

ABSTRACT
Proteasomes are large, multisubunit complexes that support normal cellular activities by executing the bulk of protein turnover. During infection, many viruses have been shown to promote viral replication by using proteasomes to degrade cellular factors that restrict viral replication. For example, the human cytomegalovirus (HCMV) pp71 protein induces the proteasomal degradation of Daxx, a cellular transcriptional repressor that can silence viral immediate early (IE) gene expression. We previously showed that this degradation requires both the proteasome catalytic 20S core particle (CP) and the 19S regulatory particle (RP). The 19S RP associates with the 20S CP to facilitate protein degradation but also plays a 20S CP-independent role promoting transcription. Here, we present a nonproteolytic role of the 19S RP in HCMV IE gene expression. We demonstrate that 19S RP subunits are recruited to the major immediate early promoter (MIEP) that directs IE transcription. Depletion of 19S RP subunits generated a defect in RNA polymerase II elongation through the MIE locus during HCMV infection. Our results reveal that HCMV commandeers proteasome components for both proteolytic and nonproteolytic roles to promote HCMV lytic infection.
IMPORTANCE Proteasome inhibitors decrease or eliminate 20S CP activity and are garnering increasing interest as chemotherapeutics. However, an increasing body of evidence implicates 19S RP subunits in important proteolytic-independent roles during transcription. Thus, pharmacological inhibition of the 20S CP as a means to modulate proteasome function toward therapeutic effect is an incomplete capitalization on the potential of this approach. Here, we provide an additional example of nonproteolytic 19S RP function in promoting HCMV transcription. These data provide a novel system with which to study the roles of different proteasome components during transcription, a rationale for previously described shifts in 19S RP subunit localization during HCMV infection, and a potential therapeutic intervention point at a pre-immediate early stage for the inhibition of HCMV infection.
INTRODUCTION
Proteasomes mediate the majority of cellular protein turnover, promoting multiple signaling pathways, cell cycle progression, and general cellular homeostasis (1). These large, multisubunit complexes also permit immune detection of virally infected cells by generating the foreign peptides displayed by major histocompatibility complexes (MHCs) (2). In addition to this prominent antiviral role, proteasomes promote viral infection through the degradation of cellular factors that restrict viral infection. The dichotomous roles of proteasomes during viral infection and their general requirement for cellular health necessitate that viruses modulate proteasomal activities in a precise manner to promote viral infection (3–5).
The barrel-like 20S catalytic core particle (CP) of the proteasome, consisting of two sets of seven distinct alpha (α1 to α7) and beta (β1 to β7) subunits, contains the proteolytic components responsible for protein degradation (including the β1 caspase-, β2 trypsin-, and β5 chymotrypsin-like subunits) (6). One or both ends of the 20S CP can associate with an assortment of proteasome activators that vary proteasome CP accessibility and function (7). For example, the 20S CP associates with the 19S regulatory particle (RP) to form the 26S proteasome. The 19S RP is a proteasome activator made up of at least 18 distinct subunits that are classified as either base or lid components (8). The base of the 19S RP directly contacts the 20S CP and contains six ATPase subunits (Rpt1 to Rpt6), as well as two non-ATPase subunits (Rpn1 and Rpn2). The lid consists of eight subunits (Rpn3, -5, -6, -7, -8, -9, -11, and -12), one of which (Rpn11) contains deubiquitinase activity (9). Rpn10 and Rpn13 are ubiquitin receptors that show association with the 19S RP (10, 11). The 26S proteasome complex mediates the majority of ubiquitin-dependent protein degradation within cells but catalyzes ubiquitin-independent degradation as well (12, 13).
At the start of lytic infection, human cytomegalovirus (HCMV), a ubiquitous herpesvirus that causes significant morbidity and mortality in individuals with compromised immune systems, subverts 26S proteasome function for the degradation of key cellular factors capable of restricting viral replication (14–20). Upon entry, HCMV virions deposit the viral tegument protein pp71 into the cell, where it commandeers the 26S proteasome to degrade cellular transcriptional corepressors Daxx, BclAF-1, retinoblastoma protein (Rb), p107, and p130 to promote viral gene expression (16, 18, 19, 21–22). Interestingly, in the only two cases analyzed (Daxx and Rb), these degradation events do not require prior substrate ubiquitination but still utilize the 19S RP, a proteasome activator predominantly associated with ubiquitin-dependent processes (13, 19, 23).
In addition to their roles in ubiquitin-dependent (1) and ubiquitin-independent (24) proteasomal degradation events, 19S RP subunits are emerging as transcriptional regulators independent of their association with the 20S CP. In vitro transcription with human RNA polymerase II (RNAPII) requires the 19S RP, while 19S RP subunits promote transcriptional elongation and are found associated with active promoters in Saccharomyces cerevisiae (25–28). Furthermore, Tat-mediated transcription from an integrated HIV-1 long terminal repeat requires 19S RP function (29). During the late stages of HCMV infection, 19S RP subunits relocalize to the periphery of replication compartments where there are robust levels of transcription (30), but the significance of this phenomenon is not understood. While small-molecule inhibitor studies have demonstrated a role for the 20S CP in HCMV gene expression (18, 30, 31), here we explore a role for 19S subunits in that process. We found that 19S RP subunits are recruited to and required for efficient transcriptional initiation and elongation of the major immediate early (MIE) locus at the early stages of HCMV infection. In contrast, the 20S CP is neither recruited to the promoter nor required for transcriptional elongation but does promote RNA polymerase II (RNAPII) occupancy of the MIE promoter (MIEP), as expected. We thus define a nonproteolytic role for proteasome components in promoting HCMV immediate early (IE) gene expression.
MATERIALS AND METHODS
Cells and viruses.
Human foreskin fibroblasts (HFs), telomerase-immortalized human foreskin fibroblasts (tHFs), and cells depleted of Daxx by short hairpin RNA targeting Daxx (shDaxx) (32) were cultured as previously described (18). Cells were infected with HCMV-AD169 as previously described (18). For small interfering RNA (siRNA) experiments, the cell count of a companion scrambled (Scr) well was used to determine viral inocula for the desired multiplicity of infection (MOI) for all other wells. Cell cycle analysis by flow cytometry has been previously described (33).
Inhibitors and antibodies.
Trichostatin A (TSA); 100 ng/ml in dimethyl sulfoxide ([DMSO]) (Millipore) was added 20 h prior to infection; lactacystin (20 μM in DMSO; Calbiochem) was added at the start of infection. Antibodies used for immunoblotting and chromatin immunoprecipitation (ChIP) assays are listed in Table 1. Goat anti-mouse and goat anti-rabbit secondary antibodies conjugated with horseradish peroxidase were purchased from Chemicon and used for visualizing immunoblots with film as previously described (18). Immunoblots for semiquantitative analysis were also analyzed using a Li-Cor Odyssey Fc imaging system as previously described (13). All quantitative immunoblotting graphs represent the means ± standard errors (SE) of at least three biological replicates and have been analyzed with two-tailed paired t tests.
TABLE 1.
Antibodies and siRNA sequences
| Protein | Antibody or siRNA (source or reference) | siRNA sequence |
|---|---|---|
| Rpn1 | SC-68352 (Santa Cruz) | 5′-GACAAAGACAAGAAGGAAAUU-3′ |
| Rpt2 | PW8305 (Enzo) | 5′-AGAAGGAUGACAAGGACAAUU-3′ |
| Rpn11 | S2324 (Sigma) | 5′-GGACAUGAACCAAGACAAAUU-3′ |
| Rpn7 | PW8225 (Enzo) | |
| β5 | PW8895 (Enzo) | 5′-CCAACAUGGUGUAUCAGUAUU-3′ |
| β7 | PW9150 (Enzo) | |
| IgG | I5381 (Sigma) | |
| RNAPII, unphosphorylated | MMS-126R (Covance), 8WG16 monoclonal | |
| RNAPII, Ser2 phosphorylated | MMS-129R (Covance), H5 monoclonal | |
| p53 | OP43 (Calbiochem) | |
| Daxx | D7810 (Sigma) | |
| β-actin | ab8226 (Abcam) | |
| pp71 | 2H10-9 (33) | |
| IE1 | 1B12 (50) | |
| Scrambled | D-001810-01-20 (Dharmacon) |
RNA interference.
Cells were transfected with small interfering RNA oligonucleotides from Dharmacon (Table 1). Transfections were carried out using Lonza nucleofection reagents (VPI-1002; Lonza) or Lipofectamine RNAiMax (13778150; Invitrogen), according to the manufacturers' protocols. In all experiments, cells were serum starved for 24 h after transfection with medium containing 0.1% fetal bovine serum and infected at 72 h posttransfection. When cells were transfected with Lonza reagents, an equal number of HFs were transfected with 80 pmol of siRNA/106 cells (for Rpn1 and β5) and equally distributed among culture dishes. When Lipofectamine RNAiMax was used, an equal number of HFs were transfected with 20 pmol (for Rpn1 and β5), 40 pmol (Rpn11), or 80 pmol (Rpt2) of siRNA/106 cells. In all experiments, concentrations were balanced with scrambled siRNA to equalize siRNA delivery.
Chromatin immunoprecipitations.
Equal numbers of cells were fixed with 1% formaldehyde at room temperature for 8 min. Cross-linking reactions were then quenched with 125 mM glycine for 5 min, and cells were washed with phosphate-buffered saline (PBS). Nuclei were isolated with ChIP lysis/wash buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.4], 5 mM EDTA [pH 8.0], 0.5% NP-40, 1% Triton X-100, protease inhibitors) and then lysed with ChIP lysis buffer (50 mM Tris [pH 8.1], 10 mM EDTA, 1% SDS, protease inhibitors) and subjected to sonication. Cellular debris was removed by centrifugation, and supernatants were diluted with ChIP dilution buffer (16.7 mM Tris [pH 8.1], 167 mM NaCl, 1.2 mM EDTA, 1.1% Triton X-100, 0.01% SDS, protease inhibitors) for overnight incubation with ChIP antibodies at 4°C. Magnetic protein A/G beads (Millipore) were added for 1 h, and beads were washed once with a low-salt buffer (20 mM Tris [pH 8.1], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), a high-salt buffer (20 mM Tris [pH 8.1], 0.5 M NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), and a LiCl buffer (10 mM Tris [pH 8.1], 0.25 M LiCl, 1 mM EDTA, 1% NP-40, 1% DOC); they were washed twice with TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA). Beads were then resuspended in water and incubated at 95°C for 10 min. The reaction mixtures were then reverse cross-linked for at least 30 min with ∼80 mM NaCl and 10 μg of proteinase K at 55°C. Reverse cross-linking was terminated at 95°C for 10 min, and DNA was isolated using a QIAquick PCR purification kit (28104; Qiagen) according to the manufacturer's protocol. Isolated DNA was analyzed by real-time PCR.
Real-time PCR.
DNA and RNA were isolated using DNA and RNA extraction kits (IB47201 and IB47322; IBI Scientific) according to the manufacturer's protocol. To make cDNA, isolated nucleic acids were quantified, and equal amounts of extracted RNA were treated with DNase (M6101; Promega) and converted into first-strand cDNA using a SuperScript III first-strand synthesis system (18080-051; Invitrogen). Primer and probe sets for real-time analysis are listed in Table 2. All real-time graphs represent the means ± SE of at least three independent experiments and have been analyzed for statistical significance using a permutation test (Mstat, version 6.1.2) that utilizes each independent experiment analyzed in triplicate by quantitative PCR (qPCR).
TABLE 2.
Primer and probe sets for real-time PCR
| Target | Primer or probe sequencea | Commentb |
|---|---|---|
| MIEP | Forward, 5′-CTTATGGGACTTTCCTACTTG-3′ | Used with SYBR green; primers adapted from 51 |
| Reverse, 5′-CGATCTGACGGTTCACTAA-3′ | ||
| IE exon 3 | Forward, 5′-CGACGTTCCTGCAGACTATG-3′ | |
| Reverse, 5′-TCCTCGGTCACTTGTTCAAA-3′ | ||
| Probe, 5′-FAM-TGGGAGACCCGCTGTTTCCA-TAMRA-3′ | ||
| IE exon 4 | Forward, 5′-TTAAGGTTCGAGTGGACATGG-3′ | |
| Reverse, 5′-TGAATTTCTCTTCCGTCTGGG-3′ | ||
| Probe, 5′-FAM-CATGTGCTCCTTGATTCTATGCCGC-TAMRA-3′ | ||
| IE exon 5 | Forward, 5′-CCCTTCACGATTCCCAGTATG-3′ | |
| Reverse, 5′-CACCTCACTCTTCACCTCATG-3′ | ||
| Probe, 5′-FAM-TCTGGATGCCCTTGTTGTTCACCT-TAMRA-3′ | ||
| UL47 5′ end | Forward, 5′-ATGGCGAGGCGCACGGTA-3′ | Primer and probe sequences (L0) from 39 |
| Reverse, 5′-GCCGATCTCCAATTGGCT-3′ | ||
| Probe, 5′-FAM-GATAGAGCAGCTGCGGGCAC-TAMRA-3′ | ||
| UL48 3′ end | Forward, 5′-CTGCTCCAGGACACGTGGAC-3′ | Primer and probe sequences (L3) from 39 |
| Reverse, 5′-GGTCATACAGCGGGAAGGTG-3′ | ||
| Probe, 5′-FAM-ACGGCTACGCGATTACCTGCGTTTC-TAMRA-3′ | ||
| 16S mitochondrial rRNA | Forward, 5′-CCGCAAGGGAAAGATGAAAAAT-3′ | Used with SYBR green |
| Reverse, 5′-TCGTCTGGTTTCGGGGGTCT-3′ |
Primer and probe sequences for β-actin, which was used as a control, were from reference 52. FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
SYBR green was from Bio-Rad (catalog no. 172-5120).
RESULTS
19S RP subunit knockdown inhibits both Daxx degradation and IE1 transcript accumulation.
Depletion of the Daxx protein during HCMV infection, either naturally through degradation by tegument-delivered pp71 or artificially through RNA interference, stimulates viral IE gene expression (18, 34, 35). Correspondingly, stabilization of Daxx through genetic alteration of pp71 or proteasome inhibition inhibits viral IE gene expression (18). Because the 19S RP is required for Daxx degradation (13), we asked if it was also required for viral IE gene expression. Independent knockdown of three distinct 19S RP subunits (Rpn1, Rpt2, and Rpn11) stabilized Daxx and impaired IE1 protein accumulation during HCMV infection (Fig. 1A to D). 19S RP subunit knockdown was confirmed by Western blotting, and inhibition of 19S RP function was confirmed by observing the stabilization of p53, a constitutively degraded 26S proteasome substrate (36). Consistent with the known role for Daxx in suppressing IE1 transcription (18, 32, 37), 19S RP knockdown suppressed the accumulation of IE1 transcripts (Fig. 2A and B). We found no defects in HCMV genome delivery (Fig. 2C and D) or cell cycle synchronization by serum starvation (Fig. 2E) in 19S RP subunit knockdown cells that could explain the observed inhibition of IE gene expression. Thus, we conclude that 19S RP subunit depletion inhibits HCMV IE1 transcription.
FIG 1.
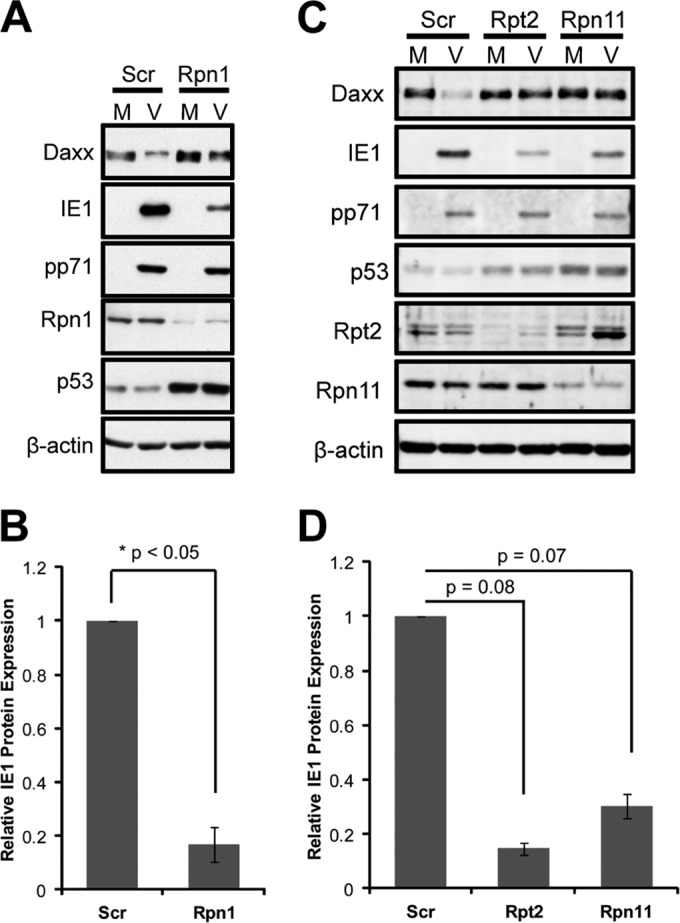
Daxx stabilization in 19S knockdown cells correlates with a defect in IE protein expression during HCMV infection. (A) HFs were transfected with Rpn1-specific or scrambled (Scr) control siRNAs for 72 h and then mock infected (M) or infected (V) at an MOI of 1. At 8 h postinfection, lysates were harvested and analyzed by immunoblotting. (B) HFs were transfected with the indicated siRNAs for 72 h and then infected at an MOI of 0.1 for 6 h. Lysates from three biological replicates were collected and analyzed by semiquantitative immunoblotting. IE1 protein expression was quantified and normalized to β-actin, and values are reported relative to the scrambled control. (C) HFs were transfected with the indicated siRNAs for 72 h and were infected and analyzed by immunoblotting as described for panel B. (D) Three biological replicates from the experiment shown in panel C were analyzed by semiquantitative immunoblotting and reported as described for panel B.
FIG 2.
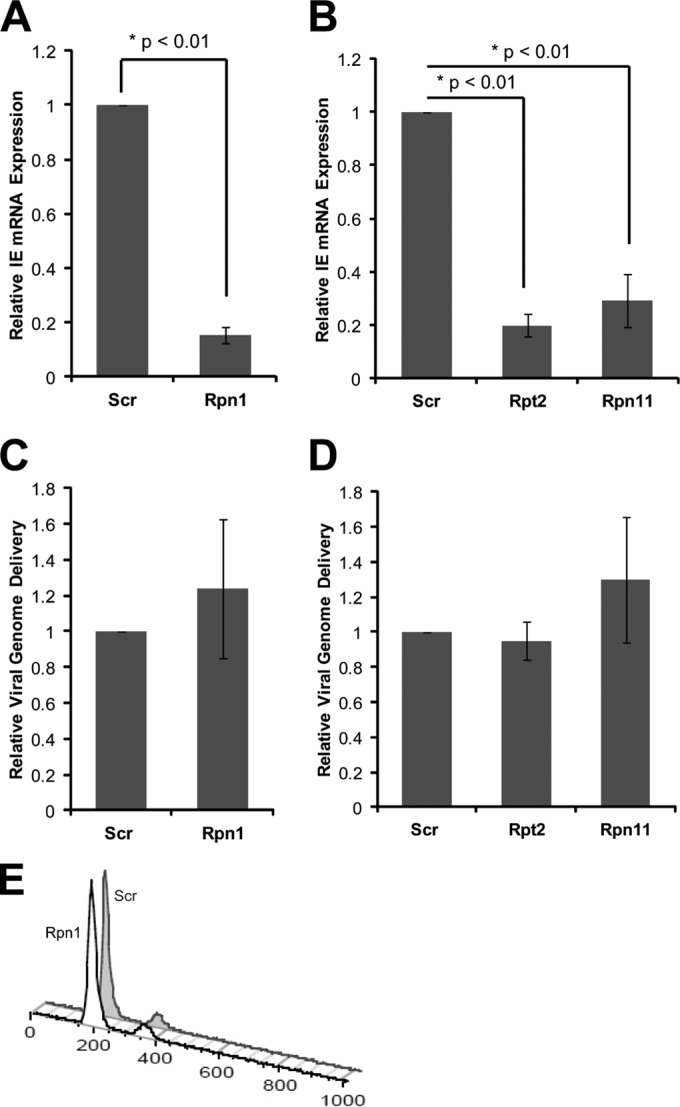
19S subunit depletion inhibits IE transcript accumulation during HCMV infection. (A) HFs transfected with scrambled (Scr) or Rpn1-targeting siRNAs for 72 h were infected at an MOI of 0.1 for 6 h. RNA and DNA were isolated from three biological replicates and analyzed by reverse transcription and real-time PCR. IE transcript levels (IE exon 3 primers) were normalized to both 16S mitochondrial rRNA transcripts and the number of HCMV genomes delivered per cell, and IE gene expression is reported relative to the scrambled control. (B) HFs were transfected with the indicated siRNAs and analyzed as described for panel A. (C) Lysates from the experiment shown in panel A were also analyzed with real-time PCR for genome delivery. The number of HCMV genomes (IE exon 3 primers) was normalized to cellular genomes (16S mitochondrial rRNA gene) and reported relative to scrambled controls. (D) Lysates from the experiment shown in panel B were also analyzed as described for panel C. (E) For 72 h, HFs were transfected with scrambled or Rpn1-specific siRNAs. Cells were then harvested, fixed, stained with propidium iodide, and analyzed by flow cytometry. Data were analyzed using FlowJo software.
Maintenance of the intrinsic defense through 19S RP knockdown is not the sole reason for depressed IE1 transcription.
Daxx institutes an intrinsic defense against HCMV that silences viral IE gene expression. It is inactivated through Daxx degradation by tegument-delivered pp71 or pharmacological histone deacetylase (HDAC) inhibition (18) and is maintained by proteasome impairment through small-molecule inhibition of the 20S CP or 19S RP subunit knockdown (13, 18). The silencing of IE gene expression observed upon 20S CP inhibition can be reversed by Daxx knockdown or HDAC inhibition (18), indicating that the intrinsic defense is the sole or major impediment to IE gene expression in 20S CP-inhibited cells. In contrast, Daxx knockdown (Fig. 3A) or HDAC inhibition (Fig. 3B) failed to rescue IE1 protein accumulation in 19S RP knockdown cells. While HDAC inhibition could rescue IE1 transcription during 20S CP knockdown, it was unable to promote IE1 transcription during 19S RP knockdown (Fig. 3C). We conclude that in addition to maintaining the Daxx- and HDAC-mediated intrinsic defense by disabling pp71-mediated Daxx degradation, 19S RP knockdown triggers an additional restriction to HCMV IE1 transcription.
FIG 3.
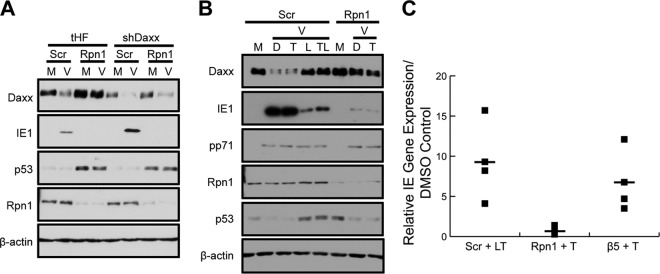
Dismantling the intrinsic immune defense is not the only role the 19S RP plays in promoting IE gene expression. (A) Telomerase-immortalized human fibroblasts (tHF) and shDaxx knockdown cells were transfected with scrambled (Scr) or Rpn1-targeting siRNA. After 72 h, HFs were mock infected (M) or infected (V) with HCMV at an MOI of 1 for 10 h. Cell lysates were collected and analyzed by immunoblotting. (B) HFs were transfected with scrambled control (Scr) or Rpn1-specific siRNAs for 72 h and then were mock infected (M) or infected (V) with HCMV at an MOI of 0.5. At 20 h prior to infection, cells were treated with DMSO (D) or TSA (T). At the start of infection, the indicated scrambled control groups were also treated with lactacystin (L). Cell lysates were collected at 4 h postinfection and analyzed by immunoblotting. (C) HFs were treated as described for panel B, with the addition of HFs transfected with siRNAs targeting the chymotrypsin-like subunit of the 20S core (β5), and nucleic acids were isolated and analyzed by real-time PCR. IE transcript levels (exon 3) were normalized to the number of HCMV genomes (exon 3) delivered per cell (β-actin). IE gene expression of TSA-treated samples is reported relative to each sample's respective DMSO control. Three biological replicates (closed squares) are reported and averaged (closed square with dash).
The 19S RP is recruited to the MIE locus at the onset of HCMV infection and promotes transcriptional elongation.
Inhibition of both the 20S CP and the 19S RP impairs protein degradation and HCMV IE gene expression. The observations that Daxx knockdown or HDAC inhibition can rescue IE gene expression only upon 20S CP but not 19S RP inhibition implies a nonproteolytic role for the 19S RP in HCMV transcription. Indeed, a nonproteolytic role for the 19S RP in cellular gene expression is emerging (38). While mechanisms remain elusive, 19S RP subunits have been observed at transcriptionally active loci (25, 29), and their presence promotes transcriptional elongation (26, 29). Likewise, using immunoprecipitation (ChIP), we found the endogenous 19S RP subunit Rpn7 but not the 20S CP subunit β7 present at the MIE locus, both at the promoter (Fig. 4A) and within exon 3 (Fig. 4B), in HCMV-infected cells. At this time during infection, the UL47 and UL48 genes are not transcribed. Encoded by the same DNA strand, these genes at late times after infection generate transcripts with unique 5′ ends but identical 3′ termini (39). Rpn7 showed a very modest recruitment to the 5′ (promoter) region of the UL47 gene (Fig. 4C) that may represent poising for future transcription, as has been postulated for cellular genes (40). Rpn7 was not recruited to the 3′ end of the UL48 gene (Fig. 4D). Thus, the 19S RP is recruited more substantially to the actively transcribed MIE locus than the transcriptionally inactive UL47/48 locus. The ability of the β7 antibody to immunoprecipitate the endogenous protein (Fig. 4E) instills confidence in the accuracy of the negative ChIP experiments.
FIG 4.
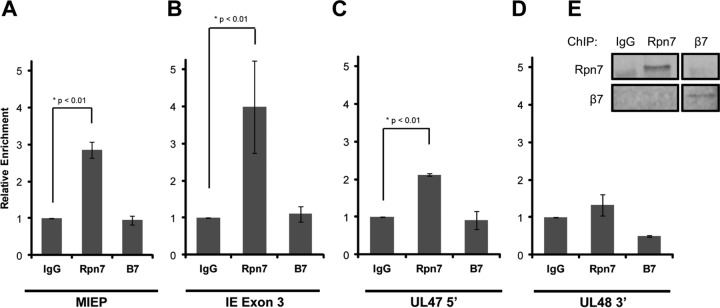
The 19S RP is recruited to the MIE locus. (A) HFs were infected with HCMV at an MOI of 3 for 3 h. Lysates were collected, fixed, and then subjected to ChIP using the indicated antibodies. Isolated DNA was quantified using real-time PCR, and enrichment at the MIEP was normalized to IgG controls. Three biological replicates are represented. β7 versus IgG, P = 0.62. (B) Samples from the experiment shown in panel A were also analyzed for enrichment at exon 3 of IE1/2. β7 versus IgG, P = 0.46. (C) Samples from the experiment shown in panel A were evaluated for enrichment at the 5′ end of UL47. β7 versus IgG, P = 0.32. (D) Samples from the experiment shown in panel A were analyzed for association with the 3′ end of UL48. Rpn7 versus IgG, P = 0.05; β7 versus IgG, P < 0.01. (E) Aliquots from the experiment shown in panel A were analyzed by immunoblotting to examine the efficacy of the immunoprecipitation.
We also utilized ChIP experiments to gauge transcriptional elongation by quantifying RNAPII occupancy at three sites within the MIE locus (Fig. 5A and 6A): the promoter, exon 4, and exon 5. Furthermore, we used an antibody that recognizes the nonphosphorylated form of RNAPII (8WG16) or one (H5) that recognizes RNAPII only when it is phosphorylated on serine 2, a mark of a transcriptionally elongating polymerase (41). Knockdown of the 19S RP subunit Rpn1 decreased the occupancy of total RNAPII at the MIEP, exon 4, and exon 5 (Fig. 5B) and of serine 2-phosphorylated RNAPII at exon 5 (Fig. 5C). In contrast, knockdown of the 20S CP subunit β5 impaired RNAPII recruitment to the MIEP but not occupancy at exon 4 or exon 5 (Fig. 6B) and had no effect on exon 5 occupancy of serine 2-phosphorylated RNAPII (Fig. 6C). Western blot controls indicated that proteasome subunit knockdown was achieved and impaired proteasome activity (detected by stabilization of p53) but that it did not affect the steady-state levels of the relevant forms of RNAPII (Fig. 7). From these data we conclude that the 19S RP, but not the 20S CP, facilitates RNAPII transcriptional elongation through the MIE locus in HCMV-infected cells. In total, our work uncovers proteolytic and nonproteolytic roles for the 19S RP in promoting viral IE gene expression at the onset of HCMV lytic infection.
FIG 5.

19S knockdown cells exhibit a defect in RNAPII elongation at the MIE locus. (A) Cartoon schematic of the MIE gene locus and the primer sets used during real-time analysis of RNAPII ChIP. Beginning nucleotides of exon 4 and exon 5 are numbered relative to the transcription start site (+1). (B) At 72 h after transfection with scrambled (Scr) or Rpn1 siRNA, HFs were infected with HCMV at an MOI of 1 for 3 h. Lysates were subjected to ChIP using the 8WG16 monoclonal antibody that recognizes the nonphosphorylated form of RNAPII. Enrichment of the indicated regions of the IE gene was analyzed by real-time PCR. Five biological replicates are represented. (C) Samples from the experiment shown in panel B were also analyzed for exon 5 occupancy with the H5 monoclonal antibody that recognizes the (elongating) Ser2-phosphorylated (Ser2P) form of RNAPII. Three biological replicates were completed.
FIG 6.

20S knockdown inhibits RNAPII recruitment to the MIEP but not RNAPII occupancy at the 3′ end of the MIE locus. (A) A cartoon representing the IE gene and the primer sets used during real-time analysis of ChIP samples. Beginning nucleotides of exon 4 and exon 5 are numbered relative to the transcription start site (+1). (B) At 72 h after transfection with scrambled (Scr) or β5 siRNA, HFs were infected with HCMV at an MOI of 1 for 3 h. Lysates were subjected to ChIP using the 8WG16 monoclonal antibody that recognizes the nonphosphorylated form of RNAPII. Enrichment of the indicated regions of the IE gene was analyzed by real-time PCR. Five biological replicates are represented. Scr versus β5, P = 0.14 (exon 4) and P = 0.44 (exon 5). (C) Samples from the experiment shown in panel B were also analyzed for exon 5 occupancy using the H5 monoclonal antibody that recognizes the Ser2-phosphorylated (the elongating) form of RNAPII. Three biological replicates are shown. Scr versus β5, P = 0.32.
FIG 7.
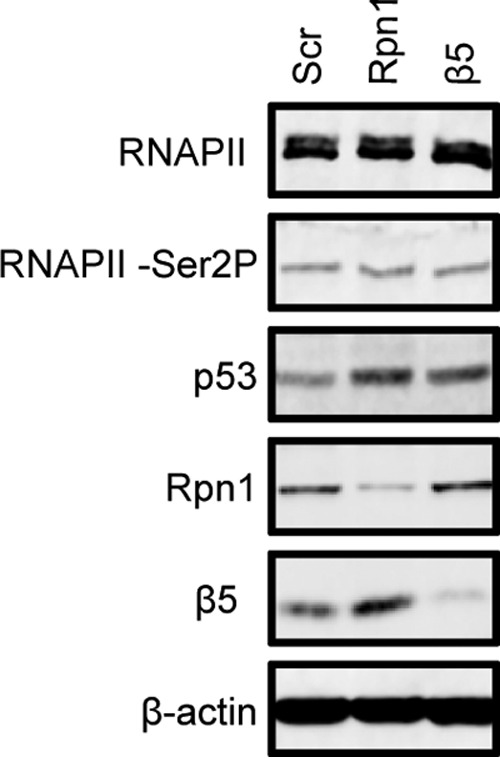
19S RP or 20S CP knockdown does not alter steady-state levels of RNAPII. HFs were transfected with the indicated siRNAs and analyzed by immunoblotting to examine steady-state levels of the indicated proteins.
DISCUSSION
The ubiquitin-proteasome pathway affects multiple cellular pathways during infections with many different viruses, with the proteolytic functions of the proteasome being the most appreciated and well studied. For example, adenovirus, human papillomavirus (HPV), and HCMV degrade substrates involved in cell cycle progression (42); HCMV, herpes simplex virus 1 (HSV-1), and HIV-1 destroy cellular restriction factors that inhibit viral replication (43–45); and HCMV stimulates MHC class I downregulation to impair adaptive immunity (46, 47). An additional growing body of evidence illustrates nonproteolytic ways in which the ubiquitin-proteasome system may be commandeered for the benefit of viral infections.
We previously reported that the monoubiquitination of histone H2B, which does not result in protein degradation, was required for efficient transcriptional elongation of HCMV genes (39). Here, we show that HCMV also utilizes the 19S RP in a nonproteolytic manner to promote efficient viral gene expression, a finding mirrored for HIV (29). Our overall data support a model whereby Daxx degradation by tegument-delivered pp71 and the 26S proteasome prevents repressive epigenetic modification of the MIEP, permitting RNAPII recruitment to the promoter and fostering transcriptional initiation. Subsequently, the 19S RP promotes efficient transcriptional elongation of RNAPII (Fig. 8). Our results illuminate a novel role that the 19S RP plays during HCMV infection and provide a mechanistic rationale for previously published results demonstrating that 19S RP subunits localized to areas of robust transcription at late times postinfection (30).
FIG 8.
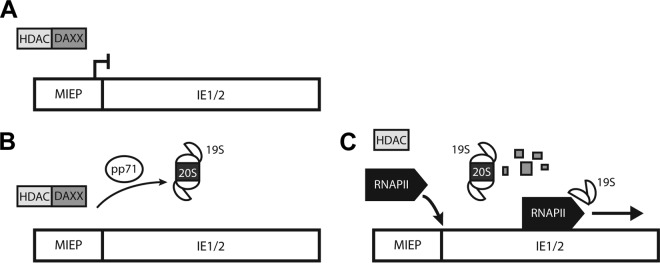
The 19S RP promotes HCMV IE gene expression through proteolytic and nonproteolytic mechanisms. (A) At the start of HCMV infection, Daxx induces a repressive chromatin structure at the MIEP, prohibiting RNAPII recruitment. (B) To induce IE gene expression, tegument-delivered pp71 degrades Daxx using both the 19S RP and the 20S CP. (C) Once Daxx has been eliminated and the local chromatin structure is relaxed, RNAPII can access the MIEP. RNAPII is then recruited, and the 19S RP promotes the efficient elongation of transcription complexes.
The inhibition of 19S RP function to impair HCMV IE gene transcriptional elongation may be a strategy for antiviral therapeutic intervention. It has recently been documented that decelerating the rate of HCMV gene expression confers a significant fitness cost to the virus (48). Dampened yet detectable IE gene expression might slow viral replication while still producing viral antigens detectable by adaptive immune responses. In this respect, slowing IE gene expression through selectively impairing 19S RP function during transcriptional elongation may be more attractive than silencing IE gene expression through 20S CP inhibition as this leads to latent virus that cannot be cleared by the host immune system (34).
How the 19S RP facilitates RNAPII elongation remains an enigma (49). Potential mechanisms include stabilizing protein interactions within the elongation complex, recruiting positively acting elongating factors, or removing factors that slow transcriptional elongation. Our demonstration that the 19S RP promotes transcriptional elongation of viral genes establishes a physiologically relevant, genetically tractable model for further investigations to decipher the mechanism of nonproteolytic yet proteasome-mediated transcriptional regulation.
ACKNOWLEDGMENTS
We thank Phil Balandyk (University of Wisconsin—Madison) for expert technical assistance, both Shelby Malone and Norman Drinkwater for help with statistical analyses, and Halena VanDeusen for illustrations.
This work was supported by National Institutes of Health grant AI074984 (to R.F.K.) and training grant T32 CA009135 (to L.L.W.). R.F.K. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1. Jung T, Catalgol B, Grune T. 2009. The proteasomal system. Mol. Aspects Med. 30:191–296. 10.1016/j.mam.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 2. Loureiro J, Ploegh HL. 2006. Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv. Immunol. 92:225–305. 10.1016/S0065-2776(06)92006-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blanchette P, Branton PE. 2009. Manipulation of the ubiquitin-proteasome pathway by small DNA tumor viruses. Virology 384:317–323. 10.1016/j.virol.2008.10.005 [DOI] [PubMed] [Google Scholar]
- 4. Choi AG, Wong J, Marchant D, Luo H. 2013. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev. Med. Virol. 23:85–96. 10.1002/rmv.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hwang J, Winkler L, Kalejta RF. 2011. Ubiquitin-independent proteasomal degradation during oncogenic viral infections. Biochim. Biophys. Acta 1816:147–157. 10.1016/j.bbcan.2011.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. 1997. Structure of 20S proteasome from yeast at 2.4A resolution. Nature 386:463–471. 10.1038/386463a0 [DOI] [PubMed] [Google Scholar]
- 7. Rechsteiner M, Hill CP. 2005. Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15:27–33. 10.1016/j.tcb.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 8. da Fonseca PC, He J, Morris EP. 2012. Molecular model of the human 26S proteasome. Mol. Cell 46:54–66. 10.1016/j.molcel.2012.03.026 [DOI] [PubMed] [Google Scholar]
- 9. Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78:477–513. 10.1146/annurev.biochem.78.081507.101607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elsasser S, Chandler-Militello D, Müller B, Hanna J, Finley D. 2004. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279:26817–26822. 10.1074/jbc.M404020200 [DOI] [PubMed] [Google Scholar]
- 11. Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. 2008. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488. 10.1038/nature06926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murakami Y, Matsufuji S, Kameji T, Hayashi S-i, Igarashi K, Tamura T, Tanaka K, Ichihara A. 1992. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360:597–599. 10.1038/360597a0 [DOI] [PubMed] [Google Scholar]
- 13. Winkler LL, Hwang J, Kalejta RF. 2013. Ubiquitin-independent proteasomal degradation of tumor suppressors by human cytomegalovirus pp71 requires the 19S regulatory particle. J. Virol. 87:4665–4671. 10.1128/JVI.03301-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang A, Hildreth RL, Colberg-Poley AM. 2013. Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J. Virol. 87:5657–5668. 10.1128/JVI.00145-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fehr AR, Gualberto NC, Savaryn JP, Terhune SS, Yu D. 2012. Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog. 8:e1002789. 10.1371/journal.ppat.1002789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SH, Kalejta RF, Kerry J, Semmes OJ, O'Connor CM, Khan Z, Garcia BA, Shenk T, Murphy E. 2012. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc. Natl. Acad. Sci. U. S. A. 109:9575–9580. 10.1073/pnas.1207496109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim Y-E, Lee J-H, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn J-H. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J. Virol. 85:11928–11937. 10.1128/JVI.00758-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate early gene expression. J. Virol. 80:3863–3871. 10.1128/JVI.80.8.3863-3871.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kalejta RF, Shenk T. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. U. S. A. 100:3263–3268. 10.1073/pnas.0538058100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barel MT, Hassink GC, Voorden Sv, Wiertz EJHJ. 2006. Human cytomegalovirus-encoded US2 and US11 target unassembled MHC class I heavy chains for degradation. Mol. Immunol. 43:1258–1266. 10.1016/j.molimm.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 21. Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799. 10.1126/science.1152095 [DOI] [PubMed] [Google Scholar]
- 22. Penkert RR, Kalejta RF. 2012. Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7:855–869. 10.2217/fvl.12.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338. 10.1016/j.virol.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 24. Jariel-Encontre I, Bossis G, Piechaczyk M. 2008. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 1786:153–177. 10.1016/j.bbcan.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 25. Gonzalez F, Delahodde A, Kodadek T, Johnston SA. 2002. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296:548–550. 10.1126/science.1069490 [DOI] [PubMed] [Google Scholar]
- 26. Ferdous A, Gonzalez F, Sun L, Kodadek T, Johnston SA. 2001. The 19S regulatory particle of the proteasome is required for efficient transcription elongation by RNA polymerase II. Mol. Cell 7:981–991. 10.1016/S1097-2765(01)00250-7 [DOI] [PubMed] [Google Scholar]
- 27. Ferdous A, Kodadek T, Johnston SA. 2002. A nonproteolytic function of the 19S regulatory subunit of the 26S proteasome is required for efficient activated transcription by human RNA polymerase II. Biochemistry 41:12798–12805. 10.1021/bi020425t [DOI] [PubMed] [Google Scholar]
- 28. Geng F, Tansey WP. 2012. Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc. Natl. Acad. Sci. U. S. A. 109:6060–6065. 10.1073/pnas.1200854109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lassot I, Latreille D, Rousset E, Sourisseau M, Linares LK, Chable-Bessia C, Coux O, Benkirane M, Kiernan RE. 2007. The proteasome regulates HIV-1 transcription by both proteolytic and nonproteolytic mechanisms. Mol. Cell 25:369–383. 10.1016/j.molcel.2006.12.020 [DOI] [PubMed] [Google Scholar]
- 30. Tran K, Mahr JA, Spector DH. 2010. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 84:3079–3093. 10.1128/JVI.02236-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sadanari H, Tanaka J, Li Z, Yamada R, Matsubara K, Murayama T. 2009. Proteasome inhibitor differentially regulates expression of the major immediate early genes of human cytomegalovirus in human central nervous system-derived cell lines. Virus Res. 142:68–77. 10.1016/j.virusres.2009.01.010 [DOI] [PubMed] [Google Scholar]
- 32. Cantrell SR, Bresnahan WA. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188–6191. 10.1128/JVI.02676-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalejta RF, Bechtel JT, Shenk T. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885–1895. 10.1128/MCB.23.6.1885-1895.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109–9120. 10.1128/JVI.00827-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ Cells. J. Virol. 84:5594–5604. 10.1128/JVI.00348-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yu ZK, Geyer RK, Maki CG. 2000. MDM2-dependent ubiquitination of nuclear and cytoplasmic P53. Oncogene 19:5892–5897. 10.1038/sj.onc.1203980 [DOI] [PubMed] [Google Scholar]
- 37. Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652–37660. 10.1074/jbc.M604273200 [DOI] [PubMed] [Google Scholar]
- 38. Kodadek T. 2010. No splicing, no dicing: non-proteolytic roles of the ubiquitin-proteasome system in transcription. J. Biol. Chem. 285:2221–2226. 10.1074/jbc.R109.077883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hwang J, Saffert RT, Kalejta RF. 2011. Elongin B-mediated epigenetic alteration of viral chromatin correlates with efficient human cytomegalovirus gene expression and replication. mBio 2(2):e00023-11. 10.1128/mBio.00023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szutorisz H, Georgiou A, Tora L, Dillon N. 2006. The proteasome restricts permissive transcription at tissue-specific gene loci in embryonic stem cells. Cell 127:1375–1388. 10.1016/j.cell.2006.10.045 [DOI] [PubMed] [Google Scholar]
- 41. Heidemann M, Hintermair C, Voß K, Eick D. 2013. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 1829:55–62. 10.1016/j.bbagrm.2012.08.013 [DOI] [PubMed] [Google Scholar]
- 42. Isaacson MK, Ploegh HL. 2009. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host. Microbe 5:559–570. 10.1016/j.chom.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nomaguchi M, Fujita M, Adachi A. 2008. Role of HIV-1 Vpu protein for virus spread and pathogenesis. Microbes Infect. 10:960–967. 10.1016/j.micinf.2008.07.006 [DOI] [PubMed] [Google Scholar]
- 44. Huthoff H, Towers GJ. 2008. Restriction of retroviral replication by APOBEC3G/F and TRIM5α. Trends Microbiol. 16:612–619. 10.1016/j.tim.2008.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169–2178. 10.1128/JVI.78.5.2169-2178.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. 1996. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384:432–438. 10.1038/384432a0 [DOI] [PubMed] [Google Scholar]
- 47. Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, Ploegh HL. 1996. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84:769–779. 10.1016/S0092-8674(00)81054-5 [DOI] [PubMed] [Google Scholar]
- 48. Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. 2012. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell 151:1569–1580. 10.1016/j.cell.2012.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Geng F, Wenzel S, Tansey WP. 2012. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 81:177–201. 10.1146/annurev-biochem-052110-120012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82:11167–11180. 10.1128/JVI.01218-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hänfler J, Kreuzer K, Laurisch K, Rayes N, Neuhaus P, Schmidt C, Oettle H. 2003. Quantitation of cytomegalovirus (hCMV) DNA and β-actin DNA by duplex real-time fluorescence PCR in solid organ (liver) transplant recipients. Med. Microbiol. Immunol. 192:197–204. 10.1007/s00430-002-0166-6 [DOI] [PubMed] [Google Scholar]