

Abstract
Aim
SRT2104 is a selective activator of SIRT1. In animal models, SRT2104 improves glucose homeostasis and increases insulin sensitivity. We evaluated the tolerability and pharmacokinetics of SRT2104, and its effects on glycaemic control, in adults with type 2 diabetes mellitus.
Method
Type 2 diabetics with glycosylated haemoglobin (HbA1c) ≥ 7.5% and ≤10.5%, fasting glucose ≥160 and ≤240 mg dl−1, and on stable doses of metformin were evenly randomized to placebo or SRT2104 0.25 g, 0.5 g, 1.0 g or 2.0 g, administered orally once daily for 28 days. Changes in fasting and post-prandial glucose and insulin were analyzed.
Results
Safety evaluation found no major differences between groups in the frequency of adverse events. SRT2104 concentrations did not increase in a dose-proportional fashion. Significant variability in exposure was observed. Treatment with SRT2104 did not lead to any consistent, dose-related changes in glucose or insulin. Day 28 change from baseline (mean (SD)): fasting glucose (mmol l−1) = −1.17 (2.42), −1.11 (3.45), −0.52 (2.60), −0.97 (2.83) and −0.15 (2.38) for placebo, 0.25 g, 0.5 g, 1.0 g and 2.0 g, respectively. Day 28 change from baseline (mean (SD)): fasting insulin (mmol l−1) = 1.0 (51.66), 8.9 (95.04), −6.9 (41.45), 4.1 (57.16) and 15.2 (138.79) for placebo, 0.25 g, 0.5 g, 1.0 g and 2.0 g, respectively) Treatment with SRT2104 was associated with improvement in lipid profiles.
Conclusion
Treatment with SRT2104 for 28 days did not result in improved glucose or insulin control which is likely due to the observed pharmacokinetics which were not dose proportional and had large between subject variability.
Keywords: glucose homeostasis, insulin, SIRT1, sirtuin, SRT2104, type 2 diabetes mellitus
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Through modulation of various substrates including PGC-1α, NF<ϕχ>κ</ϕχ>B and FOXO, SIRT1 (an NAD-dependent deacetylase) is involved in the regulation of multiple cellular processes including survival, mitochondrial biogenesis, and metabolism. SRT2104 is a novel and selective small molecule activator of SIRT1 that improves glucose homeostasis, increases insulin sensitivity, and reduces plasma triglycerides and hepatic steatosis in diet-induced obese (DIO) mice.
WHAT THIS STUDY ADDS
This study summarizes the key tolerability, clinical activity and pharmacokinetic data obtained for this compound in type 2 diabetics. Treatment with SRT2104 for 28 days resulted in highly variable pharmacokinetics and resulting exposure. Treatment with SRT2104 for 28 days did not result in improved glucose or insulin control but had a modest beneficial impact on lipids.
Introduction
During the past three decades, the number of people with diabetes has more than doubled throughout the world [1], and type 2 diabetes is increasing in prevalence in children and adolescents [2,3]. Oral agents treat type 2 diabetes through various mechanisms, such as increasing insulin secretion (sulfonylureas), reducing hepatic glucose output while increasing glucose utilization (metformin), reducing insulin resistance in multiple tissues (glitazones), or increasing endogenous incretin hormones such as glucagon-like peptide 1 (gliptins or dipeptidyl peptidase-IV inhibitors. While these treatments have demonstrated efficacy in various patient populations, there remain unmet therapeutic goals including improved insulin resistance and cardiovascular outcomes.
Given the limitations of current therapy, several novel classes of drugs are currently being explored for managing diabetes [4]. Sirtuin 1 (SIRT1) is a NAD+-dependent deacetylase and a member of the sirtuin family of proteins. Modulation of SIRT1 activity appears to regulate whole body glucose metabolism locally and systemically [5]. In mice, increased dosage of SIRT1 in pancreatic islet cells improves glucose tolerance and enhances insulin secretion in response to glucose [6], whereas deletion of SIRT1 impairs glucose-stimulated insulin secretion [7]. Modest over-expression of SIRT1 has a protective effect against high fat induced glucose intolerance [8]. Small molecule activators of SIRT1 improve insulin sensitivity, lower plasma glucose and increase mitochondrial capacity in obese mice [9].
SRT2104 is a selective activator of SIRT1 that was identified from a high throughput screen of 290 000 compounds [9,10]. In diet-induced obese mice (DIO), SRT2104 improves glucose homeostasis, increases insulin sensitivity, and reduces plasma triglycerides and hepatic steatosis [11]. Also, in obese mice, SRT2104 increases energy expenditure by enhancing carbohydrate and fatty acid oxidation [12]. We examined the tolerability and pharmacokinetics of SRT2104 dosed orally, once daily for 28 consecutive days, in subjects with type 2 diabetes managed by stable doses of metformin therapy. Also assessed were measures of glycaemic control, lipid metabolism and weight.
Methods
Subjects
Eligible subjects were 30–70 years of age and had glycosylated haemoglobin (HbA1c) ≥ 7.5% and ≤10.5%, fasting glucose ≥160 and ≤240 mg dl−1, and BMI ≥25.0 kg m−2 and ≤40.0 kg m−2. All subjects had to be on stable metformin medication for at least 3 months (≥1.0 g day−1) prior to screening. Subjects were ineligible if their liver enzymes were >2 × upper limit of normal (ULN) or if serum creatinine was ≥1.4 mg dl−1 in females or ≥1.5 mg dl−1 in males. Also ineligible were subjects taking any diabetes therapies besides metformin in the month prior to enrolment. All subjects provided written informed consent before participating in any study-related activities. The study protocol was approved by each institution's review board or independent ethics committee.
Study design
This was a phase 2, multicentre, randomized, double-blind, placebo-controlled study with multiple doses of SRT2104 or placebo administered orally once daily for 28 consecutive days (EudraCT number 2009-010720-26). Subjects were evenly randomized to placebo or SRT2104 0.25 g, 0.5 g, 1.0 g or 2.0 g daily. The 2.0 g dose was selected as the highest dose as a previous study demonstrated that higher drug concentrations were not achieved when SRT2104 was administered at doses above 2.0 g [13]. Eight SRT2104 or matching placebo capsules were to be taken every morning approximately 15 min following consumption of a standardized meal (Ensure Plus, 237 ml bottle, Abbott Nutrition), as a food effect resulting in enhanced drug absorption had been previously demonstrated for this compound [13]. Subjects stayed overnight at the clinic on days −1 and 27 and returned to the clinic on days 2, 8, 15, 22, and 29 as well as 7 days after completing dosing (follow-up). Subjects were required to monitor their fasting blood glucose and complete a diary daily between days 1 and 35. Subjects received a safety call 30 days following their final dose of SRT2104 or placebo.
Randomization was carried out in a double-blind manner using an Interactive Web-based Response System (Bilcare IVRS Solutions, Bilcare Global Clinical Supplies, Powys, UK). At each site, subjects were randomized in blocks of 10.
Safety assessments
Safety was monitored from the time of informed consent until 30 days after cessation of dosing. Safety was evaluated by using reports of medical events from informed consent to first dose administration, adverse events (AEs) (occurring after first dose administration), vital signs, physical examinations, laboratory parameters, electrocardiogram parameters and concomitant medications. AEs were coded using the Medical Dictionary for Regulatory Affairs (MedDRA) version 12.0 and summarized by system organ class and preferred term.
Pharmacokinetic assessments
Plasma samples were collected for determination of SRT2104 concentration on day 1 at pre-dose and 15 min, 30 min, 1 h, 2 h, 3 h, 4 h, 8 h and 12 h post-dose, and on day 2 at 24 h post-day 1 dose. Plasma samples were also collected on days 8, 15 and 22 at trough (t = 0, pre-dose), on day 28 at trough (t = 0, pre-dose), and at 15 min, 30 min, 1 h, 2 h, 3 h, 4 h, 8 h and 12 h post-dose and on day 29 at 24 h post-day 28 dose. SRT2104 plasma concentrations were determined by a validated liquid chromatography method with tandem mass spectrometry (LC-MS/MS) using d8-SRT2104 as a stable label, internal standard. Assay performance was evaluated using four quality control (QC) concentrations, 0.5 ng ml−1, 2.1 ng ml−1, 42.5 ng ml−1 and 425 ng ml−1. Intra- and interbatch QC samples showed accuracy ranging from 90.8% to 103.2% and the coefficient of variation did not exceed 8.0%. The upper limit of quantification was 500 ng ml−1 and the lower limit of quantification was 0.5 ng ml−1 [13]. Plasma concentrations and pharmacokinetic parameters were summarized and compared by using descriptive statistics.
Efficacy assessments
Evaluation of fasting glucose and insulin was done at screening, on days 1, 8, 15, 22, and 28 and day 35. Post-prandial glucose and insulin testing was performed on days 1 and 28 at 2 h after consumption of a standardized meal for all subjects. In addition, some subjects had post-prandial data obtained at 30 min and 60 min after meal consumption (following protocol amendment). HbA1c and fructosamine testing was done at screening and on days 1 and 28. Samples for lipid profiles and haematology panels were collected at screening on days 1, 8, 15, 22 and 28 and approximately 7 days after the last dose of study medication. Weight was documented during screening and at days 1, 28 and 35.
Endpoints and statistical analyses
The primary objectives of this study were to evaluate safety and pharmacokinetics. Therefore, the sample size was based on feasibility rather than formal statistical operating characteristics. However, with regard to glycaemic control, an evaluation of sample size indicated that 37 subjects per group would have 80% power to detect an effect size of 66.7% (effect size defined as difference in means divided by common standard deviation) at an alpha level of 0.05. Adverse events are presented as counts and percentages. Safety laboratory results are presented as means (SD).
For glycaemic endpoints, observed values and change from baseline for fasting plasma glucose, fasting plasma insulin, post-prandial glucose, post-prandial insulin, HbA1c, and fructosamine were compared with placebo using a general linear model with Dunnett's test and a post hoc unadjusted anova for each study day assessed. P values associated with the unadjusted anova are presented. Percent change for fasting plasma glucose, fasting plasma insulin, post-prandial glucose, post-prandial insulin, HbA1c and fructosamine for each active dose group was compared with placebo using Dunnett's test for day 28 only. Changes from baseline in lipid parameters and weight were analyzed by using an ancova model with treatment arm as a fixed effect and baseline as a covariate, without adjustment for multiple comparisons. An exploratory analysis was conducted to identify baseline characteristics that were predictive of improvements in lipids (general linear model). Day 28 exposure (AUC(0,t)) was dichotomized based on distribution into high (quartiles 3 + 4) and low (quartiles 1 + 2) drug exposure groups for this analysis.
Results
Study population
From August 2009, to September 2010, 227 subjects were randomized (215 treated) at 45 study centres in Bulgaria, Estonia, Hungary, Poland, Romania, Russia, Ukraine and the UK. Among all subjects, the mean (± SD) age was 57.0 ± 8.67 years, 55.5% were female and 99% were Caucasian. Mean weight was 90.7 ± 14.20 kg and mean BMI was 31.9 ± 3.97 kg m−2. Baseline fasted and post-prandial glucose and insulin concentrations, as well as HbA1c, were as expected for a population with type 2 diabetes (Table 1). Of 227 randomized subjects, 203 (89.4%) completed treatment and 24 (10.6%) discontinued (12 [5.3%] discontinued before receiving treatment) (Figure 1). The most common reasons for discontinuation were subject withdrawal or refusal (15 subjects) and AEs (seven subjects). All post-treatment discontinuations occurred in the active treatment arms.
Table 1.
Subject baseline characteristics
| SRT2104 (g day−1) | |||||
|---|---|---|---|---|---|
| Placebo (n = 46) | 0.25 (n = 45) | 0.5 (n = 46) | 1.0 (n = 45) | 2.0 (n = 45) | |
| Fasting plasma glucose | |||||
| n | 43 | 41 | 43 | 40 | 43 |
| Mean (SD) mmol l−1 | 10.8 (2.6) | 11.3 (3.0) | 11.2 (2.9) | 11.1 (3.2) | 10.3 (2.4) |
| Post-prandial glucose | |||||
| 30 min, n | 25 | 28 | 28 | 24 | 27 |
| Mean (SD) mmol l−1 | 13.6 (2.8) | 14.7 (3.8) | 14.7 (3.7) | 14.8 (4.7) | 13.4 (3.0) |
| 60 min, n | 25 | 26 | 29 | 25 | 26 |
| Mean (SD) mmol l−1 | 14.8 (3.5) | 15.7 (4.3) | 16.5 (4.2) | 17.0 (4.6) | 14.0 (3.5) |
| 120 min, n | 41 | 40 | 41 | 40 | 41 |
| Mean (SD) mmol l−1 | 13.7 (3.7) | 15.0 (4.3) | 15.2 (3.3) | 15.2 (5.2) | 13.7 (4.1) |
| Fasting plasma insulin | |||||
| n | 41 | 42 | 41 | 40 | 40 |
| Mean (SD) mmol l−1 | 83.6 (64.5) | 80.1 (83.4) | 74.2 (65.0) | 60.3 (46.4) | 72.9 (47.8) |
| Post-prandial insulin | |||||
| 30 min, n | 24 | 29 | 28 | 26 | 26 |
| Mean (SD) mmol l−1 | 204.9 (105.6) | 235.4 (278.5) | 203.4 (131.0) | 187.5 (153.3) | 216.5 (135.8) |
| 60 min, n | 24 | 28 | 29 | 26 | 26 |
| Mean (SD) mmol l−1 | 283.5 (292.9) | 274.8 (347.5) | 248.4 (152.0) | 236.5 (169.0) | 235.3 (103.9) |
| 120 min, n | 41 | 41 | 39 | 40 | 42 |
| Mean (SD) mmol l−1 | 205.0 (115.7) | 229.9 (302.8) | 224.0 (165.2) | 193.6 (175.2) | 175.3 (92.7) |
| HbA1c | |||||
| n | 43 | 41 | 43 | 40 | 43 |
| Mean (SD) percent | 8.5 (0.8) | 8.9 (1.0) | 8.7 (0.9) | 8.6 (0.9) | 8.4 (0.9) |
Figure 1.
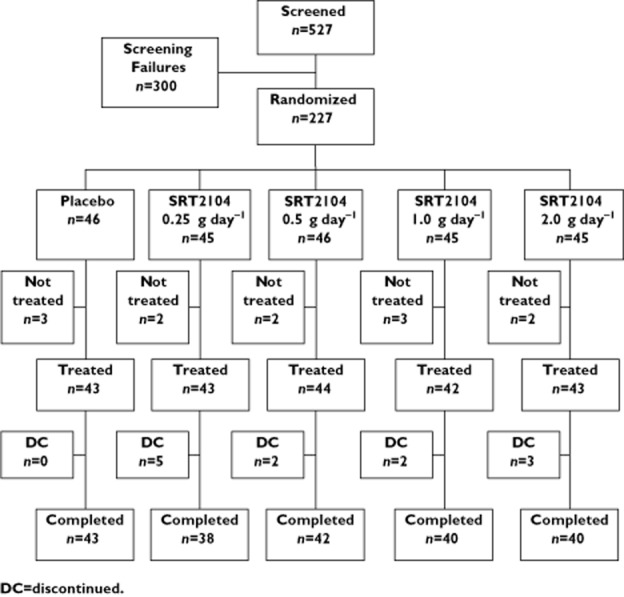
Disposition of subjects. DC discontinued
Safety assessments
SRT2104 was generally well tolerated, with a minority of subjects (35%) experiencing AEs. The most common AEs, nausea, diarrhoea, asthenia and hyperglycaemia, occurred with similar frequency for placebo and SRT2104 treatment (Table 2). Among the subjects who reported AEs, 54% reported mild events and 42% reported moderate events. Two subjects experienced serious adverse events (SAEs). One had interstitial lung disease, which resolved after treatment with acetylcysteine, aminophylline, ceftriaxone and dexamethasone. The other subject had two SAEs, 1) wound infection on the leg following major trauma associated with sepsis, which ultimately resulted in 2) death. All three SAEs were considered by the investigator to be not related to study medication. The number of subjects experiencing AEs that led to discontinuation were: three receiving 0.25 g day−1, one receiving 0.5 g day−1, one receiving 1.0 g day−1 and two receiving 2.0 g day−1 (3.9% overall). None of the subjects in the placebo group experienced AEs that led to discontinuation.
Table 2.
Summary of subjects with treatment-emergent adverse events (AEs) and AEs present in ≥5% of subjects across treatment groups
| SRT2104 (g day−1) | |||||
|---|---|---|---|---|---|
| Placebo (n = 43) | 0.25 (n = 43) | 0.5 (n = 44) | 1.0 (n = 42) | 2.0 (n = 43) | |
| Subjects with ≥1 AE, n (%) | 11 (26) | 15 (35) | 19 (43) | 11 (26) | 15 (35) |
| AEs by intensity, n (%) | |||||
| Mild | 8 (19) | 10 (23) | 11 (25) | 8 (19) | 9 (21) |
| Moderate | 7 (16) | 6 (14) | 12 (27) | 5 (12) | 7 (16) |
| Severe | 0 | 1 (2) | 1 (2) | 0 | 0 |
| Deaths | 0 | 0 | 1 (2) | 0 | 0 |
| Serious adverse events, n (%) | 0 | 1 (2) | 1 (2) | 0 | 0 |
| AEs leading to discontinuation, n (%) | 0 | 3 (7) | 2 (5) | 1 (2) | 2 (5) |
| AEs in ≥5% of subjects, n (%) | |||||
| Nausea | 2 (5) | 3 (7) | 4 (9) | 1 (2) | 2 (5) |
| Diarrhoea | 2 (5) | 1 (2) | 4 (9) | 1 (2) | 2 (5) |
| Asthenia | 1 (2) | 1 (2) | 4 (9) | 0 | 0 |
| Hyperglycaemia | 3 (7) | 1 (2) | 3 (7) | 4 (10) | 4 (9) |
Few clinically relevant laboratory abnormalities were noted; however, two subjects, each on active treatment, reported increased bilirubin (>1 × ULN in both subjects) and increased ALT (>2 × ULN in one subject, > 1 × ULN in one subject). Eosinophilia was reported as an AE in one subject receiving SRT2104 0.25 g day−1 (620 cells μl−1) and two subjects receiving 1.0 g day−1 (both with 610 cells μl−1). Leukopenia (320 cells μl−1), increased creatine phosphokinase (265 IU l−1) and hypoglycaemia were each reported once among subjects receiving SRT2104. The hypoglycaemia occurred on day 35, 7 days after treatment had ceased.
Pharmacokinetic assessments
Exposure to SRT2104 (as assessed by AUC(0,t)) increased in a less than dose-proportional fashion. On day 1, exposure to SRT2104 increased from 0.25 to 0.5 g day−1 and from 1.0 and 2.0 g day−1 but not 0.5 and 1.0 g day−1 (Table 3). A similar trend was seen on day 28.
Table 3.
Summary of day 28 vs. day 1 SRT2104 pharmacokinetic data
| SRT2104 Dose | Parameter | Geometric LS means | Day 28/Day 1(%) (90% CI) | |
|---|---|---|---|---|
| Day 1 | Day 28 | |||
| 0.25 g day−1 | Cmax (ng ml−1) | 84 | 103 | 122 (87, 172) |
| AUC(0.t) (ng ml−1 h) | 577 | 971 | 168 (124, 229) | |
| AUC(0,∞)* (ng ml−1 h) | 830 | 920 | 111 (88, 139) | |
| 0.5 g day−1 | Cmax (ng ml−1) | 178 | 168 | 94 (69, 129) |
| AUC(0,t) (ng ml−1 h) | 1172 | 1560 | 133 (101, 176) | |
| AUC(0,∞)* (ng ml−1 h) | 1655 | 1560 | 94 (70, 126) | |
| 1.0 g day−1 | Cmax (ng ml−1) | 164 | 188 | 115 (92, 144) |
| AUC(0,t) (ng ml−1 h) | 1186 | 1680 | 142 (111, 180) | |
| AUC(0,∞)* (ng ml−1 h) | 1529 | 1750 | 114 (86, 153) | |
| 2.0 g day−1 | Cmax (ng ml−1) | 194 | 233 | 120 (89, 163) |
| AUC(0,t) (ng ml−1 h) | 1523 | 2544 | 167 (121, 231) | |
| AUC(0,∞)* (ng ml−1 h) | 2104 | 2477 | 118 (85, 163) | |
AUC values included in the analysis are AUC(0,∞) on day 1 and AUC(0,τ) on day 28. Results obtained from a mixed model anova on log-transformed data with a fixed effect of study day and a random effect of subject.
Mean peak plasma concentrations of SRT2104 increased in an approximately proportional fashion between the doses of 0.25 and 0.5 g day−1, but had little further change with 1.0 or 2.0 g day−1. In addition, there was considerable inter-subject variability in pharmacokinetic parameters, with coefficients of variation (CVs >75%) for the exposure parameters AUC(0,t)) and Cmax for all groups. The mean (± SD) time at which Cmax was achieved relative to drug intake (tmax) was quantitatively similar for all dose groups: 3.0 ± 1.2 h for 0.25 g day−1, 4.0 ± 3.7 h for 0.5 g day−1, 3.6 ± 1.8 h for 1.0 g day−1 and 4.4 ± 2.5 h for 2.0 g day−1.
There was an approximate 1.5-fold (1.3–1.7) increase in exposure (AUC(0,t)) on day 28 as compared with day 1 across doses. This increase in exposure is consistent with the known half-life of the drug (t1/2 = approximately 24 h), and did not demonstrate any dose dependence [13].
Efficacy assessments
Glycaemic control
No trends in fasting plasma glucose or insulin or post-prandial glucose or insulin were observed with SRT2104 dose (Table 4). Compared with placebo, SRT2104 did not lead to any statistically significant differences in fasting plasma glucose or insulin in absolute values or changes from baseline at any time point evaluated. For post-prandial glucose, a significant difference between SRT2104 and placebo was seen at one data point: change from baseline on day 28 at 30 min after a standardized meal with SRT2104 2.0 g day−1 vs. placebo (2.0 ± 2.2 vs. 3.4 ± 2.9 mmol l−1, P = 0.042). There were no other significant differences in post-prandial glucose on day 28 compared with day 1, either in absolute values or percent change. On day 28, the increase in post-prandial insulin was significantly smaller with SRT2104 2.0 g day−1 vs. placebo at 30 min (57 ± 73 vs. 162 ± 204 mmol l−1, P = 0.010) and 60 min (121 ± 95 vs. 223 ± 207 mmol l−1, P = 0.046) after a standardized meal.
Table 4.
Day 28 change from baseline: glucose and insulin concentrations (ITT population)
| SRT2104 (g day−1) | |||||
|---|---|---|---|---|---|
| Placebo (n = 45) | 0.25 (n = 45) | 0.5 (n = 46) | 1.0 (n = 45) | 2.0 (n = 45) | |
| Day 28 vs. baseline | |||||
| Fasting plasma glucose | |||||
| n | 43 | 35 | 42 | 36 | 38 |
| Mean (SD) mmol l−1 | −1.2 (2.4) | −1.1 (3.5) | −0.5 (2.6) | −1.0 (2.8) | −0.2 (2.4) |
| Fasting plasma insulin | |||||
| n | 41 | 36 | 39 | 38 | 36 |
| Mean (SD) mmol l−1 | 1.0 (51.7) | 8.9 (95.0) | −6.9 (41.5) | 4.1 (57.2) | 15.2 (138.8) |
| Change from Day 28 baseline | |||||
| Post-prandial glucosea | |||||
| 30 min, n | 24 | 26 | 28 | 25 | 26 |
| Mean (SD) mmol l−1 | 3.4 (2.9) | 2.8 (2.4) | 3.4 (2.5) | 3.1 (1.8) | 2.0 (2.2)b |
| 60 min, n | 24 | 26 | 29 | 25 | 26 |
| Mean (SD) mmol l−1 | 4.5 (2.9) | 3.9 (3.6) | 5.1 (2.6) | 4.7 (2.3) | 3.7 (3.1) |
| 120 min, n | 43 | 35 | 41 | 38 | 38 |
| Mean (SD) mmol l−1 | 3.6 (3.4) | 2.9 (3.3) | 4.2 (2.9) | 3.7 (2.5) | 3.5 (3.4) |
| Post-prandial insulina | |||||
| 30 min, n | 24 | 26 | 28 | 28 | 26 |
| Mean (SD) mmol l−1 | 162 (204) | 109 (136) | 119 (157) | 95 (110) | 57 (73)c |
| 60 min, n | 24 | 26 | 28 | 27 | 26 |
| Mean (SD) mmol l−1 | 223 (207) | 177 (279) | 154 (147) | 128 (106) | 121 (95)d |
| 120 min, n | 43 | 37 | 40 | 40 | 39 |
| Mean (SD) mmol l−1 | 142 (114) | 123 (254) | 129 (107) | 121 (130) | 117 (127) |
Not all subjects had samples for post-prandial glucose and insulin collected at 30 min and 1 h after the meal.
P = 0.042 unadjusted anova vs. placebo.
P = 0.034 anova with Dunnett's test vs. placebo; P = 0.010 unadjusted anova vs. placebo.
P = 0.046 unadjusted anova vs. placebo.
Neither HbA1c nor fructosamine levels exhibited SRT2104 dose-related trends. However, for HbA1c, there were statistically significant differences at day 28 between SRT2104 0.25 g day−1 (8.7% ± 1.00, P =0.036) and 0.5 g day−1 (8.6% ± 0.93, P = 0.037) and placebo (8.2% ± 0.95). There was also a statistically significant difference in change from baseline HbA1c between SRT2104 1.0 g day−1 and placebo (0.01% ± 0.78 vs. −0.31% ± 0.55, respectively, P = 0.029). The decrease in HbA1c in the placebo arm was more substantial than would be expected for a treatment interval of 28 days, while the SRT2104 arms in general had little change. By day 28, fructosamine levels decreased in the placebo group (−14.4 ± 41.2 mmol l−1) and in all SRT2104 groups except 0.5 g day−1. In this group, fructosamine was increased 9.8 ± 62.8 mmol l−1 relative to baseline (P = 0.024).
Lipids
In general, improvements in lipid parameters were noted for subjects treated with SRT2104 compared with placebo. A statistically significant increase from baseline in the HDL : LDL ratio was seen with SRT2104 0.25 g day−1 vs. placebo on days 15 (P = 0.0445, Table 5) and 22 (P = 0.0228). SRT2104 1.0 g day−1 was associated with a statistically significant decrease from baseline in total cholesterol on days 22 (mean −19 mg dl−1, P = 0.046) and 28 (−21 mg dl−1, P = 0.0478) (Table 6) and in triglycerides on days 15 (−44 mg dl−1, P = 0.0365), 22 (−73 mg dl−1, P = 0.0332) and 35 (−80 mg dl−1, P = 0.0293). SRT2104 1.0 g day−1 resulted in increases from baseline in HDL compared with placebo on day 15 (3 mg dl−1, P = 0.0277). Whether SRT2104 had a dose-dependent effect on lipid levels was unclear. Modelling of day 28 Cmax and AUC(0,t), which included adjustment for baseline levels, indicated a trend of association between higher Cmax and AUC(0,t) and decreased LDL and total cholesterol and increased HDL and HDL : LDL ratio. A trend of association between exposure and a decrease in triglycerides was also noted. Baseline LDL concentrations (P < 0.0001), male gender (P = 0.0472) and BMI (P = 0.0992) appeared to have had an independent effect on decrease in LDL after SRT2104 treatment. Adjusted mean percent change (decrease) in LDL in the high exposure group was greater for males than females (−12.7 vs. 2.2, respectively). Percent change in the high exposure group was greater for subjects with BMI ≥30 kg m−2 than for BMI <30 kg m−2 (−8.3 vs. −2.2, respectively). These effects were additive (−11.8% change in males with BMI ≥30 kg m−2). Total cholesterol behaved similarly, as expected. No concentration or baseline characteristic effects were observed for HDL or triglycerides in this analysis.
Table 5.
HDL : LDL ratio by visit
| Visit | Placebo (n = 45) | SRT2104 (g day−1) | |||
|---|---|---|---|---|---|
| 0.25 (n = 45) | 0.5 (n = 46) | 1.0 (n = 45) | 2.0 (n = 45) | ||
| HDL : LDL ratio | |||||
| Mean (SD) | |||||
| Day 1 | 0.42 (0.162) | 0.42 (0.198) | 0.44 (0.190) | 0.39 (0.153) | 0.46 (0.189) |
| Day 15 | 0.44 (0.167) | 0.50 (0.267)1 | 0.47 (0.129) | 0.43 (0.135) | 0.51 (0.196) |
| Day 22 | 0.43 (0.173) | 0.52 (0.284)2 | 0.48 (0.159) | 0.43 (0.133) | 0.48 (0.207) |
| Day 28 | 0.44 (0.198) | 0.50 (0.353) | 0.47 (0.193) | 0.43 (0.152) | 0.48 (0.191) |
P = 0.0445.
P = 0.0228.
Table 6.
Changes in lipid parameters and weight
| Day 28 change from baseline | Placebo (n = 45) | SRT2104 (g day−1) | |||
|---|---|---|---|---|---|
| 0.25 (n = 45) | 0.5 (n = 46) | 1.0 (n = 45) | 2.0 (n = 45) | ||
| Cholesterol | |||||
| n | 41 | 35 | 41 | 38 | 40 |
| Mean (SD) mg dl−1 | −5.8 (37.5) | −8.9 (35.9) | −9.7 (34.4) | −21.2 (31.3)* | −8.9 (35.5) |
| HDL | |||||
| n | 42 | 35 | 41 | 38 | 40 |
| Mean (SD) mg dl−1 | −1.5 (9.3) | −0.8 (7.7) | −1.5 (7.7) | 0.4 (6.9) | −1.9 (7.7) |
| LDL | |||||
| n | 41 | 36 | 39 | 38 | 36 |
| Mean (SD) mg dl−1 | −2.7 (26.6) | −6.7 (29.7) | −11.2 (29.0) | −10.8 (22.8) | −8.5 (33.2) |
| Triglycerides | |||||
| n | 42 | 35 | 41 | 37 | 39 |
| Mean (SD) mg dl−1 | −7.1 (93.8) | −8.0 (141.6) | 19.5 (84.0) | −66.4 (169.0) | 2.7 (133.6) |
| Weight | |||||
| n | 42 | 37 | 42 | 40 | 39 |
| Mean (SD) kg | −0.2 (1.3) | −0.8 (2.2)* | −1.3 (2.1) | −0.6 (1.7) | −1.5 (3.8)** |
P ≤ 0.05.
P ≤ 0.01.
Weight
Weight tended to decrease in all SRT2104 active treatment arms on day 28 compared with baseline (Table 5). A statistically significant decrease from baseline in weight was seen with SRT2104 0.5 g day−1 (P = 0.0237) and 2.0 g day−1 (P = 0.0092) vs. placebo. Mean decrease from baseline to day 28 ranged from 0.56–1.54 kg across SRT2104 treatment arms. As with lipids, after adjusting for baseline, a trend of association between higher Cmax and AUC(0,t) and decrease in weight was observed.
Discussion
SRT2104 administered orally to type 2 diabetics on stable background metformin for 28 days, at doses ranging from 0.25 g to 2.0 g, was well tolerated. AEs in general were mild and self-limited. The most common AEs, nausea, diarrhea, asthenia and hyperglycaemia, occurred similarly on active treatment and placebo. No clear dose response was noted for any parameter. In addition, there were no safety signals for laboratory assessments or cardiac/ECG monitoring.
SRT2104 drug concentrations were variable between subjects and dose proportionality across the doses tested was not achieved. There was significant inter-subject variability with regard to pharmacokinetic parameters, with coefficients of variation (CVs) > 75% for the exposure parameters AUC(0,t) and Cmax for all treatment groups, despite providing a standard meal to be consumed at the time of drug administration. Therefore, the pharmacokinetic properties were suboptimal and limited the ability to identify dose responses in this study.
In this 28 day study, SRT2104 doses of 0.25 g day−1 to 2.0 g day−1 did not have a significant impact on glycaemic control. Although some statistically significant differences in individual data points were observed, no consistent, dose-related changes in fasting concentrations of glucose or insulin, post-prandial glucose or insulin, HbA1c, or fructosamine were observed with SRT2104. The significant difference in HbA1c was due to a decrease in the placebo arm vs. little change in the SRT2104 arm; this level of decrease in the placebo arm (0.31 percentage points) in a 28 day period was not expected and is not well understood.
Inadequate exposure may have contributed to the lack of improvement in glycaemic control observed in this study. SRT2104 exposure increased in a less than dose-proportional fashion, creating broad overlap in exposures between dosing groups. In this study, the highest dose of SRT2104 (2.0 g day−1) did not achieve exposure levels equivalent to those that are efficacious in the murine model of type 2 diabetes (DIO). The mean Cmax plasma exposure at the 2.0 g day−1 dose was 434 ng ml−1 and AUC(0,t) was 4380 ng ml−1 h. These exposures are 10-fold and 13-fold lower, respectively, than had been observed in mice at the minimally efficacious dose for this indication (unpublished data). Indeed, the exposure observed in this study was lower than anticipated based on previous clinical studies [13], where a substantial increase in exposure was reported when subjects were administered SRT2104 following a standardized solid meal. In the current study, in an effort to standardize meals across multiple international sites, a liquid dietary supplement was chosen as the pre-dose meal because it was similar in caloric content to solid meals used in earlier studies. However, the anticipated food effect associated with higher absorption was not observed. Exposure was essentially similar to what had previously been observed in the fasted state [13], thus leading to sub-optimal SRT2104 drug concentrations. The reasons for this apparent lack of anticipated food effect are unclear, but could be a function of the difference in a liquid vs. solid meal, suggesting that gastric emptying time may be a critical factor affecting SRT2104 absorption. Alternately, it may be attributable to a characteristic of the study population (type 2 diabetes subjects) where changes in GI motility and absorption are known to occur, since previous pharmacokinetic analyses had been done in healthy volunteers [13]. There is also a possibility that not all subjects adhered to consuming the liquid dietary supplement as their standard meal at breakfast.
Despite the lower than anticipated drug exposures, mean improvements in most lipid parameters were observed (approximately 5–10%) for subjects treated with SRT2104 compared with placebo. Weight reductions were also observed in subjects treated with SRT2104. At the 2.0 g day−1 dose, mean weight loss was 1.5 kg after 28 days of SRT2104 treatment (P = 0.0092). These are interesting observations given that improvements in fatty acid metabolism and weight loss are also observed when SRT2104 is administered to DIO mice [11]. One target for SIRT1 is PGC-1α, a transcriptional co-activator that regulates the genes involved in energy metabolism [14]. Through deacetylation of PGC-1α, SIRT1 is thought to impact on mitochondrial efficiency, leading to an improved metabolic state, which may mechanistically help to explain benefit on lipids and weight [15]. As dyslipidaemia and obesity are common co-morbidities in type 2 diabetes mellitus, these properties of SRT2104 may be of added benefit, if efficacy in glucose and insulin management can be achieved with higher and more consistent SRT2104 blood concentrations. In conclusion, treatment with SRT2104 for 28 days did not result in improved glucose or insulin control in our trial. Further studies with more potent SIRT1 activators and/or with SIRT1 activators that achieve greater exposure in humans are indicated to explore further the metabolic effects of SIRT1 activators in type 2 diabetes.
Acknowledgments
This study was funded by Sirtris, a Division of GSK. This trial was sponsored and financially supported by Sirtris, a Division of GSK, Cambridge, MA, USA.
The authors thank the subjects and the investigators for their participation in this study.
Competing Interests
EWJ, GPV, PE, EH, JH were employees of Sirtris, a GSK Company during the conduct of the study and the preparation of the manuscript.
References
- 1.Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, Lin JK, Farzadfar F, Khang YH, Stevens GA, Rao M, Ali MK, Riley LM, Robinson CA, Ezzati M. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. Global Burden of Metabolic Risk Factors of Chronic Diseases Collaborating Group (Blood Glucose) [DOI] [PubMed] [Google Scholar]
- 2.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus-present and future perspectives. Nat Rev Endocrinol. 2011;8:228–236. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 3.Pinhas-Hamiel O, Zeitler P. The global spread of type 2 diabetes mellitus in children and adolescents. J Pediatr. 2005;146:693–700. doi: 10.1016/j.jpeds.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 4.Unger J. Current strategies for evaluating, monitoring, and treating type 2 diabetes mellitus. Am J Med. 2008;121(Suppl. 6):S3–8. doi: 10.1016/j.amjmed.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Kazgan N. Mammalian sirtuins and energy metabolism. Int J Biol Sci. 2011;7:575–587. doi: 10.7150/ijbs.7.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Méneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, MaDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin S, Guarente L. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nukes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pui YN, Jean EB, Jeremy SD, Chi BV, Christopher JO, Amy VL, David PC, Thomas VR, Jeffrey S, Jesse JS, Siva L, Angela T, Meghan D, Marie Y, Kristina K, Akanksha G, Vipin S, Peter JE, Jill CM, Joseph JN, Michael RJ, George PV, James LE, Robert BP. The identification of the SIRT1 activator SRT2104 as a clinical candidate. Lett Drug Des Discov. 2013;10:793–797. [Google Scholar]
- 11.Qi Y, Davis ML, Hirsch ML, Cote AM, Lainez EO, Johnson MO, Gagne DJ, Vlasuk GP, Ellis JL. Activation of sirtuin1 (SIRT1) by the novel small molecule SRT2104 promotes body weight loss, increases exercise capacity and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2010;59(Suppl. 1) 390-PP. [Google Scholar]
- 12.Qi Y, Davis ML, Lainez EO, Cote AM, Johnson MO, Gagne DJ, Vlasuk GP, Ellis JL, Suri V. SRT2104, a novel small molecule SIRT1 activator ameliorates insulin resistance and promotes glucose utilization measured under a hyperinsulinemic-euglycemic clamp by enhancing both glycolysis and carbohydrate oxidation in mice fed a high fat diet. Diabetes. 2011;60(Suppl. 1) 1007-P. [Google Scholar]
- 13.Hoffmann E, Wald J, Lavu S, Roberts J, Beaumont C, Haddad J, Elliott P, Westphal C, Vlasuk GP, Jacobson EW. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br J Clin Pharmacol. 2013;75:186–196. doi: 10.1111/j.1365-2125.2012.04340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 15.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]