

Significance
B lymphocytes are crucial cells in immune responses. Their activity is regulated by signaling pathways involving reactive oxygen species (ROS). Voltage-gated proton channels modulate B-cell responses by facilitating production of ROS. Here we compare the full-length proton channel HVCN1L with a shorter protein isoform, HVCN1S, which lacks the first 20 amino acids. Cells with HVCN1S display enhanced proton currents upon stimulation. In addition, HVCN1S is internalized to a lesser extent by interactions with the B-cell receptor, resulting in greater plasma membrane expression. Finally, HVCN1S expression results in greater proliferation and migration. Compared with normal B lymphocytes, HVCN1S expression is higher in B-cell lines and in B cells from patients with chronic lymphocytic leukemia, where it may contribute to disease pathogenesis.
Keywords: proton currents, chronic lymphocytic leukemia, Hv1, gating kinetics, phosphorylation
Abstract
HVCN1 (Hydrogen voltage-gated channel 1) is the only mammalian voltage-gated proton channel. In human B lymphocytes, HVCN1 associates with the B-cell receptor (BCR) and is required for optimal BCR signaling and redox control. HVCN1 is expressed in malignant B cells that rely on BCR signaling, such as chronic lymphocytic leukemia (CLL) cells. However, little is known about its regulation in these cells. We found that HVCN1 was expressed in B cells as two protein isoforms. The shorter isoform (HVCN1S) was enriched in B cells from a cohort of 76 CLL patients. When overexpressed in a B-cell lymphoma line, HVCN1S responded more profoundly to protein kinase C-dependent phosphorylation. This more potent enhanced gating response was mediated by increased phosphorylation of the same residue responsible for enhanced gating in HVCN1L, Thr29. Furthermore, the association of HVCN1S with the BCR was weaker, which resulted in its diminished internalization upon BCR stimulation. Finally, HVCN1S conferred a proliferative and migratory advantage as well as enhanced BCR-dependent signaling. Overall, our data show for the first time, to our knowledge, the existence of a shorter isoform of HVCN1 with enhanced gating that is specifically enriched in malignant B cells. The properties of HVCN1S suggest that it may contribute to the pathogenesis of BCR-dependent B-cell malignancies.
The voltage-gated proton channel HVCN1 (or HV1 or VSOP) is a small protein that conducts protons across membranes selectively (1, 2) and in a regulated manner. Previously, we described its function in B lymphocytes, where proton channels sustain B-cell receptor (BCR) signaling via regulation of reactive oxygen species production by the NADPH oxidase enzyme complex (3). In addition, we found HVCN1 to be directly associated with the BCR. Upon receptor stimulation, the BCR and HVCN1 were cointernalized to late endosomal/lysosomal organelles called “MIICs,” or MHC class II-containing compartments, where antigens bound to the BCR are digested into small peptides and loaded onto MHC class II molecules for presentation to T cells (3).
HVCN1 is expressed not only by normal but also by malignant B cells, such as those in chronic lymphocytic leukemia (CLL) (3). CLL cells are characterized by their reliance on BCR signaling for survival and growth (4), so it is possible that they maintain or upregulate HVCN1 expression to sustain their growth. Other tumor cells, such as those in breast (5) and colorectal cancer (6), have been found to rely on HVCN1 for survival. In these tumor cells, proton channels prevent excessive acidification of the cytoplasm and allow increased cell migration. In malignant B cells, HVCN1 may regulate intracellular pH and at the same time sustain BCR signaling. However, its precise roles remain to be elucidated.
We show here that CLL cells and other B-cell lines specifically express higher levels of a shorter isoform of HVCN1, HVCN1S. We identified the existence of two distinct isoforms of relatively similar size when immunoblotting B-cell lysates with an HVCN1-specific antibody (3). HVCN1S is only weakly expressed in normal B cells, and in light of its apparent upregulation in tumor cells, we set out to characterize its function. We show that HVCN1S responds more strongly to phosphorylation by protein kinase C (PKC) and identify the phosphorylation site. We provide evidence that HVCN1S in B cells is preferentially expressed at the plasma membrane, even upon BCR stimulation and subsequent internalization, due to a weaker association with the BCR. Finally, we show that HVCN1S expression results in stronger BCR signaling, increased proliferation, and augmented chemokine-dependent migration. Overall, our data indicate that HVCN1S is an alternative protein isoform that mediates stronger currents upon PKC phosphorylation, is more highly expressed at the plasma membrane, and can confer a growth advantage to malignant B cells.
Results
Identification of an HVCN1 Isoform Enriched in Malignant B Cells.
We investigated HVCN1 expression in a panel of B-cell lines and noted the presence of two bands of similar molecular weight. Some cell lines appeared to express more of the shorter isoform, whereas the Burkitt cell line Raji expressed it exclusively (Fig. 1A, Left). Peripheral blood B cells expressed this short isoform (here called HVCN1S) at much lower levels compared with CLL cells (Fig. 1A, Center and Right).
Fig. 1.

HVCN1S is an alternative isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. (A, Left and Center) Immunoblots showing expression of two isoforms of HVCN1, HVCN1L (“L”), and HVCN1S (“S”) in B-cell lines, primary B cells, and CLL samples. R (RI1), U (U2932), and H (HBL1) are diffuse large B-cell lymphoma cell lines; E (EJM) is a multiple myeloma cell line, and Ra (RAJI) is a Burkitt lymphoma cell line. (Right) Densitometry analysis of protein expression of HVCN1S in B cells from healthy donors (n = 7) and CLL patients (n = 76). Protein expression levels as determined by Western blot, relative to loading control (β-actin or α-tubulin) and normalized to a positive control used across different blots (cell line HBL1). Statistical analysis carried out with Mann–Whitney U test. (B, Left and Center) In vitro transcription-translation assays with recombinant HVCN1L and mutated ttgHVCN1L. (Right) Expression of ttgHVCN1L transduced in LK35.2 cells. (C) PCR on HBL1 mRNA to identify the three mRNA sequences reported for human HVCN1 (SI Appendix, Fig. S1). (Left) Duplex PCR with primers designed for mRNA variant 2, which amplifies a band of 432 bp, and primers recognizing all variants (228 bp). (Right) PCR performed with different annealing temperatures with primers designed to recognize HVCN1 mRNA variants 1 and 3. The expected bands for isoforms 1 and 3 are 433 and 393 bp, indicated by arrows. (D) Immunoblots of a CLL sample and LK35.2 cells overexpressing HVCN1L and HVCN1S with an anti-HVCN1 that recognizes residues 26–46 (Left) and residues 1–20 (HVCN1L-specific, Right).
When we started this study, the GenBank database reported only one viable splicing variant for human HVCN1 (SI Appendix, Fig. S1, variant 1). Given the presence of an ATG 60 base pairs (bp) downstream from the first ATG, we set out to investigate if a shorter isoform could be the result of translation from this alternative start site. To this end, we expressed recombinant HVCN1 Long (HVCN1L) in an in vitro translation assay (Fig. 1B, Left). We then mutated the first ATG to TTG, which resulted in significantly reduced translation from this codon (Fig. 1B, Center). A shorter protein was expressed from the mutated plasmid (Fig. 1B, Center) when expressed in whole cells also (Fig. 1C, Right), indicating that the second ATG functioned as a start codon.
More recently, three splicing variants for human HVCN1 have been reported (SI Appendix, Fig. S1). Variant 1 and 2 differ in the use of alternative 5′-untranslated regions (UTRs) but code for the same full-length HVCN1 protein, whereas variant 3 uses the same 5′-UTR as variant 1 but it does not possess the first coding exon. Therefore, variant 3 lacks the first ATG and consequently translation can start only from the second ATG 60 bp downstream. To test the existence of the three mRNA variants, we designed specific PCR primers and tested them with mRNA from the cell line HBL1. We used the same reverse primer and specific forward primers for variants 1/3 and 2, annealing on their respective 5′-UTRs. To distinguish variants 1 and 3, we relied on differences in the band size of the amplified PCR product (433 for variant 1 and 393 for variant 3). As Fig. 1C, Left, shows, the PCR with primers for variant 2 and all variants produced bands of the expected sizes. For variants 1 and 3, we ran a gradient PCR (Fig. 1C, Right), which showed the expected bands of 433 and 393 bp (Fig. 1C, arrows). Overall, our results indicate the existence of three distinct mRNA variants and therefore suggest that HVCN1S is the result of alternative mRNA splicing, which produces a protein identical to the long isoform but lacking the first 20 amino acids.
To confirm the nature of this shorter isoform, we raised an antibody toward the first 20 amino acids of HVCN1, which should recognize the long isoform only. Our original anti-HVCN1 antibody, which recognizes a region in the N terminus closer to the first transmembrane domain (amino acids 26–46), detected both isoforms in CLL cells (Fig. 1D, Left). Also, recombinant HVCN1L and HVCN1S were both detected. In contrast, an immunoblot with the anti-HVCN1L–specific antibody detected only the long isoform, both recombinant and endogenously expressed (Fig. 1D, Right). These results confirmed that the shorter isoform of HVCN1 is a protein with a shorter N terminus domain, lacking the first 20 amino acids.
Both Isoforms Are Voltage-Gated Proton Channels.
Biophysical properties of HVCN1L and HVCN1S were compared in transduced LK-35.2 cells using the whole-cell voltage-clamp configuration over a wide range of pH. The H+ currents generated by the two isoforms appeared generally similar. SI Appendix, Fig. S2A, confirms that both isoforms were proton selective because the reversal potential for current (Vrev) was close to the Nernst potential for H+ (EH). This result is expected because the selectivity filter is in the middle of the S1 transmembrane helix (1), far from the N terminus where the two isoforms differ.
A unique property of all known voltage-gated proton channels crucial to their function is ΔpH-dependent gating (2, 7). The position of the proton conductance–voltage (gH–V) relationship depends strongly on both pHo and pHi, with the consequence that the channel opens only when there is an outward electrochemical gradient, and the open channel will extrude H+ from the cell. HVCN1L and HVCN1S exhibited similar ΔpH-dependent gating, both activating at potentials about 10 mV positive to Vrev at symmetrical pH (SI Appendix, Fig. S2B).
To compare gating kinetics, we used the perforated-patch voltage-clamp method, a more physiological configuration that preserves cytoplasmic contents. Comparison of the gating kinetics of HVCN1L and HVCN1S in unstimulated LK-35.2 cells (SI Appendix, Table S1) reveals that HVCN1S channels opened more slowly, with a time constant of current turn-on, τact, more than double that of HVCN1L. Closing kinetics (τtail) and the position of the gH-V relationship did not differ significantly.
HVCN1S Responds More Strongly to PKC-Dependent Phosphorylation.
Proton currents in phagocytes and other cells are greatly augmented by phosphorylation of the channel by PKC (8). The enhanced gating response is stimulated effectively by the PKC activator PMA (phorbol myristate acetate) and is best studied using the perforated-patch configuration that preserves intracellular signaling pathways (9). Fig. 2 illustrates families of proton currents in cells expressing HVCN1L and HVCN1S before and after PMA stimulation. In response to PMA, the currents turn on more rapidly and at more negative voltages and turn off more slowly, and the current amplitude is increased. Although HVCN1L responds distinctly, the response of HVCN1S was consistently more profound. Because there is a tendency early in each experiment for proton currents to become larger and activate at more negative voltages as the amphotericin in the pipette solution improves electrical access to the cell membrane, and as pHi is clamped to 7.0 by the applied NH4+ gradient (9, 10), the PMA response may be exaggerated if measurements are made before complete equilibration. A crucial quantitative control is to reverse the effects of PMA using the PKC inhibitor GF 109203X (GFX). The reversal of enhanced gating by GFX in both representative cells in Fig. 2 was complete, validating the responses.
Fig. 2.

HVCN1S responds more strongly to PMA stimulation than HVCN1L. Perforated-patch voltage clamp was used to evaluate the electrophysiological properties of the two HVCN1 isoforms. Families of currents in 10-mV increments up to 70 mV from Vhold = −40 mV are shown in representative LK35.2 cells expressing HVCN1L (A) or HVCN1S (B) before stimulation, after application of the PKC activator PMA, and after inhibition of PKC by GF 109203X (GFX). Between the families are superimposed currents obtained during test pulses to 60 mV (for HVCN1L) or 40 mV (for HVCN1S) applied at 30-s intervals before and after addition of PMA or GFX to the bath solution. (C and D) Proton conductance–voltage relationships, gH–V. The current amplitude was determined by extrapolating a single exponential fitted to the rising current, and gH was calculated from the current using Vrev measured in each solution. Measurements were made in symmetrical pH 7.0 solutions containing 50 mM NH4+ to clamp pHi (9).
SI Appendix, Fig. S3, shows normalized gH-V relationships after the PMA response and after GFX treatment in all cells studied expressing HVCN1L or HVCN1S. This comparison is more informative than control vs. PMA for reasons just discussed and because some cells were spontaneously active, as judged by GFX reversal being greater than the initial response to PMA. A possible spurious explanation for the greater PMA responsiveness of HVCN1S is that HVCN1L might have a greater tendency to activate spontaneously. SI Appendix, Fig. S3, does not support this view, verifying that HVCN1S is indeed more responsive.
SI Appendix, Fig. S4, summarizes the magnitude of the changes in four kinetic parameters of HVCN1 gating resulting from PMA and GFX treatment of cells expressing the two isoforms. For each parameter, the PMA response was significantly greater in HVCN1S than in HVCN1L. Also for each parameter, GFX reversed the PMA response completely. In summary, HVCN1S channels exhibit a more profound response to phosphorylation than do HVCN1L channels.
Because B cells from CLL patients have higher-than-normal levels of HVCN1S, their responsiveness might be enhanced. B cells from CLL patients and normal controls were studied by perforated-patch voltage clamp (n = 4 and 3, respectively), confirming that both CLL and normal B cells had proton currents that responded to PMA and GFX. Interestingly, CLL cells, which have a variable mixture of HVCN1S and HVCN1L (SI Appendix, Table S3), exhibited enhanced gating responses comparable with those of LK35.2 cells expressing HVCN1S exclusively (SI Appendix, Fig. S5), indicating that HVCN1S prevails in the response.
Identification of HVCN1S Phosphorylation Site.
Two predicted high-probability PKC-δ phosphorylation sites exist in the N terminus of HVCN1L (11), Thr29 and Ser97. We reported previously that both sites were phosphorylated after PMA exposure, but only Thr29 contributed detectably to the enhanced gating response of HVCN1L channels (11). Because the lack of the first 20 amino acids of the N terminus might result in different protein folding or architecture, it was important to examine the contribution of both corresponding sites in HVCN1S, namely Thr9 and Ser77. We generated mutants that lacked one (T9A and S77A) or both (T9A/S77A) putative phosphorylation sites.
Cells with the T9A mutation (n = 9) and the double mutant T9A/S77A (n = 5) did not respond detectably to either PMA or GFX (SI Appendix, Fig. S6), implicating Thr9 as crucial to the responses. In contrast, cells expressing the S77A mutant did respond to PMA and to GFX. Mean changes in parameter values are given in SI Appendix, Table S2. Therefore, the key residue in the PMA response is Thr9 (HVCN1S), the equivalent of Thr29 in HVCN1L channels (11). Any participation of Ser77 (HVCN1S) or Ser97 (HVCN1L) in the enhanced gating response was below our ability to detect it.
Intriguingly, T9A cells studied before stimulation exhibited ninefold faster activation and a 24-mV more negative Vthreshold than did unstimulated HVCN1S cells (SI Appendix, Table S1), resembling enhanced gating mode behavior. We reanalyzed data from analogous mutations studied previously in the full-length channel (11) and found that T29A also exhibited “enhanced gating” in unstimulated cells (SI Appendix, Table S1). Unstimulated “phosphomimetic” T29D cells exhibited weaker enhanced gating than T29A. Evidently, this Thr position is a potent determinant of gating kinetics. Together, these results suggest that phosphorylation of Thr29 in HVCN1L or Thr9 in HVCN1S produces enhanced gating by a mechanism not simply involving the negative charge provided by the phosphate group. However, we cannot rule out the possibility that these mutations alter gating through a mechanism unrelated to that elicited by phosphorylation.
Because HVCN1S responded more strongly to PKC phosphorylation and the effect was mediated by phosphorylation of Thr9 exclusively, we speculated that it could reflect increased phosphorylation of this residue. Indeed, our in vitro kinase assay showed that overall phosphorylation of wild-type HVCN1S was significantly higher than for wild-type HVCN1L (Fig. 3, Left, WT lanes). To assess phosphorylation of the critical PKC site mediating the enhanced-gating response, we carried out the same experiment with T29A (T9A for HVCN1S), S97A (S77A for HVCN1S), and T29A/S97A (T9A/S77A for HVCN1S) mutants. As shown in Fig. 3, phosphorylation of the T9A mutant was reduced as much as for the T29A one, confirming that this Thr is phosphorylated by PKC in both isoforms. The analysis of the other PKC site mutant, S97A (S77A for HVCN1S), and double mutants indicated a further reduction in overall phosphorylation, as we reported previously for HVCN1L (11), suggesting that Ser is also phosphorylated by PKC. However, as before, the patch-clamp experiments (SI Appendix, Fig. S6 and Table S1) indicated that phosphorylation of Thr9 alone is responsible for the enhancement of HVCN1S proton currents.
Fig. 3.
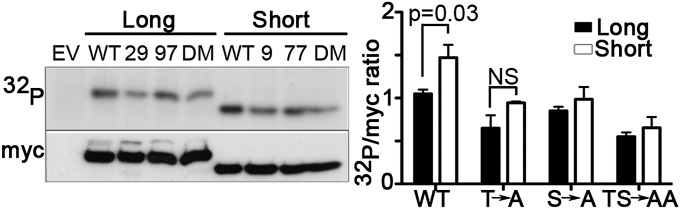
HVCN1S is phosphorylated more by PKC-δ than HVCN1L. PKC-δ in vitro kinase assay showing phosphorylation of HVCN1L and HVCN1S wild type (“WT”), single mutants T29A (“29”) or T9A for HVCN1S (“9”), S97A (“97”) or S77A for HVCN1S (“77”), and double mutants T29A/S97A and T9A/S77A for HVCN1S (“DM”). The assay was carried out with recombinant HVCN1L and HVCN1S expressed in HEK293T cells and immunoprecipitated with an anti-myc antibody. Cells transfected with an empty vector (“EV”) were used as negative control. The myc immunoblot indicates loading. Bars represent the densitometry analysis of the 32P-HVCN1 versus myc-HVCN1 of three independent experiments (mean ± SEM). NS, not significant.
HVCN1S Has a Weaker Association with the BCR than Does HVCN1L.
We found previously that HVCN1 was associated with the BCR in both normal and malignant B cells (3). The association causes HVCN1 to be internalized together with the BCR after its stimulation, an event that downregulates HVCN1 expression at the cell surface (3). Because the intracellular domains are likely to be the regions mediating the association with the BCR, we investigated if HVCN1S behaved differently from HVCN1L. We overexpressed both isoforms in the B lymphoma cell line A20 D1.3 and assessed their expression at steady state and after BCR stimulation with a F(ab′)2 anti-IgM. Cells expressed similar levels of HVCN1, IgM, and the BCR coreceptor CD79B (SI Appendix, Fig. S7 A–C). We did not note significant differences in the pattern of expression of the two isoforms at steady state. Importantly, however, upon BCR stimulation, the extent of HVCN1 internalization for the two isoforms appeared different (Fig. 4A). Quantification of the immunofluorescence staining showed a significantly larger percentage of internalized HVCN1 in cells expressing HVCN1L compared with HVCN1S (36 vs. 25%, Fig. 4A, Upper graph). Furthermore, the extent of colocalization with IgM was also reduced from 0.562 to 0.407 (Fig. 4A, Lower graph). Diminished HVCN1S internalization was not due to reduced IgM internalization, which was actually increased, compared with cells overexpressing HVCN1L (SI Appendix, Fig. S7D). These data indicate that HVCN1S association with the BCR is different and that this results in increased localization of proton channels at the plasma membrane.
Fig. 4.

HVCN1S is associated and internalized with the BCR to a lesser extent compared with HVCN1L. (A, Left) Confocal images of A20 D1.3 cells overexpressing HVCN1L or HVCN1S at steady state (“resting” panels) or 30 min after activation with an anti-IgM crosslinking F(ab')2 fragment (“activated” panels). Merge panels represent IgM and HVCN1 staining. White arrows indicate cointernalized HVCN1 and IgM. (Right) Quantification of percentage of internalized HVCN1 (Upper graph) and colocalization coefficient for HVCN1 and IgM (Lower graph). Each symbol represents a single cell [n = 78 (Upper); n = 67 (Lower)], and horizontal lines indicate the mean. Pearson’s colocalization coefficient (0, no colocalization; 1, total colocalization). (B) Coimmunoprecipitation of overexpressed HVCN1L or HVCN1S and endogenous CD79B (Ig-associated-β or Ig-β). Proteins were coimmunoprecipitated from A20 D1.3 cells and analyzed by immunoblot in nonreducing conditions. EV, cells transduced with empty vector; IN, input cell lysate (2% of the cell lysate used for immunoprecipitation); Ig, negative control beads conjugated to mouse or rat IgG; IP, immunoprecipitation; IB, immunoblot. Graphs represent densitometry analysis of both co-IP experiments (mean ± SEM, three independent experiments).
To determine the reason for the different association of HVCN1 isoforms with the BCR, we carried out a coimmunoprecipitation (co-IP) with an anti-CD79B and the reciprocal experiment with an anti-myc (for the myc-tagged HVCN1 isoforms). As shown in Fig. 4B, interaction of HVCN1S with CD79B was indeed significantly weaker with both co-IP assays. The differences between the two antibodies are likely due to differences in the efficiency of pull down (with the anti-myc being more efficient than the anti-CD79B). Overall, these data show that the first 20 amino acids present in HVCN1L are critical for the association with the BCR and that their absence in HVCN1S results in greater expression of this isoform at the plasma membrane after BCR stimulation.
HVCN1S Expression Is Enriched in CLL Cells.
Given the increased expression of HVCN1S in B-cell lines and in CLL samples (Fig. 1A), combined with its ability to mediate stronger proton currents (Fig. 2), we investigated whether its expression could have an impact on CLL disease progression. To this end, we analyzed the expression of HVCN1S in a cohort of 76 samples of peripheral blood CLL cells with annotated clinical data. Details of the patients’ characteristics are reported in SI Appendix, Table S3. The samples investigated showed variable expression of both isoforms, with an average ratio of HVCN1S to HVCN1L of 0.52 ± 0.034 (mean ± SE). As shown in Fig. 1A, HVCN1S was markedly higher in CLL than in primary B cells from healthy donors, in which HVCN1S is barely detectable. Given the wide range of HVCN1S expression in CLL cells, we split samples in two groups, one with a ratio of HVCN1S/HVCN1L below the median value of 0.477 (i.e., with lower expression of HVCN1S) and one with higher HVCN1S expression (above the median). A Kaplan–Meier curve for overall survival showed that patients with higher expression of HVCN1S had reduced overall survival, although differences did not reach statistical significance (SI Appendix, Fig. S8). We further analyzed the data specifically for the two main subgroups of CLL patients: those presenting a mutated variable region in the BCR heavy chain (IGHV) and those with an unmutated IGHV. Given the small number of samples for which we knew the mutation status (SI Appendix, Fig. S8 and Table S3), it was not surprising that differences did not reach statistical significance; nonetheless, higher expression of HVCN1S tended to correlate with a poorer outcome in both groups.
HVCN1S Expression Results in Stronger BCR-Dependent Signaling, Proliferation, and Chemokine-Dependent Migration.
To establish if HVCN1S could confer a growth advantage to malignant B cells, we first assessed BCR-dependent signaling in A20 D1.3 cells transduced with empty vector (EV), HVCN1L, and HVCN1S. As shown in Fig. 5A, the presence of HVCN1S resulted in more prolonged activation of the extracellular signal-regulated kinase (Erk), which is responsible for the upregulation of several antiapoptotic proteins in CLL cells (12). Furthermore, to determine if the two proton channel isoforms regulated cell proliferation, we measured the extent of EdU incorporation, a nucleoside analog included in nascent DNA. Interestingly, only HVCN1S provided a significant advantage compared with EV cells (Fig. 5B).
Fig. 5.

HVCN1S expression modulates BCR signaling, cell proliferation, and migration. (A) BCR stimulation with 20 μg/mL F(ab')2 anti-IgM in A20 D1.3 cells overexpressing empty vector (EV), HVCN1L, and HVCN1S. Immunoblot showing phosphorylated Erk (p-Erk) and total Erk. Bars indicate the ratio of the densitometry analysis of p-Erk versus total Erk from three independent experiments (mean ± SEM). (B) Proliferation of A20 D1.3 cells assessed by EdU incorporation after 3 h of incubation. Results are shown as percentage of Edu+ cells versus total number of GFP+ cells of two independent experiments (mean ± SEM). (C) Transwell chamber assay of migration toward the chemokine CXCL12. Migrated cells were counted after 4 h. Results are shown as fold increase of migrated cells in the presence of CXCL12 versus media alone. Data represent the average of two experiments (mean ± SEM).
Another important property of CLL cells is the ability to respond to the chemokine CXCL12 [chemokine (C-X-C motif) ligand 12], which affects their migration, homing, and survival (13). Because HVCN1 was shown to regulate migration of breast (5) and colorectal cancer cells (6), we set out to characterize if HVCN1 isoforms affected chemokine-dependent migration in a B-cell setting. As Fig. 5C shows, both HVCN1L and HVCN1S expression resulted in increased migration to CXCL12; however, only HVCN1S resulted in a significant advantage. Taken together, these data indicate that HVCN1S promotes malignant B-cell survival through enhanced proliferation and migration.
Discussion
Only one proton channel gene has been identified in any species. However, the human gene can generate two different isoforms, HVCN1L and HVCN1S (3). In this paper, we confirmed the existence of alternative splicing variants as reported in the GenBank database, presenting evidence that translation of HVCN1S starts at an alternative ATG. The resulting protein is 20 amino acids shorter at the N terminus, as confirmed here by immunoblotting with an antibody raised against the first 20 amino acids of full-length HVCN1 (HVCN1L). Compared with peripheral B cells from healthy donors, B-cell lines and CLL cells showed increased expression of total HVCN1 due to an upregulation of HVCN1S. Higher levels of HVCN1S tended to correlate with decreased overall survival in a cohort of 76 blood samples from CLL patients. Given the wide range of expression of HVCN1S in CLL and the limited number of samples analyzed, it would be necessary to screen a much larger panel of samples to determine if this trend is significant. This would be particularly interesting for the mutated CLL subgroup because these patients have a more favorable prognosis overall; however, some still present more aggressive disease, and markers to identify this subpopulation are lacking.
Comparison of their electrophysiological properties revealed that HVCN1S channels open about twice as slowly as HVCN1L channels. A more profound difference was seen in response to stimulation by PMA. Agonists that activate PKC strongly amplify the proton conductance in many human and mammalian cells (8, 9, 14–18), a phenomenon called “enhanced gating.” Although both isoforms responded to PMA, the HVCN1S response was significantly greater. This differential responsiveness enables cells to modulate proton channel activity by preferential expression of HVCN1S or HVCN1L isoforms. That the PMA response of CLL cells resembled that of LK35.2 cells expressing exclusively HVCN1S reveals that, in a mixture of isoforms, HVCN1S will dominate due to its lower Vthreshold.
The distinct gating of the two isoforms HVCN1S and HVCN1L that differ only in the first 20 amino acids of the N terminus emphasizes the importance of this intracellular domain in modulating gating kinetics. Proton channels are thought to open as a result of outward movement of the S4 transmembrane helix (19–21). Being an extension of the S4 helix, the C terminus affects gating directly (22). Nevertheless, gating kinetics is modulated drastically (i) in the enhanced gating mode by phosphorylation of Thr29 (11), (ii) by the point mutation T9A in HVCN1S or T29A in HVCN1L (SI Appendix, Table S1), and (iii) in the naturally occurring mutation M91T (23), all of which are localized to the N terminus.
Because T9A and T9A/S77A mutants failed to respond to PMA or GFX, enhanced gating of HVCN1S is evidently mediated entirely by phosphorylation of Thr9. This residue is equivalent to Thr29, the key phosphorylation site producing enhanced gating in HVCN1L (11). Here we show that phosphorylation of Thr9 and overall phosphorylation is greater in HVCN1S than in HVCN1L, suggesting that the loss of the first 20 amino acids facilitates phosphorylation of this residue. Additionally, the mechanism by which phosphothreonine orchestrates enhanced gating may be modified in HVCN1S. Whether the two isoforms are phosphorylated at steady state in tumor B cells will depend upon expression and activation of PKC in these cells. Because PKC was expressed and activated in the great majority of CLL samples investigated (SI Appendix, Table S3), we believe constitutive phosphorylation of proton channels in CLL cells to be highly likely.
Furthermore, we present evidence that HVCN1S has a weaker association with the BCR, which results in greater expression at the plasma membrane and reduced internalization upon BCR stimulation. Evidently, the first 20 amino acids of HVCN1 are important for association with the BCR. It will be interesting to investigate which residues in HVCN1 are involved in this interaction and whether there is a direct association with the BCR complex (either the Ig or the coreceptors CD79A and CD79B) or if additional scaffolding proteins are involved. Malignant B cells such as CLL cells are stimulated through their BCR in the tumor microenvironment, and each round of stimulation normally results in BCR internalization (4). That tumor B cells upregulate an isoform of HVCN1 that remains preferentially at the plasma membrane after BCR stimulation suggests that these cells might find plasma membrane expression of HVCN1S beneficial to their growth. This is corroborated by the advantage in BCR-dependent Erk activation, proliferation, and chemokine-dependent migration conferred by HVCN1S. Higher levels of HVCN1 expression correlate with metastatic tendency and poor prognosis in breast cancer (5) and colorectal cancer (6) although the isoform involved has not been reported.
Overall, our data show the existence of a shorter isoform of HVCN1 with enhanced gating responses that is specifically enriched in malignant B cells. The enhanced gating of HVCN1S produces larger proton currents in CLL cells that may contribute to the pathology, as suggested by stronger BCR signaling, increased cell proliferation, and chemokine response provided by HVCN1S expression.
Materials and Methods
Cell Lines and Plasmids.
The mouse B lymphoma cell lines LK35.2 HyHEL10 (IgG2a, κ-chain; H-2kxd) and A20 D1.3 (IgG2a, κ-chain; H-2d) overexpressing recombinant IgM receptors were a gift from F. Batista (London Research Institute, London). The cells were transduced with MigRI retroviral vectors coding for myc-tagged HVCN1L or HVCN1S. T29A (T9A for HVCN1S), S97A (S77A for HVCN1S), and T29A/S97A (T9A/S77A for HVCN1S) mutants were generated using a QuikChange site-directed mutagenesis kit (Stratagene). The retroviral particles were produced in the Phoenix α packaging cell line as described elsewhere (11).
Phosphorylation Assay.
HEK293T cells were transfected by Ca2+ phosphate with myc-tagged HVCN1L, HVCN1S, and mutant plasmids. Forty-eight hours after transfection, cells were lysed in 20 mM Hepes, 1% Triton X-100, 137 mM NaCl, 2.5 mM β-glycerophosphate, 1 mM Na3VO4, 2 mM EDTA, and proteases inhibitors (Sigma Aldrich). One milligram of proteins was immunoprecipitated with anti-myc tag antibody (9B11, Cell Signaling Technology) conjugated to protein-G Sepharose beads for 1 h. After washing with lysis buffer, beads were incubated in 40 μL kinase assay buffer [20 mM Hepes, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.1 mM CaCl2, 0.05 mM DTT, 0.1 mg/mL phosphatidylserine, 0.01 mg/mL diacylglycerol, 2.5 mM β-glycerophosphate, 1 μM PMA, 100 nM PKC-δ (Millipore), 100 μM cold ATP, and 10 μCi of [γ-32P]ATP (Perkin-Elmer)] for 20 min at 30 °C. The reaction was stopped by resuspending beads in 2× Laemmli sample buffer. Samples were then separated by SDS/PAGE, transferred to a nitrocellulose membrane, and exposed to X-ray films. Membranes were immunoblotted with anti-myc antibody to determine loading.
Electrophysiology.
Whole-cell or perforated-patch variants of the patch-clamp technique were carried out as described in detail previously (24). Perforated-patch studies included ∼225 μM amphotericin B in the pipette to permeabilize the patch membrane, and 50 mM NH4+ to clamp pHi to pHo. For whole-cell studies, the main pipette solution (also used externally) contained (in mM) 130 TMACH3SO3, 2 MgCl2, 2 EGTA, and 80 MES, titrated to pH 5.5 with ∼20 tetramethylammonium hydroxide (TMAOH). Bath solutions at pH 7.0 had (in mM) 90 TMACH3SO3, 3 CaCl2, 1 EGTA, 100 N,N-Bis-(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), and 36–40 TMAOH. When pH was varied, the following buffers with pKa near the desired pH were used: Homopipes for pH 4.6, MES for pH 5.5–6.0, BisTris for pH 6.5, Hepes for pH 7.5, and Tricine for pH 8.0. Experiments were done at 21 °C or at room temperature (20–25 °C). No leak correction has been applied to current records.
Patch-Clamp Studies of CLL and Healthy B Cells.
Cells were shipped frozen on dry ice. After thawing, cells were suspended in RPMI with serum, centrifuged at 1,100 × g for 5 min, and resuspended in a small volume (1–2 mL) of RPMI with serum.
Statistics.
Statistical analysis was carried out by Student’s unpaired t test. Further materials and methods are provided in SI Appendix.
Supplementary Material
Acknowledgments
M.C. is the recipient of a Bennett Fellowship from Leukaemia and Lymphoma Research (ref. 12002). M.A.B. is supported by a GlaxoSmithKline Oncology–Biotechnology and Biological Sciences Research Council Collaborative Awards in Science and Engineering PhD studentship. This work was supported by National Institutes of Health Grants GM087507 and GM102336 (to T.E.D.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411390111/-/DCSupplemental.
References
- 1.Musset B, et al. Aspartate 112 is the selectivity filter of the human voltage-gated proton channel. Nature. 2011;480(7376):273–277. doi: 10.1038/nature10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeCoursey TE. Voltage-gated proton channels: Molecular biology, physiology, and pathophysiology of the HV family. Physiol Rev. 2013;93(2):599–652. doi: 10.1152/physrev.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capasso M, et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat Immunol. 2010;11(3):265–272. doi: 10.1038/ni.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313–4320. doi: 10.1182/blood-2011-06-338855. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Li SJ, Wu X, Che Y, Li Q. Clinicopathological and biological significance of human voltage-gated proton channel Hv1 protein overexpression in breast cancer. J Biol Chem. 2012;287(17):13877–13888. doi: 10.1074/jbc.M112.345280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Wu X, Li Q, Zhang S, Li SJ. Human voltage-gated proton channel Hv1: A new potential biomarker for diagnosis and prognosis of colorectal cancer. PLoS ONE. 2013;8(8):e70550. doi: 10.1371/journal.pone.0070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherny VV, Markin VS, DeCoursey TE. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J Gen Physiol. 1995;105(6):861–896. doi: 10.1085/jgp.105.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan D, et al. Sustained activation of proton channels and NADPH oxidase in human eosinophils and murine granulocytes requires PKC but not cPLA2 α activity. J Physiol. 2007;579(Pt 2):327–344. doi: 10.1113/jphysiol.2006.124248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeCoursey TE, Cherny VV, Zhou W, Thomas LL. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci USA. 2000;97(12):6885–6889. doi: 10.1073/pnas.100047297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grinstein S, Romanek R, Rotstein OD. Method for manipulation of cytosolic pH in cells clamped in the whole cell or perforated-patch configurations. Am J Physiol. 1994;267(4 Pt 1):C1152–C1159. doi: 10.1152/ajpcell.1994.267.4.C1152. [DOI] [PubMed] [Google Scholar]
- 11.Musset B, et al. Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. J Biol Chem. 2010;285(8):5117–5121. doi: 10.1074/jbc.C109.082727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Efremov DG, Gobessi S, Longo PG. Signaling pathways activated by antigen-receptor engagement in chronic lymphocytic leukemia B-cells. Autoimmun Rev. 2007;7(2):102–108. doi: 10.1016/j.autrev.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 13.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43(3):461–466. doi: 10.1080/10428190290011921. [DOI] [PubMed] [Google Scholar]
- 14.Bánfi B, et al. A novel H+ conductance in eosinophils: Unique characteristics and absence in chronic granulomatous disease. J Exp Med. 1999;190(2):183–194. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeCoursey TE, Cherny VV, DeCoursey AG, Xu W, Thomas LL. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J Physiol. 2001;535(Pt 3):767–781. doi: 10.1111/j.1469-7793.2001.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mori H, et al. Regulatory mechanisms and physiological relevance of a voltage-gated H+ channel in murine osteoclasts: Phorbol myristate acetate induces cell acidosis and the channel activation. J Bone Miner Res. 2003;18(11):2069–2076. doi: 10.1359/jbmr.2003.18.11.2069. [DOI] [PubMed] [Google Scholar]
- 17.Musset B, Cherny VV, DeCoursey TE. Strong glucose dependence of electron current in human monocytes. Am J Physiol Cell Physiol. 2012;302(1):C286–C295. doi: 10.1152/ajpcell.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musset B, et al. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc Natl Acad Sci USA. 2008;105(31):11020–11025. doi: 10.1073/pnas.0800886105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez C, Rebolledo S, Perez ME, Larsson HP. Molecular mechanism of voltage sensing in voltage-gated proton channels. J Gen Physiol. 2013;141(3):275–285. doi: 10.1085/jgp.201210857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kulleperuma K, et al. Construction and validation of a homology model of the human voltage-gated proton channel hHV1. J Gen Physiol. 2013;141(4):445–465. doi: 10.1085/jgp.201210856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takeshita K, et al. X-ray crystal structure of voltage-gated proton channel. Nat Struct Mol Biol. 2014;21(4):352–357. doi: 10.1038/nsmb.2783. [DOI] [PubMed] [Google Scholar]
- 22.Fujiwara Y, Kurokawa T, Okamura Y. Long α helices projecting from the membrane as the dimer interface in the voltage-gated H+ channel. J Gen Physiol. 2014;143(3):377–386. doi: 10.1085/jgp.201311082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iovannisci D, Illek B, Fischer H. Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J Gen Physiol. 2010;136(1):35–46. doi: 10.1085/jgp.200910379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan D, Cherny VV, Murphy R, Katz BZ, DeCoursey TE. The pH dependence of NADPH oxidase in human eosinophils. J Physiol. 2005;569(Pt 2):419–431. doi: 10.1113/jphysiol.2005.094748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.