

Abstract
The regulation of vascular endothelial growth factor A (VEGF) is critical to neovascularization in numerous tissues under physiological and pathological conditions. VEGF has multiple isoforms, created by alternative splicing or proteolytic cleavage, and characterized by different receptor-binding and matrix-binding properties. These isoforms are known to give rise to a spectrum of angiogenesis patterns marked by differences in branching, which has functional implications for tissues. In this review, we detail the extensive extracellular regulation of VEGF and the ability of VEGF to dictate the vascular phenotype. We explore the role of VEGF-releasing proteases and soluble carrier molecules on VEGF activity. While proteases such as MMP9 can ‘release’ matrix-bound VEGF and promote angiogenesis, for example as a key step in carcinogenesis, proteases can also suppress VEGF’s angiogenic effects. We explore what dictates pro- or anti-angiogenic behavior. We also seek to understand the phenomenon of VEGF gradient formation. Strong VEGF gradients are thought to be due to decreased rates of diffusion from reversible matrix binding, however theoretical studies show that this scenario cannot give rise to lasting VEGF gradients in vivo. We propose that gradients are formed through degradation of sequestered VEGF. Finally, we review how different aspects of the VEGF signal, such as its concentration, gradient, matrix-binding, and NRP1-binding can differentially affect angiogenesis. We explore how this allows VEGF to regulate the formation of vascular networks across a spectrum of high to low branching densities, and from normal to pathological angiogenesis. A better understanding of the control of angiogenesis is necessary to improve upon limitations of current angiogenic therapies.
Keywords: angiogenesis, systems biology, mathematical model, computational model, protease, receptor, extracellular matrix, microenvironment, gradient
1. INTRODUCTION
VEGF-A is a key member of the VEGF family of cytokines, along with VEGF-B, -C, -D, and PlGF (1, 2). VEGF-A mediates angiogenesis, the expansion of an existing vascular bed by sprouting of new blood vessels (3). Angiogenesis typically occurs as a response to a stimulus such as tissue hypoxia, and results in improved perfusion and increased oxygen delivery. Other stimuli can induce angiogenesis, including shear stress (4) and genetic transformation in tumor cells (3). Angiogenesis is important for organ development (5) as well as for physiological processes including wound closure and exercise training (6, 7). It is upregulated but disorganized in pathological processes such as diabetic retinopathy and solid organ tumorigenesis (8–10), where vasculature is needed to supply the tumor’s rapid consumption of glucose and oxygen beyond the limits of diffusion.
The vegfa gene is translated into a number of splice isoforms, the most notable in humans being VEGF121, VEGF165, and VEGF189 (Fig. 1). These isoforms have differences in biochemical properties such as their affinities for VEGF receptors and heparan sulfate proteoglycans (HSPGs), resulting in strikingly different effects on vessel growth. A major focus of the current review is the extracellular regulation of VEGF (Sections 3, 4). In normal healthy situations, VEGF isoforms are differentially sequestered by heparan sulfate proteoglycans (HSPGs) in the ECM (Section 3.1) and are subject to various VEGF inhibitors (Section 3.2), e.g. sVEGFR1, a secreted isoform of the membrane VEGF receptor VEGFR1 (11); these inhibitors are involved in establishing vascular quiescence (12). During inflammation and tumorigenesis, sequestered VEGF can be released by proteases, such as the zinc-dependent matrix metalloproteinases (MMPs). Extracellular proteases can act on VEGF in several ways (Section 3.3) including cleavage of the ECM, cleavage of VEGF generating new isoforms such as VEGF114, and also cleavage of the soluble inhibitors of VEGF. These can lead to different biological outcomes. Proteases such as MMP9 are typically thought to release VEGF and induce angiogenesis, but in other situations can reduce angiogenesis activity, e.g. by cleavage of VEGF (13). We will explore what dictates whether proteolytic release of VEGF is pro- or anti-angiogenic, and the roles of specific proteases.
Figure 1. Properties of VEGF isoforms and proteolytic cleavage sites.
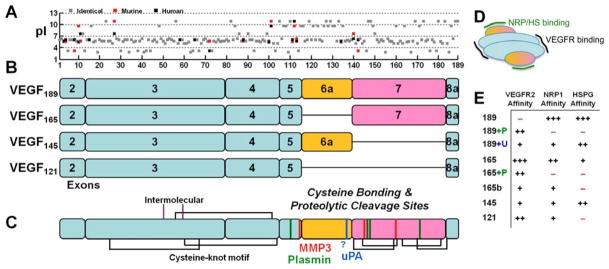
A, The acidity of the individual amino acids (pI) for human (black) and murine (red) VEGF shows the basic residues responsible for the heparin-binding domains of exons 6 and 7. Murine VEGF contains a deletion of Gly-8 found in human VEGF and is frame-shifted for comparison. The overall sequence identity between murine and human VEGF189 orthologs is 89%. B, Exon structure of the predominant VEGF isoforms in humans, scaled to the 189 amino acids shown in panel A. Note that VEGF165/164 replace the last residue in exon 5, Lys, with Asp. Exon 1 is not present in processed VEGF, it is removed by signal peptidase. Of the anti-angiogenic VEGFxxxb isoforms, which use exon 8b instead of 8a, VEGF165b is most common. C, Disulfide bonding structure (black and purple lines) (223) and known proteolytic cleavage sites for the serine proteases (plasmin, green; uPA, blue) and MMPs (red). The uPA cleavage site has not been specifically mapped but is thought to reside in the C-terminal portion of exon 6a. Exon 7 is linked to the first amino acid in exon 8 in all isoforms except VEGF121. Processing of exon 7 by the MMPs results in a single fragment due to linkage by disulfide bonds, whereas complete processing by plasmin may yield a second two-amino-acid Arg147-Lys148 (in VEGF189) fragment. D, VEGF receptors (VEGFR1, R2, R3) bind to the bivalent dimeric ligand in the region encoded by exons 3 and 4 (blue). Neuropilins (NRP) and heparan sulfates (HS) bind VEGF in the region encoded by exons 6 and 7 (yellow/purple). E, VEGF isoforms differentially bind to Neuropilin-1, VEGFR2, and heparin/heparan sulfate. Cleavage by plasmin (P) and uPA (U) can activate VEGFR2 binding in VEGF189 and decreases NRP1 and HSPG binding.
The spatial distribution of VEGF is a key regulator of angiogenesis and is itself regulated by both matrix binding and proteolytic release (Section 4). For example, VEGF isoforms that bind strongly to the ECM, such as VEGF165 and VEGF189, have a steep gradient (14, 15) and tight pericellular sequestration (15–18). Gradient formation has been commonly thought to be due to a restriction of the rate of diffusion by ECM binding (Section 4.2). However, using computational modeling, we have shown that HSPG binding alone cannot explain most aspects of VEGF gradients (19). This and other differences between experimental and theoretical results require us to revisit the underlying mechanics of VEGF transport in vivo (Sections 4.3, 4.4). Recent advances have indicated that soluble VEGF inhibitors also play an important role in VEGF patterning (20–22).
Different tissues express different ratios of the VEGF isoforms (Fig. 2) and this may serve to produce vascular networks that match the specific needs of each tissue (23). Mice expressing only VEGF120 instead of the full range of VEGF isoforms have significant defects in cardiac and pulmonary development due to defective angiogenesis (24, 25). On the other hand, tumor growth appears to be most rapid in tumors that express VEGF164 (16, 26). We review how VEGF, its spatial distribution and receptor signaling, regulates angiogenesis. Heparin-binding VEGF isoforms produce a branching network with narrow vessels, while VEGF120 (the murine equivalent of VEGF121) results in poorly branching, tortuous, leaky vessels (14, 15, 27, 28) (Section 5.2). We explore the specific mechanisms by which VEGF isoforms can cause these different vascularization states (Section 5.3)? VEGF is a mediator of sprouting angiogenesis, but in some situations high levels of VEGF can result in a highly proliferative, dysregulated state and lack of sprouting (14, 29). We explore how these pathological angiogenesis states can arise (Section 5.4).
Figure 2. Relative expression level of the VEGF isoforms in normal tissues and tumors.

This ternary diagram depicts relative isoform expression based on mRNA RT-PCR of cell culture, or of embryonic, adult, and tumor tissues (see Supplemental Table S5). The location of each point on this ternary diagram denotes the relative expression of VEGF121, of VEGF165, and of the exon 6-containing isoforms grouped together; VEGF145 typically has low levels of expression and VEGF183 seems to be functionally equivalent to VEGF189 (62). At vertices, all VEGF expression is comprised only of that isoform, while the midpoint of the opposite edge would indicate 0% expression of that isoform and 50% expression each of other isoforms. Each point represents specific organs from individual studies; points with the same color denote the same organ of origin; circles: tumors, squares: normal embryonic or adult tissue. The dashed line represents a line of equal VEGF121:VEGF189 expression, as discussed in Section 5.2; the VEGF164-only and VEGF120/188 mice would be expected to fall on this line. Note that most tumors fall below this line (i.e. high VEGF120/121 expression).
This review aims to provide a comprehensive overview of the biochemistry, physical transport, and biology of the splice and proteolytic isoforms of VEGF. We highlight several uncertainties in our understanding of VEGF, which may be avenues for future research.
2. SPLICE ISOFORMS AND PROTEOLYTIC ISOFORMS OF VEGF
The vegfa gene encodes several splice isoforms of VEGF-A, each of which may be processed by a variety of proteases to produce yet more isoforms (Fig. 1). Detailed reviews of VEGF splicing, VEGF receptor binding and intracellular signaling are available (1, 30–32); here we discuss how the structure of the native and proteolytically-processed isoforms determine binding to receptors, co-receptors and extracellular matrix proteoglycans.
2.1 Alternate splicing results in multiple VEGF isoforms that bind differently to receptors, co-receptors, and HSPGs
The human VEGF gene (located on chromosome 6) consists of several exons that can be alternatively spliced (30, 31) to encode several protein isoforms, including: VEGF121, VEGF145, VEGF162, VEGF165, VEGF183, VEGF189, and VEGF206. These isoforms are denoted by their length (number of amino acids) and differ in incorporation of exons 6 and 7, which convey HSPG- and neuropilin-1 (NRP1)-binding motifs (Fig. 1B). Compared to human VEGF isoforms, murine isoforms have the same functional components and biological function but are one amino acid shorter, e.g. VEGF120, VEGF164, VEGF188. With ~90% sequence identity between human and murine forms (Fig. 1A), slight structural variations seem to exist in terms of proteolytic processing and antibody recognition (33). Human VEGF isoforms have weakly-angiogenic or anti-angiogenic counterparts, VEGFxxxb, in which exon 8b is substituted for exon 8a (30, 34). There is some debate over whether the antiangiogenic VEGFxxxb isoforms are expressed in the mouse (35, 36).
The primary receptor tyrosine kinases (RTKs) for VEGF isoforms are VEGFR1 (Flt-1), VEGFR2 (Flk-1 or KDR), and VEGFR3 (Flt-4). Receptor binding of VEGF depends on the presence of isoform-selective co-receptors such as NRP1, NRP2, and cell-surface glycosaminoglycans (GAGs). VEGFR2 is the primary receptor responsible for endothelial cell mitogenic and migratory responses to VEGF (14), while VEGFR1 is thought to function primarily as a decoy receptor during development, and to modulate signaling of VEGFR2 in the adult (37, 38). Although it is considered a weaker kinase, VEGFR1 does have direct signaling functions (37, 38). NRP1, in contrast, is not thought to directly transduce VEGF signals but selectively enhances VEGF165 binding to VEGFR2 (by presenting NRP1-bound VEGF to bind VEGFR2) (39, 40) and alters VEGFR2 intracellular trafficking (41). NRP1-enhanced VEGF signaling has been shown to be important for p38/MAPK activation, and thus is central to vessel branching (27, 42).
VEGF isoforms are typically secreted as cysteine-linked antiparallel dimers (Fig. 1D). Binding to VEGFR1 and VEGFR2 monomers at the cell surface is mediated by the sequence encoded by exons 3 and 4 respectively, epitopes located at either end of the bivalent dimeric ligand (Fig. 1C–D). Binding to HSPG is mediated in a sequence- and charge- specific manner by the basic amino acids from exons 6a and 7 (“heparin-binding domain”), while NRP1 binding is mediated by exon 7 regions. Exon 8a imparts important structural stability and function to exon 7 through disulfide bonding (Fig. 1C–D). Exon 6 has a poorly understood role in VEGF structure, and is discussed further in section 2.3, below.
2.2 Differential cellular effects of VEGF121 and VEGF165 are mediated by binding to NRP1 and HSPGs
While both VEGF121 and VEGF165 are capable of signaling through VEGFR2, VEGF165 binding to (and activation of) VEGFR2 is potentiated by cell-surface NRP1 (40, 43, 44), cell-surface HSPGs such as glypican-1 (45, 46), and exogenous GAGs such as heparin (47, 48). Unlike VEGFR2, VEGFR1 shows intrinsically greater affinity towards VEGF165 than VEGF121 especially in the absence of heparin (47); however, the presence of NRP1 may block the VEGF165-VEGFR1 interaction, leading VEGFR1 to be a preferential receptor for VEGF121 (49, 50).
NRP1 is a co-receptor for several VEGF isoforms, notably VEGF165 (40). It facilitates formation of a stable ternary NRP1-VEGF165-VEGFR2 complex, increasing the overall avidity of the isoform for VEGFR2 (27, 39). VEGF121 may also bind NRP1, despite the absence of the canonical exon 7, via exon 8 sequences (51, 52). However, unlike for VEGF165, NRP1 cannot bridge VEGF121 and VEGFR2 (44, 52). GAGs similarly potentiate heparin-binding isoforms such as VEGF165. Interestingly, heparin and solubilized glypican-1 can stabilize VEGF/VEGFR2 complexes even after pre-treatment with exogenous heparinase (45, 47, 53). NRP2, another member of the neuropilin family, also serves as a co-receptor for VEGFR2, but with slightly different isoform specificity (54, 55).
VEGF165b (an anti-angiogenic isoform mentioned above) lacks HS and NRP1 binding (30, 43, 44, 56). Unlike VEGF121, however, it assumes a reduced signaling state once bound to VEGFR2 that is resistant to rescue by exogenous VEGF165 (34, 43, 44). This difference is thought to arise due to the lack of the stabilizing influence of exon 8a (Fig. 1B), rather than the presence of exon 8b itself (43, 56).
2.3 Exon 6a may function as an intrinsic VEGF inhibitor and its cleavage activates VEGF
Exon 6a-containing isoforms such as VEGF145 and VEGF189 continue to be poorly understood. Exon 6a itself is directly inhibitory for VEGF activity (57). Exon 6a is heavily dominated by basic amino acids, encodes a nuclear localization sequence (58), and interferes with VEGFR2 binding (59). Full-length VEGF189 and VEGF145 are weaker mitogens and chemotactic agents than even VEGF121 (44, 60, 61). In contrast, VEGFR1 activity is unaffected by the presence of exon 6, being similar to that of VEGF165 (60, 62), and this allows exon 6-containing isoforms to induce vascular permeability (62, 63) or neutrophil cell migration (64).
Binding of these exon 6a isoforms to VEGFR2 seems to require proteolytic processing by urokinase plasminogen activator (uPA) or plasmin (60, 62), or binding to ECM/heparin (17, 61). For example, uncleaved VEGF189 applied directly to bovine adrenal cortex-derived ECs showed no activity (60). However, cleavage by uPA, which only partially removes exon 6a and all of exon 7, activates VEGF189 to the level of VEGF145, while cleavage by plasmin, which entirely removes exon 6a and 7, activates VEGF189 to the level of VEGF121 (60) (Fig. 1E). VEGF189 activity may depend on cell type as it seems to have similar activity as VEGF165 towards HUVECs (65).
VEGF145 has low affinity for NRP1 (44, 54) and does not bridge VEGFR2 and NRP1 (44). VEGF189 has been shown to have a 10-fold greater NRP1 affinity than does VEGF165 (66, 67); however, given that VEGF189 is a weak mitogen like VEGF145 (60), it is likely unable to form or support a functional ternary complex with NRP1 and VEGFR2.
2.4 Proteolytic processing results in additional VEGF isoforms
Proteases can have several distinct effects on VEGF, including cleavage, activation, liberation from extracellular stores, and degradation. Each of these has consequences for VEGF activity. The most-studied VEGF proteases are plasmin (47, 60, 62, 68) and the matrix metalloproteinases (MMPs), including MMPs 1, 3, 9, 7, 12, 16, and 19 (13, 69). Others include uPA (60), elastase (70, 71), and tissue kallikrein (72, 73).
VEGF165 cleavage by plasmin significantly reduces its overall bioactivity (over 100-fold increase in EC50 (47)) and removes NRP1- and heparin-binding abilities (47, 52). Plasmin-, MMP-, and tissue kallikrein-mediated cleavage of VEGF occurs in exon 5 (Fig. 1C) (13, 47, 56, 72) leading to an N-terminal active fragment of VEGF that has an electrophoretic mobility of 13 kDa (via plasmin) or 16 kDa (via MMP) (13, 70). In contrast, uPA-mediated digestion, as discussed above, likely occurs in the C-terminal portion of exon 6 (60), allowing the main cleavage product to preserve some of its ECM-binding character (60, 61). Elastase cleavage occurs at both N- and C- termini (71). Since VEGF is homodimeric, proteolysis proceeds through a heterodimer intermediate with only one chain cleaved (47, 74). While cleaving fragments on the N- and C- termini, these proteases do not seem to digest the core sequences mediating receptor binding (exons 3, 4) (47), with the exception of elastase which abrogates VEGFR2 binding (71). The effective cleavage rate of VEGF165 by plasmin is kcat/Km = 328 M−1s−1 with Km > 1 mM, at 25°C (74), which is similar to that observed by MMP3 (13), but is lower than typical ECM proteolysis reactions, e.g. fibrin/plasmin (kcat/Km = 8,100 M−1s−1 at 37°C), or Type 1 collagen/MMP2 (kcat/Km = 5,300 M−1s−1 at 25°C) (74). A significant portion (40–80%) of VEGF has been shown to have been proteolyzed in pathological systems (13, 69, 75).
While murine VEGF isoforms can be cleaved by MMPs, this may not be true for human VEGF isoforms (76, 77). While Lee et al. have shown that murine VEGF can be cleaved by MMPs (13), the available evidence that human VEGF is similarly cleaved is a 16 kDa VEGF band in human ovarian cancer ascites fluid which may represent a glycosylation variant of plasmin-cleaved VEGF (13). On the other hand, numerous studies have shown the absence of direct cleavage of human VEGF by MMPs (70, 71, 76–78). This may be due to the fact that murine and human VEGF are highly dissimilar at the proteolytic cleavage sites (amino acids 110–114) (Fig. 1A), and VEGF-cleaving proteases exhibit a high degree of substrate specificity; for example, replacing the plasmin cleavage site in murine VEGF, R109-T110, with K109-P110 makes murine VEGF not only plasmin resistant, but also MMP resistant at the MMP cleavage site, amino acids 113–114 (13). It should be noted that MMP-cleaved human VEGF should numerically correspond to VEGF114, not VEGF113, due to the extra amino acid present in exon 2 not found in murine VEGF. To our knowledge, this has not been demonstrated. A VEGF114 isoform proposed by Mintz et al. assumes a 27 instead of 26 amino acid signal sequence (79), and thus should correspond to human VEGF115, which lacks the entirety of exon 8.
3. LOCAL AVAILABILITY AND ACTIVITY OF VEGF: ECM, PROTEASES AND INHIBITORS
The rate of VEGF secretion is a key driver of VEGF-induced angiogenesis (80). However, once secreted, numerous processes regulate VEGF activity in vivo; for example, in the cornea, the activity of secreted VEGF is repressed by co-secretion of sVEGFR1 (12). Along with interstitial diffusion and convection, several distinct processes affect local VEGF availability and activity: sequestration of VEGF by stationary molecules in the ECM or on cell surfaces; VEGF inhibition or activation by other soluble molecules; enzymatic release from the ECM and from soluble carriers; and loss due to clearance or degradation.
3.1 ECM sequestration of VEGF affects diffusion and modulates VEGF bioactivity
VEGF binds to the ECM near cells secreting VEGF, as has been shown in retina, brain, various tumors, and other tissues (14, 16, 17). VEGF sequestration is not restricted to matrix binding sites; VEGF binding to cell surface VEGF receptors also reversibly immobilizes VEGF, and before secretion or after internalization may be sequestered intracellularly (81, 82). Sequestration in the ECM and on cell surfaces seems to restrict VEGF gradients (15, 16) and guide endothelial cell migration (14, 28). Sequestered VEGF seems to function haptotactically as migrating ECs extend filopodia to explore sites of deposited VEGF, even towards that found on other cells (14, 28, 69, 83, 84). Matrix-bound VEGF is capable of binding and activating VEGF receptors, serving as a biochemical as well as a mechanical signal; matrix-bound VEGF induces different signaling patterns than soluble VEGF (28, 85). Sequestration by the matrix limits VEGF diffusion and decreases clearance from the tissue, which may facilitate autocrine signaling (64). Sequestration also allows the storage of VEGF that can be released to locally amplify a pro-angiogenic signal (86), and may confer resistance to certain types of proteolytic processing; binding to GAGs or fibronectin may protect against plasmin and MMP9, respectively (13, 87).
Among the many matrix binding sites for VEGF, GAGs are thought to predominate based on heparin elution (17, 62, 68); GAGs can be ECM-associated, e.g. perlecan, or membrane-associated, e.g. syndecan. All known VEGF-A isoforms bind heparin/HS except VEGF121, VEGFxxxb, and the proteolytic isoforms produced after processing by MMPs, plasmin, or elastase (13, 43, 47, 71) (Fig. 1E). The exon 6a-containing isoforms, including VEGF145, show greater affinity to the HSPGs than does VEGF165; this binding may be partially independent of GAGs (44, 61, 68). Other matrix binding sites include collagen (28), fibrin (88), and fibronectin (28, 84, 89), while Type IV collagen and vitronectin are incapable of binding VEGF (89). VEGF may also indirectly bind to ECM through another ECM-binding soluble mediator, such as sVEGFR1 (21, 90) or ADAMTS1 (91). VEGF cell surface receptors also sequester VEGF until VEGF dissociates or is internalized. This receptor binding induces signaling in endothelial and other cells, but also alters the local gradients of VEGF in the interstitial space (92).
3.2 Soluble carriers of VEGF play a role in vascular quiescence and gradient formation
Despite active secretion of VEGF, some tissues including the cornea (12) and certain precancerous lesions (76, 78), are held in an anti-angiogenic or angiostatic state due to the simultaneous secretion of soluble inhibitors of VEGF, which in binding VEGF prevent VEGF-induced dimerization and activation of VEGFR2 (91). Parallel to ECM sequestration, soluble inhibitors create a diffusible reservoir of VEGF, accessible through unbinding or proteases (76). For example, corneal infection by the herpes simplex virus 1 leads to corneal stromal keratitis through the infiltration of new blood vessels, but requires either overcoming the endogenous sVEGFR1 barrier by upregulation of VEGF, mediated by the virus, or by MMP-mediated degradation of sVEGFR1, mediated by the virus-induced acute neutrophilic response (12, 93). Other soluble VEGF inhibitors include alpha-2-macroglobulin (94) and thrombospondin (TSP) domain-containing molecules, which preferentially bind VEGF165 (91): connective tissue growth factor (CTGF) (70, 76) contains one such domain, while thrombospondin-1 (TSP1) (95) and ADAMTS1 (91) each contain three. TSP1 can block VEGF-HSPG association at the cell surface (95) as well as interfere with VEGFR2 signaling (96). Finally, soluble proteins can inhibit VEGF binding to receptors more indirectly, such as platelet factor 4 (PF4) (97). Just as there are soluble VEGF inhibitors, other carriers (soluble molecules that bind VEGF in solution) can permit VEGF to retain its activity. Soluble activators of VEGF include heparin and proteolytically-released heparan sulfates (e.g. syndecan), which can enhance VEGFR2 activation by co-activation of integrins (98), and fibronectin, which upon release by platelets binds VEGF and can crosslink α5β1 and VEGFR2, enhancing endothelial activity (87).
Unlike the stationary ECM, soluble VEGF carriers can mediate the transport and clearance of VEGF. Heparin can release and disperse FGF2 and FGF10, enabling activation of cells at a distance and altering gradients (99, 100). Similarly, VEGF release by proteases and heparanases produces diffusible VEGF that can escape subsequent rebinding to the matrix and is resistant to degradation (13, 101). Carriers may also enhance the spatial complexity of the local VEGF gradients (21, 102). sVEGFR1 secreted by a blood vessel is thought to shape the VEGF concentration field near the vessel, guiding nascent vascular sprouts to migrate perpendicularly from the parent vessel (20).
3.3 Distinct mechanisms of VEGF release promote pro- or anti- angiogenic effects
Among the many roles for proteolytic enzymes in angiogenesis (103), they are important modulators of extracellular VEGF, which is important in numerous pathologies including pancreatic islet carcinogenesis, breast cancer, and oxygen-induced retinopathy (Table 1, Table S1). Proteolytic release of VEGF from the matrix is typically viewed as being pro-angiogenic (76, 86), however in other situations, protease action on VEGF may inhibit angiogenesis (13, 104). Similarly, protease inhibitors have been shown to both decrease angiogenic sprouting (86) and increase sprouting (13). This dichotomy seems to be explained by distinct mechanisms of action that proteases have on VEGF (Fig. 3).
Table 1.
Multiple roles of proteases in VEGF-induced angiogenesis
| System | Host* | Enzyme | VEGF mRNA† | VEGF Protein† | VEGF Processing† | Angiogenic Response† | Pathogenesis † | Ref |
|---|---|---|---|---|---|---|---|---|
| Proteases hinder vessel growth | ||||||||
| Breast tumor, T47D, subcutaneous | m/h | n.a. | n.a. | soluble VEGF ↑ relative to total VEGF | + implied | ↓ (instead enlargement) | ↓ | (13) |
|
| ||||||||
| Oxygen-induced retinopathy | m | MMP12 | Unch. | binding to vasculature ↑ | + | ↑ malformation | ↑ | (69) |
|
| ||||||||
| Wound, chronic leg ulcer | h | Plasmin | ↑ | ↓ VEGF165 | + | ↓ | ↑ | (104) |
| Wound, skin | m | Plasmin | n.a. | ↓ VEGF165 implied | + | ↓ | ↑ | (105) |
|
| ||||||||
| Proteases induce patent vessel growth | ||||||||
| Breast tumor | m | MMP9 | Unch. | total VEGF Unch. ↓ gradient total
VEGF VEGF-VEGFR2 ↑ |
– | ↑ | ↑ | (111) |
| m/h | MMP9 | n.a. | total VEGF Unch. VEGF-VEGFR2 ↑ |
n.a. | ↑ | ↑ | (137) | |
|
| ||||||||
| Cervical cancer | m | MMP9 | n.a. | VEGF-VEGFR2 ↑ | n.a. | ↑ | ↑ | (114) |
|
| ||||||||
| Colon carcinoma (HT29) (ex vivo) | h | MMP9, 2, 8 | n.a. | soluble VEGF165 ↑ ↓ total HSPG | – | ↑ | Implied ↑ | (77) |
| Colorectal cancer | h | MMP7 | ↑ | n.a. | n.a. | ↑ | ↑ | (76) |
|
| ||||||||
| Cornea | m | MMP7, 9, 2 | ↑ | n.a. | n.a. | ↑ | ↑ | (93) |
| Cornea | m | MMP9,2 | ↑ | VEGF ↑ | n.a. | ↑ | Implied ↑ | (140) |
|
| ||||||||
| Glioblastoma | m | MMP9 | ↑ | % soluble VEGF ↑ total VEGF
Unch. VEGF-VEGFR2 ↑ |
– | ↑ | Implied ↓ | (113) |
|
| ||||||||
| HUVEC migration and tube formation (in vitro) | MMP7 | n.a. | VEGF165 ↑ | – | ↑ | n.a. | (78) | |
|
| ||||||||
| Ovarian tumor ascites (in vitro) | m/h | MMP2,9 | n.a. | soluble VEGF ↑ | – | ↑ permeability | ↑ | (222) |
|
| ||||||||
| Pancreatic islet | m | MMP9 | Unch. | (ex vivo) soluble VEGF ↑ (in vivo) VEGF-VEGFR2 ↑ | n.a. | ↑ | ↑ | (86) |
| m | Heparanase | Unch. | VEGF-VEGFR2 ↑ | n.a. | ↑ | ↑ | (109) | |
In vivo except where indicated.
Host organism; m = mouse; h = human; m/h = human tumor in mouse
Arrows denote effect of protease-dependent state vs. control or protease-KO state; Unch = unchanged; n.a. = data not available.
Figure 3. Mechanisms of proteolytic regulation of extracellular VEGF.
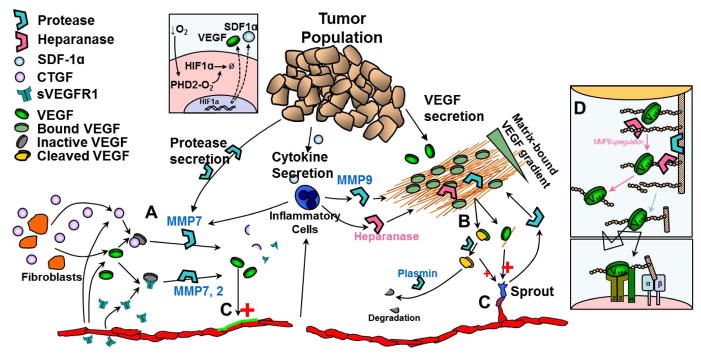
Proteases play a key role in determining the fate of VEGF and its detection by endothelial cells. Proteases can degrade soluble VEGF inhibitors (e.g. CTGF, sVEGFR1) (A) or release matrix-sequestered VEGF (B); both allow free VEGF to escape inactive states and bind to endothelial cell receptors (C). VEGF gradients are altered by release of matrix-sequestered VEGF. In tumor angiogenesis, cancer cells, endothelial cells and inflammatory cells can all contribute to proteolytic activity. VEGF164 has two heparin-binding domains; cleavage of either domain results in a VEGF164/113 intermediate (B), with lower overall affinity for the ECM. Subsequent cleavage of the second domain results in freely diffusing VEGF113. Some MMPs can cleave VEGF bound to heparin/HSPGs (e.g. MMP3) while others cannot (e.g. MMP9). Heparanase activity on cell-surface HSPGs can lead to upregulation of MMP9 leading to cleavage of both GAG chains and core protein (D) and enhanced signaling at the cell surface.
Several extracellular and membrane-bound proteases can directly cleave VEGF. Plasmin and MMP3 cleave only the C-terminal domain (13, 47), while elastase and plasmin are associated with complete degradation involving cleavage at both N-terminal and C-terminal sites (71, 104). In non-healing wounds, interstitial fluid is very proteolytic, resulting in rapid degradation of VEGF, preventing the recruitment of a vascular supply (104, 105). In contrast, cleavage of the C-terminus facilitates VEGF release and increases the level of soluble VEGF; the overall balance still seems to be anti-angiogenic as the released VEGF molecule is less bioactive (13, 73). Breast cancer xenografts engineered to express a C-terminal truncated isoform, VEGF113, demonstrate decreased tumor growth rates, lower vascular density, and dilated tortuous vessels compared to VEGF164 expressing tumors (13). Despite this, VEGF release by cleavage may be pro-angiogenic in the short term if the release occurs rapidly enough (106), but it is not certain that the pro-angiogenic effects can be sustained. In oxygen-induced retinopathy (OIR), proteolytic VEGF release seems to produce similar irregular endothelial proliferation as in the VEGF113-only tumors, apparently mediated by macrophage-secreted MMP12, however the angiogenesis is chaotic and hyperproliferative (69).
Enzymes can also degrade soluble VEGF inhibitors (and presumably soluble activators), altering VEGF transport and/or activity. Cleavage of VEGF inhibitors has been directly associated with increased angiogenesis, and in vivo, seems to be primarily mediated by MMP7. For example, in a Capan-1 xenograft model of pancreatic adenocarcinoma, stromal fibroblasts secrete VEGF inhibited with CTGF; and cancer cells can secrete MMP7 or other MMPs to cleave CTGF (76) (Fig. 3A). Other VEGF carriers can be cleaved: TSP-1 by plasmin and elastase (107); CTGF by plasmin, elastase, MMPs 1, 3, 7, 13, and ADAM28 (70, 108); and sVEGFR1 by MMP7 or weakly by MMP2 and MMP9 (78, 93).
Enzymes can also release VEGF from sequestration by cleaving the ECM, i.e. the GAGs, HSPG core proteins, and other associated ECM molecules, without processing VEGF. This released VEGF may remain complexed with an ECM fragment (98, 99). In vivo studies demonstrating this release mechanism are limited, but it is associated with increased angiogenic potential (109). The primary molecular mediators are heparanase, which cleaves the heparan sulfate chains and possibly MMP9, which can cleave HSPG core proteins (77, 98). Cleavage of the core proteins in a glycoprotein such as perlecan can also occur via MMP1, MMP13, and plasmin (110) and MMP2 or MMP8 (77).
MMP9 is implicated as a major player in VEGF release and the angiogenic switch (86, 111), however its predominant mechanism of action on VEGF is unknown. MMP9 is thought to be primarily secreted by infiltrating macrophages and neutrophils (112–115) stimulated by tumor-secreted cytokines (Fig. 3). MMP9 is thought to raise the concentration of active VEGF in tissues, increasing VEGF-VEGFR2 association (86). While MMP9 has been shown to cleave murine VEGF (13), studies where MMP9 was implicated in release of murine VEGF have not shown evidence of VEGF cleavage (111, 113). MMP9-mediated cleavage of VEGF164 may even be inhibited by heparan sulfates (13). Most evidence points to MMP9 releasing VEGF through HSPG cleavage (77, 98). For example, heparanase shows similar pro-angiogenic behavior as MMP9 in the RIP1-Tag2 pancreatic islet model (109) and heparanase action on cell-surface HSPGs can induce MMP9 expression to further cleave HSPGs (98) (Fig. 3D). However, MMP9 is also able to cleave soluble VEGF inhibitors and thus this alternate mechanism of VEGF activation cannot be excluded (70, 93).
3.4 Loss of VEGF activity from the tissue by clearance and degradation
Loss of VEGF via clearance and degradation plays a key role in controlling VEGF activity and in shaping VEGF gradients (19). Relevant mechanisms include lymphatic drainage, transvascular transport into the blood stream, proteolytic degradation, and cellular endocytosis; the relative importance of these mechanisms is not clearly understood.
Endothelial cells can degrade VEGF through internalization of the VEGF-VEGFR complex and through alternate pathways such as via low density lipoprotein receptor-related protein-1 (LRP1) in conjunction with TSP1 (116). In chronic wounds and some tumors, VEGF loss appears to be more dependent on proteolytic degradation (104, 117). This seems to require an initial cleavage by plasmin (13, 104, 117, 118), however the extent of degradation seems inconsistent between studies (47, 104) and it does not seem to occur with MMPs (13). VEGF experiences loss of activity in vitro under cellular conditions shown not be due to cellular uptake, with a rate constant of 2.3–2.8 ·10−4 s−1 (τ1/2 ~40 min) (119, 120). Whether this represents inactivation by secreted soluble factors (78) or degradation in solution is not known; factors present in the serum commonly added in many of these studies (120) may be involved. Finally, VEGF also has been demonstrated to degrade in isolation, with a half-life of ~96 min in acellular conditions at 37°C (28, 101). Matrix-sequestered VEGF may be protected from this intrinsic degradation (85, 101).
4. IN VIVO SPATIAL PATTERNING OF VEGF ISOFORM GRADIENTS
Spatial gradients of VEGF regulate vessel activation and sprout guidance (14, 86), and may be shaped by numerous mechanisms in vivo including diffusion, matrix sequestration, competitive binding, and proteolytic release (121). Heavier VEGF isoforms show increased matrix sequestration and steeper spatial gradients, suggesting that heparin binding, by slowing diffusion, directly leads to sharper gradients. However, theoretical models show that the heparin binding alone is not the source of observed molecular gradients, and thus we must delve deeper to find the mechanism.
The overall VEGF gradient not only reflects the diffusion of VEGF from secreting cells to where it is bound or consumed, but also reflects heterogeneities in the concentration of the binding partners such as receptors and matrix binding sites. As an example of the latter, VEGF188, or VEGF in the presence of diminished MMP9, can show amplified staining near or at the cell surface relative to VEGF164 or VEGF120, e.g. (18, 111) (Fig. 4A, VEGF189); this effect would likely exist even if the underlying soluble VEGF was uniform and hence there were no gradient for diffusion (92). In this section, we discuss how VEGF diffusion shapes gradients.
Figure 4. In vivo patterning and biological effects of VEGF isoforms.

Heparin-binding affinity and proteolytic susceptibility modulate the VEGF patterning in tissues and the subsequent vascular phenotype. A, Angiogenesis in tumor xenografts expressing VEGF120 or VEGF113 only (left); VEGF164 only (middle); VEGF188 or the non-cleavable VEGF164Δ108–118 only (right). Note differences in spatial distribution with increase in cell-surface associated VEGF (green outlines) in the absence of proteases or in presence of VEGF188/189 (16, 17, 111). Heavier VEGF isoforms result in networks with greater capillary density, lower caliber, and increased pericyte coverage. B, Hindbrain VEGF distribution and angiogenesis in mice secreting (i) VEGF120 only or (ii) wildtype VEGF, which is predominantly VEGF164 (15). VEGF localizes closer to the source in wildtype relative to VEGF120-secreting hindbrain; note that the two VEGF gradients will intersect if superimposed. C, Postnatal murine retina (14) showed diffuse VEGF distribution (green dots) with a lack of association of VEGF to astrocytes (yellow), plus greater intercellular VEGF in VEGF120-only mice (left) compared to wildtype mice or VEGF164-only mice (right). Note the enlarged vessels and apparent increase in total VEGF staining in VEGF120-secreting retina.
4.1 VEGF120 demonstrates greater dispersion and lower pericellular binding in vivo than VEGF164
The clearest examples of VEGF gradients are seen in images from the mouse hindbrain, retina, and cerebellum (14, 15, 18). In the hindbrain, mice engineered to secrete only VEGF120 display a broad, diffuse immunostaining pattern, whereas wild-type mice, secreting predominantly VEGF164, instead show a higher VEGF level at the midline that falls off more rapidly, i.e. a steeper gradient and shorter propagation distance (Fig. 4B) (15). In the retina, VEGF120 secretion from astrocytes results in a dispersed VEGF distribution lacking obvious cellular localization; conversely, VEGF in wildtype mice predominantly associates with the astrocyte cell surface but is lacking in the interstitium (Fig. 4C) (14). VEGF188 shows the greatest levels of matrix sequestration and/or pericellular localization (16–18). In support of these observations, expression of VEGF120 alone results in greater levels of soluble VEGF (122) and lower levels of matrix deposition (16) in vivo, compared to systems expressing only VEGF164 or VEGF188.
The key feature of the VEGF gradient is that matrix-binding isoforms displays a higher total VEGF concentration at the source of secretion and at the surface of nearby cells, while VEGF120 has a higher concentration than matrix-binding isoforms distant from the source (15, 18). Note that the flatter nature of the VEGF120 distribution seems to be due to both the lack of amplification provided by matrix and cell-surface binding as well as VEGF120’s ability to travel farther. The VEGF distributions in vivo also appear to be static (or only slowly evolving), indicating that they are not transient dissipative phenomena, but are continually and actively maintained. In the developing mouse hindbrain, the VEGF gradient, as well as the differences between VEGF120 and VEGF164 gradients, persists for at least three days (E10.5–E13.5), until vascularization of the subventricular zone is complete (15). Similarly, in the retina, the zone of greatest VEGF secretion appears to move with the circumferential expanding vascular front during the first postnatal week (14).
There are limitations to interpreting images of VEGF spatial gradients in vivo. First, the extent to which intracellular VEGF contributes to the immunostaining is not clear (82, 123); a large intracellular fraction of matrix-binding isoforms (17, 58) may make it difficult to determine the quantity of extracellularly diffusing VEGF. On the other hand, VEGF120 is thought to elicit biological effects at a longer range than heparin-binding isoforms (13, 16, 124), suggesting that the immunostaining indeed reflects the true extracellular VEGF gradient. There may also be unaccounted factors associated with the biological models themselves. For example, dispersed VEGF gradients in the retina and hindbrain of VEGF120/120 mice might be due to defective arteriolar development of neural-derived tissues, leading to more extensive hypoxia and hence VEGF secretion (125); global hypoxia can result in a similar retinal vascular phenotype as the VEGF120/120 mice (126). Constitutive expression models could be informative, but clear examples of VEGF gradients have not been visualized in such systems (16). In contrast to developmental systems, which demonstrate vivid VEGF120 staining interstitially (14, 15, 18), VEGF120-expressing tumors show little VEGF staining (16, 17). A summary of VEGF isoform patterning is provided in Tables S2–S4.
4.2 HSPG binding cannot solely account for differences in VEGF gradients between isoforms
To study the diffusion and patterning of VEGF isoforms, we developed mathematical models of the microenvironment (19). In the models, VEGF is secreted into the interstitial space and diffuses through the ECM and basement membranes where it can reversibly bind to HSPGs; ultimately the VEGF in solution is either cleared away or degraded (Fig 5). Assuming an initial state of no VEGF, simulations show that upon secretion, diffusing soluble VEGF slowly loads the matrix until HSPGs reach an equilibrium binding capacity at steady state; the tighter an isoform binds the matrix, the more it can load in the matrix, and the longer this process takes (19, 50, 127). Comparing secretion of different isoforms, the models predict the spatial distribution of the unbound soluble fraction at steady state to be the same, regardless of the isoform’s affinity for the matrix. The matrix-bound fraction in each case had the same relative curvature as the soluble VEGF fraction, but with its concentration amplified in proportion to the HSPG-binding affinity (Fig. 5A, Sequestration). Matrix-binding isoforms would thus result in higher VEGF concentrations than VEGF120 regardless of the distance from the secretion source, assuming sequestration only. In the model, HSPGs do hinder the rate and time course of diffusion as is commonly thought, but this only has an impact on gradients during the transient period, and not at steady state.
Figure 5. Model-predicted effects of degradation and sequestration on VEGF patterning.
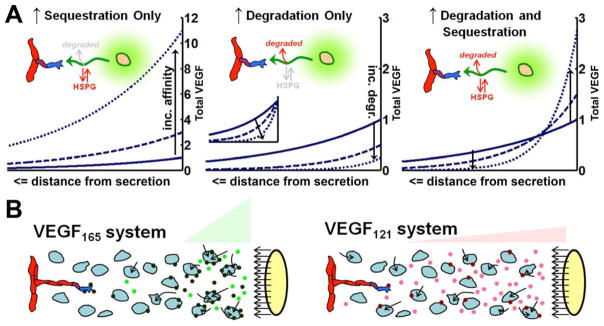
A, Isoform-specific VEGF patterning as seen in vivo relies on isoform-specific differences in both degradation and sequestration (19). The graphs show the spatial distribution of the total VEGF (soluble + sequestered) for three VEGF isoforms (representing increasing matrix binding affinities and/or degradation). All conditions have identical secretion rates. Graphs are scaled to maximum concentration of the lowest affinity isoform (VEGF121 – solid line, identical in each case), except the inset in the middle graph which shows each distribution normalized to its maximum concentration, to show relative steepness. Arrows indicate effect of increasing degradation and/or sequestration. B, In vivo, different rates of uptake of VEGF isoforms by surrounding tissues may account for experimentally observed gradients. We hypothesize that VEGF165 (left) shows greater pericellular accumulation and localization because of dual effects of greater sequestration by HSPGs or cell-surface receptors, and greater degradation (loss from the system) by the resultant cellular internalization. Secretion of VEGF121 (right) has lower binding to cells and lower degradation, and thus increased levels in solution and dispersed spatial gradients. Internalization (arrows) may be due to interstitial cells as well as the endothelium, because both cell types can express VEGF receptors.
The observation that reversible matrix binding would not impact the VEGF gradient at steady state has a biophysical explanation: only soluble VEGF diffuses, and at steady state, for every soluble VEGF molecule that binds to the matrix, an equivalent molecule dissociates from the matrix, at each point in space (19). Thus, for equal secretion rates of each VEGF isoform, the same amount of soluble (diffusing) VEGF would be present at steady state. Furthermore, since the effective time spent by each isoform in solution, diffusing, would be identical, the underlying soluble VEGF gradient is also identical. VEGF120 typically results in high levels of soluble VEGF relative to other isoforms; this is typically attributed to its lack of sequestration by the matrix, but the above analysis shows that this is not the case (19).
4.3 Combined sequestration and degradation can explain VEGF gradient formation
If isoform-dependent sequestration alone cannot explain experimentally-observed VEGF isoform gradients, an alternate mechanism must be responsible. Degradation has an important role in morphogen gradient formation in developmental Drosophila systems (127–129). Unlike the effects of sequestration, degradation is able to make VEGF gradients steeper (Fig. 5A, Degradation, Inset). Biophysically, degradation results in shortened lifespan of VEGF molecules, decreasing the amount of time spent in solution and, thus, the distance the molecules can diffuse. More rapid degradation results in steeper VEGF distributions and reduced soluble VEGF levels, however as a consequence, the total amount of VEGF is also significantly reduced (Fig. 5A, Degradation). Thus, degradation by itself is also insufficient to explain experimental data.
It is only when the effects of both sequestration and degradation are combined, such that VEGF isoforms are degraded at a rate that increases with their sequestration binding affinity, that the model predicts the observed behavior of VEGF isoforms in vivo (Fig. 5A, Degradation & Sequestration; note the intersection of VEGF concentration curves). In this scenario, VEGF189 has a steeper distribution than VEGF121 for two reasons: first, greater sequestration resulting in a high concentration near the source of secretion; second, increased degradation and therefore decreased time for diffusion in solution and a steeper fall-off in concentration. The in vivo observation that VEGF120/1 has higher concentration is due to the decreased rate of degradation that it experiences.
The simplest mechanism that accounts for this behavior is sequestration and degradation being coupled. We posit that in vivo, bound VEGF, sequestered in an isoform-dependent manner, is subject to degradation, and that this accounts for patterning differences between isoforms (19). Matrix binding with subsequent intrinsic or proteolytic degradation is a possibility, however, matrix-binding is thought to stabilize VEGF (85, 101) and there do not seem to be indications of VEGF cleavage in the absence of pathology (69). Alternatively, VEGF may bind to the surface of cells in an NRP1- or HSPG-dependent fashion, both of which have progressively greater affinity for the longer isoforms, and be subsequently internalized and degraded (Fig. 5B). In zebrafish, knockout of perlecan has been shown to increase total VEGF levels with associated dispersal of the VEGF spatial gradient, indicating that perlecan may potentiate degradation or loss of VEGF (130). VEGF gradients are observed near the vascular front/avascular border in tissue such as retina and tumors (14, 15), which may be due to high levels of VEGF receptors and NRP1 at the endothelial cell surface (131, 132). A recent study shows that the endothelial cells at the sprouting front exhibit a higher rate of VEGFR2 internalization (133), which not only increases VEGF signaling at the front but could also directly shape the extracellular VEGF gradient. VEGF gradients may also be patterned in the stroma/parenchyma as NRP1 and HSPG co-receptors can also be found on stromal and parenchymal cells (18, 40, 66, 134, 135).
VEGF120 gradients are observed in developmental systems but not tumor systems (16, 136), and it may be possible that different tissues operate at different ratios of sequestration to degradation. Gradients observed in developmental systems suggest that sequestration and degradation take place (e.g. Fig. 5A, Degradation & Sequestration), while tumors may have lower degradation of sequestered isoforms (e.g. Fig. 5A, Sequestration). A prediction of the sequestration-dependent degradation model is that total VEGF in the system is relatively fixed with respect to changes in the secreted isoform (or enzymatic release, discussed in Section 4.4) (Fig. 5A). Protein expression studies (western blots) may support this finding (111); while other studies do not (58, 80). A more direct method of validation would be to compare the gradients or half-lives of VEGF164 with different VEGF isoforms, e.g. VEGF-E (binds NRP1 but not HSPG), VEGF145 (binds HSPGs but not NRP1), or VEGF164Δ108–118, a modified isoform which resists proteolytic degradation (13).
Other growth factor systems where spatial gradients are attributed to sequestration may also operate based on the principle of sequestration-dependent degradation. The ex vivo epithelial bud model branches in the presence of matrix-binding FGF10 but elongates in the presence of non-matrix-binding FGF7 (100). Interestingly, FGF10 appears to induce strong radially-directed autologous gradients (i.e. gradients generated by the cell), while FGF7 does not. While the original authors attributed these gradients to diffusion, we note that in their study, contrary to the authors’ interpretations, cleavage of cell-surface HS diminished elongation while cleavage of HSPGs in surrounding matrix did not, suggesting that autologous gradients may be due to cell surface HS-mediated internalization. Similarly formed autologous gradients may explain differences in morphogenic behavior observed between different VEGF isoforms in an embryoid model of PAE expressing VEGFR2, which lack NRP1 or sVEGFR1-based countergradients (13).
4.4 Proteolytic release of VEGF increases VEGF spatial range by reducing degradation
In models of carcinogenesis, inflammatory cells are thought to secrete proteases that release matrix-bound VEGF into solution, promoting diffusion and endothelial cell binding, and thereby inducing the angiogenic switch (86). Due to proteolytic release, VEGF is redistributed: matrix-binding and peritumoral VEGF localization is lost and VEGF binding to VEGFR2 on endothelial cells increases, while the total VEGF level is unaffected (69, 111, 137) (Table 1). In pancreatic islets of the RIP1-Tag2 mice, redistribution occurs without upregulation of VEGF or VEGFR2, and without a shift in VEGF isoform expression (86, 138); this permits exclusive study of the extracellular regulation of VEGF. Even in studies where protease expression may be associated with VEGF upregulation (93, 113, 139), redistribution is still evident by an increase in the soluble:total VEGF ratio (113, 137).
Since the angiogenic switch is irreversible, this implies that the proteolytic release of stored VEGF is not a one-time burst but is rather continuously maintained to keep a positive angiogenic balance. Increased vascular binding and VEGF-VEGFR2 association seen upon MMP9-, MMP12-, or heparanase-mediated release appears to be unchanging for weeks (86, 111, 115), a steady state scenario. Computational modeling of proteolytic VEGF release (via cleavage) suggests that, at steady state, VEGF release cannot occur any faster than soluble VEGF binds to the matrix, with the result that proteolytic release, like matrix binding, also does not affect soluble VEGF levels by itself (19). Thus, the observed increase in soluble VEGF due to proteases is not a result of more rapid solubilization of matrix-bound VEGF. While ex vivo experiments where proteases are applied to VEGF-containing matrices can show increased soluble VEGF concentration (77, 86, 140), this may be due to the absence of convective and other mechanisms for VEGF loss that are found in vivo.
Interestingly, the sequestration-dependent degradation model that gives rise to isoform-specific VEGF gradients can also reproduce VEGF redistribution due to proteolytic release. Increased soluble VEGF and increased VEGF-VEGFR2 association in the presence of protease suggests that the protease releases VEGF from sequestration and degradation (19); the released VEGF molecule effectively exhibits both decreased sequestration and degradation. This suggests that the proteases either release a VEGF bound to an ECM fragment that impairs further sequestration (99, 100), or that these enzymes cleave HSPG sequestration sites (77), preventing HSPG-associated VEGF degradation or cellular uptake. Similarly, cleaved VEGF113/4 satisfies both requirements as it is not able to subsequently bind to either HSPGs or NRP1. When proteases cleave VEGF inhibitors, VEGF may be released and activated, avoiding both its sequestration and its clearance by the inhibitor.
Tip cells are also known to secrete proteases, which have been implicated in local release of VEGF, enhancing spatial gradients and morphogenesis (141). Our model argues against this occurring in vivo. If proteolytic release results in a decrease in VEGF degradation, gradients would be expected to be flatter: absolute gradients can be enhanced far away, but locally they would be reduced. Furthermore, the tip cell by itself has little proteolytic potential in isolation and is unable to alter soluble VEGF levels (74). Instead, we hypothesize that proteases can alter cellular behavior by acting at the cell surface and modulating active, cell-surface associated VEGF. Indeed, VEGF cleavage has been shown to primarily occur at the cell surface in which case it results in decreased morphogenetic behavior (13).
4.5 Tissue degradation impacts VEGF transport throughout the body
In previous sections, we have focused primarily on local VEGF availability within a tissue, such as a tumor or muscle. The VEGF in those tissues comes primarily from the parenchymal and stromal cells of that tissue (134, 142, 143). VEGF in the bloodstream is primarily a result of clearance and extravasation from tissues and plasma clearance; we have predicted the role of luminal VEGF secretion by the endothelium in recent models (144).
Loss of VEGF in tissues explains the counterintuitive result that serum levels of VEGF increase following administration of bevacizumab, an antibody-derived VEGF sequestering molecule (142, 145). Based on experimental (146) and theoretical studies (50, 143), a fraction of VEGF from parenchymal cells is either captured and internalized by endothelial cells or degraded in the interstitium. Pharmacokinetic models show that anti-VEGF agents have a dispersive effect, similar to that of proteolytic release of VEGF. Upon injection, bevacizumab can extravasate into tissues, bind VEGF and inhibit receptor-mediated internalization. Depending on the tumor microenvironment, this may divert a part of the flux of tissue VEGF into the blood stream resulting in an increase in total plasma VEGF (142). Plasma free VEGF may also increase if the VEGF-bevacizumab complex preferentially dissociates in plasma, based on mass action principles, and may explain the counterintuitive observation of VEGF increase following administration of a VEGF sequestering agent (142, 147, 148). Other agents blocking receptor-mediated internalization of VEGF could elicit a similar phenomenon, e.g. sVEGFR1 (143) or isoform-specific agents such as anti-NRP1 antibodies (39, 149). We predict that this phenomenon is not observed with VEGF-Trap (aflibercept) due to its very strong affinity for VEGF (1000 times higher than bevacizumab), resulting in little dissociation of VEGF-Trap–VEGF complex in the blood stream (144).
5. VEGF ISOFORM CONTROL OF VASCULAR PATTERNING
VEGF isoforms, with their differences in biotransport, sequestration, and NRP-1 binding, induce a spectrum of vascular phenotypes, from the malformed, edematous, hypovascular networks of VEGF120, to the stable, thin, and branching vessels of VEGF188 (Fig. 4). While in normal tissues vascular networks are organized hierarchically and adequately meet the needs of tissues, numerous disease states are characterized by exuberant, highly disturbed phenotypes, the result of pathological angiogenesis. Angiogenesis is regulated by numerous molecular families such as MMPs, FGFs, BMPs and VEGFs (2, 126, 150, 151). In this section, we focus on how VEGF signals shape the phenotypes of normal and pathological angiogenesis.
5.1 Tissue VEGF expression and role of VEGF isoforms in development and adult
VEGF levels must be highly regulated during development, as either a two-fold gain or 50%-loss of VEGF expression results in lethal cardiovascular complications (152, 153). However, across tissues, total VEGF expression has a wide range. In the adult, highest expression rates are found in the omental adipose tissue, lung, retina, ovary; moderate amounts in the kidney, heart, skeletal muscle, and adrenals; and low levels expressed in liver, brain, and breast (25, 79, 154–156). VEGF has numerous roles in the adult organism (157, 158). VEGF seems to be most highly expressed where it is actively being used either for angiogenesis, such as the ovary, or for maintenance of basal permeability such as in the lung, kidney, and heart (154). In contrast, the brain has low VEGF expression and exceptionally low basal permeability (159).
VEGF isoform expression also varies across organs as well as in pathology. VEGF164/5 is the predominant isoform expressed in normal adult tissues (Fig. 2, Table S5), with VEGF188/9 also high in tissues such the lungs, heart, and liver which are initially vascularized by vasculogenesis (25). In the lungs, alveolarization and alveolar vascularization are coupled, and loss of heparin binding isoforms results in reduced alveolarization and vascularization. During development, alveolar vasculature begins with sprout formation from the primitive plexus, which then undergoes extensive intussusceptive angiogenesis, the in situ division of a vessel by transcapillary pillars of stroma, to mature and reach vascular complexity (160–162). VEGF120 is able to support initial vessel outgrowth, but heparin-binding VEGF isoforms are thought to be needed for continued vascularization and maturation – VEGF188 can maintain strong localization and is upregulated by Type II alveolar epithelial cells during primitive alveolar formation (25). Intussusception plays an important role in developmental angiogenesis, however the involvement of VEGF in intussusception is uncertain (162). VEGF seems to be overexpressed in the sprouting phase of network formation and down-regulated during intussusception (160), however other studies show that VEGF can induce splitting forms of angiogenesis (29, 163).
VEGF isoforms may also have other roles such as maintenance of permeability (164) or promotion of inflammation (165). For example, the high VEGF expression in the lung, specifically of VEGF165 and VEGF189, may also have an additional function in recruiting immune cells to provide basal alveolar immunity (165). Pathological states of increased neovascularization, such as tumors, consistently seem to have an increased relative expression of non-heparin-binding isoforms such as VEGF120/121 (Fig. 2), the significance of which is currently unknown and which seems non-optimal for tumor growth (145), since the most rapid tumor growth is usually found with expression of VEGF164/5 (16).
5.2 VEGF splice and cleavage isoforms induce a spectrum of vascular phenotypes
The connection between VEGF isoforms and vascular morphology has been investigated using both in vivo animal experiments and ex vivo explant cultures (23). Particularly useful are the VEGF isoform-specific mice, generated by replacing the multiple splice-site vegfa gene with cDNA for a specific isoform, resulting in VEGF secretion rates for that single isoform equal to the total of all isoforms in wildtype mice (14, 15, 24, 166).
Transgenic mice that express only VEGF120 display widespread perfusion defects from impaired angiogenesis. Severe ischemic cardiomyopathy in these mice results in death soon after birth or within two weeks (24, 25); the heart almost exclusively expresses heparin-binding isoforms in wildtype mice (Fig. 2) (24, 25). However, vascular defects were found in most organs studied including renal glomeruli (decreased glomerular arteries), pulmonary alveoli, retina, bones, and brain (14, 15, 25, 125, 167, 168). Expression only of VEGF120 resulted in fragile, leaky vessels because of poor pericyte coverage, decreased vessel density and poor outgrowth in organs, and impaired arterial and venous development (24, 125). Specifically, sprouting angiogenesis suffered from filopodial disorganization, decreased migration and branching, and formation of closed, blind-ended loops resembling glomeruli (14, 15, 125). In contrast, transgenic mice that express only VEGF188 exhibited higher vascular density than wildtype mice, normal pericyte coverage, and normal coronary and glomerular architecture; however these mice had severe deficits in arterial development in the retina (125) and half of these mice died in utero. VEGF164-only mice were viable and displayed vascularization that was phenotypically similar to wildtype mice (125).
Specific vascular phenotypes resulting from specific VEGF isoforms have also been observed in tumors. Tumors expressing only VEGF120 implanted subcutaneously show poor internal vascularization and poor branching, and peritumoral vessels are enlarged and edematous (16, 80, 169). In contrast, VEGF188-expressing tumors exhibit a high density of low caliber vessels, with significant branching, rich pericyte coverage, and low permeability (16, 80, 170).
Thus, there appears to be a spectrum of vascular behavior dictated by VEGF isoforms, with VEGF120 and VEGF188 at opposite ends. The severity of vascular defects in particular organs, e.g. in the heart and lungs, correlates with endogenous patterns of VEGF isoform expression in wildtype mice (24) (Fig. 2). Furthermore, vascular phenotype seems to be dependent on functional properties such as VEGF gradients, HSPG binding, or NRP1 binding, and as a result, isoforms can be substituted as long as overall function is maintained. For example, dual expression of VEGF120 and VEGF188 in VEGF120/188 heterozygote mice or tumors mimics wildtype and VEGF164-specific vascular phenotypes despite not expressing VEGF164 (15, 16, 23). This functional equivalence of one allele each of VEGF120 and VEGF188 and two alleles of VEGF164 defines a boundary between isoform ratios that yield typical vascular patterns (Fig. 2, gray dashed line). The heart has the closest overall isoform profile to that of VEGF188 only, and this organ showed the greatest defects in VEGF120-expressing mice. We also note that U87MG (human glioblastoma cell line), despite having a lower fraction of VEGF164/5 expression relative to normal mouse brain, may have similar net VEGF properties due to the ratio of VEGF120 to VEGF188.
VEGF120, VEGF164, and VEGF188 have a monotonic relationship between isoform length and properties such as matrix binding and NRP1 binding affinity, and these properties have been suggested as possible key drivers of the above spectrum of vascular phenotypes. VEGF cleavage, producing VEGF113/4, has a similar phenotypic effect as moving the isoform balance towards VEGF120; proteolysis-resistant VEGF164 (VEGF164Δ108–118) results in a similar vascular phenotype to VEGF188 (13). This was interpreted by Lee et al to indicate that endothelial phenotypes are driven by the levels of matrix-bound VEGF in tissues. In contrast, VEGF145 binds HS more strongly than VEGF165 but does not bind NRP1 (44), and this isoform shows weak angiogenic activity, leading Kawamura et al to conclude that NRP1-binding is the key driver of angiogenic phenotype (44).
Not all studies are consistent with the above described monotonic spectrum of vascular phenotypes (Table S6). In specific cases, VEGF189 has been observed to enlarge vessels relative to VEGF165 (66) while VEGF121 can result in stronger angiogenesis than VEGF165 (58, 124, 171). In addition, implantation of myoblasts transfected with any one of the three main isoforms displayed similar vascular malformations such as glomeruloid-like proliferations in skeletal muscle (172).
5.3 VEGF control of the sprouting branching phenotype through gradients, matrix-sequestration, and NRP1-dependent sensing
The vascular phenotypes resulting from different VEGF isoforms is related to the ability of isoforms to induce the sprouting, migratory phenotype. Sprouting angiogenesis starts with VEGF activation of a nascent vessel, sprout formation, basement membrane degradation, sprout extension, lumen formation, and finally anastomosis to another sprout or vessel to complete a flow circuit (for excellent reviews, see (173, 174)). On completion of angiogenesis, the new capillaries can be stabilized by pericytes or undergo pruning (by cellular apoptosis) based on local metabolic demands.
Angiogenic sprouts consist of a distal, VEGFR2-positive migratory tip cell and one or more trailing stalk cells that are highly proliferative and mediate lumen formation (14); the tip and stalk cells can re-order and be dynamically re-assigned based on local VEGF cues (175). Sprouts extend in the direction of VEGF gradients by stabilization of filopodia, dynamic mechanical and sensory apparatuses primarily found on tip cells (14), possibly assisted by collective shifting of roles (175, 176). Branching density (high for VEGF188, low for VEGF120) is thought to reflect the rate of new sprout initiation (15); alternately it may reflect decreased vessel pruning due to increased stabilization by pericytes (125). VEGF, particularly VEGF120, can induce vessel enlargement (14, 177, 178), possibly due to stalk cell proliferation without accompanying tip cell migration. This can cause tortuosity and coiling of sprouts, and the formation of blind-ended tufts as is seen with VEGF120 alone, or in the absence of NRP1 (15, 179).
VEGF initiates endothelial cell activation and induces sprouting (14, 20). VEGF activation through VEGFR2 on an endothelial cell in the parent vessel leads to promotion of the tip cell phenotype (20), with simultaneous inhibition of the tip cell phenotype in adjacent cells via Dll4-Notch-1 signaling. In a vessel with cells undergoing such local mutual competition, the outcome is thought to be dictated by local VEGF and stochasticity in VEGFR2 expression (175). VEGF spatial heterogeneity and gradients are thought to increase the frequency of sprouting (15, 141, 180, 181) and to enhance the migratory phenotype (83, 182, 183). Dynamic filopodial extension causes local variation in VEGFR2 density, and coupled with VEGF heterogeneity, this enhances variability in VEGFR2 activation between cells, leading to more effective distinction of cell fates. The strong, heterogeneous VEGF gradient seen in VEGF188 can thus lead to numerous tip cells and sprouts, ultimately resulting in a high branching density (15). The vascular phenotypes resulting from VEGF188 expression and from reduced Dll4/Notch signaling (Dll4+/− mice or Dll4-neutralizing antibodies) are similar, characterized by increased tip cell formation, high branching density, decreased vessel caliber, and overall poor vascular function (16, 80, 184). However, these phenotypes are not exactly the same: with partial loss of Dll4, poor vascular function is due to immature vessels (185), while in VEGF188, vessels are pericyte rich (80) but possibly poorly connected to external vessels (16). The high sprouting density may be due to higher variation in tip:stalk cell signal for VEGF188, and due to insufficient lateral inhibition in the case of Dll4+/− (186).
Manipulations that disrupt existing VEGF gradients, e.g. the VEGF120/120 genotype, hypoxemia, or injection of VEGF into the eye, result in decreased frequency of sprouting, decreased migration with disorganized filopodia, and enlargement of existing vessels (14, 126). Loss of sVEGFR1, which is thought to form a counter-gradient that enhances the local VEGF gradient (20, 21), similarly results in fewer tip cells and increased but disorganized filopodial protrusion (20). In this situation, vessels are more likely to enlarge through proliferation rather than by sprouting, due to the combined effect of fewer tip cells (due to mutual lateral inhibition (180)) and defective migration (14). Interestingly, other experiments suggest the opposite – that high levels of VEGF, expressed from the lens or injected into the eye, result in exuberant tip cell formation though with abnormal sprouting angles (14, 20). How high levels of VEGF can lead to both states of increased or decreased sprouting is not known (180), but sufficiently high levels seem to suppress sprouting behavior according to theoretical models (187).
While branching and migration due to VEGF188 may be driven primarily by the intensity and heterogeneity of VEGF gradients, the role of distinct signaling outcomes mediated by NRP1-dependent or matrix-bound VEGF cannot be excluded. The role of NRP1 was not initially obvious, as neither VEGF120 nor VEGF188 were thought to bind NRP1, which yet when co-secreted displayed a similar phenotype to VEGF164 (15). It has since been shown that VEGF188 has stronger NRP1 binding than VEGF164 (66, 67). Furthermore, injection of VEGF164 into the retina, which disrupts endogenous gradients but increases matrix-bound VEGF and NRP1-dependent VEGFR2 signaling, disrupted the sprouting/migratory phenotype which was taken to imply that HSPGs and NRP1 were by themselves insufficient to mediate sprouting (14). That experiment, however, cannot rule out the possibility that VEGF gradients are themselves sensed in a NRP1-dependent or bound VEGF-dependent manner. Subsequent studies comparing VEGF121, VEGF165, VEGF-E, and VEGF145 show that both NRP1 and HSPGs enhance the frequency of sprouting, through increased p38/MAPK signaling (27, 44).
Furthermore, in spheroids of porcine aortic endothelial cells expressing VEGFR2 (PAE-VEGFR2), which do not express NRP1, uncleavable VEGF164 (VEGF164Δ108–118) produced strong sprouting as well as proliferation while pre-cleaved VEGF (i.e. VEGF113) resulted only in proliferation (13). This suggests that while both matrix-sequestered and soluble VEGF enable proliferation, matrix-sequestered VEGF additionally provides cues necessary for organized sprouting morphogenesis. There may be a role for isoform-specific autologous VEGF gradients in this system which cannot be excluded, as noted in Section 4.3. Subsequent studies showed that whereas soluble VEGF165 leads to prolonged activation of Akt involved in proliferation and survival, matrix bound VEGF165 resulted in a distinct signaling state characterized by VEGFR2 clustering and prolonged activation of p38/MAPK (28). Even matrix-tethered VEGF121 was shown to induce a sprouting migratory phenotype, whereas soluble VEGF121 induced proliferation and vessel malformations (106, 188). Simulations further suggest that individual tip cells are unable to appreciably alter local soluble VEGF levels; instead, proteases can act on active, cell-surface associated VEGF to alter VEGF signaling (74). In support of this, loss of HS on endothelial cells result in decreased vessel outgrowth in a spheroid model (189) and decreased vessel branching density in tumor xenografts (46). In retinal angiogenesis, loss of HS and fibronectin binding sites for VEGF on astrocytes results in decreased vascular plexus outgrowth similar to the secretion of VEGF120 (84).
5.4 VEGF levels control the spectrum of normal to pathological angiogenesis
While the study of VEGF isoforms helps elucidate mechanisms of sprouting angiogenesis, sprouting does not explain some clinically important vascular behavior. For example, tumor vasculature is characterized by a highly pro-angiogenic balance, leading to an exuberant, irregularly branching, destabilized network (3, 190, 191). Tumor vessels are enlarged, tortuous, leaky, and contain glomeruloid-like swellings, features which lead to stasis/coagulation, edema, resulting in acidic hypoxic microenvironments. Similar dysfunction is also found in retinopathy and neovascular macular degeneration (69, 192) and controlling these excessively angiogenic states is currently a major therapeutic goal.
Tissues can rely on alternate mechanisms of vascularization, e.g. intussusceptive angiogenesis and intraluminal bridging (192–195). Unlike intussusceptive angiogenesis, intraluminal bridging (also known as longitudinal division) divides a parent vessel by intraluminal extension of endothelial processes; it is disputed whether they represent distinct mechanisms (161, 195). Here, we collectively refer to them as splitting angiogenesis. In exercising skeletal muscle, both sprouting and splitting angiogenesis have been shown to occur (29, 196). Splitting is more immediately functional than sprouting angiogenesis as it does not require cellular proliferation, invasion or anastomosis, it retains patency throughout the process, and does not affect the permeability of the original vessels (29, 193). It results in better oxygen transport at high oxygen consumption rates (193, 197). Tumors may switch from sprouting to intussusception following anti-VEGF therapy (198).
What leads to a sprouting response as opposed to these alternative vascularization patterns? Sprouting is primarily a mechanism to vascularize avascular tissues and thus is thought to be dominant when strong VEGF gradients are present, e.g. at the edge of the avascular retina (14). In contrast, in regions with high existing vascular density, such as in muscle (199) or in experimental settings where VEGF is introduced directly into tissues (200), vascularization can proceed more efficiently through splitting of existing vessels. This is consistent with developmental angiogenesis where sprouting creates an initial vessel that can expand into a more complex network through intussusception (160). Which surface of the vessel – luminal or abluminal – receives stimulation may also dictate vascularization (196). Similar to abluminal VEGF gradients, exercise-induced mechanical stretch in muscles and interstitial flow enhance sprouting (141, 181, 196, 201); in contrast, intraluminal shear stress strongly induces intussusceptive angiogenesis (196) and suppresses sprouting (181). Finally, VEGF concentrations play a role. Sprouting is thought to occur best with hypoxia-induced, modest increases in VEGF expression, which preserve existing gradients, whereas splitting angiogenesis occurs more at moderate-to-high VEGF concentrations, which may result in vessel dilation or enlargement (178, 199) and enhanced luminal flow (29).
In a quiescent venule or capillary, appropriate concentrations of VEGF can induce sprouting or splitting angiogenesis without resulting in abnormal vascular structures (29, 202). VEGF increases vessel diameter (178), but at high concentrations, VEGF induces a strong morphogenetic and proliferative response, with pericytes detaching and the vessel enlarging to form an abnormal, highly permeable sinus called a mother vessel (199, 200). Mother vessels can subsequently undergo splitting to form capillaries, can re-associate with pericytes to form stable enlarged vessels called vascular malformations, or can form glomeruloid microvascular proliferations (191). Glomeruloid proliferations arise through continued proliferation of the vessel wall intraluminally and abluminally, subsuming the original mother vessel lumen and forming small channels (203). Other mechanisms of formation of glomeruloid bodies have also been proposed (204). Glomeruloid bodies are loosely associated with perivascular cells, are leaky, and can become hemangiomas in the presence of continued high VEGF (202).
Pathological angiogenesis results from vascular destabilization. VEGF induces EC proliferation, and loss of pericyte coverage occurs through disruption of PDGFR-β activity and an increase in Ang-2 signaling, while NRP1 may promote pericyte coverage (205). Among the VEGF isoforms, VEGF120 has the highest propensity for unstable angiogenesis due to its shallow gradients and low NRP1 association; VEGF188 induces significant pericyte coverage and is protective (206, 207). Despite this, high levels of any of the VEGF isoforms can give rise to glomeruloid bodies (172). Isoforms also vary in terms of their matrix binding affinities. Soluble VEGF121-loaded fibrin gels placed on the chorioallantoic membrane released VEGF in a diffusive burst, and induced splitting angiogenesis and hemangioma formation; gels formulated with fibrin-tethered VEGF121, which allowed for low levels of release over time in a cell-demanded manner, resulted in stable, branched vessels with pericyte coverage (106, 188). The release of VEGF from matrix sequestration into solution is associated with a pro-angiogenic and hence destabilized state (86), but matrix-bound VEGF appears to have similar proliferative activity as soluble VEGF (13, 28, 208). It is unknown whether matrix-bound VEGF achieves its increased vessel stability relative to soluble VEGF due to differences in VEGF localization or to differences in stability-promoting signaling pathways. Numerous other biological factors are also involved in vascular stability, including hypoxia, which induces endothelial HIF2α, pericytes which induce Ang-1 or Ang-2 signaling, and perivascular NO gradients, reviewed in (3).
The goal of anti-angiogenic therapy has typically been to prune unwanted vasculature, however this can be difficult because pathological angiogenesis, especially in tumors, can result in leaky, low-flow vessels, e.g. mother vessels and glomeruloid proliferations, make the surrounding tumor or tissue a poor target for drug delivery. Furthermore, the underlying acidosis and hypoxia, while reducing tumor growth rate, favor long term tumor expansion by promoting epithelial-to-mesenchymal transition and malignancy potential, and hindering the action of immune cells, radiation, and chemotherapy (209). Glomeruloid proliferations, indicative of a highly angiogenic phenotype and of vascular disorganization, are associated with poor patient prognosis (210, 211). Anti-angiogenic therapy has shown poor clinical success, possibly because a large subset of tumor vessels may be VEGF-independent (191). An alternate therapeutic strategy is to use anti-angiogenic therapy for a short period to enable normalization of pathological structures, e.g. mother vessels and glomeruloid proliferations (212), thereby enabling normal blood flow patterns and delivery of cytotoxic agents (3, 191, 209, 213). An alternate hypothesis proposes that vascular normalization occurs by intussusception (198). Normalization may be more successful in tumors showing signs of vascular disorganization, such as the presence of glomeruloid proliferations, or those that secrete higher levels of VEGF121; for example, VEGFR2 blockade specifically increased blood flow in VEGF120-expressing but not VEGF188-expressing tumors (206, 214).
6. PERSPECTIVE
Modulating angiogenesis via control of the VEGF family is a promising therapeutic approach for numerous diseases. In this review, we have detailed important aspects of VEGF extracellular regulation, including the effects of proteases and the formation of VEGF gradients. The different VEGF isoforms play key roles in the resultant vascular morphology, and there is still much that remains to be learned, especially regarding the control of alternative splicing, the role of soluble VEGF inhibitors in VEGF gradients, vascular heterogeneity in tissues and tumors, and optimal intervention strategies. Here we summarize the major points and include potential future areas of study.
1. Regulation of angiogenesis by VEGF isoforms
Vascularization is controlled by overall VEGF concentrations and by the balance of the various VEGF splice and proteolytic isoforms. These isoforms regulate the balance of branching/migratory and proliferative behavior. The endothelial cell decisions are based on isoform-specific differences in: spatial patterning and gradients; binding to cell surface receptors, the VEGFRs and NRPs; binding to the ECM. Recently, the role of matrix-bound VEGF in directly binding to VEGF receptors has been recognized. Clinically, control of vessel morphogenesis through the VEGF family would be of use in a wide range of diseases.
2. Role of VEGF isoforms and vascular morphology in perfusion, function, and in tumor growth
Stimulation with different VEGF isoforms results in structurally different vasculature, and this can lead to different vascular architecture in different tissues, as well as in pathological situations. It is not clear in all cases what functional differences – for example, in perfusion and oxygen delivery – result from these differences. For example, tumors seem to be characterized by relatively high expression of VEGF121; however, single-isoform xenograft studies suggest that VEGF164/5-tumors grow most efficiently. It is unclear whether VEGF121, which causes deranged vasculature, poor tumor growth, and hypoxia, may result in greater malignant potential in the long run, for example through hypoxia-related hypermutation.
3. VEGF gradients arise from a balance of sequestration and degradation in the microenvironment
VEGF isoforms, like other growth factors, show increasing localization with increasing affinity for ECM proteoglycans. We hypothesize that in order to give rise to the observed VEGF isoform gradients, isoforms with increased matrix-binding affinity are also lost from the microenvironment at higher rates (19). This suggests that VEGF sequestered in an isoform-dependent manner (either in the ECM or via cell surface co-receptors such as NRP1 or HSPGs) is subsequently lost via proteolytic degradation or internalization. It is difficult to verify which (or both) of these mechanisms holds in vivo without more detailed observations of VEGF gradients. This also explains observations following VEGF release by proteases, which we hypothesize disperse VEGF by interfering with sequestration-dependent degradation. The balance of sequestration and degradation may be tissue specific. Our analysis assumes a dynamic equilibrium or pseudo-steady state for VEGF, which seems to hold true in vivo.
4. Different enzymes involved in VEGF release have different effects on angiogenic potential
VEGF-cleaving proteases seem to suppress angiogenesis and vascularity, while VEGF release by heparanase is pro-angiogenic. The mechanism by which MMP9 releases VEGF is still unknown, but its pro-angiogenic role tends to support a HSPG-cleaving activity. An interesting question is whether rapid release of cleaved VEGF is able to overcome diffusible VEGF’s intrinsically lower activity compared to matrix-bound VEGF and induce angiogenesis in vivo. Furthermore, if cleaved VEGF levels subsequently return to baseline, would the pro-angiogenic effect be sustained? It has previously been shown that even a temporary burst of VEGF can induce stable angiogenesis, which has been termed the Spike Hypothesis (215).
5. Soluble VEGF inhibitors play an important role in VEGF patterning
The role of soluble VEGF inhibitors in altering VEGF gradients is only recently being recognized. Similar to HSPGs, soluble VEGF inhibitors seem to have multiple effects, from maintenance of quiescence to control of VEGF gradients and sprouting morphogenesis. Understanding how proteases cleave VEGF inhibitors and thereby induce VEGF redistribution and exert control on angiogenesis is an emerging area of study.
6. Computational models in coordination with experimental studies can provide deeper insight into complex biological mechanisms
There have been significant attempts to simulate angiogenesis in computational models based on VEGF transport and endothelial cell responses (183, 216–221); however, it is clear from experimental studies that our knowledge of VEGF transport in tissues is incomplete. The strength of computational models lies in their ability to simulate complex interactions and determine non-intuitive relationships (183). Models can be used to study biological mechanisms, and to generate new hypotheses that can then be tested experimentally.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants R01 HL101200 and R01 CA138264 (ASP) and R00 HL093219 (FMG). The authors thank Dr. David Noren, Dr. Elena Rosca, and Dr. Marianne O. Engel-Stefanini and other members of the Popel laboratory for useful discussions and critical comments.
Biographies

Prakash Vempati, M.Sc., is a medical student at the Vanderbilt University School of Medicine in Nashville, TN. He completed a master’s degree under the direction of Dr. Aleksander S. Popel in biomedical engineering at The Johns Hopkins University in 2009 studying the extracellular regulation of VEGF and matrix metalloproteinases in angiogenesis. He is pursuing a career in internal medicine and is interested in the application of plasma biomarkers and pharmacogenetic information towards clinical practice in vascular diseases, hemostasis, and oncology.

Aleksander S. Popel, Ph.D. is a Professor of Biomedical Engineering at the Johns Hopkins University School of Medicine. He holds joint appointments as Professor of Oncology in the School of Medicine, and Professor of Chemical & Biomolecular Engineering in the Johns Hopkins Whiting School of Engineering. He is a member of the Institute for Nanobiotechnology, In Vivo Cellular Molecular Imaging Center, and the Sydney Kimmel Comprehensive Cancer Center. He has published over 250 scientific papers in the areas of angiogenesis and microcirculation, systems biology, computational medicine & biology. He is the recipient of the Eugene M. Landis Award from the Microcirculatory Society. He is a Fellow of the American Institute of Medical and Biological Engineering, American Heart Association, American Physiological Society, and American Society of Mechanical Engineers, and an Inaugural Fellow of the Biomedical Engineering Society. He has been a member of editorial boards of biological and biomedical engineering journals, and has served in an advisory role to biotech and pharmaceutical companies. He regularly serves on grant review boards and advisory panels at the National Institutes of Health, National Science Foundation, and other US and international funding agencies.

Feilim Mac Gabhann, Ph.D., joined Johns Hopkins University as an Assistant Professor in 2009, with an appointment in Biomedical Engineering and in the Institute for Computational Medicine. He completed his PhD in Biomedical Engineering in 2007, also at Johns Hopkins University, working with Aleksander S. Popel to create mathematical models of growth factor networks in peripheral artery disease and cancer. During postdoctoral work with Shayn M. Peirce and Thomas C. Skalak at the University of Virginia, he conducted experimental research on microvascular remodeling in mouse skeletal muscle. The Mac Gabhann lab creates molecularly-detailed mathematical models of human physiology and disease, including peripheral artery disease, cancer, ALS, pre-eclampsia and HIV. The models have a particular focus on the development and testing of therapeutics. Dr. Mac Gabhann is a Sloan Research Fellow and recipient of a K99/R00 NIH Pathway to Independence Award, the 2010 August Krogh Young Investigator Award from the Microcirculatory Society, and the 2012 Arthur C. Guyton Award for Excellence in Integrative Physiology from the American Physiology Society. He is the author of 44 peer-reviewed papers, and is an Associate Editor for PLoS Computational Biology and BMC Physiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2:a006502. doi: 10.1101/cshperspect.a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mac Gabhann F, Popel AS. Systems biology of vascular endothelial growth factors. Microcirculation. 2008;15:715–738. doi: 10.1080/10739680802095964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egginton S. In vivo shear stress response. Biochem Soc Trans. 2011;39:1633–1638. doi: 10.1042/BST20110715. [DOI] [PubMed] [Google Scholar]
- 5.Chung AS, Ferrara N. Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol. 2011;27:563–584. doi: 10.1146/annurev-cellbio-092910-154002. [DOI] [PubMed] [Google Scholar]
- 6.Liu G, Qutub AA, Vempati P, Mac Gabhann F, Popel AS. Module-based multiscale simulation of angiogenesis in skeletal muscle. Theor Biol Med Model. 2011;8:6. doi: 10.1186/1742-4682-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustafsson T. Vascular remodelling in human skeletal muscle. Biochem Soc Trans. 2011;39:1628–1632. doi: 10.1042/BST20110720. [DOI] [PubMed] [Google Scholar]
- 8.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 9.Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273:114–127. doi: 10.1111/joim.12019. [DOI] [PubMed] [Google Scholar]
- 10.Kinnunen K, Yla-Herttuala S. Vascular endothelial growth factors in retinal and choroidal neovascular diseases. Ann Med. 2012;44:1–17. doi: 10.3109/07853890.2010.532150. [DOI] [PubMed] [Google Scholar]
- 11.Wu FT, Stefanini MO, Mac Gabhann F, Kontos CD, Annex BH, Popel AS. A systems biology perspective on sVEGFR1: its biological function, pathogenic role and therapeutic use. J Cell Mol Med. 2010;14:528–552. doi: 10.1111/j.1582-4934.2009.00941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber HP, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grunstein J, Masbad JJ, Hickey R, Giordano F, Johnson RS. Isoforms of vascular endothelial growth factor act in a coordinate fashion To recruit and expand tumor vasculature. Mol Cell Biol. 2000;20:7282–7291. doi: 10.1128/mcb.20.19.7282-7291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–1326. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz de Almodovar C, Coulon C, Salin PA, Knevels E, Chounlamountri N, Poesen K, Hermans K, Lambrechts D, Van Geyte K, Dhondt J, Dresselaers T, Renaud J, Aragones J, Zacchigna S, Geudens I, Gall D, Stroobants S, Mutin M, Dassonville K, Storkebaum E, Jordan BF, Eriksson U, Moons L, D’Hooge R, Haigh JJ, Belin MF, Schiffmann S, Van Hecke P, Gallez B, Vinckier S, Chedotal A, Honnorat J, Thomasset N, Carmeliet P, Meissirel C. Matrix-binding vascular endothelial growth factor (VEGF) isoforms guide granule cell migration in the cerebellum via VEGF receptor Flk1. J Neurosci. 2010;30:15052–15066. doi: 10.1523/JNEUROSCI.0477-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vempati P, Popel AS, Mac Gabhann F. Formation of VEGF isoform-specific spatial distributions governing angiogenesis: computational analysis. BMC Syst Biol. 2011;5:59. doi: 10.1186/1752-0509-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chappell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17:377–386. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashambhoy YL, Chappell JC, Peirce SM, Bautch VL, Mac Gabhann F. Computational modeling of interacting VEGF and soluble VEGF receptor concentration gradients. Front Physiol. 2011;2:62. doi: 10.3389/fphys.2011.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kappas NC, Zeng G, Chappell JC, Kearney JB, Hazarika S, Kallianos KG, Patterson C, Annex BH, Bautch VL. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J Cell Biol. 2008;181:847–858. doi: 10.1083/jcb.200709114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng YS. The Biology of Vascular Endothelial Cell Growth Factor Isoforms. In: Ruhrberg C, editor. VEGF in Development. Landis Bioscience; 2008. [Google Scholar]
- 24.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard JC, Dewerchin M, Flameng W, Nagy A, Lupu F, Moons L, Collen D, D’Amore PA, Shima DT. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 25.Ng YS, Rohan R, Sunday ME, Demello DE, D’Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 26.Yuan A, Lin CY, Chou CH, Shih CM, Chen CY, Cheng HW, Chen YF, Chen JJ, Chen JH, Yang PC, Chang C. Functional and structural characteristics of tumor angiogenesis in lung cancers overexpressing different VEGF isoforms assessed by DCE- and SSCE-MRI. PLoS One. 2011;6:e16062. doi: 10.1371/journal.pone.0016062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawamura H, Li X, Goishi K, van Meeteren LA, Jakobsson L, Cebe-Suarez S, Shimizu A, Edholm D, Ballmer-Hofer K, Kjellen L, Klagsbrun M, Claesson-Welsh L. Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood. 2008 doi: 10.1182/blood-2007-12-125856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 2010;188:595–609. doi: 10.1083/jcb.200906044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gianni-Barrera R, Trani M, Fontanellaz C, Heberer M, Djonov V, Hlushchuk R, Banfi A. VEGF over-expression in skeletal muscle induces angiogenesis by intussusception rather than sprouting. Angiogenesis. 2013;16:123–136. doi: 10.1007/s10456-012-9304-y. [DOI] [PubMed] [Google Scholar]
- 30.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grunewald FS, Prota AE, Giese A, Ballmer-Hofer K. Structure-function analysis of VEGF receptor activation and the role of coreceptors in angiogenic signaling. Biochim Biophys Acta. 2010;1804:567–580. doi: 10.1016/j.bbapap.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Eichmann A, Simons M. VEGF signaling inside vascular endothelial cells and beyond. Curr Opin Cell Biol. 2012;24:188–193. doi: 10.1016/j.ceb.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu L, Wu X, Cheng Z, Lee CV, LeCouter J, Campa C, Fuh G, Lowman H, Ferrara N. Interaction between bevacizumab and murine VEGF-A: a reassessment. Invest Ophthalmol Vis Sci. 2008;49:522–527. doi: 10.1167/iovs.07-1175. [DOI] [PubMed] [Google Scholar]
- 34.Catena R, Larzabal L, Larrayoz M, Molina E, Hermida J, Agorreta J, Montes R, Pio R, Montuenga LM, Calvo A. VEGF(1)(2)(1)b and VEGF(1)(6)(5)b are weakly angiogenic isoforms of VEGF-A. Mol Cancer. 2010;9:320. doi: 10.1186/1476-4598-9-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dokun AO, Annex BH. The VEGF165b “ICE-o-form” puts a chill on the VEGF story. Circ Res. 2011;109:246–247. doi: 10.1161/CIRCRESAHA.111.249953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris S, Craze M, Newton J, Fisher M, Shima DT, Tozer GM, Kanthou C. Do anti-angiogenic VEGF (VEGFxxxb) isoforms exist? A cautionary tale. PLoS One. 2012;7:e35231. doi: 10.1371/journal.pone.0035231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, Hicklin DJ, Persico G, Herbert JM, Communi D, Shibuya M, Collen D, Conway EM, Carmeliet P. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9:936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- 38.Mac Gabhann F, Popel AS. Model of competitive binding of vascular endothelial growth factor and placental growth factor to VEGF receptors on endothelial cells. Am J Physiol Heart Circ Physiol. 2004;286:H153–164. doi: 10.1152/ajpheart.00254.2003. [DOI] [PubMed] [Google Scholar]
- 39.Mac Gabhann F, Popel AS. Differential binding of VEGF isoforms to VEGF receptor 2 in the presence of neuropilin-1: a computational model. Am J Physiol Heart Circ Physiol. 2005;288:H2851–2860. doi: 10.1152/ajpheart.01218.2004. [DOI] [PubMed] [Google Scholar]
- 40.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 41.Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molina F, Speichinger K, Prahst C, Zhang J, Wang Y, Davis G, Toomre D, Ruhrberg C, Simons M. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev Cell. 2013;25:156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fantin A, Vieira JM, Plein A, Denti L, Fruttiger M, Pollard JW, Ruhrberg C. NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood. 2013;121:2352–2362. doi: 10.1182/blood-2012-05-424713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cebe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, Ballmer-Hofer K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci. 2006;63:2067–2077. doi: 10.1007/s00018-006-6254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- 45.Gengrinovitch S, Berman B, David G, Witte L, Neufeld G, Ron D. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J Biol Chem. 1999;274:10816–10822. doi: 10.1074/jbc.274.16.10816. [DOI] [PubMed] [Google Scholar]
- 46.Fuster MM, Wang L, Castagnola J, Sikora L, Reddi K, Lee PH, Radek KA, Schuksz M, Bishop JR, Gallo RL, Sriramarao P, Esko JD. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keyt BA, Berleau LT, Nguyen HV, Chen H, Heinsohn H, Vandlen R, Ferrara N. The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J Biol Chem. 1996;271:7788–7795. doi: 10.1074/jbc.271.13.7788. [DOI] [PubMed] [Google Scholar]
- 48.Krilleke D, DeErkenez A, Schubert W, Giri I, Robinson GS, Ng YS, Shima DT. Molecular mapping and functional characterization of the VEGF164 heparin-binding domain. J Biol Chem. 2007;282:28045–28056. doi: 10.1074/jbc.M700319200. [DOI] [PubMed] [Google Scholar]
- 49.Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem. 2000;275:26690–26695. doi: 10.1074/jbc.M003955200. [DOI] [PubMed] [Google Scholar]
- 50.Mac Gabhann F, Popel AS. Interactions of VEGF isoforms with VEGFR-1, VEGFR-2, and neuropilin in vivo: a computational model of human skeletal muscle. Am J Physiol Heart Circ Physiol. 2007;292:H459–474. doi: 10.1152/ajpheart.00637.2006. [DOI] [PubMed] [Google Scholar]
- 51.Cebe-Suarez S, Grunewald FS, Jaussi R, Li X, Claesson-Welsh L, Spillmann D, Mercer AA, Prota AE, Ballmer-Hofer K. Orf virus VEGF-E NZ2 promotes paracellular NRP-1/VEGFR-2 coreceptor assembly via the peptide RPPR. FASEB J. 2008;22:3078–3086. doi: 10.1096/fj.08-107219. [DOI] [PubMed] [Google Scholar]
- 52.Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, Bagri A, Tessier-Lavigne M, Koch AW, Watts RJ. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem. 2007;282:24049–24056. doi: 10.1074/jbc.M703554200. [DOI] [PubMed] [Google Scholar]
- 53.Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem. 1992;267:6093–6098. [PubMed] [Google Scholar]
- 54.Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165 [corrected] J Biol Chem. 2000;275:18040–18045. doi: 10.1074/jbc.M909259199. [DOI] [PubMed] [Google Scholar]
- 55.Favier B, Alam A, Barron P, Bonnin J, Laboudie P, Fons P, Mandron M, Herault JP, Neufeld G, Savi P, Herbert JM, Bono F. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood. 2006;108:1243–1250. doi: 10.1182/blood-2005-11-4447. [DOI] [PubMed] [Google Scholar]
- 56.Delcombel R, Janssen L, Vassy R, Gammons M, Haddad O, Richard B, Letourneur D, Bates D, Hendricks C, Waltenberger J, Starzec A, Sounni NE, Noel A, Deroanne C, Lambert C, Colige A. New prospects in the roles of the C-terminal domains of VEGF-A and their cooperation for ligand binding, cellular signaling and vessels formation. Angiogenesis. 2012 doi: 10.1007/s10456-012-9320-y. [DOI] [PubMed] [Google Scholar]
- 57.Lee TY, Folkman J, Javaherian K. HSPG-binding peptide corresponding to the exon 6a-encoded domain of VEGF inhibits tumor growth by blocking angiogenesis in murine model. PLoS One. 2010;5:e9945. doi: 10.1371/journal.pone.0009945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang HT, Scott PA, Morbidelli L, Peak S, Moore J, Turley H, Harris AL, Ziche M, Bicknell R. The 121 amino acid isoform of vascular endothelial growth factor is more strongly tumorigenic than other splice variants in vivo. Br J Cancer. 2000;83:63–68. doi: 10.1054/bjoc.2000.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jia H, Jezequel S, Lohr M, Shaikh S, Davis D, Soker S, Selwood D, Zachary I. Peptides encoded by exon 6 of VEGF inhibit endothelial cell biological responses and angiogenesis induced by VEGF. Biochem Biophys Res Commun. 2001;283:164–173. doi: 10.1006/bbrc.2001.4761. [DOI] [PubMed] [Google Scholar]
- 60.Plouet J, Moro F, Bertagnolli S, Coldeboeuf N, Mazarguil H, Clamens S, Bayard F. Extracellular cleavage of the vascular endothelial growth factor 189-amino acid form by urokinase is required for its mitogenic effect. J Biol Chem. 1997;272:13390–13396. doi: 10.1074/jbc.272.20.13390. [DOI] [PubMed] [Google Scholar]
- 61.Poltorak Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodavsky I, Keshet E, Neufeld G. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- 62.Jingjing L, Srinivasan B, Roque RS. Ectodomain shedding of VEGF183, a novel isoform of vascular endothelial growth factor, promotes its mitogenic activity in vitro. Angiogenesis. 2001;4:103–112. doi: 10.1023/a:1012214931986. [DOI] [PubMed] [Google Scholar]
- 63.Ancelin M, Buteau-Lozano H, Meduri G, Osborne-Pellegrin M, Sordello S, Plouet J, Perrot-Applanat M. A dynamic shift of VEGF isoforms with a transient and selective progesterone-induced expression of VEGF189 regulates angiogenesis and vascular permeability in human uterus. Proc Natl Acad Sci U S A. 2002;99:6023–6028. doi: 10.1073/pnas.082110999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ancelin M, Chollet-Martin S, Herve MA, Legrand C, El Benna J, Perrot-Applanat M. Vascular endothelial growth factor VEGF189 induces human neutrophil chemotaxis in extravascular tissue via an autocrine amplification mechanism. Lab Invest. 2004;84:502–512. doi: 10.1038/labinvest.3700053. [DOI] [PubMed] [Google Scholar]
- 65.Herve MA, Buteau-Lozano H, Mourah S, Calvo F, Perrot-Applanat M. VEGF189 stimulates endothelial cells proliferation and migration in vitro and up-regulates the expression of Flk-1/KDR mRNA. Exp Cell Res. 2005;309:24–31. doi: 10.1016/j.yexcr.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 66.Herve MA, Buteau-Lozano H, Vassy R, Bieche I, Velasco G, Pla M, Perret G, Mourah S, Perrot-Applanat M. Overexpression of vascular endothelial growth factor 189 in breast cancer cells leads to delayed tumor uptake with dilated intratumoral vessels. Am J Pathol. 2008;172:167–178. doi: 10.2353/ajpath.2008.070181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vintonenko N, Pelaez-Garavito I, Buteau-Lozano H, Toullec A, Lidereau R, Perret GY, Bieche I, Perrot-Applanat M. Overexpression of VEGF189 in breast cancer cells induces apoptosis via NRP1 under stress conditions. Cell Adh Migr. 2011;5:332–343. doi: 10.4161/cam.5.4.17287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- 69.Lundkvist A, Lee S, Iruela-Arispe L, Betsholtz C, Gerhardt H. Growth factor gradients in vascular patterning. Novartis Found Symp. 2007;283:194–201. doi: 10.1002/9780470319413.ch15. [DOI] [PubMed] [Google Scholar]
- 70.Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288–36295. doi: 10.1074/jbc.M201674200. [DOI] [PubMed] [Google Scholar]
- 71.Kurtagic E, Jedrychowski MP, Nugent MA. Neutrophil Elastase Cleaves VEGF to Generate a VEGF Fragment with Altered Activity. Am J Physiol Lung Cell Mol Physiol. 2009 doi: 10.1152/ajplung.90505.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakamura S, Morimoto N, Tsuruma K, Izuta H, Yasuda Y, Kato N, Ikeda T, Shimazawa M, Hara H. Tissue kallikrein inhibits retinal neovascularization via the cleavage of vascular endothelial growth factor-165. Arterioscler Thromb Vasc Biol. 2011;31:1041–1048. doi: 10.1161/ATVBAHA.111.223594. [DOI] [PubMed] [Google Scholar]
- 73.Fukuhara J, Noda K, Murata M, Namba S, Kinoshita S, Dong Z, Ando R, Lennikov A, Kanda A, Ishida S. Tissue Kallikrein Attenuates Choroidal Neovascularization via Cleavage of Vascular Endothelial Growth Factor. Invest Ophthalmol Vis Sci. 2013;54:274–279. doi: 10.1167/iovs.12-10512. [DOI] [PubMed] [Google Scholar]
- 74.Vempati P, Mac Gabhann F, Popel AS. Quantifying the proteolytic release of extracellular matrix-sequestered VEGF with a computational model. PLoS One. 2010;5:e11860. doi: 10.1371/journal.pone.0011860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gutierrez J, Konecny GE, Hong K, Burges A, Henry TD, Lambiase PD, Lee Wong W, Meng YG. A new ELISA for use in a 3-ELISA system to assess concentrations of VEGF splice variants and VEGF(110) in ovarian cancer tumors. Clin Chem. 2008;54:597–601. doi: 10.1373/clinchem.2007.096099. [DOI] [PubMed] [Google Scholar]
- 76.Ito TK, Ishii G, Chiba H, Ochiai A. The VEGF angiogenic switch of fibroblasts is regulated by MMP-7 from cancer cells. Oncogene. 2007;26:7194–7203. doi: 10.1038/sj.onc.1210535. [DOI] [PubMed] [Google Scholar]
- 77.Hawinkels LJ, Zuidwijk K, Verspaget HW, de Jonge-Muller ES, van Duijn W, Ferreira V, Fontijn RD, David G, Hommes DW, Lamers CB, Sier CF. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur J Cancer. 2008;44:1904–1913. doi: 10.1016/j.ejca.2008.06.031. [DOI] [PubMed] [Google Scholar]
- 78.Ito TK, Ishii G, Saito S, Yano K, Hoshino A, Suzuki T, Ochiai A. Degradation of soluble VEGF receptor-1 by MMP-7 allows VEGF access to endothelial cells. Blood. 2009;113:2363–2369. doi: 10.1182/blood-2008-08-172742. [DOI] [PubMed] [Google Scholar]
- 79.Mintz L, Savitzky K, Engel S. VEGF nucleic acid and amino acid sequences. 6783954. US Patent. 2004
- 80.Tozer GM, Akerman S, Cross NA, Barber PR, Bjorndahl MA, Greco O, Harris S, Hill SA, Honess DJ, Ireson CR, Pettyjohn KL, Prise VE, Reyes-Aldasoro CC, Ruhrberg C, Shima DT, Kanthou C. Blood vessel maturation and response to vascular-disrupting therapy in single vascular endothelial growth factor-A isoform-producing tumors. Cancer Res. 2008;68:2301–2311. doi: 10.1158/0008-5472.CAN-07-2011. [DOI] [PubMed] [Google Scholar]
- 81.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoier B, Prats C, Qvortrup K, Pilegaard H, Bangsbo J, Hellsten Y. Subcellular localization and mechanism of secretion of vascular endothelial growth factor in human skeletal muscle. FASEB J. 2013 doi: 10.1096/fj.12-224618. [DOI] [PubMed] [Google Scholar]
- 83.Liu L, Ratner BD, Sage EH, Jiang S. Endothelial cell migration on surface-density gradients of fibronectin, VEGF, or both proteins. Langmuir. 2007;23:11168–11173. doi: 10.1021/la701435x. [DOI] [PubMed] [Google Scholar]
- 84.Stenzel D, Lundkvist A, Sauvaget D, Busse M, Graupera M, van der Flier A, Wijelath ES, Murray J, Sobel M, Costell M, Takahashi S, Fassler R, Yamaguchi Y, Gutmann DH, Hynes RO, Gerhardt H. Integrin-dependent and -independent functions of astrocytic fibronectin in retinal angiogenesis. Development. 2011;138:4451–4463. doi: 10.1242/dev.071381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anderson SM, Shergill B, Barry ZT, Manousiouthakis E, Chen TT, Botvinick E, Platt MO, Iruela-Arispe ML, Segura T. VEGF internalization is not required for VEGFR-2 phosphorylation in bioengineered surfaces with covalently linked VEGF. Integr Biol (Camb) 2011;3:887–896. doi: 10.1039/c1ib00037c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wijelath ES, Murray J, Rahman S, Patel Y, Ishida A, Strand K, Aziz S, Cardona C, Hammond WP, Savidge GF, Rafii S, Sobel M. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91:25–31. doi: 10.1161/01.res.0000026420.22406.79. [DOI] [PubMed] [Google Scholar]
- 88.Sahni A, Francis CW. Vascular endothelial growth factor binds to fibrinogen and fibrin and stimulates endothelial cell proliferation. Blood. 2000;96:3772–3778. [PubMed] [Google Scholar]
- 89.Wijelath ES, Rahman S, Murray J, Patel Y, Savidge G, Sobel M. Fibronectin promotes VEGF-induced CD34 cell differentiation into endothelial cells. J Vasc Surg. 2004;39:655–660. doi: 10.1016/j.jvs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 90.Sela S, Natanson-Yaron S, Zcharia E, Vlodavsky I, Yagel S, Keshet E. Local retention versus systemic release of soluble VEGF receptor-1 are mediated by heparin-binding and regulated by heparanase. Circ Res. 2011;108:1063–1070. doi: 10.1161/CIRCRESAHA.110.239665. [DOI] [PubMed] [Google Scholar]
- 91.Luque A, Carpizo DR, Iruela-Arispe ML. ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J Biol Chem. 2003;278:23656–23665. doi: 10.1074/jbc.M212964200. [DOI] [PubMed] [Google Scholar]
- 92.Mac Gabhann F, Ji JW, Popel AS. Multi-scale computational models of pro-angiogenic treatments in peripheral arterial disease. Ann Biomed Eng. 2007;35:982–994. doi: 10.1007/s10439-007-9303-0. [DOI] [PubMed] [Google Scholar]
- 93.Suryawanshi A, Mulik S, Sharma S, Reddy PB, Sehrawat S, Rouse BT. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J Immunol. 2011;186:3653–3665. doi: 10.4049/jimmunol.1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bhattacharjee G, Asplin IR, Wu SM, Gawdi G, Pizzo SV. The conformation-dependent interaction of alpha 2-macroglobulin with vascular endothelial growth factor. A novel mechanism of alpha 2-macroglobulin/growth factor binding. J Biol Chem. 2000;275:26806–26811. doi: 10.1074/jbc.M000156200. [DOI] [PubMed] [Google Scholar]
- 95.Gupta K, Gupta P, Wild R, Ramakrishnan S, Hebbel RP. Binding and displacement of vascular endothelial growth factor (VEGF) by thrombospondin: effect on human microvascular endothelial cell proliferation and angiogenesis. Angiogenesis. 1999;3:147–158. doi: 10.1023/a:1009018702832. [DOI] [PubMed] [Google Scholar]
- 96.Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J Biol Chem. 2010;285:38923–38932. doi: 10.1074/jbc.M110.172304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sulpice E, Contreres JO, Lacour J, Bryckaert M, Tobelem G. Platelet factor 4 disrupts the intracellular signalling cascade induced by vascular endothelial growth factor by both KDR dependent and independent mechanisms. Eur J Biochem. 2004;271:3310–3318. doi: 10.1111/j.1432-1033.2004.04263.x. [DOI] [PubMed] [Google Scholar]
- 98.Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–2457. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Flaumenhaft R, Moscatelli D, Rifkin DB. Heparin and heparan sulfate increase the radius of diffusion and action of basic fibroblast growth factor. J Cell Biol. 1990;111:1651–1659. doi: 10.1083/jcb.111.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Makarenkova HP, Hoffman MP, Beenken A, Eliseenkova AV, Meech R, Tsau C, Patel VN, Lang RA, Mohammadi M. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci Signal. 2009;2:ra55. doi: 10.1126/scisignal.2000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kleinheinz J, Jung S, Wermker K, Fischer C, Joos U. Release kinetics of VEGF165 from a collagen matrix and structural matrix changes in a circulation model. Head Face Med. 2010;6:17. doi: 10.1186/1746-160X-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ekker SC, Bedell VM. The ins and outs of VEGF signaling. Blood. 2009;113:2123–2124. doi: 10.1182/blood-2008-11-189746. [DOI] [PubMed] [Google Scholar]
- 103.Ghajar CM, George SC, Putnam AJ. Matrix metalloproteinase control of capillary morphogenesis. Crit Rev Eukaryot Gene Expr. 2008;18:251–278. doi: 10.1615/critreveukargeneexpr.v18.i3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lauer G, Sollberg S, Cole M, Flamme I, Sturzebecher J, Mann K, Krieg T, Eming SA. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol. 2000;115:12–18. doi: 10.1046/j.1523-1747.2000.00036.x. [DOI] [PubMed] [Google Scholar]
- 105.Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, Davidson JM, Eming SA. Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am J Pathol. 2006;168:670–684. doi: 10.2353/ajpath.2006.050372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ehrbar M, Djonov VG, Schnell C, Tschanz SA, Martiny-Baron G, Schenk U, Wood J, Burri PH, Hubbell JA, Zisch AH. Cell-demanded liberation of VEGF121 from fibrin implants induces local and controlled blood vessel growth. Circ Res. 2004;94:1124–1132. doi: 10.1161/01.RES.0000126411.29641.08. [DOI] [PubMed] [Google Scholar]
- 107.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res. 2000;98:323–332. doi: 10.1016/s0049-3848(99)00242-x. [DOI] [PubMed] [Google Scholar]
- 108.Mochizuki S, Tanaka R, Shimoda M, Onuma J, Fujii Y, Jinno H, Okada Y. Connective tissue growth factor is a substrate of ADAM28. Biochem Biophys Res Commun. 2010;402:651–657. doi: 10.1016/j.bbrc.2010.10.077. [DOI] [PubMed] [Google Scholar]
- 109.Joyce JA, Freeman C, Meyer-Morse N, Parish CR, Hanahan D. A functional heparan sulfate mimetic implicates both heparanase and heparan sulfate in tumor angiogenesis and invasion in a mouse model of multistage cancer. Oncogene. 2005;24:4037–4051. doi: 10.1038/sj.onc.1208602. [DOI] [PubMed] [Google Scholar]
- 110.Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem. 1996;271:10079–10086. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- 111.Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 2001;98:12485–12490. doi: 10.1073/pnas.171460498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104:20262–20267. doi: 10.1073/pnas.0706438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Giraudo E, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest. 2004;114:623–633. doi: 10.1172/JCI22087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Greenaway J, Lawler J, Moorehead R, Bornstein P, Lamarre J, Petrik J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1) J Cell Physiol. 2007;210:807–818. doi: 10.1002/jcp.20904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mineur P, Colige AC, Deroanne CF, Dubail J, Kesteloot F, Habraken Y, Noel A, Voo S, Waltenberger J, Lapiere CM, Nusgens BV, Lambert CA. Newly identified biologically active and proteolysis-resistant VEGF-A isoform VEGF111 is induced by genotoxic agents. J Cell Biol. 2007;179:1261–1273. doi: 10.1083/jcb.200703052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lauer G, Sollberg S, Cole M, Krieg T, Eming SA. Generation of a novel proteolysis resistant vascular endothelial growth factor165 variant by a site-directed mutation at the plasmin sensitive cleavage site. FEBS Lett. 2002;531:309–313. doi: 10.1016/s0014-5793(02)03545-7. [DOI] [PubMed] [Google Scholar]
- 119.Chen RR, Silva EA, Yuen WW, Brock AA, Fischbach C, Lin AS, Guldberg RE, Mooney DJ. Integrated approach to designing growth factor delivery systems. FASEB J. 2007;21:3896–3903. doi: 10.1096/fj.06-7873com. [DOI] [PubMed] [Google Scholar]
- 120.Serini G, Ambrosi D, Giraudo E, Gamba A, Preziosi L, Bussolino F. Modeling the early stages of vascular network assembly. EMBO J. 2003;22:1771–1779. doi: 10.1093/emboj/cdg176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ferrara N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell. 2010;21:687–690. doi: 10.1091/mbc.E09-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mitchell CA, Rutland CS, Walker M, Nasir M, Foss AJ, Stewart C, Gerhardt H, Konerding MA, Risau W, Drexler HC. Unique vascular phenotypes following over-expression of individual VEGFA isoforms from the developing lens. Angiogenesis. 2006;9:209–224. doi: 10.1007/s10456-006-9056-7. [DOI] [PubMed] [Google Scholar]
- 123.Kut C, Mac Gabhann F, Popel AS. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br J Cancer. 2007;97:978–985. doi: 10.1038/sj.bjc.6603923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morbidelli L, Birkenhaeger R, Roeckl W, Granger HJ, Kaerst U, Weich HA, Ziche M. Distinct capillary density and progression promoted by vascular endothelial growth factor-A homodimers and heterodimers. Angiogenesis. 1997;1:117–130. doi: 10.1023/A:1018361217467. [DOI] [PubMed] [Google Scholar]
- 125.Stalmans I, Ng YS, Rohan R, Fruttiger M, Bouche A, Yuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes HP, Moons L, Dewerchin M, Collen D, Carmeliet P, D’Amore PA. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–246. doi: 10.4161/org.4.4.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eldar A, Rosin D, Shilo BZ, Barkai N. Self-enhanced ligand degradation underlies robustness of morphogen gradients. Dev Cell. 2003;5:635–646. doi: 10.1016/s1534-5807(03)00292-2. [DOI] [PubMed] [Google Scholar]
- 128.Lander AD. Morpheus unbound: reimagining the morphogen gradient. Cell. 2007;128:245–256. doi: 10.1016/j.cell.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 129.Kicheva A, Pantazis P, Bollenbach T, Kalaidzidis Y, Bittig T, Julicher F, Gonzalez-Gaitan M. Kinetics of morphogen gradient formation. Science. 2007;315:521–525. doi: 10.1126/science.1135774. [DOI] [PubMed] [Google Scholar]
- 130.Zoeller JJ, Whitelock JM, Iozzo RV. Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 2009;28:284–291. doi: 10.1016/j.matbio.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Akeson AL, Greenberg JM, Cameron JE, Thompson FY, Brooks SK, Wiginton D, Whitsett JA. Temporal and spatial regulation of VEGF-A controls vascular patterning in the embryonic lung. Dev Biol. 2003;264:443–455. doi: 10.1016/j.ydbio.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 132.Jubb AM, Strickland LA, Liu SD, Mak J, Schmidt M, Koeppen H. Neuropilin-1 expression in cancer and development. J Pathol. 2012;226:50–60. doi: 10.1002/path.2989. [DOI] [PubMed] [Google Scholar]
- 133.Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, Itoh N, Hirose T, Breier G, Vestweber D, Cooper JA, Ohno S, Kaibuchi K, Adams RH. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249–260. doi: 10.1038/ncb2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Finley SD, Engel-Stefanini MO, Imoukhuede PI, Popel AS. Pharmacokinetics and pharmacodynamics of VEGF-neutralizing antibodies. BMC Syst Biol. 2011;5:193. doi: 10.1186/1752-0509-5-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rosenstein JM, Krum JM. New roles for VEGF in nervous tissue--beyond blood vessels. Exp Neurol. 2004;187:246–253. doi: 10.1016/j.expneurol.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 136.Stollman TH, Scheer MG, Franssen GM, Verrijp KN, Oyen WJ, Ruers TJ, Leenders WP, Boerman OC. Tumor accumulation of radiolabeled bevacizumab due to targeting of cell- and matrix-associated VEGF-A isoforms. Cancer Biother Radiopharm. 2009;24:195–200. doi: 10.1089/cbr.2008.0574. [DOI] [PubMed] [Google Scholar]
- 137.Mira E, Lacalle RA, Buesa JM, de Buitrago GG, Jimenez-Baranda S, Gomez-Mouton C, Martinez AC, Manes S. Secreted MMP9 promotes angiogenesis more efficiently than constitutive active MMP9 bound to the tumor cell surface. J Cell Sci. 2004;117:1847–1857. doi: 10.1242/jcs.01035. [DOI] [PubMed] [Google Scholar]
- 138.Christofori G, Naik P, Hanahan D. Vascular endothelial growth factor and its receptors, flt-1 and flk-1, are expressed in normal pancreatic islets and throughout islet cell tumorigenesis. Mol Endocrinol. 1995;9:1760–1770. doi: 10.1210/mend.9.12.8614412. [DOI] [PubMed] [Google Scholar]
- 139.Zetser A, Bashenko Y, Edovitsky E, Levy-Adam F, Vlodavsky I, Ilan N. Heparanase induces vascular endothelial growth factor expression: correlation with p38 phosphorylation levels and Src activation. Cancer Res. 2006;66:1455–1463. doi: 10.1158/0008-5472.CAN-05-1811. [DOI] [PubMed] [Google Scholar]
- 140.Ebrahem Q, Chaurasia SS, Vasanji A, Qi JH, Klenotic PA, Cutler A, Asosingh K, Erzurum S, Anand-Apte B. Cross-talk between vascular endothelial growth factor and matrix metalloproteinases in the induction of neovascularization in vivo. Am J Pathol. 2010;176:496–503. doi: 10.2353/ajpath.2010.080642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Helm CL, Fleury ME, Zisch AH, Boschetti F, Swartz MA. Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proc Natl Acad Sci U S A. 2005;102:15779–15784. doi: 10.1073/pnas.0503681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stefanini MO, Wu FT, Mac Gabhann F, Popel AS. Increase of plasma VEGF after intravenous administration of bevacizumab is predicted by a pharmacokinetic model. Cancer Res. 2010;70:9886–9894. doi: 10.1158/0008-5472.CAN-10-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu FT, Stefanini MO, Mac Gabhann F, Kontos CD, Annex BH, Popel AS. Computational kinetic model of VEGF trapping by soluble VEGF receptor-1: effects of transendothelial and lymphatic macromolecular transport. Physiol Genomics. 2009;38:29–41. doi: 10.1152/physiolgenomics.00031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Finley SD, Dhar M, Popel AS. Compartment model predicts VEGF secretion and investigates the effects of VEGF trap in tumor-bearing mice. Front Oncol. 2013;3:196. doi: 10.3389/fonc.2013.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Finley SD, Popel AS. Effect of tumor microenvironment on tumor vegf during anti-VEGF treatment: systems biology predictions. J Natl Cancer Inst. 2013;105:802–811. doi: 10.1093/jnci/djt093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rudge JS, Holash J, Hylton D, Russell M, Jiang S, Leidich R, Papadopoulos N, Pyles EA, Torri A, Wiegand SJ, Thurston G, Stahl N, Yancopoulos GD. VEGF Trap complex formation measures production rates of VEGF, providing a biomarker for predicting efficacious angiogenic blockade. Proc Natl Acad Sci U S A. 2007;104:18363–18370. doi: 10.1073/pnas.0708865104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Segerstrom L, Fuchs D, Backman U, Holmquist K, Christofferson R, Azarbayjani F. The anti-VEGF antibody bevacizumab potently reduces the growth rate of high-risk neuroblastoma xenografts. Pediatr Res. 2006;60:576–581. doi: 10.1203/01.pdr.0000242494.94000.52. [DOI] [PubMed] [Google Scholar]
- 148.Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, Kozin SV, Petit L, Jain RK, Chung DC, Sahani DV, Kalva SP, Cohen KS, Scadden DT, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Shellito PC, Mino-Kenudson M, Lauwers GY. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- 149.Gu C, Limberg BJ, Whitaker GB, Perman B, Leahy DJ, Rosenbaum JS, Ginty DD, Kolodkin AL. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002;277:18069–18076. doi: 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- 150.Blanco R, Gerhardt H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 2013;3:a006569. doi: 10.1101/cshperspect.a006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ziyad S, Iruela-Arispe ML. Molecular mechanisms of tumor angiogenesis. Genes Cancer. 2011;2:1085–1096. doi: 10.1177/1947601911432334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 153.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 154.Berse B, Brown LF, Van de Water L, Dvorak HF, Senger DR. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell. 1992;3:211–220. doi: 10.1091/mbc.3.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Saint-Geniez M, Maldonado AE, D’Amore PA. VEGF expression and receptor activation in the choroid during development and in the adult. Invest Ophthalmol Vis Sci. 2006;47:3135–3142. doi: 10.1167/iovs.05-1229. [DOI] [PubMed] [Google Scholar]
- 156.Zhang QX, Magovern CJ, Mack CA, Budenbender KT, Ko W, Rosengart TK. Vascular endothelial growth factor is the major angiogenic factor in omentum: mechanism of the omentum-mediated angiogenesis. J Surg Res. 1997;67:147–154. doi: 10.1006/jsre.1996.4983. [DOI] [PubMed] [Google Scholar]
- 157.Lazarus A, Keshet E. Vascular endothelial growth factor and vascular homeostasis. Proc Am Thorac Soc. 2011;8:508–511. doi: 10.1513/pats.201102-021MW. [DOI] [PubMed] [Google Scholar]
- 158.Maharaj AS, D’Amore PA. Roles for VEGF in the adult. Microvasc Res. 2007;74:100–113. doi: 10.1016/j.mvr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci U S A. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Makanya AN, Stauffer D, Ribatti D, Burri PH, Djonov V. Microvascular growth, development, and remodeling in the embryonic avian kidney: the interplay between sprouting and intussusceptive angiogenic mechanisms. Microsc Res Tech. 2005;66:275–288. doi: 10.1002/jemt.20169. [DOI] [PubMed] [Google Scholar]
- 161.Makanya AN, Hlushchuk R, Djonov VG. Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis. 2009;12:113–123. doi: 10.1007/s10456-009-9129-5. [DOI] [PubMed] [Google Scholar]
- 162.De Spiegelaere W, Casteleyn C, Van den Broeck W, Plendl J, Bahramsoltani M, Simoens P, Djonov V, Cornillie P. Intussusceptive angiogenesis: a biologically relevant form of angiogenesis. J Vasc Res. 2012;49:390–404. doi: 10.1159/000338278. [DOI] [PubMed] [Google Scholar]
- 163.Baum O, Suter F, Gerber B, Tschanz SA, Buergy R, Blank F, Hlushchuk R, Djonov V. VEGF-A promotes intussusceptive angiogenesis in the developing chicken chorioallantoic membrane. Microcirculation. 2010;17:447–457. doi: 10.1111/j.1549-8719.2010.00043.x. [DOI] [PubMed] [Google Scholar]
- 164.Xu D, Fuster MM, Lawrence R, Esko JD. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J Biol Chem. 2011;286:737–745. doi: 10.1074/jbc.M110.177006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Usui T, Ishida S, Yamashiro K, Kaji Y, Poulaki V, Moore J, Moore T, Amano S, Horikawa Y, Dartt D, Golding M, Shima DT, Adamis AP. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368–374. doi: 10.1167/iovs.03-0106. [DOI] [PubMed] [Google Scholar]
- 166.Ng IO, Poon RT, Lee JM, Fan ST, Ng M, Tso WK. Microvessel density, vascular endothelial growth factor and its receptors Flt-1 and Flk-1/KDR in hepatocellular carcinoma. Am J Clin Pathol. 2001;116:838–845. doi: 10.1309/FXNL-QTN1-94FH-AB3A. [DOI] [PubMed] [Google Scholar]
- 167.Mattot V, Moons L, Lupu F, Chernavvsky D, Gomez RA, Collen D, Carmeliet P. Loss of the VEGF(164) and VEGF(188) isoforms impairs postnatal glomerular angiogenesis and renal arteriogenesis in mice. J Am Soc Nephrol. 2002;13:1548–1560. doi: 10.1097/01.asn.0000013925.19218.7b. [DOI] [PubMed] [Google Scholar]
- 168.Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73. doi: 10.1016/s0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 169.Kusters B, de Waal RM, Wesseling P, Verrijp K, Maass C, Heerschap A, Barentsz JO, Sweep F, Ruiter DJ, Leenders WP. Differential effects of vascular endothelial growth factor A isoforms in a mouse brain metastasis model of human melanoma. Cancer Res. 2003;63:5408–5413. [PubMed] [Google Scholar]
- 170.Cheng SY, Nagane M, Huang HS, Cavenee WK. Intracerebral tumor-associated hemorrhage caused by overexpression of the vascular endothelial growth factor isoforms VEGF121 and VEGF165 but not VEGF189. Proc Natl Acad Sci U S A. 1997;94:12081–12087. doi: 10.1073/pnas.94.22.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fenton BM, Paoni SF, Liu W, Cheng SY, Hu B, Ding I. Overexpression of VEGF121, but not VEGF165 or FGF-1, improves oxygenation in MCF-7 breast tumours. Br J Cancer. 2004;90:430–435. doi: 10.1038/sj.bjc.6601539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Springer ML, Banfi A, Ye J, von Degenfeld G, Kraft PE, Saini SA, Kapasi NK, Blau HM. Localization of vascular response to VEGF is not dependent on heparin binding. FASEB J. 2007;21:2074–2085. doi: 10.1096/fj.06-7700com. [DOI] [PubMed] [Google Scholar]
- 173.Geudens I, Gerhardt H. Coordinating cell behaviour during blood vessel formation. Development. 2011;138:4569–4583. doi: 10.1242/dev.062323. [DOI] [PubMed] [Google Scholar]
- 174.Logsdon EA, Finley SD, Popel AS, Mac Gabhann F. A systems biology view of blood vessel growth and remodeling. J Cell Mol Med. 2013 doi: 10.1111/jcmm.12164. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12:943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 176.Arima S, Nishiyama K, Ko T, Arima Y, Hakozaki Y, Sugihara K, Koseki H, Uchijima Y, Kurihara Y, Kurihara H. Angiogenic morphogenesis driven by dynamic and heterogeneous collective endothelial cell movement. Development. 2011;138:4763–4776. doi: 10.1242/dev.068023. [DOI] [PubMed] [Google Scholar]
- 177.Nakatsu MN, Sainson RC, Perez-del-Pulgar S, Aoto JN, Aitkenhead M, Taylor KL, Carpenter PM, Hughes CC. VEGF(121) and VEGF(165) regulate blood vessel diameter through vascular endothelial growth factor receptor 2 in an in vitro angiogenesis model. Lab Invest. 2003;83:1873–1885. doi: 10.1097/01.lab.0000107160.81875.33. [DOI] [PubMed] [Google Scholar]
- 178.von Degenfeld G, Banfi A, Springer ML, Wagner RA, Jacobi J, Ozawa CR, Merchant MJ, Cooke JP, Blau HM. Microenvironmental VEGF distribution is critical for stable and functional vessel growth in ischemia. FASEB J. 2006;20:2657–2659. doi: 10.1096/fj.06-6568fje. [DOI] [PubMed] [Google Scholar]
- 179.Gerhardt H, Ruhrberg C, Abramsson A, Fujisawa H, Shima D, Betsholtz C. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev Dyn. 2004;231:503–509. doi: 10.1002/dvdy.20148. [DOI] [PubMed] [Google Scholar]
- 180.Bentley K, Gerhardt H, Bates PA. Agent-based simulation of notch-mediated tip cell selection in angiogenic sprout initialisation. J Theor Biol. 2008;250:25–36. doi: 10.1016/j.jtbi.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 181.Song JW, Munn LL. Fluid forces control endothelial sprouting. Proc Natl Acad Sci U S A. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Barkefors I, Le Jan S, Jakobsson L, Hejll E, Carlson G, Johansson H, Jarvius J, Park JW, Li Jeon N, Kreuger J. Endothelial cell migration in stable gradients of vascular endothelial growth factor A and fibroblast growth factor 2: effects on chemotaxis and chemokinesis. J Biol Chem. 2008;283:13905–13912. doi: 10.1074/jbc.M704917200. [DOI] [PubMed] [Google Scholar]
- 183.Bentley K, Jones M, Cruys B. Predicting the future: towards symbiotic computational and experimental angiogenesis research. Exp Cell Res. 2013;319:1240–1246. doi: 10.1016/j.yexcr.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 184.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032–1037. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 185.Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley EJ, Duarte A, Gill PS. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood. 2007;109:4753–4760. doi: 10.1182/blood-2006-12-063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Benedito R, Hellstrom M. Notch as a hub for signaling in angiogenesis. Exp Cell Res. 2013;319:1281–1288. doi: 10.1016/j.yexcr.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 187.Carlier A, Geris L, Bentley K, Carmeliet G, Carmeliet P, Van Oosterwyck H. MOSAIC: a multiscale model of osteogenesis and sprouting angiogenesis with lateral inhibition of endothelial cells. PLoS Comput Biol. 2012;8:e1002724. doi: 10.1371/journal.pcbi.1002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ehrbar M, Zeisberger SM, Raeber GP, Hubbell JA, Schnell C, Zisch AH. The role of actively released fibrin-conjugated VEGF for VEGF receptor 2 gene activation and the enhancement of angiogenesis. Biomaterials. 2008;29:1720–1729. doi: 10.1016/j.biomaterials.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 189.Jakobsson L, Kreuger J, Holmborn K, Lundin L, Eriksson I, Kjellen L, Claesson-Welsh L. Heparan sulfate in trans potentiates VEGFR-mediated angiogenesis. Dev Cell. 2006;10:625–634. doi: 10.1016/j.devcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 190.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36:321–331. doi: 10.1055/s-0030-1253454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nagy JA, Chang SH, Dvorak AM, Dvorak HF. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. 2009;100:865–869. doi: 10.1038/sj.bjc.6604929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ribatti D, Djonov V. Intussusceptive microvascular growth in tumors. Cancer Lett. 2012;316:126–131. doi: 10.1016/j.canlet.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 194.Gianni-Barrera R, Trani M, Reginato S, Banfi A. To sprout or to split? VEGF, Notch and vascular morphogenesis. Biochem Soc Trans. 2011;39:1644–1648. doi: 10.1042/BST20110650. [DOI] [PubMed] [Google Scholar]
- 195.Egginton S. Invited review: activity-induced angiogenesis. Pflugers Arch. 2009;457:963–977. doi: 10.1007/s00424-008-0563-9. [DOI] [PubMed] [Google Scholar]
- 196.Egginton S, Zhou AL, Brown MD, Hudlicka O. Unorthodox angiogenesis in skeletal muscle. Cardiovasc Res. 2001;49:634–646. doi: 10.1016/s0008-6363(00)00282-0. [DOI] [PubMed] [Google Scholar]
- 197.Ji JW, Tsoukias NM, Goldman D, Popel AS. A computational model of oxygen transport in skeletal muscle for sprouting and splitting modes of angiogenesis. J Theor Biol. 2006;241:94–108. doi: 10.1016/j.jtbi.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 198.Hlushchuk R, Makanya AN, Djonov V. Escape mechanisms after antiangiogenic treatment, or why are the tumors growing again? Int J Dev Biol. 2011;55:563–567. doi: 10.1387/ijdb.103231rh. [DOI] [PubMed] [Google Scholar]
- 199.Springer ML, Chen AS, Kraft PE, Bednarski M, Blau HM. VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell. 1998;2:549–558. doi: 10.1016/s1097-2765(00)80154-9. [DOI] [PubMed] [Google Scholar]
- 200.Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, Carter R, Krieger JE, Manseau EJ, Harvey VS, Eckelhoefer IA, Feng D, Dvorak AM, Mulligan RC, Dvorak HF. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 201.Rivron NC, Vrij EJ, Rouwkema J, Le Gac S, van den Berg A, Truckenmuller RK, van Blitterswijk CA. Tissue deformation spatially modulates VEGF signaling and angiogenesis. Proc Natl Acad Sci U S A. 2012;109:6886–6891. doi: 10.1073/pnas.1201626109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ozawa CR, Banfi A, Glazer NL, Thurston G, Springer ML, Kraft PE, McDonald DM, Blau HM. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J Clin Invest. 2004;113:516–527. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF. Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am J Pathol. 2001;158:1145–1160. doi: 10.1016/S0002-9440(10)64062-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dome B, Hendrix MJ, Paku S, Tovari J, Timar J. Alternative vascularization mechanisms in cancer: Pathology and therapeutic implications. Am J Pathol. 2007;170:1–15. doi: 10.2353/ajpath.2007.060302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J, Plowman G, Peale F, Koch AW, Wu Y, Bagri A, Tessier-Lavigne M, Watts RJ. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007;11:53–67. doi: 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 206.Akerman S, Fisher M, Daniel RA, Lefley D, Reyes-Aldasoro CC, Lunt SJ, Harris S, Bjorndahl M, Williams LJ, Evans H, Barber PR, Prise VE, Vojnovic B, Kanthou C, Tozer GM. Influence of soluble or matrix-bound isoforms of vascular endothelial growth factor-A on tumor response to vascular-targeted strategies. Int J Cancer. 2013 doi: 10.1002/ijc.28281. [DOI] [PubMed] [Google Scholar]
- 207.Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood. 2011;118:2906–2917. doi: 10.1182/blood-2011-01-331694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Zisch AH, Schenk U, Schense JC, Sakiyama-Elbert SE, Hubbell JA. Covalently conjugated VEGF--fibrin matrices for endothelialization. J Control Release. 2001;72:101–113. doi: 10.1016/s0168-3659(01)00266-8. [DOI] [PubMed] [Google Scholar]
- 209.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Straume O, Chappuis PO, Salvesen HB, Halvorsen OJ, Haukaas SA, Goffin JR, Begin LR, Foulkes WD, Akslen LA. Prognostic importance of glomeruloid microvascular proliferation indicates an aggressive angiogenic phenotype in human cancers. Cancer Res. 2002;62:6808–6811. [PubMed] [Google Scholar]
- 211.Tanaka F, Oyanagi H, Takenaka K, Ishikawa S, Yanagihara K, Miyahara R, Kawano Y, Li M, Otake Y, Wada H. Glomeruloid microvascular proliferation is superior to intratumoral microvessel density as a prognostic marker in non-small cell lung cancer. Cancer Res. 2003;63:6791–6794. [PubMed] [Google Scholar]
- 212.Sitohy B, Nagy JA, Jaminet SC, Dvorak HF. Tumor-surrogate blood vessel subtypes exhibit differential susceptibility to anti-VEGF therapy. Cancer Res. 2011;71:7021–7028. doi: 10.1158/0008-5472.CAN-11-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 214.Akerman S, Reyes-Aldasoro CC, Fisher M, Pettyjohn KL, Bjorndahl MA, Evans H, Tozer GM. Microflow of fluorescently labelled red blood cells in tumours expressing single isoforms of VEGF and their response to vascular targeting agents. Med Eng Phys. 2011;33:805–809. doi: 10.1016/j.medengphy.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 215.Indraccolo S, Favaro E, Amadori A. Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle. 2006;5:1751–1755. doi: 10.4161/cc.5.16.2985. [DOI] [PubMed] [Google Scholar]
- 216.Milde F, Bergdorf M, Koumoutsakos P. A hybrid model for three-dimensional simulations of sprouting angiogenesis. Biophys J. 2008;95:3146–3160. doi: 10.1529/biophysj.107.124511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Qutub AA, Mac Gabhann F, Karagiannis ED, Vempati P, Popel AS. Multiscale models of angiogenesis. IEEE Eng Med Biol Mag. 2009;28:14–31. doi: 10.1109/MEMB.2009.931791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Qutub AA, Popel AS. Elongation, proliferation & migration differentiate endothelial cell phenotypes and determine capillary sprouting. BMC Syst Biol. 2009;3:13. doi: 10.1186/1752-0509-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Travasso RD, Corvera Poire E, Castro M, Rodriguez-Manzaneque JC, Hernandez-Machado A. Tumor angiogenesis and vascular patterning: a mathematical model. PLoS One. 2011;6:e19989. doi: 10.1371/journal.pone.0019989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Amyot F, Small A, Boukari H, Camphausen K, Gandjbakhche A. Topology of the heterogeneous nature of the extracellular matrix on stochastic modeling of tumor-induced angiogenesis. Microvasc Res. 2009;77:87–95. doi: 10.1016/j.mvr.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bauer AL, Jackson TL, Jiang Y. Topography of extracellular matrix mediates vascular morphogenesis and migration speeds in angiogenesis. PLoS Comput Biol. 2009;5:e1000445. doi: 10.1371/journal.pcbi.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, Giavazzi R. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res. 2003;63:5224–5229. [PubMed] [Google Scholar]
- 223.Gaspar NJ, Jue RA, Hu J, Puchacz E, deForest NL, Schellenberger U. Cysteine 116 participates in intermolecular bonding of the human VEGF(121) homodimer. Arch Biochem Biophys. 2002;404:126–135. doi: 10.1016/s0003-9861(02)00239-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.