

Abstract
Knowledge of the pathobiology of pulmonary hypertension continues to accelerate. However, fundamental gaps remain in our understanding of the underlying pathological changes in pulmonary arteries and veins in the different forms of this syndrome. Although pulmonary hypertension primarily affects the arteries, venous disease is increasingly recognized as an important entity. Moreover, prognosis in pulmonary hypertension is determined largely by the status of the right ventricle, rather than the levels of pulmonary artery pressures. It is increasingly clear that while vasospasm plays a role, pulmonary hypertension is an obstructive lung panvasculopathy. Disordered metabolism and mitochondrial structure, inflammation, and dysregulation of growth factors lead to a proliferative, apoptosis-resistant state. These abnormalities may be acquired, genetically mediated as a result of mutations in bone morphogenetic protein receptor (BMPR)2 or activin-like kinase (Alk)-1 or epigenetically-inherited (as a result of epigenetic silencing of genes such as superoxide dismutase 2). There is a pressing need to better understand how the pathobiology leads to severe disease in some patients versus mild pulmonary hypertension in others. Recent recognition of a potential role of acquired abnormalities of mitochondrial metabolism in the right ventricular myocytes and pulmonary vascular cells suggests new therapeutic approaches, diagnostic modalities, and biomarkers. Finally, dissection of role of pulmonary inflammation in the initiation and promotion of pulmonary hypertension has revealed a complex yet fascinating interplay with pulmonary vascular remodeling, promising to lead to novel therapeutics and diagnostics. Emerging concepts are also relevant to the pathobiology of pulmonary hypertension, including a role for bone marrow and circulating progenitor cells and microRNAs. Continued interest in the interface of the genetic basis of pulmonary hypertension and cellular and molecular pathogenetic links should expand further our understanding of the disease.
Keywords: inflammation, metabolism, pulmonary arteries, pulmonary veins
The specific field of pathobiology of pulmonary hypertension (PH) has undergone an impressive growth since the last world meeting in Dana Point in 2008 (1, 2). Building on a remarkable list of significant accomplishments realized in the past 110 years (3), several important paradigms have taken center stage, including the role of metabolic reprogramming and inflammation, with promising insights that may lead to novel therapeutic and diagnostic targets. Notwithstanding this remarkable progress, central questions remain related to the fundamental aspects of pulmonary vascular pathology in PH.
When choosing the particular areas of emphasis in the context of the World Symposium, the Pathology and Pathobiology working group opted to continue focusing on the pathologic alterations of pulmonary veins in PH, largely inspired by the pressing questions in this particular area that arose from the Dana Point meeting (1). This was complemented by three additional topics, due to their timeliness and overall importance: the molecular determinants of mild versus severe PH; the role of metabolic reprogramming underlying cellular responses in pulmonary vascular disease; and, recent insights into the role of inflammation as a trigger and modifier of PH. Furthermore, many pathogenetic processes operational in mild disease appear to have roles in more severe PH, but how molecular processes determine the progression and ultimately the severity of the disease remains unclear. To start addressing this complex yet central question, the integration of pathology and pathogenetic mechanisms should provide insights in this topic in the years to come – perhaps one of the most vexing challenges in the field.
The group members acknowledge that the chosen discussion topics did not allow for an all-inclusive dissection of established and emerging areas of investigation. These include, among others, specific signaling pathways shown to affect pulmonary vasoconstriction and remodeling, such as epidermal growth factor (EGF) (4), fibroblast growth factor (FGF) (5), platelet derived growth factor (PDGF) (6, 7), and transforming growth factor (TGF)-β (8, 9), or are protective (such as bone morphogenetic proteins) and evolving knowledge on the involvement of pathogenic (10) or protective (11) micro RNAs in pulmonary arterial hypertension (PAH). In addition, emerging areas of investigation including the potential role of progenitor or stem cells in pulmonary vascular remodeling (12) were not addressed. Notwithstanding these limitations, when fitting, we emphasize the potential role of circulating cells derived from the bone marrow, as documented in recent human studies (13).
Does the pulmonary venous system play an important role in PAH and to what extent is PAH and PVOD part of the same spectrum of disease?
It is apparent that the different forms of PH present with either a predominance of pulmonary arterial remodeling or vein remodeling or a variable contribution of both. Paradigmatic of the former is idiopathic pulmonary arterial hypertension (IPAH), while pure pulmonary venoocclusive disease and PH due to left heart dysfunction are characterized predominantly by venous remodeling. Virtually all forms of PH, including those caused by interstitial lung disease, thromboembolic, hypoxia, and sarcoid may involve elements of both arterial and venous remodeling. However, more precise documentation, including morphometric analysis of pulmonary veins remodeling is still lacking in most of these conditions.
The difficulty in studying the pathology of pulmonary venous remodeling is compounded by the lack of distinct molecular markers that allow for their identification (as compared with the pulmonary arterial bed) when immersed in alveolar tissue. Normal veins can be recognized by the lack of a double elastic lamina and relatively thin muscular media; however, when remodeled, pulmonary veins become “arterialized”, making their distinction from pulmonary arteries more difficult. The only distinctive feature, which is often not present in biopsies, is the presence of pulmonary veins in interlobular septae. Moreover, molecular markers of veins, such as the ephrin B4 receptor, are often difficult to identify in human (and rat) lungs. The venous coaxial structure includes layers of cardiomyocytes arrayed externally around a subendothelial layer of typical smooth muscle cells, thus forming sphincter-like structures. In disease states this cardiac muscle layer extends further into the lung and its role in Group 2 PH merits investigation.
Given the limitations in recognizing veins histologically, most studies rely on their identification in selected regions of explant specimens. In subsets of PAH patients with scleroderma-associated PAH, pulmonary veno occlusive disease-like remodeling may predominate over arterial remodeling. The venous remodeling, if present, entails worse prognosis associated with decreased 6 minute-walk distance, PaO2, and diffusion capacity (14). However, in a recent pathologic study of PAH, including lungs with scleroderma associated PAH, no correlations between vein remodeling and intima and media thickness in arteries could be identified (15). Recent studies have highlighted the pattern of pulmonary vein remodeling in patients with left heart failure (LHF). Lung samples obtained from patients with LHF placed on a ventricular assist device showed increased pulmonary vein and artery media thickening. Of interest, some patients who improved on the assist device and underwent repeat lung biopsy had a significant improvement of pulmonary vein remodeling (16). The molecular drivers of these processes remain largely unknown.
In summary, we continue to lack the availability of unique molecular markers that allow the specific identification of pulmonary veins. Consequently, the prevalence of venous pathology in the various forms of PH remains unknown. This is despite the potential for deleterious effects of prostacyclin and agents that increase bioavailability of nitric oxide in the context of elevated capillary pressures. Finally, little is known about pathogenetic pathways involved in pulmonary vein remodeling and how they correlate with arterial remodeling in both LHF and in forms of PH with precapillary predominance.
Are there distinct pathways in vascular cells in mild versus severe PH?
Although PH is defined as a mean pulmonary arterial pressure at right heart catheterization of higher than 25 mmHg with a normal pulmonary capillary wedge pressure, it is well known that, on the basis of clinical and hemodynamic findings, PH can range from mild to severe, even in the presence of the same stimulus (e.g. hypoxia). This spectrum of severity raises several critical pathogenetic and clinical questions in terms of the distinctive feature that differentiate the severity stages.
Increased pulmonary artery pressures occur due to sustained vasoconstriction, excessive pulmonary vascular remodeling, and in situ thrombosis, which ultimately lead to increased pulmonary vascular resistance. However, the factors responsible for the aggravation or acceleration of PH remain poorly defined (Figure 1). The contributing factors likely involve accumulation of multiple events on a background of genetic predisposition. These factors involve the action of vasoconstrictive and pro remodeling processes, including the action of inflammatory, pro-coagulant, anti-apoptotic, and autoimmune mediators, cell-cell and cell-matrix interactions and environmental factors over time (Figure 2. Although the pulmonary artery bed appears unreactive to vasodilators in advanced disease, vasoreactivity and remodeling possibly interact in disease evolution (17).
Figure 1. Proposed multifactorial factors influencing progression of pulmonary hypertension.
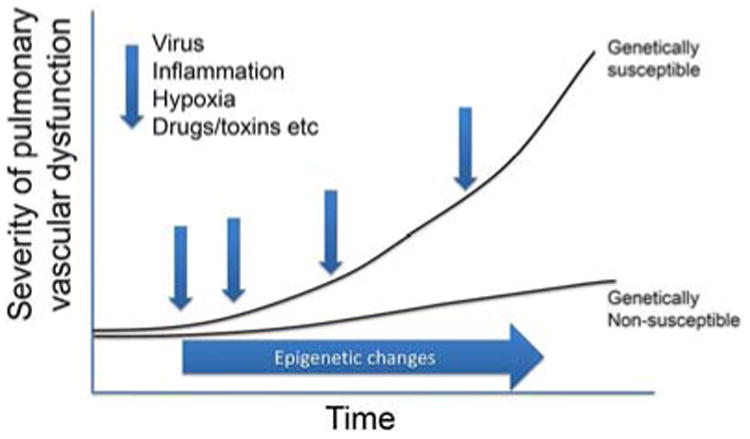
In a suitable genetic background, the interplay of epigenetics and pathobiological injurious events may amplify the severity of the disease, often associated with more pronounced remodeling and worse clinical outcome.
Figure 2. Emerging paradigms in pulmonary hypertension research, involving the broad effects of metabolic programming of intima and media pulmonary vascular cells (endothelial and smooth muscle cells) and the immediate perivascular microenvironment.
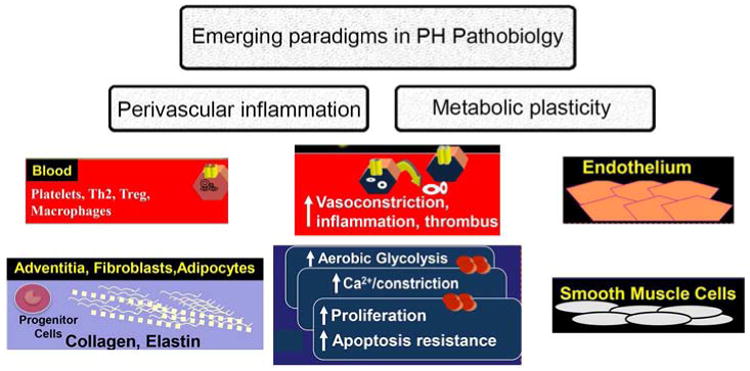
The perivascular region is dominated by fibroblasts and migrating circulating cells, including inflammatory and progenitor cells. Shown in the center are the impact of these factors in the intima and media of pulmonary arteries. The metabolic plasticity involves all cells involved in the PH panvasculopathy and is itself modified by inflammation and infiltrating progenitor cells (these paradigms are examined in more detail in Ref. (100)).
Whether the severity of pulmonary vascular disease involves a constellation of pathobiologic processes or is defined pathologically, with the hallmark finding of reduction of the pulmonary vascular lumen remains unclear. The definition of the severity of PH based on histopathological findings is complicated by the lack information on what constitutes “normal”. Surprisingly, recent analysis of unused donor “control” lungs revealed substantial neointimal formation, inflammation and venous changes (15), features usually judged to be “pathological”. This suggests a spectrum from pristine vessels (which are observed primarily in the younger controls) to vascular changes reminiscent of PH that may be present as a function of normal aging, including inflammation and left ventricular stiffening. Since a description of the pathology in mild forms of PH is largely unavailable, it is difficult to discern whether the pathological features we observe in ‘controls’ are similar but still less severe than in patients with ‘mild PH. Perhaps a better definition of severity would also incorporate the extent of the reduction in cross-sectional area of the pulmonary vascular bed. In this summary, we confine our discussion of severity to the extent of the pulmonary vascular remodeling process, though in clinical practice, the assessment of severity will also consider the function of the right ventricle. Below, we expand on how genetic factors influence with cellular and molecular pathogenetic processes to possibly account for the severity of pulmonary vascular disease (Figure 1).
Mutations in BMPR2) or ALK-1 receptor are emerging as determinants of severity of PAH. Mutations in BMPR2 (18) have been reported in more than 70% of subjects with one or more affected relatives (heritable PAH) and 11-40% of idiopathic PAH (19, 20). Mutations in several other genes have been found including mutations in the ALK1 gene (21), the endoglin gene (22), the SMAD9 gene (23), the Caveolin-1 gene (24), and recently, the KCNK3 gene (25). Patients with BMPR2 or ALK-1 mutations present with higher pulmonary vascular resistance (26). There is also evidence that these patients present and die at a younger age with more severe disease compared PAH patients without mutations, and are less likely to respond acutely to vasodilators (27). Recent evidence suggests that the degree of pulmonary vascular remodeling is greater in patients with BMPR2 mutations compared with non-BMPR2 related disease at the time of transplantation (4). In addition to these causal rare sequence variants, variable expressivity of PAH might be explained by genetic modifiers. For example, single nucleotide polymorphisms (SNPs) in several genes, such as the angiotensin-converting enzyme gene, the KCNA5 gene, the serotonin transporter gene, the serotonin 5-HT2B receptor gene have been reported to influence disease penetrance or progression. In addition, microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in idiopathic PAH (28) as well as somatic chromosome abnormalities (29) have been described. Most cases of PH do not have known genetic triggers; moreover vascular cells isolated from experimental PH maintain their in vivo characteristics when placed in culture, suggesting epigenetic abnormalities. An example of such a contributing factor is the silencing of mitochondrial superoxide dismutase 2 (SOD2), which alters redox signaling and therefore activates hypoxia inducible factor (HIF-1) α, leading to the aerobic glycolysis seen in PH (30, 31). However, further studies, particularly focused on epigenetic control of additional key genes, are required to verify their contribution to the pathogenesis of PH.
Apart from major effects of rare sequence variants in heritable forms of PAH, inter individual differences in response to the same stimulus are well documented in subjects exposed to environmental hypoxia at high altitude, or in the context of high pulmonary blood flow or pulmonary venous hypertension. This variable expressivity most certainly involves the impact of unknown genetic influences regulating the pulmonary vascular response; this variation in response to hypoxia is also seen in different strains of rat (32) and bovines exposed to high altitude (33). Elegant studies demonstrated that this response is inherited (34), though the identification of the genetic basis is the subject of ongoing research (35).
The variable expressivity of PH might also be due to exposure to different environmental influences or comorbidities, including: inflammation, auto-immunity, viral infection, and hormonal mediators. The specific topic of inflammation is addressed below. Furthermore, accumulating evidence indicates that metabolic dysfunction may also contribute to variability in susceptibility and expressivity of PH (Figure 2).
Although in general, animal models of PH do not recapitulate the severe pathology of human disease, these experimental models support the idea that PH can be triggered by different stimuli and that the structural changes observed in the pulmonary vasculature vary depending on the nature of the injurious stimulus (36). Several studies have underlined the importance of different signaling pathways in PH, including elastase inhibitors (37), antagonists of EGF (4), or dietary copper restriction (38). The lack of animal model fidelity to the human pathology needs to be critically analyzed. The obliterative changes in the most distal precapillary arteries in the SU5416/hypoxia rat model (36, 39), which evolves over time (39), do not fully recapitulate the large plexogenic lesions seen in patients – its potential reversibility may also indicate the model may not be fully reflective of the largely irreversible nature of the human disease. Indeed, recent studies indicate that the long telomeres present in mice may limit the extent to which animal model can phenocopy human disease.
In summary, several genetic, molecular, biochemical and environmental factors may explain the variable expressivity of PH and may contribute to its aggravation or acceleration. Severe PAH seems to represent the far end of a spectrum in which there is an augmented burden of vascular injury and dysregulated repair (Figure 1). Further studies, which likely require a “systems biology-like” approach, are needed to establish which key signaling pathways in pulmonary vascular cells which lead to severe disease. Factors that contribute to this accelerated pathology include genetic abnormalities that may affect: (1) The exuberance of the cellular response; (2) The chronicity of an inflammatory stimulus; (3) The severity of the altered metabolic state and cumulative DNA damage.
What are differences and similarities between cell proliferation in PAH when compared with traditional neoplastic disease?
The concept of a neoplastic-like pathobiology of PH (in particular in PAH) (40) has its origins in the finding that endothelial cell in IPAH plexiform lesions are clonal when compared with similar lesions in lungs of patients with congenital heart disease (41), coupled with somatic instability in PAH lesions (28) and cultured lung endothelial cells (29). Similar to the concept of an uncontrolled cell growth in PAH, early data emphasized the upregulation of HIF-1α in plexiform lesions and pulmonary arteries in PAH (42) and survivin in IPAH (43). These findings, largely centered on the angle of disorganized cell growth, favored the emergence of an apoptosis-resistant phenotype, a selection process that characterizes neoplastic processes (44). In the past 8 years, further experimental evidence emerged of metabolic plasticity and altered cell energetics in the pathogenesis of PAH (45, 46). Metabolic reprogramming (Figure 2) constitutes, with genomic instability and mutations, immune escape, and cell-growth promoting inflammation, the so called “emerging hallmarks” pertinent to the pathogenesis of cancer (47). Recent studies have delineated that cancer cells use these hallmarks as driving forces leading to cell growth and co-option of the surrounding stroma for their aggressiveness (48). Supporting documentation of metabolic adaption of pulmonary vascular cells derived from studies in rat smooth muscle cells from the fawn-hooded model of PH (45) and cultured human IPAH cells (30). In both experimental settings, the hypertensive vascular cells would preferentially use aerobic glycolysis (instead of mitochondrial metabolism). The preferential use of aerobic glycolysis by proliferating cells (including cancer cells) would endow a selective growth advantage to pulmonary vascular cells: glucose and glycolytic intermediates provide key substrates for the pentose phosphate shunt to generate reducing equivalents in the form of reduced nicotinamide adenine dinucleotide phosphate (NADPH) and nucleotides, essential for cell growth (49).
The preferential utilization of aerobic glycolysis appears to be related to upregulation of several glycolytic genes and increased expression of the subunit 4.2 of cytochrome oxidase in the mitochondrial respiratory chain, all events driven by expression of HIF-1α (50). In human PAH lungs, HIF-1α appears to be expressed by endothelial cells (42) and smooth muscle cells (45). The source of stabilization of HIF-1α remains unclear, but it appears to be related to altered cellular oxidant/antioxidant balance (45, 51) present in hypertensive cells. Moreover, HIF-1α could also account for the increased mobilization of hematopoietic precursors in PAH (52-54), which might promote further injury to the pulmonary endothelium (13) and possibly to contribute to the remodeled cell population (Figure 2).
In addition to playing roles in the metabolic adaption and mobilization of bone marrow precursors, HIF-1α also participates in the regulation of mitochondria dynamics. HIF-1α accounted for decreased numbers of mitochondria and decreased nitric oxide availability in IPAH cells when compared with control cells (42); of interest, these alterations also correlate with increased IPAH endothelial cell proliferation when compared with normal endothelial cells in culture (55). HIF-1α-dependent mitochondria plasticity also pertains to PH smooth muscle cells. Activation of HIF-1α activity with cobalt or desferrioxamine, leads to mitochondria fission (mediated by activation of dynamin related protein 1) in human PAH and rodent PH smooth muscle cells, driven by activation of dynamin related-protein (DRP)-1 by active cyclin B1 (56). Finally, consistent with the role of mitochondria fission in PH smooth muscle cell growth, inhibition of DRP-1 with the peptide Mdiv-1 blocks cell proliferation in cultured cells as well as PH caused by in vivo administration of cobalt and chronic hypoxia (56). Conversely, overexpression of mitofusin-1, which leads to mitochondria fusion, counteracts the mitochondria phenotype in pulmonary hypertensive smooth muscle cells (57).
The paradigm of metabolic plasticity and adaption of PH cells has wider implications, interfacing with controls of cellular stresses and protein misfolding, and protein sorting in cytoplasmic compartments (58). In line with these paradigms, there is evidence of an altered endoplasmic reticulum (ER) stress response in PH (59), which may ultimately contribute to both adverse remodeling and elevation of pulmonary artery pressures (60). Moreover, ER stress can lead to activation of the stress-related kinase activating transcription factor (ATF)-6, which increases the expression of Nogo-B. This contributes to an abnormal distancing and signaling coupling (via calcium fluxes) between the ER and the mitochondria (61). These events would lead to an increase in glycolysis, favoring vascular cell proliferation and ultimately PH.
The inroads made by the metabolic related studies in PH are starting to bear translational fruits. There is increased metabolic labeling of glucose uptake with 18-Fluorodeoxyglucose of IPAH lungs and right ventricle when compared with controls (30, 62), which have been also observed in animal models of PH (63, 64). Pharmacologic targeting of the preferential use of glycolysis by PH cells is being tested in humans with the small molecule dichloroacetate (clinicaltrials.gov NCT01083524), which improves PH in animals by blocking pyruvate dehydrogenase kinase, a negative regulator of pyruvate import into the mitochondria (65).
We are at the beginning of deciphering the role of cellular metabolism in the pathogenesis of PH and how this fundamental process relates to inflammation, remodeling, therapeutic response, etc. Genetic modifications that favor glycolysis in multiplying cells would certainly contribute to part of the process of remodeling. However, many of the vascular cells in established human PH are quiescent, long-lived, and probably resistant to apoptosis. These quiescent cells would have substantially different metabolic requirements from those of proliferating cells. Clarifying this metabolic heterogeneity and how it contributes to remodeling and vasoconstriction will be critical moving forward. The interface of cell metabolism with the genetic basis of PAH and inflammation will certainly constitute key areas of future investigation.
What is the role of inflammation in the initiation and progression of different PAH types?
Inflammation has been long recognized as an important pathogenetic element in PH (66). Expanding on prior observations of accumulation of inflammatory cells in PAH, it was documented for the first time that the amount of perivascular inflammatory infiltrate, largely of lymphocytes, correlated with parameters of pulmonary vascular remodeling and hemodynamics in PAH (15). Inflammation consists of a complex series of interactions among soluble factors and immunologically specialized cells triggered in response to traumatic, infectious, post-ischemic, toxic, or autoimmune injury (67). It is increasingly clear that early and persistent inflammation is present and contributes to pulmonary vascular disease, as documented in tissue-based studies and studies of circulating inflammatory cells and chemical mediators.
The presence of elevated levels of circulating inflammatory cytokines, such as interleukin-1α in PAH is well recognized (68). More recently, a wide range of cytokines has been shown to be elevated and to correlate with survival in PAH (69). At the tissue level, traditional cellular components of inflammation are described in the hypertensive pulmonary circulation (70, 71). More specifically, cellular inflammation involves increases in the number of perivascular macrophages (CD68+), macrophages/monocytes (CD14+), mast cells, dendritic cells (CD209+), T cells (CD3+), cytotoxic T cells (CD8+), and helper T cells (CD4+) in the walls of PAH vessels compared with control subjects (72). FoxP3(+) cells have been shown to be significantly decreased, suggesting a reduction in the local number of T regulatory cells (72). Although increased numbers of circulating Treg cells has been reported in patients with idiopathic PAH (73, 74), the reduced numbers of Treg in lung tissue may reflect decreased tissue recruitment of these cells. Given their immune soothing role, a diminished negative regulation on other active immune / inflammatory cells may trigger or amplify pulmonary vascular remodeling and PH. The innate inflammatory system appears to also participate in PAH. Natural killer (NK) cells, which target stressed, virally infected, or oncogenically transformed cells independent of antigen recognition, are dysfunctional, with reduced number and cytolytic capacity, in patients with idiopathic PAH and in mouse and rat models of PH (75). Neutrophils may have potential unrecognized roles as sources of powerful proteases, including elastases, which have recognised roles in pulmonary vascular remodelling (37, 76). Since hypoxia exerts profound effects on neutrophil function (e.g. resistance to apoptosis (77)) further evaluation of the role of the neutrophil in PAH is overdue. The complement system, which bridges innate and adaptive immunity, is also activated in PAH and deficiency of complement C3 protects mice from hypoxia induced PH (78). Mechanistic studies focusing on the innate immune system are needed for a better appreciation of its role in PH.
However, recent insights highlighted a potential role for acquired immunity, suggesting that the pathogenesis of PH may involve specific and targeted immune cellular responses. A recent study identified a large number of tertiary lymphoid follicles in lungs from patients with idiopathic PAH when compared with control lungs (79). These lymphoid follicles in idiopathic PAH patients displayed the canonical cellularity and structure of bona fide tertiary follicles, while lymphoid organogenic chemokines such as CXCL13 and CCL19/CCL21 were overexpressed. These data provide a structural basis for a local autoimmune response in this disease (80-82). Finally, inflammation is closely associated with pulmonary vascular disease in the setting of auto-immune diseases, such as scleroderma, and paradigmatic of the interplay of inflammation and PH, is its link with the infection with the parasite Schistosoma mansoni (the most frequent cause of PAH worldwide (83)) and HIV infection (84).
Vascular inflammation in general and PH in particular, has traditionally been considered an “inside-out” response centered on leukocyte/monocyte recruitment to the intima of blood vessels driven by the endothelium-expressed adhesion molecules. Growing experimental evidence, however, supports an alternative paradigm of an “outside-in” hypothesis, in which vascular inflammation is initiated in the adventitia and progresses inward toward the media and intima, coupled with the activation of the remodeling process. In support of the “outside-in” hypothesis of adventitial regulation of inflammation are observations that, in a wide variety of vascular injuries, including PH, there is a rapid influx of leukocytes into the adventitial compartment (85). The immediate perivascular environment in PH becomes populated by inflammatory/progenitor cells, in a niche dominated by adventitia fibroblasts (36, 70, 85-89). These adventitial fibroblasts and recruited monocytes, particularly under hypoxia, express in a time-dependent and pulmonary artery-specific manner several cytokines/chemokines, their receptors, and adhesion molecules; these cells therefore appear to initiate and perpetuate the inflammatory response in an “outside-in” fashion, possibly mediated by NF-kB signaling (90, 91). Therefore, temporal-spatial dysregulation and/or failure in the normal “switch-off signal” in fibroblasts and/or macrophages/DCs may directly contribute to the persistence of a chronic inflammatory immune response. These adventitia processes are more evident in non-mouse models and in lung of patients with PH possibly due to the presence of bronchial circulation.
The action of secreted cytokines, including TGF-β, in the adventitia mediates specific homing for leukocytes in this vascular compartment, leading to their inappropriate/pathologic retention and survival (91). These homing/recruitment cytokines include upregulated adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), and stromal derived factor (SDF)-1/CXCR4, which promotes adhesion of leukocytes and bone marrow precursor cells, respectively. The adventitia is therefore suited to harbor canonical innate immune cells, specifically macrophages and dendritic cells (DCs), which with adventitial fibroblasts, are all equipped with the necessary machinery, (e.g. toll-like receptors (TLRs), inflammasome components (like the nod-like receptors (NLRs)), to potently respond to a variety of exogenous and endogenous danger signals.
Importantly, there is growing evidence that epigenetic marks may “lock” innate immune cells into a distinct functional phenotype, with loss of functional plasticity and failure to respond to regulatory signals. The inflammatory microenvironment in the pulmonary circulation promotes epigenetic marks in macrophages, locking their functional phenotype into a pro-fibrogenic and pro-remodeling macrophage (92-96). Different signaling processes, mediated largely by signal transducers and activators of transcription (STAT)1, 3, and 6 may drive macrophage-promoted remodeling (97); it is likely that all these macrophages are present in PH and play complex and time dependent, sometimes overlapping or dedicated roles in the disease. Moreover, it is also likely that individual macrophages are plastic, changing their function and signaling according to the disease stage and specific molecular triggers.
Finally, emerging concepts involve the potential of organ-specific microbiota and their metabolic products to engage the inflammasome pathways and to influence pulmonary vascular responses, as shown in the liver (98). A further pathway by which remote signalling might target the pulmonary vasculature is by the release of cell-derived exosomes from distant sources, exerting anti- or pro-inflammatory depending on their cargo. Exosomes derived from mesenchymal stem cells were found to inhibit the hypoxic activation of the STAT3 pathway in hypoxic mice and prevent the development of PH (99).
In conclusion cytokines and immune cells appear to play significant but complex roles in the initiation and progression of PH. Major unanswered questions include: (1) What determines the abnormal host response to inflammation that leads to the initiation and progression of PH? (2) Is the inflammatory response in PAH caused by autoimmunity or infection? (3). Which specific features of the inflammatory response can be enhanced (if protective) or blocked (if detrimental) for therapeutic intervention?
Acknowledgments
CMREF and RC1 HL 10084 to RMT; NIH-RO1-HL071115, 1RC1HL099462SLAHL115008 and HL60917 to SCE; French National Agency for Research ANR_12_JSV1_0004_01 to CG; UGMLC-LOEWE; ECCPS; DZL to RS; NIH/NHLBI Axis Grant, 1R01HL114887, NIH PPG 5P01HL014985, and NIH/NHLBI SCCOR 5P50HL084923 to KRS; NIHR Cambridge Biomedical Research Centre and BHF to NWM
Abbreviations
- ALK-1
activin receptor-like kinase-1
- ATF-6
activating transcription factor 6
- BMPR2
bone morphogenetic protein type II receptor
- DCs
dendritic cells
- DRP-1
dynamin related protein-1
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- HIF-1
hypoxia inducible factor-1
- HIV
human immunodeficiency virus
- ICAM1
Intercellular adhesion molecule 1
- IPAH
idiopathic pulmonary arterial hypertension
- LHF
left heart failure
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NF-κB
nuclear factor kappa B
- NK
natural killer cells
- NLRs
Nod-like receptors
- PAH
pulmonary arterial hypertension
- PH
pulmonary hypertension
- SDF-1
Stromal derived factor 1
- SNPs
single nucleotide polymorphisms
- SOD2
superoxide dismutase 2
- STAT
signal transducers and activators of transcription
- TGF-β
transforming growth factor-β
- TLRs
toll-like receptors
- VCAM-1
Vascular cell adhesion molecule-1
Footnotes
Disclosures: RMT has no conflict of interest to declare
SLA has no conflict of interest to declare
PD declares what follows: Speaker fees for meetings supported by Actelion Pharmaceuticals, France.
SCE has no conflict of interest to declare
CG has no conflict of interest to declare
EM declares what follows: Medtelligence Steering Committee (young invesitgators selection committee); Bayer (clinical trial Steering Committee); Both < USD 10.000,00 total in the past 2 years
MR has no conflict of interest to declare
RTS declares what follows: Unrestricred research grants from Actelion, Bayer-Healthcare, Novartis, Noxxon, Pfizer
KRS has no conflict of interest to declare
NM has no conflict of interest to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Tuder RM, Abman SH, Braun T, et al. Development and pathology of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S3–S9. doi: 10.1016/j.jacc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 2.Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol. 2005;33:425–31. doi: 10.1165/rcmb.F307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–31. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 5.Izikki M, Guignabert C, Fadel E, et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–23. doi: 10.1172/JCI35070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perros F, Montani D, Dorfmuller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–8. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 7.Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115:2811–21. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long L, Crosby A, Yang X, et al. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119:566–76. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- 9.Zaiman AL, Podowski M, Medicherla S, et al. Role of TGF-b/ALK5 kinase in Monocrotaline-Induced Pulmonary Hypertension. Am J Respir Crit Care Med. 2008;177:896–905. doi: 10.1164/rccm.200707-1083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Kang Y, Kojima Y, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. 2013;19:74–82. doi: 10.1038/nm.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Courboulin A, Paulin R, Gigu+¿re NJ, et al. Role for miR-204 in human pulmonary arterial hypertension. Jornal of Experimental Medicine. 2011;208:535–48. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavoie JR, Stewart DJ. Genetically modified endothelial progenitor cells in the therapy of cardiovascular disease and pulmonary hypertension. Curr Vasc Pharmacol. 2012;10:289–99. doi: 10.2174/157016112799959413. [DOI] [PubMed] [Google Scholar]
- 13.Asosingh K, Farha S, Lichtin A, et al. Pulmonary vascular disease in mice xenografted with human BM progenitors from patients with pulmonary arterial hypertension. Blood. 2012;120:1218–27. doi: 10.1182/blood-2012-03-419275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunther S, Jais X, Maitre S, et al. Computed tomography findings of pulmonary venoocclusive disease in scleroderma patients presenting with precapillary pulmonary hypertension. Arthritis Rheum. 2012;64:2995–3005. doi: 10.1002/art.34501. [DOI] [PubMed] [Google Scholar]
- 15.Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:261–72. doi: 10.1164/rccm.201201-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunt JM, Bethea B, Liu X, et al. Pulmonary veins in the normal lung and pulmonary hypertension due to left heart disease. American journal of physiology Lung cellular and molecular physiology. 2013 doi: 10.1152/ajplung.00186.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oka M, Homma N, Taraseviciene-Stewart L, et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res. 2007;100:923–9. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 18.Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) Is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nature genetics. 2000;26 doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 20.Machado RD, Aldred MA, James V, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Human mutation. 2006;27 doi: 10.1002/humu.20285. [DOI] [PubMed] [Google Scholar]
- 21.Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–34. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 22.Chaouat A, Coulet F, Favre C, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59:446–8. doi: 10.1136/thx.2003.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. Journal of Medical Genetics. 2009 doi: 10.1136/jmg.2008.062703. jmg. [DOI] [PubMed] [Google Scholar]
- 24.Austin ED, Ma L, Leduc C, et al. Whole Exome Sequencing to Identify a Novel Gene (Caveolin-1) Associated with Human Pulmonary Arterial Hypertension. Circulation Cardiovascular genetics. 2012:336–43. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–61. doi: 10.1056/NEJMoa1211097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181:851–61. doi: 10.1164/rccm.200908-1284OC. [DOI] [PubMed] [Google Scholar]
- 27.Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation. 2006;113:2509–15. doi: 10.1161/CIRCULATIONAHA.105.601930. [DOI] [PubMed] [Google Scholar]
- 28.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88:e8–e11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 29.Aldred MA, Comhair SA, Varella-Garcia M, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:1153–60. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu W, Koeck T, Lara AR, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Archer SL, Marsboom G, Kim GH, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–71. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguirre JI, Morrell NW, Long L, et al. Vascular remodeling and ET-1 expression in rat strains with different responses to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2000;278:L981–L987. doi: 10.1152/ajplung.2000.278.5.L981. [DOI] [PubMed] [Google Scholar]
- 33.Weir EK, Tucker A, Reeves JT, Will DH, Grover RF. The genetic factor influencing pulmonary hypertension in cattle at high altitude. Cardiovasc Res. 1974;8:745–9. doi: 10.1093/cvr/8.6.745. [DOI] [PubMed] [Google Scholar]
- 34.Cruz JC, Reeves JT, Russell BE, Alexander AF, Will DH. Embryo transplanted calves: the pulmonary hypertensive trait is genetically transmitted. Proc Soc Exp Biol Med. 1980;164 doi: 10.3181/00379727-164-40837. [DOI] [PubMed] [Google Scholar]
- 35.Newman JH, Holt TN, Hedges LK, et al. High-altitude pulmonary hypertension in cattle (brisket disease): Candidate genes and gene expression profiling of peripheral blood mononuclear cells. Pulm Circ. 2011;1:462–9. doi: 10.4103/2045-8932.93545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. American journal of physiology Lung cellular and molecular physiology. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 37.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6:698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 38.Bogaard HJ, Mizuno S, Guignabert C, et al. Copper-dependence of angioproliferation in pulmonary arterial hypertension in rats and humans. Am J Respir Cell Mol Biol. 2011:582–91. doi: 10.1165/rcmb.2011-0296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abe K, Toba M, Alzoubi A, et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–54. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 40.Voelkel NF, Cool CD, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1999;114:225S–30S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 41.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101:927–34. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fijalkowska I, Xu W, Comhair SA, et al. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176:1130–8. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMurtry MS, Archer SL, Altieri DC, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115:1479–91. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 45.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1a}-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 46.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med. 2012;185:260–6. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanahan D, Weinberg RA. Hallmarks of Cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 50.Shimoda LA, Semenza GL. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011;183:152–6. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–8. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 52.Asosingh K, Aldred MA, Vasanji A, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172:615–27. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toshner M, Voswinckel R, Southwood M, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–7. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farha S, Asosingh K, Xu W, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–93. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masri FA, Xu W, Comhair SA, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 56.Marsboom G, Toth PT, Ryan JJ, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–97. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryan JJ, Marsboom G, Fang YH, et al. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:865–78. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sehgal PB, Mukhopadhyay S. Pulmonary arterial hypertension: a disease of tethers, SNAREs and SNAPs? Am J Physiol Heart Circ Physiol. 2007;293:H77–H85. doi: 10.1152/ajpheart.01386.2006. [DOI] [PubMed] [Google Scholar]
- 59.Yeager ME, Reddy MB, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Activation of the unfolded protein response is associated with pulmonary hypertension. Pulm Circ. 2012;2:229–40. doi: 10.4103/2045-8932.97613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.112.133413. In Press. [DOI] [PubMed] [Google Scholar]
- 61.Sutendra G, Dromparis P, Wright P, et al. The role of nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3:88ra55. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lundgrin EL, Park MM, Sharp J, et al. Fasting 2-Deoxy-2-[18F]fluoro-D-glucose Positron Emission Tomography to Detect Metabolic Changes in Pulmonary Arterial Hypertension Hearts over 1 Year. Ann Am Thorac Soc. 2013;10:1–9. doi: 10.1513/AnnalsATS.201206-029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsboom G, Wietholt C, Haney CR, et al. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–9. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sutendra G, Dromparis P, Paulin R, et al. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl) 2013 doi: 10.1007/s00109-013-1059-4. In press. [DOI] [PubMed] [Google Scholar]
- 65.McMurtry MS, Bonnet S, Wu X, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–40. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 66.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. Journal of Laboratory & Clinical Medicine. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 67.Nathan C. Points of control in inflammation. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 68.Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 69.Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 70.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 71.Tuder RM, Groves BM, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144:275–85. [PMC free article] [PubMed] [Google Scholar]
- 72.Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. doi: 10.1164/rccm.201202-0335OC. [DOI] [PubMed] [Google Scholar]
- 73.Huertas A, Tu L, Gambaryan N, et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;40:895–904. doi: 10.1183/09031936.00159911. [DOI] [PubMed] [Google Scholar]
- 74.Ulrich S, Nicolls MR, Taraseviciene L, Speich R, Voelkel N. Increased regulatory and decreased CD8+ cytotoxic T cells in the blood of patients with idiopathic pulmonary arterial hypertension. Respiration. 2008;75:272–80. doi: 10.1159/000111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ormiston ML, Chang C, Long LL, et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation. 2012;126:1099–109. doi: 10.1161/CIRCULATIONAHA.112.110619. [DOI] [PubMed] [Google Scholar]
- 76.Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105:21–34. doi: 10.1172/JCI6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walmsley SR, Cowburn AS, Clatworthy MR, et al. Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood. 2006;108:3176–8. doi: 10.1182/blood-2006-04-018796. [DOI] [PubMed] [Google Scholar]
- 78.Bauer EM, Zheng H, Comhair S, Erzurum S, Billiar TR, Bauer PM. Complement C3 deficiency attenuates chronic hypoxia-induced pulmonary hypertension in mice. PLoS ONE. 2011;6:e28578. doi: 10.1371/journal.pone.0028578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perros F, Dorfmuller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–21. doi: 10.1164/rccm.201105-0927OC. [DOI] [PubMed] [Google Scholar]
- 80.Dib H, Tamby MC, Bussone G, et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Resp J. 2011 doi: 10.1183/09031936.00181410. [DOI] [PubMed] [Google Scholar]
- 81.Tamby MC, Chanseaud Y, Humbert M, et al. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60:765–72. doi: 10.1136/thx.2004.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Terrier B, Tamby MC, Camoin L, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:1128–34. doi: 10.1164/rccm.200707-1015OC. [DOI] [PubMed] [Google Scholar]
- 83.Graham BB, Bandeira AP, Morrell NW, Butrous G, Tuder RM. Schistosomiasis-associated pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest. 2010;137:20S–9S. doi: 10.1378/chest.10-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Almodovar S, Knight R, Allshouse AA, et al. Human Immunodeficiency Virus nef signature sequences are associated with pulmonary hypertension. AIDS Res Hum Retroviruses. 2012;28:607–18. doi: 10.1089/aid.2011.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li M, Riddle SR, Frid MG, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–22. doi: 10.4049/jimmunol.1100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. vii. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006;21:134–45. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 89.Sahara M, Sata M, Morita T, Nakamura K, Hirata Y, Nagai R. Diverse contribution of bone marrow-derived cells to vascular remodeling associated with pulmonary arterial hypertension and arterial neointimal formation. Circulation. 2007;115:509–17. doi: 10.1161/CIRCULATIONAHA.106.655837. [DOI] [PubMed] [Google Scholar]
- 90.Lo D, Feng L, Li L, et al. Integrating innate and adaptive immunity in the whole animal. Immunol Rev. 1999;169:225–39. doi: 10.1111/j.1600-065x.1999.tb01318.x. [DOI] [PubMed] [Google Scholar]
- 91.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends in immunology. 2001;22 doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 92.Cavasin MA, Demos-Davies K, Horn TR, et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110:739–49. doi: 10.1161/CIRCRESAHA.111.258426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukoc Biol. 2010;87:59–67. doi: 10.1189/jlb.0409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ishii M, Wen H, Corsa CA, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–54. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liao X, Sharma N, Kapadia F, et al. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–49. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Satoh T, Takeuchi O, Vandenbon A, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–44. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 97.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–55. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–85. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee C, Mitsialis SA, Aslam M, et al. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. 2012;126:2601–11. doi: 10.1161/CIRCULATIONAHA.112.114173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Archer SL, Weir EK, Wilkins MR. Basic Science of Pulmonary Arterial Hypertension for Clinicians: New Concepts and Experimental Therapies. Circulation. 2010;121:2045–66. doi: 10.1161/CIRCULATIONAHA.108.847707. [DOI] [PMC free article] [PubMed] [Google Scholar]