

Abstract
Evidence suggests that autophagy may be a new therapeutic target for stroke, but whether activation of autophagy increases or decreases the rate of neuronal death is still under debate. This review summarizes the potential role and possible signaling pathway of autophagy in neuronal survival after cerebral ischemia and proposes that autophagy has dual effects.
Keywords: nerve regeneration, autophagy, lysosome, autophagosome, neuron, cerebral ischemia, signaling pathway, apoptosis, necrosis, survival, NSFC grant, neural regeneration
Introduction
Nerve cell survival depends on the balance between the formation and degradation of cellular proteins and damaged organelles. The ubiquitin-proteasome system and autophagy pathway are the two major mechanisms for maintaining this balance. The ubiquitin-proteasome system is an important route for the degradation of short-lived proteins whereas autophagy is responsible for the degradation of long-lived proteins and damaged organelles (Pan et al., 2008). The activation of autophagy has been observed in many diseases such as cardiovascular disease (Martinet et al., 2007), cerebral hemorrhage (Hu et al., 2011), pancreatic cancer (Grasso et al., 2012) and neurodegenerative disease such as Alzheimer's disease and Parkinson's disease (Shacka et al., 2008). There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (Rubinsztein et al., 2005; Levine and Kroemer, 2008). Macroautophagy occurs through autophagosomes, double-membrane-bound vesicles that fuse with lysosomes which subsequently degrade proteins. Microautophagy is the engulfment of cytoplasmic cargo directly by the lysosomal membrane through the processes of invagination, protrusion, and separation. Chaperone-mediated autophagy transports unfolded proteins via lysosomal chaperonin heat-shock constitutive protein 70 (Hsc70) and lysosomal membrane-2A (LAMP-2A), a lysosomal membrane receptor (Periyasamy-Thandavan et al., 2009). Macroautophagy is the most common and the best-studied form of autophagy. Cerebral ischemia is the leading cause of death and disability worldwide. Two major therapeutic strategies are currently being used to treat cerebral ischemia (Gabryel et al., 2012). The first is the use of thrombolytic, antithrombotic and anti-aggregation drugs. However, these drugs have a narrow therapeutic window of 3 hours, and could lead to hemorrhagic complications. The second is neuroprotection. But, most neuroprotective agents show poor efficacy or severe toxicity/side effects and thus new therapeutic agents for the treatment of ischemic stroke need to be developed. Recent studies show that macroautophagy is activated in cerebral ischemia and indicate that autophagic induction might serve as a new therapeutic target for stroke (Wen et al., 2008; Wei et al., 2012). The relationship between autophagy and cerebral ischemia is unclear; one report suggests activation of autophagy protects neurons from death (Carloni et al., 2008), whereas another indicates it has a more destructive role (Koike et al., 2008). In this review, we introduce macroautophagy (herein referred to as autophagy), and focus on its potential role and possible signaling pathways in cerebral ischemia.
The process of autophagy and its functional complexes
Autophagy plays an important physiological role in the process of cellular growth and differentiation (Gabryel et al., 2012) and is extremely important in maintaining cellular homeostasis which requires the continual turnover of nonfunctional proteins and organelles. The process of autophagy consists of several sequential steps (Klionsky and Ohsumi, 1999; Mizushima, 2007; He and Klionsky, 2009): (1) Sequestration. A unique membrane, called an isolation membrane, sequesters cytoplasmic constituents including organelles to form an autophagosome. (2) Transportation. The material sequestrated by autophagosomes is transported to the lysosome. (3) Degradation. Autophagosomes fuse with lysosomes. These structures are often called “autolysosomes” or “autophagolysosomes.” Both the inner membrane and the materials contained within the autophagosome are degraded by lysosomal hydrolases. (4) Utilization of degradative products. The degraded material is exported to the cytoplasm for reuse.
Autophagy depends on the interaction of different complexes that are composed of several different autophagy-related (Atg) proteins. The complexes include (1) two ubiquitin-like conjugation systems, Atg12-Atg5 and Atg8/LC3-PE (microtubule-associated protein 1A/1B-light chain 3 -phosphatidylethanolamine) conjugate systems (Klionsky and Emr, 2000; Suzuki et al., 2005; Periyasamy-Thandavan et al., 2009); (2) a phosphatidylinositol 3-kinase (PI3K) complex; (3) the Atg1/Unc-51-like kinase (ULK) complexes; (4) mAtg9; and (5) the Atg2-Atg18 complexes (Longatti and Tooze, 2009; Chen and Klionsky, 2011; Mizushima et al., 2011).
Apoptosis, necrosis and autophagy in cerebral ischemia
Stroke is the third most common cause of death worldwide and the major cause of adult neurological disability. It can be caused by a number of different disorders and results in temporary or permanent disruption of blood supply to the brain. Approximately 80% of stroke cases are due to primary cerebral ischemia resulting in infarction, whilst 20% are attributed to cerebral hemorrhage (Markus, 2012). When blood flow to the brain is interrupted, cells undergo a series of molecular events which include excitotoxicity, mitochondrial dysfunction, acidotoxicity, ionic imbalance, oxidative stress and inflammation. These molecular events can lead to cell death and irreversible tissue injury (Ouyang and Giffard, 2012). There is no doubt that neurons have a vital role in the nervous system and so it is particularly important to protect them from insult and death in cerebral ischemia.
There are two morphological types of cell death following cerebral ischemia, necrosis and apoptosis (Puyal and Clarke, 2009). Morphological characteristics of necrosis include vacuolation of the cytoplasm, breakdown of the plasma membrane and an induction of inflammation around the dying cell by release of cellular contents and proinflammatory molecules (Edinger and Thompson, 2004). Apoptosis is also termed type I programmed cell death (type I PCD) (Shintani and Klionsky, 2004) and is characterized by nuclear condensation and fragmentation, cleavage of chromosomal DNA into internucleosomal fragments and the formation of apoptotic bodies, which are removed by phagocytosis without the breakdown of the plasma membrane (Edinger and Thompson, 2004). Recently, studies have identified that aside from necrosis and apoptosis, a third type of cell death that occurs during ischemic stroke, autophagy (Adhami et al., 2006; Rami et al., 2008) which can also be termed type II PCD (Ouyang et al., 2012).
The correlation between apoptosis, autophagy and necrosis and their pathologic processes in ischemic stroke is not clear. Some studies have shown that neurons at the core of ischemia tend to undergo necrosis, while neurons in the penumbra (surrounding region) are subjected to apoptosis (Sabri et al., 2013). However, some in vivo experiments indicate that both apoptosis and autophagy are activated in the ischemic penumbra (Pamenter et al., 2012). Terminal deoxynucleotidyl transferase-mediated dUTP-biotin in situ nick end labeling (TUNEL) and propidium iodide (PI) are used as markers of apoptosis and necrosis, respectively (Unal Cevik and Dalkara, 2003). According to a study by Balduini et al. (2012), autophagy-positive neurons that were also TUNEL-positive were mainly found in superficial layers of the cerebral cortex 24 hours after hypoxia-ischemia, with only scattered necrotic cells observed in the same area. They also reported that after ischemia, necrotic cells (PI-positive) were mainly detected in the hippocampus and the deep layers of the cerebral cortex, and most cells in the superficial layers of the cortex were PI-positive (necrotic) at later time points (48–72 hours after hypoxia-ischemia). When 3-methyladenine (3-MA) and wortmannin, two type III PI3K inhibitors), were used to inhibit autophagy in a model of neonatal hypoxia-ischemia, activation of the autophagic pathway was significantly reduced and there was a switch from apoptosis to necrosis in cell death. In contrast, it could be observed that necrotic cell death and brain injury were reduced when rapamycin, the inhibitor of the mammalian target of rapamycin (mTOR), was administered to increase autophagy (Balduini et al., 2009). The activated pathways of autophagy and apoptosis also share some common components. For example, BCL2L11 (also known as BIM), a BH3-only protein, is considered a novel molecular link between autophagy and apoptosis. BCL2L11 recruits Beclin-1, the homologue of yeast Atg6, to microtubules by bridging Beclin-1 and dynein light chain 1 (DYNLL1, also known as LC8), and it can inhibit autophagy and promote apoptosis (Luo and Rubinsztein, 2013). In summary, the crosstalk among the three types of cell death, especially between autophagy and apoptosis, is very complex and not understood.
Autophagy in cerebral ischemia
Autophagy is the process by which a membrane engulfs organelles and cytosolic macromolecules to form an autophagosome, with the engulfed materials being delivered to the lysosome for degradation (Yoshimori, 2007). Neuronal autophagy has two unique features. Autophagy was not observed in the brain of mice that had been deprived of food for 48 hours and it is thought that brain nutrients could be compensated from other organs under conditions of starvation (Mizushima et al., 2004). Second, localization of autophagosomes and lysosomes is different; autophagosomes are located throughout the cytoplasm but lysosomes are mainly located in the juxtanuclear cytoplasm of the cell body in the neuron. To form autolysosomes, autophagosomes which are formed in dendrites and synaptic terminal regions need be transported to lysosomes in the cell body. This means that when dendrites or axons are damaged, autophagosomes that have formed in dendrites and synaptic terminal regions cannot fuse with lysosomes to degrade sequestrated material (Komatsu et al., 2007).
Autophagy can be subclassified into basal and induced autophagy. Basal autophagy is considered a “housekeeping” process in neurons (Mizushima and Komatsu, 2011), whereas induced autophagy may be a promising neuroprotective strategy in neurodegenerative diseases. Induction of autophagy may serve to rid neurons of aberrant protein aggregates – a common hallmark of neurodegenerative diseases. Many studies have suggested that either the absence of autophagy or inadequate autophagy may be an underlying cause of neurodegenerative diseases (Puyal et al., 2012). Although currently there is no unified theory as to the role autophagy plays in cerebral ischemia, based on studies over past few years, the following five viewpoints ischemia have emerged.
(1) Activation of autophagy in cerebral ischemia protects neurons from death. Carloni et al. (2010) suggested that in neonatal hypoxia-ischemia, autophagy may be part of an integrated pro-survival signaling complex that includes PI3K-Akt-mTOR. When either autophagy or PI3K-Akt-mTOR pathways were interrupted, cells underwent necrotic cell death. Wang et al. (2012) reported that neuronal survival was promoted during cerebral ischemia when autophagy was induced by nicotinamide phosphoribosyltransferase (Nampt, also known as visfatin), which is the rate-limiting enzyme in mammalian NAD+ biosynthesis and regulates the TSC2-mTOR-S6K1 signaling pathway. These studies suggest that autophagy may be a potential target for post-ischemic neuronal protection.
(2) Activation of autophagy in cerebral ischemia has a destructive role. Mice deficient in Atg7, the gene essential for autophagy induction, showed nearly complete protection from both hypoxia-ischemia-induced caspase-3 activation and neuronal death, indicating autophagy is essential in triggering neuronal death after hypoxia-ischemia injury (Koike et al., 2008). Wen et al. (2008) confirmed autophagy was activated in a permanent middle cerebral artery occlusion (MCAO) model. In their paper, the infarct volume, brain edema and motor deficits could be significantly reduced by administration of 3-MA (an autophagy inhibitor). The neuroprotective effects of 3-MA were associated with an inhibition of ischemia-induced upregulation of LC3-II, a marker of active autophagosomes and autophagolysosomes. Moreover, it was observed that the inhibition of autophagy, either by direct inhibitor 3-MA or by indirect inhibitor 2ME2 (an inhibitor of hypoxia inducible factor-1α; HIF-1α) might prevent pyramidal neuron death after ischemia in the study of Xin et al. (2011).
(3) The degree of autophagy determines the fate of cells in cerebral ischemia. Kang and Avery (2008) proposed that levels of autophagy were critical for the survival or death of cells: physiological levels of autophagy promote survival, whereas insufficient or excessive levels of autophagy promote death. This hypothesis was confirmed in an oxygen and glucose deprivation model that observed dual roles of the autophagy inhibitor 3-MA in different stages of re-oxygenation (Shi et al., 2012). Twenty-four hours prior to reperfusion, 3-MA triggered a high rate of neuronal death. However, during 48–72 hours of reperfusion, 3-MA significantly protected neurons from death. It is possible that prolonged oxygen and glucose deprivation/reperfusion triggers excessive autophagy, switching its role from protection to deterioration.
(4) After cerebral ischemia, the time at which autophagy is induced determines its role. Autophagy could play a protective role in ischemic preconditioning but have a different effect once ischemia/reperfusion has occurred (Ravikumar et al., 2010). Infarct volume, brain edema and motor deficits induced by permanent focal ischemia were significantly reduced after ischemic preconditioning treatment. 3-MA suppressed neuroprotection induced by ischemic preconditioning, while rapamycin reduced infarct volume, brain edema and motor deficits induced by permanent focal ischemia (Sheng et al., 2010). This hypothesis was supported by a study by Yan et al. (2011) in which 3-MA administrated through intracerebroventricular injection before hyperbaric oxygen preconditioning, attenuated the neuroprotection of hyperbaric oxygen preconditioning against cerebral ischemia. Moreover, 3-MA treatment before middle cerebral artery occlusion aggravated subsequent cerebral ischemic injury. In contrast, pretreatment with rapamycin mimicked the neuroprotective effect of hyperbaric oxygen preconditioning (Yan et al., 2011). Carloni et al. (2008, 2010) also showed that when 3-MA and rapamycin were injected 20 minutes before hypoxia-ischemia, 3-MA inhibited autophagy, significantly reduced beclin-1 expression and caused neuronal death, while rapamycin increased autophagy and decreased brain injury. In addition, 3-MA administrated by intracerebroventricular injections strongly reduced the lesion volume (by 46%) even when given 4 hours after the beginning of the ischemia (Puyal et al., 2009). Gao et al. (2012) found that rapamycin applied at the onset of reperfusion might attenuate the neuroprotective effects of ischemic postconditioning. Conversely, 3-MA administered before reperfusion significantly reduced infarct size and abolished the increase of brain water content after ischemia. Targeting autophagy either pre- or post-treatment has different results and this may reflect the different effects of autophagy at early and late stages (He et al., 2012). The time of intervention could be related to the degree of autophagy at different stages of ischemia and further studies are necessary to confirm this.
(5) Autophagy may be interrupted in cerebral ischemia. A common feature of many neurodegenerative diseases is the accumulation of an abnormally large number of autophagic vacuoles (autophagosomes and autolysosomes) or the frequent appearance of irregularly shaped autophagic vacuoles. Enhanced autophagosome formation seems to be reflected by increased density of autophagic vacuoles, but these increased autophagic vacuoles may also imply impaired autolysosomal degradation (Komatsu et al., 2007). Rami et al. (2008) also observed a dramatic up-regulation of Beclin-1 and LC3 in rats after cerebral ischemia. These results indicate that autophagy was activated in the brain following ischemia. Recently, however, it has been hypothesized that the increase in proteins may reflect a failure in lysosomal function leading to an accumulation of autophagosomes, or an improvement in the activity of autophagy (Xu et al., 2012). Liu et al. (2010) found that accumulation of LC3-II was observed in sham-operated rats after treatment with lysosomal inhibitor-chloriquine, but the further change of LC3-II levels in post-ischemic brain tissues was not observed. The results indicated that accumulation of autophagy-associated protein following ischemia could be the result of failure of the autophagy pathway. Puyal and Clarke (2009) demonstrated that lysosomal activity detected by LAMP-1 and cathepsin D was increased in neurons with punctate LC3 expression in neonatal focal cerebral ischemia model. The failure of autophagosome and lysosome fusion caused an increase of autophagosomes. The deficiency of acid phosphatase activity in the lysosome could lead to the increase of autophagosomes and autolysosomes. Further studies are required to verify whether the activity of autophagy is enhanced in cerebral ischemia.
Possible autophagy signaling pathways in cerebral ischemia
Cerebral ischemia can activate multiple signaling pathways that subsequently feed into the autophagy pathway. Mammalian target of rapamycin (mTOR) is a 289 kDa serine/threonine protein kinase that regulates transcription, cytoskeleton organization, cell growth and cell survival. By binding to different co-factors, mTOR can form two distinct protein complexes, mTORC1 (mTOR complex 1) and mTORC2 (mTOR complex 2) (Jung et al., 2010). The classification of mTORC1 and mTORC2 are based on their components and sensitivity to rapamycin. mTORC1 is responsible for the inhibitory effect of rapamycin, moreso than mTORC2. Recent studies suggest that the PI3K/Akt/mTOR pathway could regulate acute nervous system injury in cerebral hypoxia-ischemia (Chong et al., 2012).
PI3K consists of class I, class II and class III. Class I PI3K plays an important role in the PI3K-Akt-mTOR pathway. PI3K phosphorylates and activates Akt which in turn phosphorylates and inactivates tuberous sclerosis complex (TSC) 1/2. Inactivated TSC1/2 increases the activation of Rheb which is part of the Ras family GTP-binding protein, and mTOR is subsequently activated. Autophagy is inhibited by activating mTOR (Glick et al., 2010). Beclin-1, a component of the class III PI3K, is essential for the initial steps of autophagy and could also induce autophagy via the interaction with other components of the class III PI3K pathway in cerebral ischemia (Xingyong et al., 2013). Peroxisome proliferator-activated receptor-γ (PPAR-γ), a member of nuclear hormone receptor superfamily, is a ligand-activated transcription factor. PPAR-γ activation antagonizes beclin-1-mediated autophagy via upregulation of Bcl-2/Bcl-xl which interact with beclin-1 in cerebral ischemia/reperfusion (Xu et al., 2013).
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase and consists of three subunits: a catalytic α-subunit and regulatory β and γ-subunits. Each subunit appears to have distinct functions. The most studied is the catalytic α-subunit which contains a threonine phosphorylation site that when phosphorylated, activates AMPK.. AMP which binds to sites located on the γ-subunit can enhance the phosphorylation of AMPK by LKB1, and AMPK activation could subsequently inhibit the activity of mTOR to induce autophagy (Poels et al., 2009; Li and McCullough, 2010).
Nuclear factor kappa B (NF-κB) is a transcription factor that regulates expression of multiple genes (Chen et al., 2011). Recent experiments have demonstrated that the knockout of p50 (NF-κB1) enhanced autophagy by repression of mTOR in cerebral ischemic mice (Li et al., 2013). NF-κB-dependent p53 signal transduction pathway is also associated with autophagy and apoptosis in the rat hippocampus after cerebral ischemia/reperfusion insult (Cui et al., 2013). Mitogen-activated protein kinases (MAPKs) include extracellular signal-related kinase (ERK), Jun NH2 terminal kinase (JNK) and p38 (Lien et al., 2013). MAPK is one upstream regulator of mTORC1 and autophagy could also be induced via MAPK-mTOR signaling pathway in cerebral ischemia/reperfusion (Wang et al., 2013).
Hypoxia is a common cause of cell death and occurs in ischemic stroke. Hypoxia-inducible factor 1 (HIF-1) is a key transcriptional factor that is activated in response to hypoxia during cerebral ischemia (Althaus et al., 2006). HIF-1 is composed of a constitutively expressed HIF-1β subunit and an inducibly expressed HIF-1α subunit. Since ubiquitination is inhibited under hypoxic conditions, HIF-1α can accumulate and dimerize with HIF-1β. This dimer activates transcription of a number of downstream hypoxia-responsive genes, including vascular endothelial growth factor (VEGF), erythropoietin (EPO), glucose transporter 1, and glycolytic enzymes (Xin et al., 2011). Bcl-2 and adenovirus E1B 19 kDa interacting proteins 3 (BNIP3) with a single Bcl-2 homology 3 (BH3) domain is a subfamily of Bcl-2 family proteins and also serves as an important target gene of HIF-1α (Cho et al., 2012). BNIP3 can compete with beclin-1 for binding to Bcl-2 and beclin-1 is released to trigger autophagy (He and Klionsky, 2009; Glick et al., 2010). BNIP3 also binds and inhibits Rheb, an upstream activator of mTOR, so it could activate autophagy by inhibiting mTOR activity. However, these models still require additional genetic testing in vivo (Zhang and Ney, 2011). The induced p53 stabilization by up-regulation of HIF-1α also plays an important role in post-ischemic autophagy activation (Xin et al., 2011).
Autophagic cell death is activated in the nervous system in response to oxidative stress (Kubota et al., 2010). Oxidative stress can occur in cerebral ischemia and could increase reactive oxygen species such as superoxide, hydroxyl radical and hydrogen peroxide. Recent studies have reported that selenium provides neuroprotection through preserving mitochondrial function, decreasing reactive oxygen species production and reducing autophagy (Mehta et al., 2012). Autophagy can also be induced under conditions of excitotoxicity which can also occur in cerebral ischemia (Puyal et al., 2012). Although excitotoxic glutamate blocks autophagic flux, it could also induce autophagy in hippocampal neurons (Kulbe et al., 2014). Sustained elevations of Ca2+ in the mitochondrial matrix are a major feature of the intracellular cascade of lethal events during cerebral ischemia. Recently, it was reported that endoplasmic reticulum stress is one of the effects of excitotoxicity (Ouyang and Giffard, 2012). When endoplasmic reticula were exposed to toxic levels of excitatory neurotransmitters, Ca2+ was released via the activation of both ryanodine receptors and IP3R, leading to mitochondrial Ca2+ overload and activation of apoptosis. During endoplasmic reticulum stress, Ca2+ increase seems to be required for activating autophagy. The signaling pathways mentioned above and their relationship to cerebral ischemia and auto phagy are shown in Figure 1.
Figure 1.
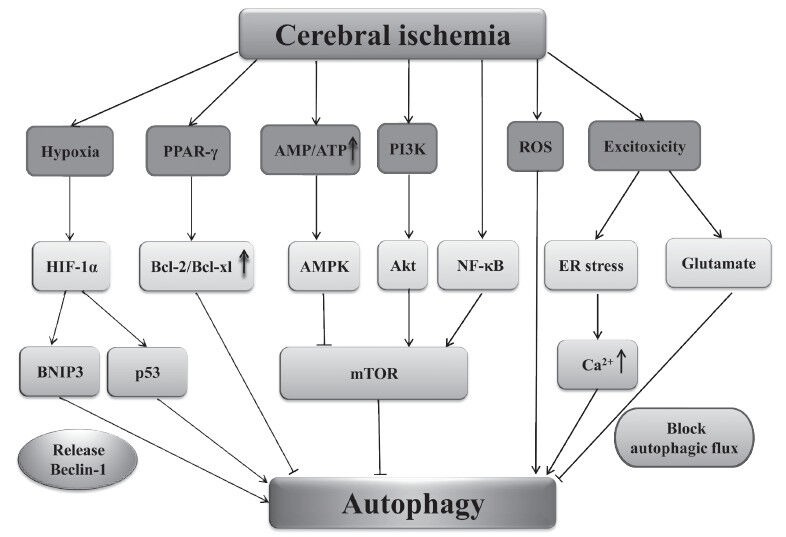
Possible autophagy signaling pathways in cerebral ischemia.
The figure shows the many different signaling pathways involved in the activation of autophagy during cerebral ischemia. When activated, Akt and NF-κB activate mTOR to inhibit autophagy in cerebral ischemia. However, the activation of AMPK could inhibit the activity of mTOR and induce autophagy. Hypoxia caused by cerebral ischemia activates HIF-1α and induces autophagy through BNIP3 and p53. Excitotoxicity could induce autophagy by ER stress and block autophagic flux by glutamate in cerebral ischemia. Autophagy could also be induced through ROS and inhibited through PPAR-γ. PPAR-γ: Peroxisome proliferator-activated receptor-γ; AMP: Adenosine 5’-monophosphate; PI3K: phosphatidylinositol 3-ki-nase; ROS: reactive oxygen species; HIF-1α: hypoxia inducible factor 1α; Bcl-2: B cell lymphoma/leukmia-2; Bcl-xL: B-cell lymphoma-extra large; AMPK: AMP-activated protein kinase; AMPK: AMP-activated protein kinase; Akt/PKB: protein kinase B; NF-κB: nuclear factor kappa B; ER: endoplasmic reticulum; BNIP3: Bcl-2 and adenovirus E1B 19 kDa interacting proteins 3; mTOR: mammalian target of rapamycin.
Conclusions
Although autophagy has no unified role in cerebral ischemia, increasing evidence supports the notion that autophagy is a double-edged sword. We believe that the degree of autophagy is critical to its role (neuroprotective or deteriorative) in ischemic stroke. Physiological levels of autophagy are favorable to neuronal survival, but excessive or inadequate levels could be harmful and cause injury. The mechanisms of autophagy underlying cerebral ischemia and whether autophagy activity is really enhanced in cerebral ischemia should be further investigated. It is also important to study the time point at which autophagy inhibitors and activators should be administered. Tian et al. (2010) have used green fluorescent protein (GFP)-fused LC3 transgenic mice to detect autophagy in vivo and this could be helpful in monitoring autophagic processes in live stroke patients as well as clarifying the detailed roles of autophagy in the ischemic brain. The idea that the time point of autophagy induction determines the fate of neurons during cerebral ischemia provides a new treatment strategy for ischemic stroke patients.
Footnotes
Funding: This work was supported by grants from the project of National Natural Science Foundation of China, No. 31171014 and 31371065; the project of Science and Technology Commission of Board of Health of Shanghai, China, No. 20134125; the Key Specialty (disease) Declaration of Pudong New Area's Health System.
Conflicts of interest: None declared.
Copyedited by Paul P, Raye W, Li CH, Song LP, Zhao M
References
- 1.Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, Rami A. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1alpha) protein following focal cerebral ischemia in rats. Neurochem Int. 2006;48:687–695. doi: 10.1016/j.neuint.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy. 2009;5:221–223. doi: 10.4161/auto.5.2.7363. [DOI] [PubMed] [Google Scholar]
- 4.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury. J Matern Fetal Neonatal Med 25 Suppl. 2012;1:30–34. doi: 10.3109/14767058.2012.663176. [DOI] [PubMed] [Google Scholar]
- 5.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32:329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 6.Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 2010;6:366–377. doi: 10.4161/auto.6.3.11261. [DOI] [PubMed] [Google Scholar]
- 7.Chen AC, Arany PR, Huang YY, Tomkinson EM, Sharma SK, Kharkwal GB, Saleem T, Mooney D, Yull FE, Blackwell TS, Hamblin MR. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS One. 2011;6:e22453. doi: 10.1371/journal.pone.0022453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124:161–170. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho B, Choi SY, Park OH, Sun W, Geum D. Differential expression of BNIP family members of BH3-only proteins during the development and after axotomy in the rat. Mol Cells. 2012;33:605–610. doi: 10.1007/s10059-012-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong ZZ, Shang YC, Wang S, Maiese K. A Critical Kinase Cascade in Neurological Disorders: PI 3-K, Akt, and mTOR. Future Neurol. 2012;7:733–748. doi: 10.2217/fnl.12.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui DR, Wang L, Jiang W, Qi AH, Zhou QH, Zhang XL. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-κB/p53 signaling pathway. Neuroscience. 2013;246:117–132. doi: 10.1016/j.neuroscience.2013.04.054. [DOI] [PubMed] [Google Scholar]
- 12.Edinger AL, Thompson CB. Death by design:apoptosis necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Gabryel B, Kost A, Kasprowska D. Neuronal autophagy in cerebral ischemia - a potential target for neuroprotective strategies? Pharmacological Reports. 2012;64:1–15. doi: 10.1016/s1734-1140(12)70725-9. [DOI] [PubMed] [Google Scholar]
- 14.Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grasso D, Garcia MN, Iovanna JL. Autophagy in pancreatic cancer. Int J Cell Biol. 2012;2012:760498. doi: 10.1155/2012/760498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He S, Wang C, Dong H, Xia F, Zhou H, Jiang X, Pei C, Ren H, Li H, Li R, Xu H. Immune-related GTPase M (IRGM1) regulates neuronal autophagy in a mouse model of stroke. Autophagy. 2012;8:1621–1627. doi: 10.4161/auto.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu S, Xi G, Jin H, He Y, Keep RF, Hua Y. Thrombin-induced autophagy: a potential role in intracerebral hemorrhage. Brain Res. 2011;1424:60–66. doi: 10.1016/j.brainres.2011.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang C, Avery L. To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation. Autophagy. 2008;4:82–84. doi: 10.4161/auto.5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klionsky DJ, Ohsumi Y. Vacuolar import of proteins and organelles from the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:1–32. doi: 10.1146/annurev.cellbio.15.1.1. [DOI] [PubMed] [Google Scholar]
- 23.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komatsu M, Ueno T, Waguri S, Uchiyama Y, Kominami E, Tanaka K. Constitutive autophagy: vital role in clearance of unfavorable proteins in neurons. Cell Death Differ. 2007;14:887–894. doi: 10.1038/sj.cdd.4402120. [DOI] [PubMed] [Google Scholar]
- 26.Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, Takeuchi T. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulbe JR, Mulcahy Levy JM, Coultrap SJ, Thorburn A, Ulrich Bayer K. Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 2014;1542:12–19. doi: 10.1016/j.brainres.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:480–492. doi: 10.1038/jcbfm.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li WL, Yu SP, Chen D, Yu SS, Jiang YJ, Genetta T, Wei L. The regulatory role of NF-kappaB in autophagy-like cell death after focal cerebral ischemia in mice. Neuroscience. 2013;244:16–30. doi: 10.1016/j.neuroscience.2013.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lien SC, Chang SF, Lee PL, Wei SY, Chang MD, Chang JY, Chiu JJ. Mechanical regulation of cancer cell apoptosis and autophagy: roles of bone morphogenetic protein receptor, Smad1/5, and p38 MAPK. Biochim Biophys Acta. 2013;1833:3124–3133. doi: 10.1016/j.bbamcr.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 32.Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68–78. doi: 10.1111/j.1471-4159.2010.06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009;16:956–965. doi: 10.1038/cdd.2009.39. [DOI] [PubMed] [Google Scholar]
- 34.Luo S, Rubinsztein DC. BCL2L11/BIM: a novel molecular link between autophagy and apoptosis. Autophagy. 2013;9:104–105. doi: 10.4161/auto.22399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markus H. Stroke: causes and clinical features. Medicine. 2012;40:484–489. [Google Scholar]
- 36.Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med. 2007;13:482–491. doi: 10.1016/j.molmed.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 37.Mehta SL, Kumari S, Mendelev N, Li PA. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012;13:79. doi: 10.1186/1471-2202-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 39.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2004;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang YB, Giffard RG. ER-mitochondria crosstalk during cerebral ischemia: molecular chaperones and ER-mitochondrial calcium transfer. Int J Cell Biol. 2012;2012:493934. doi: 10.1155/2012/493934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pamenter ME, Perkins GA, McGinness AK, Gu XQ, Ellisman MH, Haddad GG. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 2012;7:e51469. doi: 10.1371/journal.pone.0051469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain. 2008;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 46.Periyasamy-Thandavan S, Jiang M, Schoenlein P, Dong Z. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297:F244–256. doi: 10.1152/ajprenal.00033.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poels J, Spasic MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31:944–952. doi: 10.1002/bies.200900003. [DOI] [PubMed] [Google Scholar]
- 48.Puyal J, Clarke PG. Targeting autophagy to prevent neonatal stroke damage. Autophagy. 2009;5:1060–1061. doi: 10.4161/auto.5.7.9728. [DOI] [PubMed] [Google Scholar]
- 49.Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- 50.Puyal J, Ginet V, Grishchuk Y, Truttmann AC, Clarke PG. Neuronal autophagy as a mediator of life and death: contrasting roles in chronic neurodegenerative and acute neural disorders. Neuroscientist. 2012;18:224–236. doi: 10.1177/1073858411404948. [DOI] [PubMed] [Google Scholar]
- 51.Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 53.Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 54.Sabri M, Lass E, Macdonald RL. Early brain injury: A common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat. 2013;2013:9. doi: 10.1155/2013/394036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shacka JJ, Roth KA, Zhang J. The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- 56.Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 57.Shi R, Weng J, Zhao L, Li XM, Gao TM, Kong J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther. 2012;18:250–260. doi: 10.1111/j.1755-5949.2012.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki NN, Yoshimoto K, Fujioka Y, Ohsumi Y, Inagaki F. The crystal structure of plant ATG12 and its biological implication in autophagy. Autophagy. 2005;1:119–126. doi: 10.4161/auto.1.2.1859. [DOI] [PubMed] [Google Scholar]
- 60.Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, Abe K. In vivo imaging of autophagy in a mouse stroke model. Autophagy. 2010;6:1107–1114. doi: 10.4161/auto.6.8.13427. [DOI] [PubMed] [Google Scholar]
- 61.Unal Cevik I, Dalkara T. Intravenously administered propidium iodide labels necrotic cells in the intact mouse brain after injury. Cell Death Differ. 2003;10:928–929. doi: 10.1038/sj.cdd.4401250. [DOI] [PubMed] [Google Scholar]
- 62.Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. doi: 10.4161/auto.8.1.18274. [DOI] [PubMed] [Google Scholar]
- 63.Wang PR, Wang JS, Zhang C, Song XF, Tian N, Kong LY. Huang-Lian-Jie-Du-Decotion induced protective autophagy against the injury of cerebral ischemia/reperfusion via MAPK-mTOR signaling pathway. J Ethnopharmacol. 2013;149:270–280. doi: 10.1016/j.jep.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 64.Wei K, Wang P, Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther. 2012;18:879–886. doi: 10.1111/cns.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 66.Xin XY, Pan J, Wang XQ, Ma JF, Ding JQ, Yang GY, Chen SD. 2-methoxyestradiol attenuates autophagy activation after global ischemia. Can J Neurol Sci. 2011;38:631–638. doi: 10.1017/s031716710001218x. [DOI] [PubMed] [Google Scholar]
- 67.Xingyong C, Xicui S, Huanxing S, Jingsong O, Yi H, Xu Z, Ruxun H, Zhong P. Upregulation of myeloid cell leukemia-1 potentially modulates beclin-1-dependent autophagy in ischemic stroke in rats. BMC Neurosci. 2013;14:56. doi: 10.1186/1471-2202-14-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu F, Gu JH, Qin ZH. Neuronal autophagy in cerebral ischemia. Neurosci Bull. 2012;28:658–666. doi: 10.1007/s12264-012-1268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu F, Li J, Ni W, Shen YW, Zhang XP. Peroxisome proliferator-activated receptor-gamma agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS One. 2013;8:e55080. doi: 10.1371/journal.pone.0055080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yan W, Zhang H, Bai X, Lu Y, Dong H, Xiong L. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011;1402:109–121. doi: 10.1016/j.brainres.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 71.Yoshimori T. Autophagy: paying Charon's toll. Cell. 2007;128:833–836. doi: 10.1016/j.cell.2007.02.023. [DOI] [PubMed] [Google Scholar]
- 72.Zhang J, Ney PA. Mechanisms and biology of B-cell leukemia/lymphoma 2/adenovirus E1B interacting protein 3 and Nip-like protein X. Antioxid Redox Signal. 2011;14:1959–1969. doi: 10.1089/ars.2010.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]