TCA cycle and PPP was essential for TLR agonist–induced DC activation and suggested an important role for the de novo synthesis of fatty acids from citrate, fueled by glycolysis, for LPS-driven activation of DCs.
Figure 5.
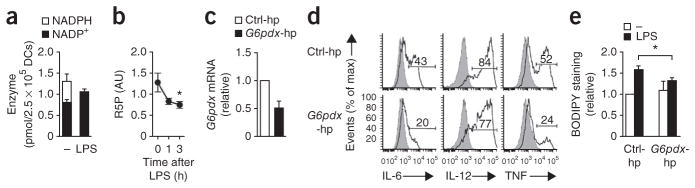
The PPP supports the accumulation of lipids in DCs stimulated via TLRs. (a) NADPH and NADP+ in GM-DCs left unstimulated (−) or stimulated for 1 h with LPS. (b) Quantification of ribose-5-phosphate (R5P) in GM-DCs stimulated for 0–3 h with LPS (horizontal axis), based on total metabolite profiling. (c–e) Expression of G6pdx (which encodes glucose-6-phosphate dehydrogenase) (c) and of the cytokines IL-6, IL-12 and TNF (d), as well as BODIPY uptake (as in Fig. 4b) (e), in GM-DCs transduced with control shRNA (Ctrl-hp) or G6pdx-specific shRNA (G6pdx-hp); mRNA results (e) are as above (Fig. 3e), and numbers above bracketed lines (e) indicate percent cells with cytokine expression. *P < 0.05 (paired Student’s t-test). Data are from one experiment representative of three (a,d; mean and s.e.m. of duplicates in a) or are from four (b), three (c) or two (e) independent experiments (mean and s.e.m.).
TBK1-IKKε controls the early increase in glycolysis
Signaling via growth factors and insulin receptors leads to Akt-dependent increases in glycolysis18. Moreover, Akt is essential for the sustained metabolic commitment to glycolysis that is apparent in iNOS-expressing DCs following activation4. We reasoned, therefore, that Akt would be essential for the early increase in glycolysis induced by TLR agonists. We found that stimulation of GM-DCs with LPS led to the rapid phosphorylation of Akt at both Thr308 and Ser473, necessary for full Akt activation19, and phosphorylation of PRAS40, a direct downstream target of Akt (Fig. 6a). Consistent with the proposal of a crucial role for Akt in the metabolic response to stimulation via TLRs, two distinct inhibitors of Akt (triciribine and Akt1/2 VIII) substantially blunted the early increase in glycolysis and concomitant DC activation induced by LPS (Fig. 6b–d and Supplementary Fig. 6a). Unexpectedly, whereas inhibition of the canonical Akt activators phosphatidylinositol-3-OH kinase (PI(3)K) and mTOR diminished the LPS-induced phosphorylation of Akt at Ser473 and of PRAS40, on the basis of both immunoblot analysis (Fig. 6e and Supplementary Fig. 6c) and flow cytometry (Supplementary Fig. 6b), that was not sufficient to impair the increase in glycolysis (Fig. 6f and Supplementary Fig. 6a). Akt has been identified as a substrate for TBK1 and IKKε20, two closely related kinases downstream of various TLRs21 that can be activated independently of PI(3)K22,23 and are known for their importance in the induction of type I interferons. We found that after stimulation with LPS, TBK1 was rapidly phosphorylated in GM-DCs in a PI(3)K-independent manner (Fig. 6g and Supplementary Fig. 6e); TBK1 was also phosphorylated in response to the TLR2 ligand Pam3CSK4 (Supplementary Fig. 6d). Moreover, GM-DCs in which TBK1 and IKKε were inhibited by BX795 showed substantial impairment in the phosphorylation of Akt at Ser473 and Thr308 and of PRAS40 and the ribosomal protein S6, and this was more profound than the inhibition observed after inhibition of PI(3)K or mTOR (Fig. 6e and Supplementary Fig. 6b,c). Consistent with that, in contrast to inhibition of PI(3)K or the metabolic checkpoint kinase complex mTORC2, inhibition of TBK1-IKKε blunted the glycolytic response (Fig. 6f) after stimulation with LPS. Those effects were recapitulated in GM-DCs derived from IKKε-deficient (Ikbke−/−) bone marrow in which TBK1 was knocked down by shRNA (Fig. 6h,i and Supplementary Fig. 6f) and in GM-DCs treated with amlexanox, a different inhibitor of TBK1-IKKε24 (Supplementary Fig. 6g). Together these data indicated TBK1 and IKKε were the main kinases upstream of Akt that regulated metabolic reprogramming in DCs after stimulation by TLR agonists.
Figure 6.

The TLR-driven glycolytic burst is dependent on TBK1-IKKε and Akt. (a) Immunoblot analysis of total Akt and of Akt phosphorylated at Thr308 (p-Akt(T308)) or Ser473 (p-Akt(S473)), and of total and phosphorylated (p-) PRAS40, in GM-DCs stimulated for 0, 15 or 30 min (above lanes) with LPS. (b) Real-time changes in the ECAR of GM-DCs left untreated (−) or given no pretreatment (LPS) or pretreated with triciribine (Tric + LPS) and then stimulated with LPS (analyzed as in Fig. 1f). (c,d) Expression of CD86 and CD40 (c) or of IL-6, IL-12 and TNF (d) in GM-DCs treated as in b (numbers in plots, as in Fig. 3f,g). (e) Immunoblot analysis of total and phosphorylated Akt (as in a), PRAS40 and S6 in GM-DCs either left untreated (far left) or given no pretreatment (second from left) or preincubated with BX795 (BX), KU0063794 (KU) or LY294002 (LY) and stimulated for 15 min with LPS. (f) Real-time changes in the ECAR of GM-DCs treated as in e (analyzed as in Fig. 1a); Inh, inhibitor (key). (g) Immunoblot analysis of total and phosphorylated TBK1 in GM-DCs stimulated for 0, 15 or 30 min (above lanes) with LPS. (h) Immunoblot analysis of TBK1 and IKKε in wild-type (WT) and Ikbke−/− GM-DCs transduced with control shRNA (Ctrl-hp) or Tbk1-specific shRNA (Tbk1-hp) and then sorted on the basis of human CD8 expression (as a reporter for transduction); β-actin serves as a loading control. (i) Real-time changes in the ECAR of GM-DCs transduced and sorted as in h and then left untreated (−) or treated with LPS (analyzed as in Fig. 1a). Data are from one experiment representative of two (a,g,i), four (b,f) or three (c,d,e) experiments (mean ± s.e.m. of triplicates in b,f,i) or are from one experiment (h).
Akt-driven activation of HK-II is critical for early glycolysis
One mechanism by which Akt has been postulated to enhance glycolytic flux is by promoting the association of HK-II with voltage-dependent anion channels located in the outer mitochondrial membrane, where presumably it can directly use mitochondrial ATP25,26. The stimulation of GM-DCs with LPS resulted in enrichment of phosphorylated HK-II in the mitochondrial fraction and increased overall hexokinase activity, effects that were prevented by the inhibition of Akt and of TBK1-IKKε (Fig. 7a,b and Supplementary Fig. 7a). That increase in hexokinase activity was functionally critical, as treatment with a cell-permeable peptide corresponding to the mitochondrial and voltage-dependent anion channel–binding motif of HK-II, which competitively dissociates HK-II from the mitochondria27,28 (Fig. 7c), prevented the LPS-induced increase in hexokinase activity (Fig. 7d) and impaired the rapid burst in glycolysis (Fig. 7e) and associated DC activation (Fig. 7f,g). Those effects were comprehensively recapitulated by clotrimazole, a drug that dissociates hexokinases from mitochondria27 (Supplementary Fig. 7b–f).
Figure 7.

The TLR-induced glycolytic burst and activation of DCs are dependent on the Akt-driven association of HK-II with mitochondria. (a) Immunoblot analysis of HK-II, β-actin (loading control), prohibitin-1 (PHB1; specific marker for mitochondria) and phosphorylated hexokinase (p-HK) in mitochondria-enriched (Mito) and cytosolic (Cyto) fractions of GM-DCs left unstimulated (0) or stimulated for 1 h with LPS (1) in the presence (+) or absence (−) of triciribine; hexokinase phosphorylation was assessed with an antibody to Akt-specific phosphorylation sites. (b) Hexokinase (HK) activity in GM-DCs left unstimulated (−) or stimulated for 1 h with LPS in the presence or absence of triciribine or BX795 (key). (c) Immunoblot analysis of HK-II and prohibitin-1 in mitochondria-enriched fractions of GM-DCs treated with control peptide (Ctrl pep) or HK-II peptide (HK-II pep; for targeted dissociation of HK-II from the mitochondria). (d) Hexokinase activity in GM-DCs left unstimulated or stimulated for 1 h with LPS in the presence of control or HK-II peptide (key). (e) Real-time changes in the ECAR of GM-DCs pretreated with control or HK-II peptide and then left unstimulated or stimulated with LPs (analyzed as in Fig. 1a). (f,g) Expression of CD86 and CD40 (f) or of IL-6, IL-12 and TNF (g) in GM-DCs pretreated with control or HK-II peptide and then stimulated with LPS (numbers in plots, as in Fig. 3f,g). Data are from one experiment representative of three (a,e,f,g) or two (c) experiments (mean ± s.e.m. of triplicates in e) or are from three independent experiments (b,d; mean and s.e.m.).
Akt can also enhance glycolytic flux directly by promoting the translocation of glucose transporters to the cell membrane29 and/or by enhancing the enzymatic activity of the glycolytic enzyme PFK1 by promoting the activity of PFK2, its allosteric activator30. However, after stimulation via TLRs, we observed neither a rapid increase in the surface expression of GLUT1 (Slc2a1), the dominant glucose transporter expressed by these cells, nor a substantial effect of the knockdown of PFK2 on the LPS-induced rapid increase in glycolysis or on DC activation (Supplementary Fig. 8a–d). In addition, Akt can enhance glycolytic flux indirectly via mTORC1 through activation of the transcription factor HIF-1α, which drives the expression of several glycolytic enzymes, including HK-II and LDHA31. However, the TLR-driven early glycolytic response was unaffected in GM-DCs that either were treated with the mTORC1 inhibitor rapamycin or were deficient in raptor, a key component of mTORC1 (Supplementary Figs. 6a,b and 8e,f). Moreover, during the rapid TLR-driven increase in glycolysis, the expression of HK-II and LDHA protein remained constant (Supplementary Fig. 8g), and neither global inhibition of transcription with actinomycin D nor knockdown of HIF-1α via shRNA had an effect on TLR-induced early metabolic reprogramming (Supplementary Fig. 8h–k), which suggested that neither mTORC1 nor HIF-1α had a prominent role in the rapid, Akt-driven changes in glycolytic metabolism that occurred in DCs stimulated by TLR agonists. Together these data suggested that TBK1-IKKε and Akt were essential for the increased glycolytic flux after stimulation by TLR agonists by directly promoting the association of HK-II with mitochondria.
Early glycolysis is important for DC function in vivo
To investigate whether DCs rapidly increased glucose uptake in response to stimulation via TLRs in vivo, we gave mice injections of 2-NBDG, a fluorescent analog of 2-DG, after intravenous challenge with LPS. Injection of LPS resulted in a reproducible albeit slight increase in the uptake of 2-NBDG by splenic CD11b+ DCs but not by CD8α+ DCs (Fig. 8a). However, both CD11b+ and CD8α+ DCs showed an increase in the ECAR in response to stimulation via TLRs ex vivo (Fig. 8b). The finding that the increase in the ECAR in response to LPS relative to the baseline ECAR was significantly lower in CD8α+ DCs than in CD11b+ DCs, coupled with the relative insensitivity of 2-NBDG labeling in vivo, may explain why we were unable to detect an increase in glucose uptake by CD8α+ DCs in vivo (Fig. 8a). As for signaling, we found that similar to results obtained for GM-DCs, the TLR-induced glycolytic response in ex vivo splenic CD11b+ DCs was dependent on Akt (Fig. 8c), TBK1-IKKε (Fig. 8d) and the association of HK-II with mitochondria (Fig. 8e) but was not dependent on mTOR or PI(3)K (Fig. 8d). Furthermore, in vivo activation of both DC subsets by LPS was significantly impaired when we gave mice simultaneous injection of 2-DG to block glycolysis (Fig. 8f). Likewise, ex vivo secretion of IL-12 in response to LPS was diminished in the presence of 2-DG in both DC subsets (Fig. 8g). Moreover, DCs activated by LPS in vivo in the presence of 2-DG were impaired in their ability to prime ovalbumin (OVA)-specific CD4+ and CD8+ T cells in vivo (Fig. 8h). In addition, DCs in which fatty acid synthase was inhibited during LPS-driven activation were considerably compromised in their ability to prime CD8+ T cell responses in vivo (Fig. 8i,j), which demonstrated an essential role for TLR-induced glycolytic metabolism and downstream fatty acid synthesis in the immunological priming function of DCs in vivo.
Figure 8.

DCs rely on a rapid increase in glycolytic metabolism and fatty-acid synthesis for their activation and function in vivo. (a) Uptake of 2-NBDG in splenic DCs obtained from mice given no injection (−) or 1 h after injection of LPS (LPS + 2-NBDG) or vehicle control (2-NBDG), analyzed by flow cytometry. (b) Real-time changes in the ECAR of naive splenic DCs left unstimulated or stimulated with LPS (key) and analyzed ex vivo (as in Fig. 1a). (c–e) Real-time changes in the ECAR of naive CD11b+ DCs either left untreated (−) or given no pretreatment or pretreated with triciribine (c), BX795, KU0063794 or LY294002 (d) or control or HK-II peptide (e) and then stimulated with LPS (analyzed as in Fig. 1a). (f) Expression of CD40 and CD86 in splenic DCs obtained from mice 6 h after no injection (−) or intravenous injection of LPS with or without 2-DG, analyzed by flow cytometry; results are presented relative to those of untreated mice, set as 1 (each symbol represents an experiment). (g) IL-12 expression in splenic DCs left unstimulated or stimulated ex vivo for 12 h with LPS or without 2-DG. Numbers adjacent to outlined areas indicate percent IL-12+CD11c+ cells. (h) Dilution of the proliferation-tracking dye CellTrace Violet (CTV) in T cells in spleen of host mice given congenically marked, CellTrace Violet–labeled OT-I or OT-II T cells and then, 1 d later, given splenic DCs activated in vivo with LPS with or without 2-DG (key) and loaded with OT-I and OT-II OVA peptide ex vivo, assessed by flow cytometry 60 h after transfer of DCs (left), and quantification of the results at left, presented as division index (right). (i,j) Flow cytometry (i) and total number (j) of OT-I or OT-II T cells in the draining lymph nodes of host mice 7 d after subcutaneous injection of GM-DCs stimulated with LPS and pulsed with OVA in vitro with or without C75, assessing the endogenous OVA-specific CD8+ T cell response. Numbers adjacent to outlined areas (i) indicate percent CD44+ cells positive for staining with the H-2Kb–OVA tetramer. Each symbol (j) represents an individual mouse; small horizontal lines indicate the mean. *P < 0.05 and **P < 0.01 (Student’s t-test (a,h,j) or paired Student’s t-test (f)). Data are from one experiment representative of three (a–e,g) or two (h–j) experiments (mean and s.e.m. of three to four mice per group (a,h–j) or triplicates (b–e)) or four experiments with three mice per group (f).
DISCUSSION
We found here that stimulation with TLR agonists led to an early rapid increase in glycolysis in both inflammatory and conventional DCs that was critical for supporting the anabolic demands of DC activation. This was functionally distinct from the delayed glycolytic switch observed in inflammatory DCs expressing iNOS that was found to support long-term survival following inhibition of oxidative phosphorylation by NO4,6.
Given that DCs are nonproliferative cells and thus presumably do not need to acquire substantial additional biomass and that cellular ATP concentrations dropped rapidly after stimulation via the TLR, we initially predicted that the main role for the TLR-induced increase in glycolysis would be to act as a rapid additional source of ATP to support DC activation. However, our experiments suggested that DCs did not require glycolytic ATP for TLR-induced activation. Moreover, blocking mitochondrial ATP production during DC activation had no effect either; this indicated flexibility of DCs in their use of either glycolysis or oxidative phosphorylation to meet the bioenergetic demands of cellular activation, which is different from T cells that do rely on ATP derived from oxidative phosphorylation for initial activation9,28. Instead, our data suggest that a chief reason for rapidly increased glucose flux is to support the de novo synthesis of fatty acids required for DC activation. TLR-induced accumulation of lipids has been reported in macrophage cell lines32,33, and both the uptake of fatty acids and de novo synthesis of fatty acids (the latter regulated by sterol regulatory element–binding protein 1a) have been suggested to contribute to the accumulation of lipids in those cells34,35. However, the functional relevance of those observations about macrophages remains poorly understood. The finding of such metabolic reprogramming toward biosynthesis by TLR stimulation may also provide new mechanistic insights into the known positive effects of TLR stimulation on tumor growth36, as it may in effect act as a growth factor. Consistent with our observations, a published study has correlated lipid content with the immunogenicity of DCs in the liver37. Notably, the immunogenicity of DCs with high lipid content was diminished when fatty acid synthesis in those cells was inhibited, in support of the view that the de novo synthesis of fatty acids is an integral part of DC activation and is required for the acquisition of an immunogenic phenotype.
Mechanistically our data support a model in which glycolysis supports the de novo synthesis of fatty acids by generating NADPH through the PPP and by providing the carbons to replenish intermediates of the TCA cycle (the process of anaplerosis) that in the form of citrate or isocitrate are being extracted from the mitochondrial TCA cycle (cataplerosis) for the synthesis of fatty acids. Furthermore, we found that glycolysis regulated DC activation at the post-transcriptional level. Our data are consistent with the view that this occurs through a pathway in which glycolysis supports the de novo synthesis fatty acids that underpins the production of membranes needed for expansion of the ER and Golgi to accommodate the increased demand for the synthesis, transport and secretion of proteins after stimulation via TLRs. Of note, the de novo synthesis of fatty acids probably has additional roles in certain aspects of DC activation independent of membrane synthesis. For instance, consistent with results obtained with macrophages38, we found that de novo synthesis of fatty acids was crucial for the generation of prostaglandin E2 by DCs in response to LPS. Finally, apart from post-transcriptional regulation, glycolysis probably additionally exerts transcriptional control on the expression of certain genes encoding molecules associated with TLR agonist–induced activation of DCs, as has been shown for IL-1β in LPS-activated macrophages39.
We found a critical role for Akt in the early increase in glycolysis in response to stimulation via TLRs. Notably, this process seemed to be dependent on Akt-driven association of HK-II with mitochondria and not on the translocation of glucose transporters to the cell membrane, enhancement of enzymatic activity of PFK2 or HIF-1α-dependent processes. That shares similarities with the dependence on mitochondrial hexokinase for glycolysis stimulated via the T cell antigen receptor in memory CD8+ T cells28. Akt has been shown to phosphorylate HK-II, which leads to binding to the mitochondrial membrane through direct interactions with the voltage-dependent anion channel26,40. We speculate that, as has been suggested before25,40, this process enhances the enzymatic activity of HK-II due to direct access to high concentrations of mitochondrial ATP, which results in an increase in glycolytic flux. In addition to being potentially important for increasing its enzymatic activity, mitochondrial localization of hexokinase has been shown to prevent disruption of the mitochondrial membrane potential41, which in DCs may ensure the integrity of mitochondria on which they rely for their activation.
Although we found contributions of signaling via PI(3)K and mTOR to the phosphorylation of Akt, we identified TBK1-IKKε as the most important kinases in regulating the phosphorylation and activation of Akt after stimulation with LPS. In line with that, we found that TBK1-IKKε, but not signaling via PI(3)K or mTOR, were important for Akt-driven rapid glycolytic reprogramming. That result was unexpected, given that PI(3)K is essential for the long-term commitment to aerobic glycolysis in response to stimulation via TLRs4, an observation consistent with the known relationship of PI(3)K and Akt. We reason that this inconsistency reflects the fact that signaling via PI(3)K through mTORC1 is essential for promoting iNOS expression and NO production42,43, which forces inflammatory DCs to commit to glycolysis in the later stages of their activation6. In addition, such long-term commitment to glycolysis would be further supported by mTORC1, through the induction of HIF-1α5. Thus, whereas TBK1 and IKKε directly induce a rapid HIF-1α-independent increase in aerobic glycolysis via Akt in response to stimulation via TLRs, PI(3)K signaling, through mTORC1, may regulate the long-term commitment to glycolysis in an indirect manner, by promoting the expression of iNOS and HIF-1α. Our data suggest that TBK1 and IKKε, in addition to being important for the induction of type I interferons21, serve a key role in bridging TLR signaling to rapid metabolic reprogramming. Given that TBK1 and IKKε are activated by most TLRs and many other pathogen-recognition receptors21,44, we speculate that glycolytic reprogramming by these kinases is not restricted to TLRs but is instead a common process in response to the recognition of pathogens.
Future studies should determine the extent to which pathogen-and host-derived factors, such as IL-10, that are known for their modulatory effects on TLR agonist–mediated activation and function of DCs, exert such effects by acting on TLR-driven rapid changes in glycolysis. Furthermore, our data indicate that glucose concentrations in local tissue are critical for the in situ activation and migration of DCs and T cell priming by DCs. While glucose homeostasis is tightly regulated, it is feasible that in particular environments, such as highly inflamed tissues, reactive lymph nodes or around tumors, the availability of nutrients, including glucose, becomes limiting to the extent that DC function is impaired3. Our findings may contribute to the identification of new metabolic targets for manipulating DC function. This knowledge could be used in the rational design of strategies to improve the immunogenicity or tolerogenicity of DCs in clinically relevant settings.
ONLINE METHODS
Mice
CD45.1+ and CD45.2+ wild-type mice, Nos2−/− mice, Ikbke−/− mice, Rptorflox/flox mice, Itgaxcre mice and C57BL/6 mice with transgenic expression of OVA-specific CD4+ T cells (OT-II) or CD8+ T cells (OT-I) were from The Jackson Laboratory. Mice were maintained at Washington University under specific pathogen–free conditions under protocols approved by the Institutional Animal Care and Use Committee and were used for experiments between 6 and 12 weeks of age.
In vitro DC differentiation
Bone marrow–derived DCs were generated as described45. Bone marrow cells were differentiated for 7 d in the presence of GM-CSF (20 ng/ml) in RPMI-1640 medium containing 10% FCS, 100 U/ml penicillin-streptomycin and 2 mM -glutamine (‘complete DC medium’) or for 8 d with Flt3L (160 ng/ml) in DMEM containing 10% FCS, 100 U/ml penicillin-streptomycin and 2 mM -glutamine. Bone marrow was cultured for 8 d with Flt3L, then CD24+SIRP1α− and SIRP1α+CD24− DCs were isolated with a FACSAria II (BD). To rule out potential secondary effects of TLR-induced iNOS expression on DC metabolism as a result of interference with oxidative phosphorylation in GM-DCs by iNOS-derived NO6, Nos2−/− GM-DCs were used for all experiments in which DCs were studied more than 6 h after activation, when expression of iNOS begins (i.e., cell death, CCR7 expression, migration, lipid staining and in vitro and in vivo T cell–priming studies).
In vivo studies
For 2-NBDG-labeling experiments, mice received 2-DG (4 g per kg body weight) or vehicle control intravenously 30 min before receiving LPS (5 μg) intravenously; 1 h later, 2-NBDG (50 μg) was injected and 15 min later mice were killed and splenic DCs were analyzed by flow cytometry (in the fluorescein isothiocyanate channel) for the uptake of 2-NBDG. For in vivo activation studies, mice were killed 6 h after of LPS and splenic DCs were analyzed by flow cytometry for the expression of activation markers (antibody identification below). Splenic DCs were isolated as described6. To increase yields of splenic DCs for in vivo T cell–priming studies and ex vivo metabolic analyses, mice were given subsequent injection of 4 × 106 Flt3L-expressing B16 tumor cells 12 d before isolation of DCs. For in vivo T cell–priming studies, splenic DCs were purified from C57BL/6 mice pretreated with LPS with or without 2-DG, then the cells were incubated for 45 min at 37 °C ex vivo with 1 μg/ml synthetic OVA peptide (amino acids 323–339 or 257–264). Then those cells were washed and injected into congenic mice that had received 1 × 106 CellTrace Violet–labeled OT-I and OT-II cells 1 d previously. The proliferation of T cells in the spleen was determined by dilution of CTV, as measured by flow cytometry, 60 h later. Endogenous responses of OVA-specific CD8+ T cells were analyzed in draining lymph nodes 7 d after footpad injection of GM-DCs that had been washed three times following treatment for 6 h with LPS and OVA (1 mg/ml of endotoxin-free egg white prepared in the laboratory) in the presence or absence of C75.
DC culture
DCs were cultured at a density of 2 × 105 cells per well in 200 μl complete DC medium and, where appropriate, were preincubated for 30 min with peptides MIASHMIACLFTELN(β-Ala)GYGRKKRRQRRG-amide (HKII), and GYGRKKRRQRRG-amide (control) (10 μM) from Selleckchem, α-cyanocinnamate (500 μM), 2-DG (10 mM), oligomycin (1 μM), etomoxir (200 μM), 6-diazo-5-oxo-L-norleucine (DON; 100 μM), clotrimazole (25 μM), amlexanox (200 μM), TOFA (5-(tetradecyloxy)-2-furoic acid; 20 μM), C75 (20 μM), actinomycin D (1 μg/ml) and rosiglitazone (5 μM) from Sigma-Aldrich; triciribine (20 μM), LY294006 (20 μM) and BX795 (5 μM) from Calbiochem, S-ethyl-isothiourea (500 μM) from Cayman Chemical; Akt1/2 VIII (5 μM), wortmannin (1 μM) and eapamycin (100 nM) from Millipore; and KU0063794 (100 μM) from Tocris Biochemicals. Subsequently, DCs were stimulated for 6 h (unless stated otherwise) with LPS (Escherichia coli serotype 0111:B4; 100 ng/ml; Sigma-Aldrich); the TLR1-TLR2 agonist Pam3CSK4 (1 μg/ml), the TLR2-TLR6 agonist Pam2CSK4 (1 μg/ml), the TLR3 agonist poly(I:C) (10 μg/ml), the TLR7-TLR8 agonist R-848 (1 μg/ml) and the TLR9 agonist CpG-B (5 μg/ml; all InvivoGen). Cells used for intracellular cytokine staining were stimulated in the presence of 10 μg/ml brefeldin A (Sigma). The concentrations of cytokines and prostaglandin E2 in supernatants of DCs cultured for 6 h with or without LPS and with or without inhibitors were determined by cytometric bead array (BD) or enzyme immunoassay (Cayman), respectively, according to the manufacturer’s instructions.
Retroviral transduction
Retroviral transduction of DCs was accomplished as described4. Sequences for shRNA were obtained from Open Biosystems and were cloned into LMP retroviral vectors expressing either human CD8 or green fluorescent protein. For gene-overexpression studies, gene sequences were cloned into MSCV IRES retroviral vectors expressing human CD8 or green fluorescent protein. Transduced cells were gated or sorted on the basis of the expression of human CD8 or green fluorescent protein.
Migration assay
CCL19 (0.2 μg/ml) in complete DC medium was added to the bottom chamber of 24-well Transwell plates (polycarbonate filter with a 5-μm pore; Costar). DCs (4 × 105 cells per Transwell) were added to the upper chamber, followed by incubation for 2 h at 37 °C. Transmigrated cells were collected from the lower chamber and counted on a flow cytometer.
Proliferation and polarization of T cells
DCs were preincubated for 30 min with the appropriate inhibitors and were subsequently treated for 6 h with LPS and OVA (1 mg/ml of endotoxin-free egg white prepared in the laboratory), then were washed three times. DCs were cultured at various ratios with 5 × 104 CD62LhiCD44lo naive OT-II or OT-I T cells labeled with CFSE (carboxyfluorescein diacetate succinimidyl ester). On day 3, T cell proliferation was assessed by flow cytometry as dilution of CFSE. Cytokine production by T cells was determined by intracellular staining (antibody identification below) on day 6 after restimulation for 5 h with PMA (phorbol 12-myristate 13-acetate) and ionomycin in the presence of brefeldin A.
Metabolism assays
For real-time analysis of the ECAR and OCR, DCs were analyzed with an XF-96 Extracellular Flux Analyzer (Seahorse Bioscience) as described6. ATP concentrations were determined with an ATP Determination Kit according to the manufacturer’s instructions (Invitrogen). The concentration of glucose and lactate in the medium, cellular ratio of NAPD+ to NADPH and hexokinase activity were assessed with a Glucose Assay Kit and Lactate Assay Kit (Eton Bioscience), NADPH quantification Kit (Biovision) and hexokinase colorimetric assay (Biovision), respectively, according to each manufacturer’s recommendations. Metabolon did the global metabolite profiling of stimulated DCs, and the Metaboanalyst 2.0 web-based pipeline was used for metabolic-pathway-enrichment analysis. For 1,2-13C-glucose–tracing experiments, DCs were left unstimulated or stimulated for 1 h with LPS, in the presence of 1,2-13C-glucose (Cambridge Isotope Laboratories) or 12C-glucose. Metabolites from cultured cells were extracted and then were analyzed by NMR and gas chromatography–mass spectrometry as described46,47. Glucose-derived lipid biosynthesis was assessed by culture of DCs (2 × 106) for 6 h in complete DC medium containing 2μM U-14C-glucose (250 mCu/mmol; PerkinElmer). Lipids were extracted with a solution of water, methanol and chloroform at a ratio of 1:1:1, and the organic layer was isolated, dried with a Speedvac and resuspended in a solution of methanol and chloroform at a ratio of 1:1, and incorporated radioactivity was measured with a MicroBeta Liquid Scintillation Counter (PerkinElmer).
Flow cytometry
Antibodies used for flow cytometry analysis were as follows: anti-CD172a (P84), anti-CD24 (M1/69), anti-CD11c (N418), anti-CD8α (53–6.7), anti-CCR7 (4B12), vTNF (TN3–19; all from eBioscience); anti–MHC class II (I-A/Eb; M5/114.15.2; Biolegend); anti-CD11b (M1/70), anti-CD86 (GL1), anti-CD40 (HM40-3), anti–MHC class I (H-2Kb; AF6-88.5), anti-CD44 (IM7), anti-IL-12p40/70 (C15.6), anti-IL-6 (MP5-20F3), antibody to Akt phosphorylated at Thr308 (J1-223.371) and antibody to Akt phosphorylated at Ser473 (M89-61; all from BD). Ovalbumin-specific CD8+ T cells were quantified by being stained with MHC–peptide tetramers of H-2Kb and OVA amino acids 257–264. For intracellular staining, DCs were fixed in 4% ultrapure paraformaldehyde, then were stained for 1 h at 20 °C in 0.2% saponin buffer. Mitochondrial membrane potential and lipid content were assessed with 20 μM DiOC6 (3,3′-dihexyloxacarbocyanine iodide; Invitrogen) and 0.5 μg/ml BODIPY 493/503 (Invitrogen) in the fluorescein isothiocyanate channel, following 20 min of incubation at 37 °C. Global transcription was assessed by a Click-it RNA HCS assay according to the manufacturers’ instructions (Invitrogen) and was analyzed by flow cytometry. Samples were analyzed on a BD Canto II Flow Cytometer.
Cell fractionation and immunoblot analysis
Mitochondria-enriched extracts were prepared from 5 × 106 cells with a Mitochondria Isolation Kit according to the manufacturer’s instructions (Mitosciences). Cell lysate preparation, SDS-PAGE, electrophoretic transfer, immunoblot analysis and development with enhanced chemiluminescence were done as described48. Immunoblot analysis used anti-β–actin (13E5), anti–hexokinase II (C64G5), anti-IKKε (D61F9), anti-TBK1 (D1B4), anti-prohibitin-1 (2426), anti-Akt (C67E7), anti-α-tubulin (11H10), antibody to TBK1 phosphorylated at Ser172 (D52C2), antibody to Akt phosphorylated at Thr308 (D25E6), antibody to Akt phosphorylated at Ser473 (D9E), antibody to S6 phosphorylated at Ser235 and Ser236 (D57.2.2E), anti-S6 (5G10), antibody to PRAS40 phosphorylated at Thr246 (C77D7), anti-PRAS40 (D23C7), anti-raptor (24C12) and anti-LDHA (2012; all from Cell Signaling), at a dilution of 1:1,000 or 1:2,000 (S6), followed by detection with horseradish peroxidase–linked antibody to rabbit IgG (1:2,000 dilution; 7074; Cell Signaling). An antibody to residues specifically phosphorylated by Akt (phospho-Akt substrate; 110B7E; Cell Signaling) was used for analysis of the phosphorylation of hexokinase by Akt.
Quantitative RT-PCR
RNA was isolated with an RNeasy Kit (Qiagen), and single-strand cDNA was synthesized with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The Taqman method was used for real-time PCR with primers from Applied Biosystems and an Applied Biosystems 7000 sequence-detection system. The expression of mRNA was normalized to that of mRNA encoding β-actin.
Immunoprecipitation
DCs (20 × 106 to 30 × 106) were harvested and then were lysed in lysis buffer (10 mM HEPES, pH 7.0, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT and 0.2 mg/ml heparin). Lysates were precleared, then were immunoprecipitated overnight with rabbit polyclonal antibody to GAPDH (G9545; Sigma). Immunocomplexes were recovered through the use of protein A agarose (Invitrogen) and were washed once with a low-salt solution, a high-salt solution and LiCl (Sigma) and twice with Tris-EDTA buffer, and were eluted with 1% SDS and 0.1 M NaHCO3. Crosslinkage of mRNA encoding GAPDH was reversed by incubation of the eluted fraction for 1 h at 42 °C, followed by an additional 2 h at 65 °C in the presence of 200 mM NaCl and 20 μg proteinase K. RNA was then extracted from the eluted fraction with TRIzol, and total RNA was quantified with Nanodrop (Thermo Scientific) or mRNA expression was measured by quantitative RT-PCR as described above.
Transmission electron microscopy
For ultrastructural analysis, cells were fixed for 1 h at 65 °C in 2% paraformaldehyde and 2.5% glutaraldehyde (Polysciences) in 100 mM sodium cacodylate buffer, pH 7.2. Samples were washed in cacodylate buffer and then were post-fixed for 1 h in 1% osmium tetroxide (Polysciences). Samples were then rinsed extensively in distilled H2O before en bloc staining for 1 h with 1% aqueous uranyl acetate (Ted Pella). After several rinses in distilled H2O, samples were dehydrated in a graded series of ethanol and were embedded in Eponate 12 resin (Ted Pella). Sections (95 nm in thickness) were cut with an Ultracut UC7 ultramicrotome (Leica Microsystems), then were stained with uranyl acetate and lead citrate and viewed on a 1200 EX transmission electron microscope (JEOL USA) equipped with an AMT eight-megapixel digital camera (Advanced Microscopy Techniques).
Statistical analysis
Data were analyzed with Graphpad Prism software (version 5). Two-group comparisons were assessed using unpaired or, where indicated, paired two-tailed Student’s t-tests. The use of these tests was justified on the basis of assessment of normality and variance of the distribution of the data. Differences were considered significant when P values were below 0.05. The identities of micrographs were removed before analysis. No randomization or exclusion of data points was used. Pilot in vivo studies were used for estimation of the sample size required to ensure adequate power.
Supplementary Material
Acknowledgments
We thank members of the Pearce laboratory for discussions; H. Virgin for support of the 14C-glucose–tracing experiments; W. Beatty and the Molecular Microbiology Imaging Facility for technical assistance with electron microscopy; and G. Bridon and B. Faubert for help with the glucose-tracing experiments. Some observations reported here were made while B.E., E.A., T.C.F., G.J.W.v.d.W., E.L.P. and E.J.P. were at the Trudeau Institute; we thank the institute for its support during that time. Supported by the US National Institutes of Health (AI53825 and CA164062 to E.J.P.; AI091965 and CA158823 to E.L.P.; and AI049823 to E.A.), the Netherlands Organisation for Scientific Research (B.E. and G.J.W.v.d.W.) and The Arthritis Society of Canada (R.G.J.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
B.E., E.A., S.C.-C.H., C.-H.C., G.J.W.v.d.W., R.G.J., E.L.P. and E.J.P. designed experiments; B.E., E.A., S.C.-C.H., C.-H.C., A.M.S., W.Y.L., V.R., T.C.F. and J.B. did experiments. B.E., E.A., S.C.-C.H., J.B., R.G.J., M.N.A., E.L.P. and E.J.P. analyzed data; and B.E. and E.J.P. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Satpathy AT, Wu X, Albring JC, Murphy KM. Re(de)fining the dendritic cell lineage. Nat Immunol. 2012;13:1145–1154. doi: 10.1038/ni.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234–247. doi: 10.1111/j.1600-065X.2008.00718.x. [DOI] [PubMed] [Google Scholar]
- 3.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krawczyk CM, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jantsch J, et al. Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 6.Everts B, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120:1422–1431. doi: 10.1182/blood-2012-03-419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 8.Grossbard L, Schimke RT. Multiple hexokinases of rat tissues. Purification and comparison of soluble forms. J Biol Chem. 1966;241:3546–3560. [PubMed] [Google Scholar]
- 9.Chang CH, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khabar KS. The AU-rich transcriptome: more than interferons and cytokines, and its role in disease. J Interferon Cytokine Res. 2005;25:1–10. doi: 10.1089/jir.2005.25.1. [DOI] [PubMed] [Google Scholar]
- 11.Wagner A. Energy constraints on the evolution of gene expression. Mol Biol Evol. 2005;22:1365–1374. doi: 10.1093/molbev/msi126. [DOI] [PubMed] [Google Scholar]
- 12.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatzivassiliou G, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 16.Jensen-Urstad AP, Semenkovich CF. Fatty acid synthase and liver triglyceride metabolism: housekeeper or messenger? Biochim Biophys Acta. 2012;1821:747–753. doi: 10.1016/j.bbalip.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagy L, Szanto A, Szatmari I, Szeles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–789. doi: 10.1152/physrev.00004.2011. [DOI] [PubMed] [Google Scholar]
- 18.Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4:a006783. doi: 10.1101/cshperspect.a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 20.Xie X, et al. IκB kinase epsilon and TANK-binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA. 2011;108:6474–6479. doi: 10.1073/pnas.1016132108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark K, Takeuchi O, Akira S, Cohen P. The TRAF-associated protein TANK facilitates cross-talk within the IκB kinase family during Toll-like receptor signaling. Proc Natl Acad Sci USA. 2011;108:17093–17098. doi: 10.1073/pnas.1114194108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo JP, Coppola D, Cheng JQ. IKBKE protein activates Akt independent of phosphatidylinositol 3-kinase/PDK1/mTORC2 and the pleckstrin homology domain to sustain malignant transformation. J Biol Chem. 2011;286:37389–37398. doi: 10.1074/jbc.M111.287433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Ou YH, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reilly SM, et al. An inhibitor of the protein kinases TBK1 and IKK-varε improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 27.Majewski N, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 28.van der Windt GJ, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mor I, Cheung EC, Vousden KH. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb Symp Quant Biol. 2011;76:211–216. doi: 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- 31.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicolaou G, Goodall AH, Erridge C. Diverse bacteria promote macrophage foam cell formation via Toll-like receptor-dependent lipid body biosynthesis. J Atheroscler Thromb. 2012;19:137–148. doi: 10.5551/jat.10249. [DOI] [PubMed] [Google Scholar]
- 33.Funk JL, Feingold KR, Moser AH, Grunfeld C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis. 1993;98:67–82. doi: 10.1016/0021-9150(93)90224-i. [DOI] [PubMed] [Google Scholar]
- 34.Feingold KR, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92:829–839. doi: 10.1189/jlb.1111537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Im SS, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13:540–549. doi: 10.1016/j.cmet.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 37.Ibrahim J, et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology. 2012;143:1061–1072. doi: 10.1053/j.gastro.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Infantino V, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J. 2011;438:433–436. doi: 10.1042/BJ20111275. [DOI] [PubMed] [Google Scholar]
- 39.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stiles BL. PI-3-K and AKT: onto the mitochondria. Adv Drug Deliv Rev. 2009;61:1276–1282. doi: 10.1016/j.addr.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 41.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 42.Jin HK, et al. Rapamycin down-regulates inducible nitric oxide synthase by inducing proteasomal degradation. Biol Pharm Bull. 2009;32:988–992. doi: 10.1248/bpb.32.988. [DOI] [PubMed] [Google Scholar]
- 43.Lisi L, Navarra P, Feinstein DL, Dello Russo C. The mTOR kinase inhibitor rapamycin decreases iNOS mRNA stability in astrocytes. J Neuroinflammation. 2011;8:1. doi: 10.1186/1742-2094-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trinchieri G. Type I interferon: friend or foe? J Exp Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutz MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 46.Faubert B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu Q, Vu H, Liu L, Wang TC, Schaefer WH. Metabolic profiles show specific mitochondrial toxicities in vitro in myotube cells. J Biomol NMR. 2011;49:207–219. doi: 10.1007/s10858-011-9482-8. [DOI] [PubMed] [Google Scholar]
- 48.Pearce EL, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.