

Abstract
With the recent clinical success of bispecific antibodies, a strategy to rapidly synthesize and evaluate bispecific or higher order multispecific molecules could facilitate the discovery of new therapeutic agents. Here we show that unnatural amino acids (UAAs) with orthogonal chemical reactivity can be used to generate site-specific antibody-oligonucleotide conjugates. These constructs can then be self-assembled into multimeric complexes with defined composition, valency and geometry. Using this approach, we generated potent bispecific antibodies that recruit cytotoxic T lymphocytes to Her2 and CD20 positive cancer cells, as well as multimeric antibody fragments with enhanced activity. This strategy should accelerate the synthesis and in vitro characterization of antibody constructs with unique specificities and molecular architectures.
INTRODUCTION
Currently there is considerable effort focused on the development of bispecific antibodies with two or more distinct specificities in a single therapeutic molecule1-3. A number of such antibodies have been engineered to simultaneously bind two differentially expressed antigens, rather than one, on a given target cell surface to achieve improved cellular selectivity. For example, a bispecific antibody that binds the epidermal growth factor receptors ErbB2 and ErbB3 is currently in clinical trials for breast cancer4. Bispecific antibodies can also be engineered to simultaneously bind a surface antigen on one cell type and a second antigen on a distinct effector cell. Indeed, a bispecific antibody that binds the B-cell antigen CD19 and the cytotoxic T-lymphocyte antigen CD3 has shown impressive activity in clinical trials for acute lymphoblastic leukemia (ALL)5. Several methods have been developed to generate bispecific antibodies, including single chain variable fragment (scFv) constructs such as BiTEs (bispecific T-cell engager)6, DARTs (dual affinity retargeting)7 and diabodies8,9, and full-length immunoglobulin G (IgG) constructs such as Triomab10, DVD-Ig (dual variable domain antibodies)11, “knobs into holes”12, and two-in-one antibodies13. In addition, chemical crosslinking methods have been developed based on relatively nonspecific electrophilic modification of lysine or cysteine residues14. However, these approaches generally require considerable engineering to optimize the biological and biophysical properties of the resulting molecules. For example, bispecific molecules synthesized as genetic fusions require flexible linkers that can lead to instability and aggregate formation, or immunogenicity. Additionally, fusion proteins can only be made in N to C or C to N terminal orientations, severely limiting the relative geometries of the individual antibody fragment subunits; such fusions are also not easily adapted to higher order structures involving three or more antibody combining sites. Chemical methods generally create heterogeneous mixtures with a range of stoichiometries and geometries that are not easily optimized. A synthetic strategy to rapidly self-assemble antibody fragments into multimers with control over composition, valency and geometry would provide a useful drug discovery tool to test a large number of combinations of antibodies for optimal activity in a given cellular system.
The selective Watson-Crick base pairing properties of oligonucleotides has been used to assemble a large number of diverse chemical structures from nanoparticle arrays and protein nanostructures to DNA knots15,16. By site-specifically coupling either oligonucleotides or peptide nucleic acids (PNAs) of defined sequences to scFv or Fab fragments of antibodies, it should be possible to similarly assemble multispecific antibody-like molecules with novel biological activities. Such a system would allow combinations of antibody fragments with distinct specificities to be rapidly generated and screened in cellular assays for a desired activity. However, assembly of antibody fragments into defined structures requires the ability to synthesize protein-oligonucleotide (or PNA) substrates with precise control of the conjugation site and stoichiometry, which is difficult to achieve using standard conjugation procedures that produce heterogeneous mixtures. Recently, we showed that one can site-specifically modify antibodies in high yields using genetically encoded unnatural amino acids with orthogonal chemical reactivity relative to the canonical 20 amino acids17. Here we apply this methodology to the generation of homodimeric, heterodimeric and multimeric antibody fragments of defined structure based on the self assembly of Fab-nucleic acid conjugates. Moreover, we show that these assemblies can efficiently modulate cell-signaling, kill cancer cells by the recruitment of cytotoxic effector cells, or directly induce cancer cell death.
EXPERIMENTAL SECTION
PNA synthesis
NovaPEG Rink Amide resin, Boc-protected aminooxy acetic acid and all peptide related reagents were purchased from Novabiochem®, and were swollen in CH2Cl2 before each reaction. PEG spacer (Fmoc-8-amino-3,6-dioxaoctanoic acid) was purchased from ASM (http://www.asm-research-chemicals.com/). PNA monomers were prepared as previously reported (Mtt-protected monomers18; Fmoc protected monomers19). Arginine-modified bases (indicated in bold) were added to increase solubility and reduce aggregation of the PNAs. Automated solid phase synthesis was carried out on an Intavis Multipep RS instrument. LC-MS analyses were carried out using an HP 1100 series or Thermo Electron Corporation HPLC with a Thermo Finnigan Surveyor MSQ Mass Spectrometer System. A Thermo Scientific column (50 × 2.1 mm) was used. MALDI spectra (2,5-dihydroxybenzoic acid matrix) were obtained using a Bruker Daltonics Autoflex II TOF/TOF spectrometer. Reverse phase chromatography was performed on a Biotage Isolera One Instrument using a H2O-MeCN (with 0.01% TFA) gradient from 100-0% to 0-100%. See Supplemental methods for detailed synthesis.
Synthesis of Fab Multimers
200 μM Fab and 100 mM methoxy aniline catalyst (final concentration) were added to a reaction containing 15-30x excess of aminooxy-modified PNA (N’-H2NO-(EG)n-ACGCAACGCGGC-C’), 20% DMSO (final concentration) and 100 mM acetate buffer (pH 4.5). After 48 hours at 37 °C, the excess PNA was removed by either Amicon filtration (30 kD MWCO) or size exclusion chromatography (GE Healthcare, Superdex 75). The reaction was buffer exchanged into PBS (pH 7.4) and analyzed by SDS-PAGE and mass spectrometry. Oligonucleotide conjugation to αHer2 Fab was carried out as previously described20. The complementary PNA strand (N’-H2NO-(EG)n-GCCGCGTTGCGT-C’) was coupled using the same procedure. The conjugates were hybridized 1:1 (~10 μM, PBS pH 7.5, 30 min at 37 °C) and the dimer was purified using size exclusion chromatography (GE Healthcare, Superdex 200). To create the Fab tetramer, four orthogonal PNA sequences were conjugated separately to either αHer2 S202 pAcF Fab or αCD20 S202 pAcF Fab. The PNA sequences are as follows: A) N’-H2NO-(EG)2-ATCCTGGAGC-(EG)-TAAGTCCGTA-C’, B) N’-H2NO-(EG)2-TACGGACTTA-(EG)-TCAATGAGGC-C’, C) N’-H2NO-(EG)2-GCCTCATTGA-(EG)-ATCATGCCTA-C’, and D) N’-H2NO-(EG)2-TAGGCATGAT-(EG)-GCTCCAGGAT-C’. After removal of excess PNA, the Fab-PNA conjugates were further purified on a hydrophobic interaction column (GE Healthcare, Buffer A: 50 mM phosphate, 10 mM (NH4)2SO4, pH 7.5, Buffer B: 50 mM phosphate, 750 mM (NH4)2SO4). The Fab-PNA conjugates were then hybridized 1:1:1:1 (~10 μM, PBS pH 7.5, 30 min at 37 °C) and the tetramer was purified using size exclusion chromatography (Superdex 200).
Her2 Phosphorylation Assay
SK-BR-3 cells (ATCC) were grown to 80% confluency in DMEM, 10% FBS and detached with 1 mL Trypsin-EDTA (Invitrogen). Cells were diluted 1:10 in media, and 100uL (~104 cells) were plated in white 96-well tissue culture plates (Corning). After overnight adherence, αHer2 Fab conjugates were added to the cells (2.5 μg/mL for DNA conjugates or 0.09-6 μg/mL for PNA conjugates) and incubated at 37 °C for 45 minutes. Media was aspirated and 100 μL lysis buffer was added to each well (IC Diluent#12, R&D Systems, 10 μg/mL Aprotinin, 10 μg/mL Leupeptin) and incubated at 4°C for 30 minutes. We prepared the ELISA plate (Nunc, maxisorp) by adsorbing 4 μg/mL phospho-ErbB2 capture antibody (DuoSet IC Human Phospho-Erbb2 ELISA, R&D Systems) in 100 μL per well overnight at room temperature. Wells were washed with PBST (PBS pH 7.4, 0.05% Tween 20) five times and blocked with 300 μL blocking buffer (1% BSA, 0.05% NaN3, PBS pH 7.4) for 1 hour. Wells were washed immediately prior to addition of the cell lysate, which was incubated at room temperature for 2 hours. The anti-phospho-tyrosine-HRP detection antibody was diluted to the working concentration specified on the vial in IC Diluent #14 (20mM Tris, 140mM NaCl, 0.05% Tween 20, 0.1% BSA, pH 7.4). The plate was washed five times and 100 uL of diluted detection antibody was added and incubated at room temperature for 2 hours. The plate was again washed five times with wash buffer prior to addition of the detection reagent, QuantaBlue (Pierce). After 10 minutes, the relative fluorescence was measured (ex. 325, em. 420) using a SpectraMax 250 plate reader (Molecular Devices Corp.). Each experimental test was performed in triplicate. Error bars represent the standard deviation.
Cytotoxicity Assays
Peripheral blood mononuclear cells (PBMCs) were purified from fresh healthy human donor blood by conventional Ficoll-Hypaque gradient centrifugation. Purified PBMCs were washed and incubated in flasks in RPMI media with 10 % FBS for 2 hours to remove adherent cells, and then transferred to αCD3 (eBioScience) and αCD28 (eBioScience) antibody coated ELISA plates at 37 °C. After 3 days, the activated PBMCs were washed once with media and transferred into a flask and incubated with 20 units/mL IL2 (R&D Systems) for T cell proliferation. HER2-transformed MDA-MB-435 or non-transformed MDA-MB-435 cells (target cells) were dissociated with 0.05 % tryspin/EDTA solution (HyClone) and washed with RPMI with 10 % FBS. 1×104 target cells were mixed with PBMCs at 1:10 ratio in 100 μL, and incubated with different concentrations (0.1 pM-10 nM) of αHER2-αCD3 PNA heterodimer or unconjugated Fabs (10 μL in media) for 16 hours at 37 °C. Cytotoxicity was measured based on LDH (lactate dehydrogenase) levels in supernatant using a Cytotox-96 non-radioactive cytotoxicity assay kit (Promega). The absorbance at 490 nm was recorded using SpectraMax 250 plate reader (Molecular Devices Corp.). Percent cytotoxicity was determined with maximum killing controls and the formula: % cytotoxicity = (Absorbanceexpt − Absorbancespontaneous average)/ (Absorbancemax − Absorbancespontaneous average). The same procedure was used for the αCD20-αCD3 PNA heterodimer cytotoxicity assay, except 5 × 104 target cells (Ramos) were mixed with PBMCs (1:10). The αCD20 tetramer assay used 1 × 105 Ramos cells per well (no PBMCs) with concentrations ranging from 0.6-150 nM. LDH levels in the supernatant (after 48 hours) were measured. Each experimental test was performed in duplicate or triplicate. Error bars represent the standard deviation.
RESULTS AND DISCUSSION
DNA-templated αHer2 Fab Dimers
As a model system, we used the Fab fragment of the monoclonal antibody trastuzumab (Herceptin; Genentech/Roche), which binds the antigen Her2 (ErbB2). Her2 is a receptor tyrosine kinase that is overexpressed in 25-30% of breast cancers and acts as a constitutively active dimerization partner with itself and other members of the ErbB family21. On the basis of the crystal structure of trastuzumab (Herceptin, PDB: 1N8Z), we separately mutated three positions (K169, S202 and S156, Figure 1A, left) on the surface-exposed constant region of the light chain (Cκ), distant from the antigen binding site, to the keto amino acid, p-acetylphenylalanine (pAcF) (Supplementary Figure 1)17. This amino acid can be efficiently and selectively modified with alkoxyamines to form stable oxime conjugates22. pAcF was site-specifically introduced into the αHer2 Fab using an orthogonal amber suppressor amino-acyl-tRNA synthetase/tRNA pair derived from M. jannaschii23. Mutants were either expressed in shake flasks (2 mg/L) or high density fermentation (>200 mg/L) in E. coli with similar yields to wild-type Fab, and purified by Protein G chromatography. We first used single-stranded DNA (ssDNA) as a template to self-assemble the αHer2 Fab to form a homodimer (Figure 1A, right). A 30 nucleotide (nt) ssDNA (TACGAGTTGAGACAGCTGATCCTGAATGCG) was designed that does not contain any significant secondary structure24, has a suitable Tm for dimerization (Tm=65 °C), and an engineered PvuII restriction site (underlined) to allow cleavage of the double stranded linker. An aminooxy functionality was coupled to a 5’-thiol ssDNA sequence using a C6 linker with a terminal maleimide group20. We then conjugated the ssDNA to αHer2 K169pAcF Fab (100 μM Fab, 3 mM ssDNA, 100 mM methoxy aniline, pH 4.5, 37 °C, 16 hours), and purified the resulting conjugate by anion exchange chromatography. The Fab-oligonucleotide conjugate migrated in SDS-PAGE at ~70 kDa, which is slightly larger than the mobility based only on molecular weight (Figure 1B, compare lanes 1 and 2). Over 90% of the αHer2 mutant Fab coupled to the aminooxy oligonucleotide as determined by gel densitometry (Supplementary Figure 1). To generate the αHer2 Fab homodimer, we also coupled the complementary oligonucleotide to the αHer2 K169pAcPhe Fab (10 μM Fab), and then exchanged the two Fab-oligonucleotide conjugates into annealing buffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 7.4). The two Fab-oligo molecules were mixed at a 1:1 molar ratio for 30 minutes at 37 °C. A single homodimer formed at the expected molecular weight (~120 kDa) in >95% yield (Figure 1B, lane 3). Upon treatment with the restriction enzyme PvuII, the homodimer was fully cleaved to generate one 60 kDa band (lane 4), confirming that the duplex DNA formed correctly.
FIGURE 1.
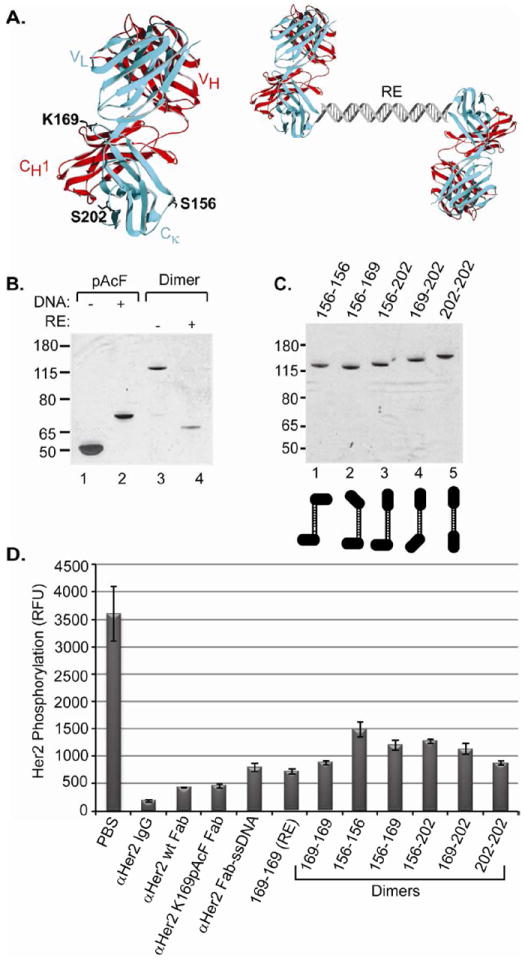
Construction of self-assembled αHer2 dimers. (A) Fab residues mutated to UAAs for site-specific conjugation (left). These mutations are in the constant region of the light chain (blue) which is paired through a disulfide bond with the heavy chain (red) of the αHer2 Fab (1N8Z). Depiction of oligonucleotide-templated αHer2 homodimer (right, RE=restriction endonuclease site). (B) Site-specific oligonucleotide conjugation and formation of the αHer2 DNA homodimer. An aminooxy-modified single-stranded DNA (ssDNA) was either omitted (lane 1) or coupled to αHer2 K169pAcF Fab (lane 2). The Fabs were analyzed by SDS-PAGE. The conjugate appeared as a lower mobility band (compare lanes 1 and 2) that migrated at a slightly higher molecular weight than the expected molecular weight of the Fab-DNA complex (~60 kDa). Monomeric Fab-ssDNA components with complementary strands were mixed (lane 3) to produce the homodimer which migrates as one band (lane 3) at the calculated molecular weight (~120kD). Treatment with PvuII results in a band with mobility similar to the monomer (lane 4). (C) The remaining five αHer2 heterodimers were created by using different UAA positions (S156, K169 and S202) in the αHer2 Fab. (D) All constructs were tested for phospho-Her2 inhibitory activity on Her2 overexpressing SK-BR-3 breast cancer cells. PBS served as the negative control. RFU indicates relative fluorescence units.
We recently showed that the site of conjugation of an oligonucleotide to an antibody can significantly affect antibody specificity17,20. The three residues mutated to pAcF (S156, K169, and S202, Figure 1A) in the aHer2 Fabs are located in different positions on the surface of the Fab constant region, and would be expected to form different orientations of the antigen binding site relative to the coupled linker. We constructed the remaining five different αHer2 dimers using the three UAA mutant positions in order to evaluate the effect of conjugation site on biological activity (Figure 1C and Supplementary Figure 2). Interestingly, these dimers migrate slightly differently on SDS-PAGE, perhaps due to differences in their tertiary structures. We only had to create three αHer2-ssDNA constructs to generate all possible combinations of αHer2 heterodimers; these dimers were constructed by simply mixing the relevant Fab-DNA monomers, then purifying by size exclusion chromatography.
To assess the ability of the αHer2 Fab homodimers to block Her2 signaling25, SK-BR-3 (Her2hi) cells were treated with each homodimer (2.5 μg/mL) and phospho-Her2 levels were determined (See Methods). Although αHer2 wild-type Fab and αHer2 K169pAcF Fab inhibited Her2 signaling in a cell-based phosphorylation assay, the DNA linked dimers were significantly less effective than the monomeric Fabs (Figure 1D). In fact, full-length trastuzumab (αHer2 IgG) had at least a 4-fold better activity than the DNA linked dimers. However, the alternate linkage positions did have significantly different activities in inhibiting phosphorylation. For example, the homodimer generated with mutant K169pAcF (169-169) was almost 2-fold better at inhibiting phosphorylation than the homodimer created with mutant S156pAcF (156-156). Of note, even the Fab linked to ssDNA (Figure 1D, αHer2 Fab-ssDNA) had lower activity than the uncoupled Fab (aHer2 K169pAcF Fab). Thus, although there were differential activities based on specific orientations of the subunits, there was also a general decrease in activity caused by conjugation of ssDNA to the Fabs, which may be due to interfering interactions of the sugar phosphate backbone and the cell membrane.
PNA-templated Self-assembly of Fab Fragments
Peptide nucleic acids (PNAs), like DNA, can form highly stable duplexes based on Watson-Crick base pairing and should also allow creation of bi- and multispecific constructs (Figure 2A and B). Unlike DNA, however, the peptide backbones of PNAs are uncharged (or positively charged) and are also resistant to serum nucleases and proteases26-28. Furthermore, PNA tagging has already been reported to combinatorially pair small molecules, glycans and peptides29-32. Because PNAs have a significantly higher Tm than their DNA counterpart, we designed a 12 base PNA sequence33 (N’-ACGCAACGCGGC-C’) with a predicted Tm exceeding 70 °C34 (Supplementary Figure 3 and Table S1). Arginine-modified bases (indicated in bold) were added to increase solubility and reduce aggregation of the PNAs35. The PNAs were synthesized through automated solid phase synthesis (See Methods and Supplementary Methods) and cleaved by 95% TFA (trifluoroacetic acid). The aminooxy functionality was introduced at the N-terminus by standard peptide coupling of Boc-protected aminooxy acetic acid, and one ethylene glycol (EG) spacer was also added for flexibility (Figure 2A). All PNAs were characterized by MALDI-TOF mass spectrometry (Supplementary Figure 3). The PNA was reacted with αHer2 S202pAcF Fab as described above, but with 20% DMSO as the solvent. After 48 hours, the reaction was analyzed by SDS-PAGE in the presence and absence of reducing agent β-mercaptoethanol (BME). A ~4 kD shift of the Fab band corresponds to the PNA conjugate in over 85% yield (Figure 2C, lanes 2 and 4). Formation of the αHer2 PNA conjugate was confirmed by electrospray ionization mass spectrometry (ESI-MS) (Expected MW: 51,649 Da; Observed MW: 51,649 Da; Supplementary Figure 4). To form a dimer we conjugated the complementary PNA to αHer2 S202pAcF Fab and hybridized the two conjugates (1:1 ratio, 10 μM) in PBS (30 min, 37 °C) to form the homodimer (Figure 2B, left). SDS-PAGE revealed a new lower mobility band that migrated at a slightly higher molecular weight than the expected molecular weight of the αHer2 PNA homodimer (~110 kDa) (Figure 2D, lane 3). Formation of the dimer was again confirmed by MALDI-TOF mass spectrometry (Expected MW: 103,298 Da; Observed MW: 103,218 Da; Supplementary Figure 5). We also asked whether we could further enhance the in vitro activity of the αHer2 Fab by creating a higher order multimer. Orthogonal PNAs36 20 bases in length with short ethylene glycol (EG) linkers were designed to form a unique cruciform structure, which when coupled to the αHer2 Fab form a Fab tetramer (Figure 2B, right and Supplementary Figure 6). Each PNA was conjugated to αHer2 Fab separately (as described above) and purified using size exclusion chromatography. In order to ensure tetramer formation, the conjugates were further purified by a hydrophobic interaction column (HIC, GE Healthcare) to remove any uncoupled Fab (Supplementary Figure 7). All 4 Fab-PNAs were simply mixed together (1:1:1:1 molar ratio; 10 μM) and incubated at 37 °C for 30 minutes. SDS-PAGE revealed the formation of the tetramer at >90% yield (Supplementary Figure 8). The tetramer was then purified by size exclusion chromatography (Supplementary Figure 8) and analyzed by SDS-PAGE (Figure 2D, lane 4)
FIGURE 2.
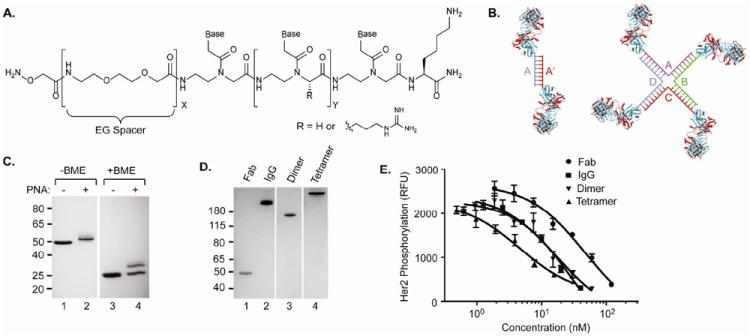
PNA mediated self-assembly and activity of anti-Her2 bispecific and multimeric antibodies. (A) Structure of the aminooxy-modified PNA linker, where x indicates number of ethylene glycol (EG) units and y indicates number of nucleobases. (B) Schematic depiction of αHer2 PNA dimer (left) and PNA tetramer (right). For the dimer, complementary oligonucleotides A and A’ are coupled to Fabs then mixed to form the dimeric Fab. For the tetramer, complementary oligonucleotides A, B, C, and D allow formation of a cruciform. B. Site-specific conjugation of aminooxy-PNA to αHer2 S202pAcF. The αHer2 S202pAcPhe Fab was left unconjugated (lanes 1 and 3) or conjugated to PNA (lanes 2 and 4) and analyzed by SDS-PAGE, in the absence (lanes 1 and 2) or presence of β-mercaptoethanol (BME) (lanes 3 and 4). C. PNA-mediated αHer2 Fab homodimer and tetramer formation. Each PNA complement was covalently coupled to αHer2 S202pAcF Fab and then hybridized to form either the homodimer (lane 3) or tetramer (lane 4) as seen by SDS-PAGE analysis. D. Phosphorylation inhibition assay of wild-type αHer2 Fab (●), αHer2 IgG (■), αHer2 Fab homodimer (▼), and αHer2 Fab tetramer (▲). ELISA analysis of phosphorylated HER2 levels in SK-BR3 cells using Human Phospho-ErBB2 DuoSet IC kit (R&D Systems).
In Vitro Activity of PNA Multimers
We then tested the ability of the αHer2 PNA multimers to inhibit phosphorylation in SK-BR-3 cancer cells. As shown in Figure 2E, the αHer2 PNA homodimer inhibits phosphorylation with a similar EC50 as the full-length trastuzumab IgG (17±7 nM vs 29±12 nM, respectively) and a 2.5-fold better EC50 than monovalent αHer2 Fab (47±15 nM). The αHer2 tetramer has an EC50 of 4.6±1.5 nM which is 6-fold lower than the αHer2 IgG, consistent with an increased avidity in binding Her2 (Figure 2E). Thus, by replacing DNA with PNA, bivalent and tetravalent Fabs were generated with equivalent or enhanced in vitro activity compared to trastuzumab (αHer2 IgG).
Synthesis and Activity of αCD3 Bispecific Antibody Fragments
To demonstrate the versatility of this self-assembly approach, we next generated a heterodimer consisting of the αHer2 Fab linked to the Fab of UCHT1, a monoclonal antibody that binds human CD3 on T lymphocytes37. This bispecific construct should recruit activated CD8+ T lymphocytes to kill tumor cells38-40. pAcF was incorporated at position K138HC (heavy chain) of the αCD3 Fab. This site is distant from the combining site, and when conjugated to the αHer2 mutant Fab should allow productive formation of a pseudo immunological synapse. The requisite aminooxy-PNA strand (N’-H2NO-(EG)3-GCCGCGTTGCGT-C’) (Supplementary Figure 9) was coupled to αCD3 K138HCpAcF, purified by filtration (Amicon concentrator 30kDa MWCO), and characterized by SDS-PAGE and mass spectrometry (Expected MW: 52,207 Da; Observed MW: 52,211 Da; Supplementary Figure 10). This Fab was then hybridized with αHer2 Fab S202pAcF-PNA (coupled to the complementary PNA strand) in a 1:1 molar ratio (10 μM, 30 min at 37 °C) and purified by size exclusion chromatography. The αHer2-αCD3 heterodimer migrates as one band on SDS-PAGE (Figure 3A, lane 3), indicating a homogeneous product, and has the expected mass by MALDI-TOF mass spectrometry (Expected MW: 104,146 Da; Observed MW: 104,076 Da; Supplementary Figure 11). We analyzed the binding properties of the αHer2–αCD3 heterodimer by flow cytometry. The heterodimer binds to both Her2+ cells (SK-BR-3) and CD3+ cells (Jurkat, ATCC), and does not have any affinity for Her2- cells (MDA-MB-435) (Supplementary Figure 12). We next assessed whether the heterodimer induces lysis in Her2+ cells in the presence of T lymphocytes. Human PBMCs (peripheral blood mononuclear cells) were combined with Her2 transformed target cells (MDA-MB-435/Her2) at a ratio of 10:141, and non-transformed MDA-MB-435 cells were used as an iso-genic Her2- negative control42. Unconjugated αHer2 Fab and αUCHT1 Fab, mixed 1:1, were used as additional negative controls. Both the unconjugated Fabs and heterodimer were added to the PBMC/target cell mixture (0.1 pM-10 nM) and incubated for 16 hours at 37 °C, and LDH (lactate dehyrogenase) released from lysed cells was measured as an indicator of cytotoxicity43. Dose dependent lysis of Her2 positive cells was observed only in the presence of the αHer2-αCD3 heterodimer (Figure 3B). In addition, the bispecific construct had no significant effect on the Her2 negative cell line, indicating specificity toward Her2+ cells. The unconjugated mixture also did not affect either cell line. The observed EC50 of the αHer2-αCD3 heterodimer is 104±22 pM, which is similar to that recently reported for a polyethylene glycol linked heterodimer41. This result further shows that PNA base pairing affords an effective conjugation strategy to generate biologically active and potent heterodimers. As discussed previously, length and/or flexibility between the two partners can have an effect on the efficiency of effector cell mediated killing44. To demonstrate this point, we compared PNA strands with either one or three EG units between the N-terminus and ami-nooxy moiety. The 3 EG construct is 3-fold better than the 1 EG construct (104±22 vs. 313±27 pM, respectively, Figure 3G, compare ■ and ●).
FIGURE 3.
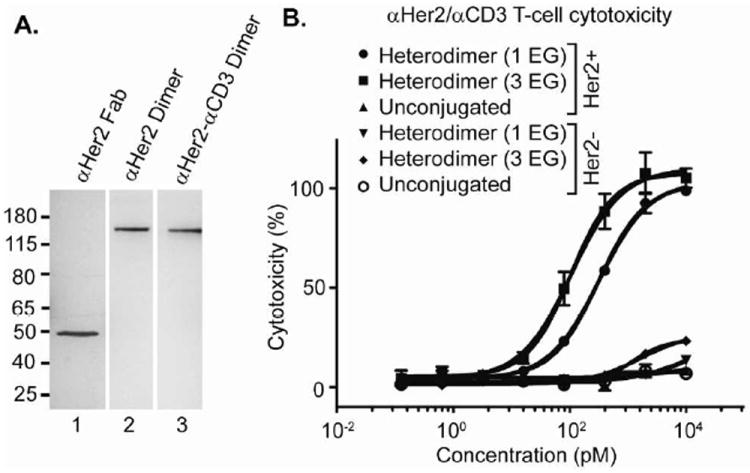
Synthesis and activity of αHer2-αCD3 heterodimer. (A) Characterization of the αHer2-αCD3 heterodimer by SDS-PAGE. After purification by size exclusion chromatography, the heterodimer (lane 3) forms one band at the same molecular weight as the αHer2 homodimer (lane 2) on SDS-PAGE. (B) PNA-linked heterodimers mediate T-cell killing of Her2+ tumor cells. Samples were incubated with PBMCs and MDA-MB-435 cells (either Her2+ or Her2-) for 16 hours at 37 °C in RPMI (10% FBS) and cell death measured by LDH release assay. Percent cytotoxicity was determined by maximum killing controls.
To evaluate the generality of this self-assembly approach, we next applied it to rituximab (Rituxan, Genentech/Roche), a monoclonal antibody against CD20, which is used to treat non-Hodgkin’s lymphoma45,46. We incorporated pAcF into the αCD20 Fab (S202pAcF) and coupled it to an aminooxy-modified PNA (N’-H2NO-(EG)3-ACGCAACGCGGC-C’), with a predicted Tm exceeding 70 °C, as described above (Expected MW: 51,522 Da; Observed MW: 51,492 Da; Supplementary Figure 13). The αCD20-αCD3 heterodimer was then synthesized by hybridizing each component (1:1 molar ratio, 10μM) and purifying the construct by size-exclusion chromatography (Figure 4A, lane 2); its composition was also confirmed by mass spectrometry (Expected MW: 103,430 Da; Observed MW: 103,527 Da; Supplementary Figure 14). We analyzed the cell binding properties of the heterodimer by flow cytometry and found that it binds to both CD20+ B-cells (Ramos) and CD3+ T-cells (Jurkat), but does not have any affinity for CD20- cells (K562) (Supplementary Figure 15). The activity of the bispecific molecule was then assessed with PBMCs and Ramos cells (10:1 ratio, respectively). The αCD20-αCD3 PNA dimer efficiently induces cell death (EC50 = 74±9 pM), while the mixture of unconjugated Fabs does not have any effect on the target cells (Figure 4B).
FIGURE 4.
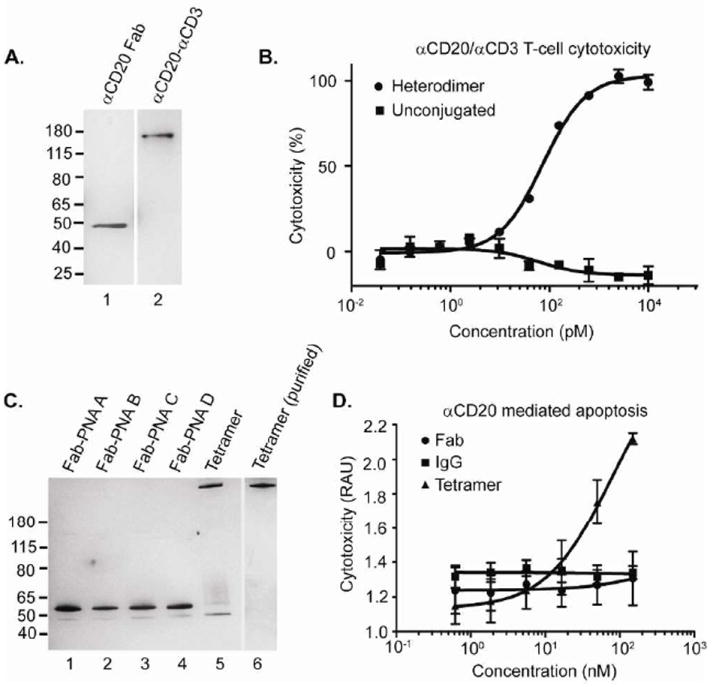
Construction and activity of αCD20 bispecific and tetrameric antibodies. (A) Characterization of the αCD20-αCD3 heterodimer (lane 2) by SDS-PAGE analysis compared to the αCD20 Fab (lane 1). B) PNA-linked heterodimers mediate T-cell killing of CD20+ target cells. The bispecifics were incubated with PBMCs and Ramos cells (10:1) for 16 hours at 37 °C and analyzed by LDH release assay. Percent cytotoxicity was determined by maximum killing controls. (C) Each PNA complement was covalently coupled to αCD20 S202pAcF Fab (lanes 1-4) and hybridized to form the αCD20 tetramer (lane 5). The αCD20 tetramer was purified by size exclusion chromatography and analyzed by SDS-PAGE (lane 6). (D) An αCD20 tetramer induces cell death. The αCD20 tetramer (▲), rituximab IgG (■), or αCD20 Fab (●) were incubated with Ramos cells (105 cells per well) for 48 hours at 37 °C in RPMI (10% FBS) and analyzed by LDH release assay. RAU indicates relative absorbance units.
Synthesis and Activity of αCD20 Tetramer
Rituximab acts primarily through ADCC, and does not directly induce lymphoma cell apoptosis45-47. Recently, however, αCD20 tetramers have been shown to induce apoptosis in lymphoma cells as either an Fab′2 homodimer48,49, or through more complex genetic fusion proteins50. Based on these observations, we assembled an αCD20 tetramer using our site-specific PNA technology. Four aminooxy-PNA sequences were conjugated to αCD20 S202pAcF Fab in an identical fashion to the construction of the αHer2 Fab tetramer (See Figure 2B, right). All four sequences couple efficiently to the αCD20 Fab, and when combined in a 1:1:1:1 molar ratio (10 μM), the αCD20 tetramer forms in over >85% yield (Figure 4C, lanes 1-5). The tetramer was then purified using size exclusion chromatography and showed a clearly defined product by SDS-PAGE (Figure 3C, lane 6). The αCD20 tetramer was tested for its ability to directly kill Ramos cells compared to the αCD20 Fab and αCD20 IgG (rituximab) controls (concentrations ranging from 0.6-150 nM). Neither the Fab nor the IgG have any effect on CD20+ cells; however, there is a clear dose dependent killing of the αCD20 tetramer (Figure 4D) which induces apoptotic activity comparable to other tetrameric constructs49.
CONCLUSION
Bispecific antibodies have substantial promise in clinical medicine, but are often challenging to generate. Fusion proteins can have substantial stability and aggregation problems, are not amenable to steric control of the binding sites, and cannot easily be made with both multivalent as well as multispecific properties. We have created a general and straightforward approach for generating homogeneous, well-defined Fab multimers of defined composition, valency, and specificity. This self-assembly technology can be easily applied to multiple antibodies to create geometrically controlled homodimers, heterodimers and higher-order multimers. To illustrate the technology, we created an αHer2 PNA-linked homodimer that exhibits comparable in vitro activity to the FDA approved drug, trastuzumab. In addition, the generality of this approach allowed us to synthesize two heterodimers with αCD3 Fab (αHer2-αCD3 and αCD20-αCD3) that were potent in targeting T-cells to tumor cells in vitro. To demonstrate that this technology can easily be applied to even higher order multimers, we created αHer2 and αCD20 tetramers. The latter contains a novel apoptotic inducing activity not present in the bivalent rituximab IgG. The Fab-PNA multimers are very stable (Tm > 70 °C) and can withstand SDS-PAGE gel, as well as long incubation times in serum without degradation. Thus, new pharmacologic activities may be identified using this approach, which would not be easily accessible using standard antibody discovery techniques. Since the multimeric constructs can be rapidly generated by mixing the relevant Fab-PNA subunits, which allows spontaneous base pairing and selective complex formation, libraries of “binders” and “effector” components can be easily “mixed and matched” to form a large series of molecules for in vitro functional testing. Components of such a library could include PNAs that enable production of several homo and heterodimers, with properties including T-cell recruitment, complement fixation, toxin activity, or imaging features. For example, one could envision the assembly of an αHer2-αHer3-αCD3 trimer which can recruit activated T-lymphocytes while targeting both ErbB2 and ErbB3 simultaneously. Finally, this technology can also be used to attach PNA-drugs and PNA-PEI probes to create site-specific antibody drug conjugates (ADCs) and imaging agents, respectively. In this regard, the PNA itself may be designed to be active as an antisense reagent, which when conjugated to an antibody, could selectively knockdown important oncogenes51.
Supplementary Material
Acknowledgments
We thank Ambrx, Inc. for helpful discussions and use of their fermentation equipment for antibody expression. This work was supported by the American Chemical Society Medicinal Chemistry Pre-doctoral Fellowship (S.A.K), National Research Foundation of Korea Grant NRF-2009-352-C00079 (C.H.K.), NIH grant R01GM062159 (P.G.S), ERC grant 201749 (N.W.), and NIH grant 1RC1EBO10745 and American Cancer Society grant RSG-09-1601 (V.V.S.). This is manuscript number 21932 of The Scripps Research Institute.
Footnotes
Author Contributions
All authors have given approval to the final version of the manuscript.
Supporting Information
Supporting figures and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Holmes D. Nat Rev Drug Discov. 2011;10:798. doi: 10.1038/nrd3581. [DOI] [PubMed] [Google Scholar]
- 2.Carter P. Nat Rev Cancer. 2001;1:118. doi: 10.1038/35101072. [DOI] [PubMed] [Google Scholar]
- 3.Lum LG, Davol PA, Lee RJ. Exp Hematol. 2006;34:1. doi: 10.1016/j.exphem.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Robinson MK, Hodge KM, Horak E, Sundberg AL, Russeva M, Shaller CC, von Mehren M, Shchaveleva I, Simmons HH, Marks JD, Adams GP. Br J Cancer. 2008;99:1415. doi: 10.1038/sj.bjc.6604700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, Einsele H, Brandl C, Wolf A, Kirchinger P, Klappers P, Schmidt M, Riethmuller G, Reinhardt C, Baeuerle PA, Kufer P. Science. 2008;321:974. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 6.Baeuerle PA, Reinhardt C. Cancer Res. 2009;69:4941. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 7.Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, Shah K, Jin L, Zhang S, He L, Zhang T, Ciccarone V, Koenig S, Bonvini E, Johnson S. Blood. 2011;117:4542. doi: 10.1182/blood-2010-09-306449. [DOI] [PubMed] [Google Scholar]
- 8.Holliger P, Winter G. Cancer Immunol Immunother. 1997;45:128. doi: 10.1007/s002620050414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cochlovius B, Kipriyanov SM, Stassar MJ, Schuhmacher J, Benner A, Moldenhauer G, Little M. Cancer Res. 2000;60:4336. [PubMed] [Google Scholar]
- 10.Jager M, Schoberth A, Ruf P, Hess J, Lindhofer H. Cancer Res. 2009;69:4270. doi: 10.1158/0008-5472.CAN-08-2861. [DOI] [PubMed] [Google Scholar]
- 11.Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L, Davis-Taber R, Kunes Y, Fung E, Schwartz A, Sakorafas P, Gu J, Tarcsa E, Murtaza A, Ghayur T. Nat Biotechnol. 2007;25:1290. doi: 10.1038/nbt1345. [DOI] [PubMed] [Google Scholar]
- 12.Ridgway JB, Presta LG, Carter P. Protein Eng. 1996;9:617. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 13.Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D, Totpal K, Wong A, Lee CV, Stawicki S, Clark R, Fields C, Lewis Phillips GD, Prell RA, Danilenko DM, Franke Y, Stephan JP, Hwang J, Wu Y, Bostrom J, Sliwkowski MX, Fuh G, Eigenbrot C. Cancer Cell. 2007;20:472. doi: 10.1016/j.ccr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 14.Wu AM, Senter PD. Nat Biotechnol. 2005;23:1137. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 15.Lin C, Liu Y, Yan H. Biochemistry. 2009;48:1663. doi: 10.1021/bi802324w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niemeyer CM. Angew Chem Int Ed Engl. 2010;49:1200. doi: 10.1002/anie.200904930. [DOI] [PubMed] [Google Scholar]
- 17.Hutchins BM, Kazane SA, Staflin K, Forsyth JS, Felding-Habermann B, Schultz PG, Smider VV. J Mol Biol. 2011;406:595. doi: 10.1016/j.jmb.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chouikhi D, Ciobanu M, Zambaldo C, Duplan V, Barluenga S, Winssinger N. Chemistry. 2012;18:12698. doi: 10.1002/chem.201201337. [DOI] [PubMed] [Google Scholar]
- 19.Pianowski Z, Gorska K, Oswald L, Merten CA, Winssinger N. J Am Chem Soc. 2009;131:6492. doi: 10.1021/ja809656k. [DOI] [PubMed] [Google Scholar]
- 20.Kazane SA, Sok D, Cho EH, Uson ML, Kuhn P, Schultz PG, Smider VV. Proc Natl Acad Sci U S A. 2012;109:3731. doi: 10.1073/pnas.1120682109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. Oncologist. 2009;14:320. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- 22.Dirksen A, Hackeng TM, Dawson PE. Angew Chem Int Ed Engl. 2006;45:7581. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 23.Wang L, Zhang Z, Brock A, Schultz PG. Proc Natl Acad Sci U S A. 2003;100:56. doi: 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storhoff JJ, Mirkin CA. Chem Rev. 1999;99:1849. doi: 10.1021/cr970071p. [DOI] [PubMed] [Google Scholar]
- 25.Mukherji M, Brill LM, Ficarro SB, Hampton GM, Schultz PG. Biochemistry. 2006;45:15529. doi: 10.1021/bi060971c. [DOI] [PubMed] [Google Scholar]
- 26.Mardirossian G, Lei K, Rusckowski M, Chang F, Qu T, Egholm M, Hnatowich DJ. J Nucl Med. 1997;38:907. [PubMed] [Google Scholar]
- 27.Buchardt O, Egholm M, Berg RH, Nielsen PE. Trends Biotechnol. 1993;11:384. doi: 10.1016/0167-7799(93)90097-S. [DOI] [PubMed] [Google Scholar]
- 28.Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. Nature. 1993;365:566. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- 29.Daguer JP, Ciobanu M, Alvarez S, Barluenga S, Winssinger N. Chem Sci. 2011;2:625. doi: 10.1039/c4sc01654h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorska K, Beyrath J, Fournel S, Guichard G, Winssinger N. Chem Commun. 2010;46:7742. doi: 10.1039/c0cc02852e. [DOI] [PubMed] [Google Scholar]
- 31.Gorska K, Huang K-T, Chaloin O, Winssinger N. Angew Chem Int Ed Engl. 2009;48:7695. doi: 10.1002/anie.200903328. [DOI] [PubMed] [Google Scholar]
- 32.Pianowski ZL, Winssinger N. Chem Soc Rev. 2008;37:1330. doi: 10.1039/b706610b. [DOI] [PubMed] [Google Scholar]
- 33.Urbina HD, Debaene F, Jost B, Bole-Feysot C, Mason DE, Kuzmic P, Harris JL, Winssinger N. Chem Bio Chem. 2006;7:1790. doi: 10.1002/cbic.200600242. [DOI] [PubMed] [Google Scholar]
- 34.Nielson PE, Egholm M. Curr Issues Mol Biol. 1999;1:89. [PubMed] [Google Scholar]
- 35.Zhou P, Wang MM, Du L, Fisher GW, Waggoner A, Ly DH. J Am Chem Soc. 2003;125:6878. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]
- 36.Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR. J Am Chem Soc. 2007;129:1959. doi: 10.1021/ja065930i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Z, Carter P. J Immunol. 1995;155:1903. [PubMed] [Google Scholar]
- 38.Staerz UD, Kanagawa O, Bevan MJ. Nature. 1985;314:628. doi: 10.1038/314628a0. [DOI] [PubMed] [Google Scholar]
- 39.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Mol Immunol. 2006;43:763. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Haas C, Krinner E, Brischwein K, Hoffmann P, Lutterbuse R, Schlereth B, Kufer P, Baeuerle PA. Immunobiology. 2009;214:441. doi: 10.1016/j.imbio.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 41.Kim CH, Axup JY, Dubrovska A, Kazane SA, Hutchins BA, Wold ED, Smider VV, Schultz PG. J Am Chem Soc. 2012;134:9918. doi: 10.1021/ja303904e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hutchins BM, Kazane SA, Staflin K, Forsyth JS, Felding-Habermann B, Smider VV, Schultz PG. Chem Biol. 2011;18:299. doi: 10.1016/j.chembiol.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, Shah K, Jin L, Zhang S, He L, Zhang T, Ciccarone V, Koenig S, Bonvini E, Johnson S. Blood. 2011;117:4542. doi: 10.1182/blood-2010-09-306449. [DOI] [PubMed] [Google Scholar]
- 44.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Cancer Immunol Immunother. 2010;59:1197. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grillo-Lopez AJ, White CA, Varns C, Shen D, Wei A, McClure A, Dallaire BK. Semin Oncol. 1999;26:66. [PubMed] [Google Scholar]
- 46.Ghesquieres H, Ferlay C, Sebban C, Chassagne C, Carausu L, Gargi T, Favier B, Philip I, Blay JY, Biron P. Hematal Oncol. 2008;26:139. doi: 10.1002/hon.850. [DOI] [PubMed] [Google Scholar]
- 47.Pescovitz MD. Am J Transport. 2006;6:859. [Google Scholar]
- 48.Ghetie MA, Podar EM, Ilgen A, Gordon BE, Uhr JW, Vitetta ES. Proc Natl Acad Sci U S A. 1997;94:7509. doi: 10.1073/pnas.94.14.7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghetie MA, Bright H, Vitetta ES. Blood. 2001;97:1392. doi: 10.1182/blood.v97.5.1392. [DOI] [PubMed] [Google Scholar]
- 50.Schultz J, Lin Y, Sanderson J, Zuo Y, Stone D, Mallett R, Wilbert S, Axworthy D. Cancer Res. 2000;60:6663. [PubMed] [Google Scholar]
- 51.Nielsen PE, Egholm M, Berg RH, Buchardt O. Anticancer Drug Des. 1993;8:53. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.