

Abstract
In several lysosomal storage disorders, including Niemann-Pick disease Type C (NP-C), sphingolipids, including glycosphingolipids, particularly gangliosides, are the predominant storage materials in the brain, raising the possibility that accumulation of these lipids may be involved in the NP-C neurodegenerative process. However, correlation of these accumulations and NP-C neuropathology has not been fully characterized. Here we derived NP-C mice with complete and partial deletion of the Siat9 (encoding GM3 synthase) gene in order to investigate the role of ganglioside in NP-C pathogenesis. According to our results, NPC mice with homozygotic deletion of GM3 synthase exhibited an enhanced neuropathological phenotype and died significantly earlier than NP-C mice. Notably, in contrast to complete depletion, NP-C mice with partial deletion of the GM3 synthase gene showed ameliorated NP-C neuropathology, including motor disability, demyelination, and abnormal accumulation of cholesterol and sphingolipids. These findings indicate the crucial role of GM3 synthesis in the NP-C phenotype and progression of CNS pathologic abnormality, suggesting that well-controlled inhibition of GM3 synthesis could be used as a therapeutic strategy.
Keywords: GM3, neuropathology, Niemann-pick type C disease, sphingolipids
INTRODUCTION
Niemann-Pick disease Type C (NP-C) is an inherited lipid storage disorder resulting from mutations in the NPC1 or NPC2 gene. The dominant feature of NP-C is severe and progressive neurodegeneration, which ultimately results in a shortened life span (Vanier, 2010). In NP-C, neurons show extensive storage of unesterified cholesterol and sphingolipids, including glycosphingolipids (GSLs) and sphingomyelin. Among the GSLs, GM2 and GM3 gangliosides are well characterized storage components of NP-C, although neuropathological influences of these accumulations are poorly understood (Zervas et al., 2001a). Gangliosides, together with cholesterol and other sphingolipids, play important roles in the normal physiological operations of the nervous system (Fishman and Brady, 1976) and constitute major lipid components of lipid rafts and related membrane microdomains in cell membranes (Sonnino et al., 2007). On one hand, accumulation of gangliosides occurs as a result of defective activity of the lysosomal proteins responsible for neuronal degradation, usually resulting in development of a rapidly progressive neurodegenerative disease (Brunetti-Pierri and Scaglia, 2008; Xu et al., 2010). These findings suggest that abnormal ganglioside patterns could play an important role in NP-C neuropathology.
Expression of individual types of gangliosides can be reduced genetically in NPC1−/− mice by crossing these animals with mice in which specific genes of GSL synthesis have been deleted. Previously, in an attempt to correct the clinical course of the disease, NPC1−/− mice were mated with GalNAcT (GM2/ GD2 synthase enzyme gene) null mice (Gondre-Lewis et al., 2009). As expected, neurons in double-deficient mice lacked intraneuronal GM2 accumulation, and, remarkably, also showed absence or dramatic reduction in the levels of free cholesterol. However, these mice showed no improvement in the clinical course of the disease. From this result, the authors concluded that accumulation of GM2 is not the primary cause of NP-C neurodegeneration. More recently, Li and colleagues reported that NPC1−/− mice with complete deletion of Siat9 (GM3 synthase enzyme gene) showed no reduction in the levels of cholesterol in visceral organs, however, lifespan was shortened, suggesting the possibility that inhibition of GM3 synthase may contribute to progression of NP-C (Li et al., 2008).
In addition to results from genetic regulation, treatment with glucosylceramide synthase inhibitor can reduce accumulation of neuronal cell ganglioside. Treatment of NPC1−/− mice with miglustat (N-butyldeoxynojirimycin) resulted in reduced accumulation of ganglioside, delayed onset of neurological dysfunction, and an expanded lifespan in this mouse model (Zervas et al., 2001b). In addition, trials of miglustat have been conducted in NP-C patients, with favorable preliminary results (Paciorkowski, 2008). This suggests that partial inhibition of ganglioside synthesis may have a positive influence on the disease course. Therefore, there remains a need to specifically define the role of gangliosides, especially GM3, in the pathogenesis of NP-C.
In this study, we evaluated the effects of complete and partial genetic deletion of the Siat9 gene on neuropathology in NPC1−/− mice in order to investigate the role of GM3 ganglioside in NP-C. Taken together, the findings of this study provide important new insights into how gangliosides contribute to the pathogenesis and/or development of NP-C.
MATERIALS AND METHODS
Establishment of Siat9−/−NPC1−/− and Siat9+/−NPC1−/− mice
Heterozygous BALB/c npcnih mice were bred with homozygous Siat9−/− mice (Yamashita et al., 2003) in order to obtain double-heterozygous mice (Fig. 1A). Siat9−/− NPC1−/− and Siat9+/−NPC1−/− mice were obtained by cross-breeding the double-heterozygotes. To minimize variations due to genetic background differences, all genotypes used in this study were derived from this cross-breeding. Mutant Siat9 and NPC1 alleles were confirmed by polymerase chain reaction (PCR) (Loftus et al., 1997; Yamashita et al., 2003). All procedures were performed in accordance with an animal protocol approved by the Kyungpook National University Institutional Animal Care and Use Committee (IACUC). Animals were housed in a room maintained under controlled temperature (20–22°C) on a 12/12-h light/dark cycle.
Fig. 1.

Partial and complete deletions of the Siat9 gene have opposite effects on pathology in NP-C mice. (A) Crossing scheme for generation of Siat9−/−NPC1−/− and Siat9+/−NPC1−/− mice. Siat9−/− mice in a C57BL/6J background were crossed with NPC1 heterozygotes (BALB/c npcnih−/+). Siat9+/−NPC1+/− offspring were then identified by PCR and crossed to yield experimental animals. (B) Kaplan-Meier survival curves of NPC1−/− (n = 33), Siat9+/−NPC1−/− (n = 12), and Siat9−/− NPC1−/− double mutant (n = 25) mice. Kaplan-Meier survival analysis showed significantly reduced survival in Siat9−/−NPC1−/− mice (p < 0.001), compared to NPC1−/− mice. No significant differences in sur-vival rate were observed between NPC1−/− mice and Siat9+/−NPC1−/− mice, however, partial deletion of the Siat9 gene in NP-C mice resulted in a slight extension of the life span. (C) Rotarod scores of individual Siat9+/−NPC1−/− (filled squares, n = 10), NPC1−/− (open squares, n = 10), and WT mice (open triangle, n = 12) were averaged and plotted beginning at five to seven weeks. Siat9+/−NPC1−/− mice were able to remain on the rotating rod for a significantly longer time than NPC1−/− mice (*p < 0.05). (D) Cerebellar sections from seven-week-old WT, Siat9+/−NPC1−/−, NPC1−/−, and Siat9−/− mice were processed for immunostaining with anti-GFAP antibody for evaluation of astrogliosis. In the cerebellum of Siat9+/−NPC1−/− and NPC1−/− mice, immunopositive activated astrocytes showed a marked increase, compared to WT mice. However, Siat9+/−NPC1−/− mice showed moderate increases in astrocyte activation, compared to NPC1−/− mice. Scale bar, 100 μm. Photos are representative of three slices per genotype. (E) Protein extracts from cerebellum of seven-week-old WT, Siat9+/−NPC1−/−, NPC1−/−, and Siat9−/− mice (n = 3 per group) were analyzed by Western blot using anti-GFAP antibody; values were normalized to actin levels. Increased expression levels of GFAP were observed in cerebellum of Siat9+/−NPC1−/− and NPC1−/− mice, compared to WT mice. However, the expression levels of GFAP in cerebellum of Siat9+/−NPC1−/− mice were significantly lower than those in NPC1−/− mice. Values are expressed as mean ± SEM. Anova, Tukey’s HSD test. ***p < 0.001, *p < 0.05 compared with WT mice. #p < 0.05 compared with NPC1−/− mice.
Glycolipid analysis
Cerebrum and cerebellum from four-week-old mice were homogenized with four volumes of ice-cold water in an all-glass Potter-Elvehjem homogenizer; 200 μl of homogenate (40 mg of wet tissue) were extracted by addition of 1.2 ml of methanol and 2 ml of chloroform. After incubation of the samples at 37°C for 1 h, 1 ml of methanol was added, and the extracts were centrifuged at 2,500 × g for 10 min. The pellet was re-ex-tracted with 2 ml of chloroform/methanol/water (1/2/0.8, v/v/v) at 37°C for 2 h. The combined supernatants were concentrated by Speed Vac, and the dried samples were dissolved in 2 ml of methanol and saponified. After neutralization, samples were diluted with 2 ml of water and applied to OASIS MAX 1 cc extraction cartridges (Waters) for separation of neutral and acidic lipids. The neutral GSL fraction was eluted with 1 ml of methanol and the acidic GSL fraction was eluted with 1 ml of 0.2 M ammonium acetate/methanol. The acidic GSL fraction was diluted with 2 ml of water and desalted using OASIS HLB 1 cc extraction cartridges (Waters). Thin layer chromatography was performed using HPTLC (Merck) and developed with chloroform/methanol/0.02 % CaCl2 (5:4:1, v/v) for acidic GSLs, and chloroform/methanol/water (65:35:8, v/v) for neutral GSLs. Lipids were visualized with orcinol-sulfuric acid for GSLs and Coomassie Brilliant Blue R-250 for sphingomyelin. After staining with orcinol-sulfuric acid, GSLs were identified by comparing their Rf to those of authentic GSL standards.
Cholesterol analysis
The concentrations of cholesterol in the samples were determined using enzymatic methods, as previously described (Simons and Ikonen, 1997). Cholesterol concentration was determined using a cholesterol determination kit, LTCII (Kyowa Medex) according to the manufacturer’s instruction.
Accelerating rotarod analysis
An accelerating rotarod apparatus (model 47600; Ugo Basile, Italy) was used. The animals were placed on the rotating rod beginning at 4 rpm and accelerated to 40 rpm over a period of 300 s. Scores were recorded once per week (at least three independent tests were performed for each time point) beginning at five to seven weeks. We performed accelerating Rotarod tests on male mice, which allowed us to exclude effects due to the hormone cycle of female mice. Uniform conditions were carefully maintained for each test, and there was a rest time of 1 h between each trial. A maximum time limit of 300 s per test was established.
Histological analysis
Four or seven-week-old mice were anesthetized with 2.5% avertin in PBS. The mice immediately underwent cardiac perfusion with 4% paraformaldehyde in PBS. Perfused brains were postfixed for 24 h and embedded in paraffin or cryoprotected in 30% sucrose for an additional 24 h. For immunostaining, the frozen sections (30 μm in thickness) were then incubated for 24 h with primary antibodies diluted in blocking solution at 4°C. The following antibodies were used for immunohistochemistry: anti-GFAP (rabbit, diluted 1:500, Dako), anti-calbindin-D 28K (rabbit, diluted 1:500, Chemicon), anti-SMI-32 (mouse, diluted 1:500, Sternberger Monoclonals), and anti-MBP (rabbit, diluted 1:500, Abcam). Sections were incubated with Alexa Fluor 488-or 594-conjugated secondary antibodies for 1 h at room temperature (RT) against the corresponding species. The sections were analyzed using a laser scanning confocal microscope equipped with Fluoview SV1000 imaging software (Olympus FV1000, Japan), or an Olympus BX51 microscope.
Western blotting
Brain samples were prepared in RIPA buffer (Cell Signaling Technology) containing 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma) and protease inhibitor cocktail (Calbiochem). Protein concentrations were determined using the Bradford technique (Bio-Rad, USA). Equal amounts of proteins (80 μg) were fractionated by SDS-PAGE, and transferred to PVDF membranes. Membranes were incubated with anti-GFAP, anti-MBP (All from Abcam), and β-actin (Santacruz). After washing, blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (anti-rabbit IgG, Cell Signaling Technology), followed by an enhanced chemiluminescence detection system (ECL; Amersham Biosciences). We performed Western blots in at least three independent experiments. We performed densitometric quantification using ImageJ software (US National Institutes of Health).
Cell culture
Dissociated cerebellar Purkinje neuron cultures were prepared from individual embryonic day (E) 18 fetuses, as previously described (Lee et al., 2010), with some minor modifications. Briefly, the entire cerebellum was removed and kept in ice-cold Ca2+/Mg2+-free Hank’s balance salt solution (HBSS) containing gentamicin (10 μg ml−1; Gibco). Each cerebellum was then dissociated into single cells using the SUMITOMO Nerve-Cell Culture System (Sumitomo Bakelite), and the cells were resuspended in seeding medium (Minimum Essential Medium; Gibco) supplemented with L-glutamine (4 mM; Fluka), D-glucose (11 mM; Sigma), sodium pyruvate (500 μM; Sigma), gentamicin (10 μg ml−1; Gibco), and 10% heat-inactivated fetal bovine serum (FBS; Gibco). The cell suspensions were seeded on poly-L-ornithine (Sigma) coated glass coverslips (12 mm) at a density of 5 × 106 cells per ml, with each coverslip in an individual well of a 24-well tissue culture plate. After incubation in a CO2 incubator (100% humidity, 37°C, 5% CO2) for 3 h, 1 ml of culture medium further supplemented with B-27 supplement (Gibco), N2 supplement (Gibco), bovine serum albumin (BSA; 100 μg ml−1; Sigma), and tri-iodothyronine (0.5 nM; Sigma) was added to each culture well. After seven days in vitro (DIV), half of the medium in each well was replaced with fresh culture medium additionally supplemented with cytosine arabinoside (4 μM; Sigma) to prevent proliferation of glia. Dissociated cortical neuron cultures were prepared from individual E 18 fetuses, as previously described (Yang et al., 2008). Cortical neurons were plated on cover slips (12 mm) coated with poly-L-lysine (Sigma) at 37°C in a humidified atmosphere of 5% CO2. For analysis of neurite outgrowth, cortical neurons were seeded in 24-well plates at a density of 2 × 104 cells per well. When cells had become attached to the substrate, the medium was replaced with serum free Neurobasal medium (Gibco) containing 2% B27 supplements, 1 mM Glutamax supplement (Gibco), and 100 U ml−1 penicillin-streptomycin (Gibco). For mouse primary oligodendroglial enriched cell cultures, cortical precursors were isolated from E 15.5 embryos, as described previously (Chen et al., 2007). Mouse cortical neural stem/progenitors were grown in Dulbecco’s Modified Eagle’s Medium/F12 (Gibco) supplemented with 2% B27, 100 U ml−1 penicillin-streptomycin, 20 ng ml−1 bFGF (PeproTech), and 20 ng ml−1 EGF (PeproTech) for four days, and then cultured in oligodendrocyte growth medium (N2 supplemented with 20 ng ml−1 bFGF and 10 ng ml−1 PDGFAA) for enrichment of O4+ oligodendrocyte progenitor cells (OPCs), as previously described (Chen et al., 2007). OPCs were adhered onto poly-D-lysine-coated glass coverslips in oligodendrocyte differentiation medium (1% N2 supplemented with 400 ng ml−1 tri-iodothyronine and 20 ng ml−1 ciliary neurotrophic factor). OPCs were then allowed to differentiate into oligodendrocytes for 14 days at 37°C in the absence or presence of 1 μM GM3 or GM2 (Alexis Biochemicals).
Immunocytochemistry
Cells were fixed with PBS containing 4% paraformaldehyde at RT for 15 min, followed by permeabilization with 0.2% Triton X-100 in PBS for 10 min. They were then preincubated for 10 min with 5% normal goat serum and 2% BSA in PBS containing 0.4% Triton X-100 to block background immunostaining, followed by incubation overnight at 4°C with primary antibodies diluted in blocking solution. The following antibodies were used for immonocytochemistry: anti-calbindin-D 28K (rabbit, diluted 1:500, Chemicon), anti-MAP2 (mouse, diluted 1:400, Sigma), and anti-MBP (rabbit, diluted 1:500, Abcam). For visualization, the primary antibody was developed by incubation with Alexa Fluor 488-conjugated secondary antibodies for 1 h at RT against the corresponding species. The cells were analyzed using a laser scanning confocal microscope equipped with Fluoview SV1000 imaging software, or an Olympus BX51 microscope.
Neurite outgrowth assay
Morphological analysis of Purkinje and cortical neurons was recorded using MetaMorph software (Universal Imaging Corp.). For each measurement, at least 80 cells per disc were counted from randomly selected fields and n = 4 discs. Each experiment was repeated at least three times. All recordings and the MetaMorph analysis were performed in a blinded manner.
Electron microscopy
A total of six mice were used for electron microscopic analysis: four-week-old WT (n = 3), Siat9−/−, NPC1−/−, and Siat9−/−NPC1−/− (n = 1 each). The mice were deeply anesthetized with sodium pentobarbital (80 mg/kg, i.p.) and underwent transcardial perfusion with heparinized normal saline, followed by a freshly prepared mixture of 1% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.4). The cerebral cortex was removed and sagittal sections were prepared using a Vibratome at 100 μm. The sections were then postfixed with 1% osmium tetroxide in PB (pH 6.0) for 1 h, dehydrated in graded alcohols, wafer-embedded in Durcupan ACM (Fluka, Switzerland) between strips of Aclar plastic film (EMS), and cured for 48 h at 58.5°C. Small pieces on the wafers cut out and glued onto blank resin blocks with cyanoacrylate. Thin sections were collected on formvar-coated single slot nickel grids and stained with uranyl acetate and lead citrate. A Hitachi H7500 electron microscope at 80 kV accelerating voltage was used for examination of grids. Digital images were captured using DigitalMicrograph software driving a CCD camera (SC1000; Gatan, USA) attached to the microscope and saved as TIFF files.
Analysis of oligodendrocyte morphology
Analysis of mature oligodendrocytes in vitro was performed using MetaMorph software. The MBP-positive myelin-like membrane sheets were outlined and the area of the sheet was reported from at least four independent experiments. All recordings and the MetaMorph analysis were performed in a blind manner.
Sphingolipid extraction and analysis
Brain tissue samples were homogenized at 4°C in nine volumes of homogenization buffer containing 50 mM HEPES, 150 mM NaCl, 0.2% Igepal, and protease inhibitors. After brief centrifugation at 13,000 rpm for 10 min for removal of large debris, the supernatant was collected and further centrifuged at 13,000 rpm at 4°C for 30 min. This second supernatant was collected. Quantification of ASM, AC, sphingomyelin, ceramide, sphingosine, and S1P was performed using a Waters Acquity Ultra Performance Liquid Chromatographic system (UPLC; Waters), as previously described (He et al., 2005).
Statistical analysis
The SPSS software package (SPSS 12.0, USA) was used for statistical analysis. The Student’s t-test was used for comparisons between two groups. In cases where more than two groups were compared, a one way analysis of variance (ANOVA) was performed, followed by Tukey’s HSD test. Statistical significance was set at p < 0.05.
RESULTS
Complete and heterozygotic deletion of GM3 synthase exerted distinct effects on the NP-C phenotype
To investigate the role of GM3 in the neuropathology of mice with NP-C, double mutant mice that were homozygous (Siat9−/−NPC1−/−) or heterozygous (Siat9+/−NPC1−/−) for the Siat9 gene and an inactivated NPC1 gene were established (Fig. 1A). We then analyzed ganglioside patterns in cerebellum (Supplementary Fig. 1A) and cerebrum (Supplementary Fig. 1B) of WT (Siat9+/+NPC1+/+), Siat9−/−, Siat9−/−NPC1−/−, Siat9+/−NPC1−/−, and NPC1−/− mice. As described previously, abnormally high levels of GM2 and GM3 were observed in brains of NPC1−/− mice, compared to brains of WT mice (Supplementary Fig. 1). However, gangliosides GM2 and GM3 were not detected in Siat9−/−NPC1−/− brains (Supplementary Fig. 1), indicating that synthesis of gangliosides GM2 and GM3 was completely blocked in these animals (Supplementary Fig. 2A). Although the ratios differed slightly, the brain ganglioside pattern of Siat9+/−NPC1−/− mice resembled that of NPC1−/− mice (Supplementary Fig. 1).
Groups of mice with various genotypes were followed over time and their body size and lifespan were compared. Of note, Siat9−/−NPC1−/− mice showed a reduced body size, compared to NPC1−/− mice. However, the body size of Siat9+/−NPC1−/− mice was indistinguishable from that of NPC1−/− mice (Supplementary Fig. 2B). In addition, Siat9−/−NPC1−/− double mutant mice died significantly earlier than NPC1−/− mice (Fig. 1B). By contrast, Siat9+/−NPC1−/− mice showed a slight extension of the mean life span, although the increase was not statistically significant (Fig. 1B). The mean life span of NPC1−/− mice was 69 days, spanning days 53–79 after birth, whereas, for Siat9−/−NPC1−/− double mutant mice, the mean life span was 50 days, with death occurring between 21–67 days after birth. The mean life span of a group of Siat9+/−NPC1−/− mice was 74 days, spanning days 64–86 after birth.
We next examined the motor coordination of WT, Siat9+/−NPC1−/−, and NPC1−/− mice using an accelerating rotarod apparatus at five, six, and seven weeks of age (Fig. 1C). Due to the severity of motor impairment, Siat9−/−NPC1−/− mice could not be subjected to motor coordination analysis. Indeed, Siat9−/−NPC1−/− mice as young as three to four weeks exhibited hind limb weakness, ataxia, and severe tremors. The length of time that the mice were able to stay on the accelerated rotarod was used as a measure of performance. We found that Siat9+/−NPC1−/− mice performed significantly better than NPC1−/− mice at weeks 6 and 7 (Fig. 1C). These results suggest that the partial genetic deletion of GM3 synthase resulted in reduced motor abnormalities in NP-C, while complete deletion resulted in exacerbation of the phenotype.
Increased glial fibrillary acidic protein (GFAP) reactivity is the hallmark of reactive gliosis, and age-dependent increased expression of GFAP is one of the most evident signs of neurodegeneration in NP-C (German et al., 2002). Markedly enhanced GFAP expression was observed in NPC1−/− mice from eight weeks of age (Supplementary Fig. 3B). At four weeks of age, no significant differences in GFAP expression were observed between NPC1−/− and WT mice, however, the levels of reactive astrocytes were had already increased in the cerebellum of Siat9−/−NPC1−/− mice, compared with NPC1−/− mice of the same age (Supplementary Figs. 3A and 3B). Of particular interest, at seven weeks of age, reactive astrocytes were observed in cerebellum of both Siat9+/−NPC1−/− and NPC1−/− mice, however, moderate increases in astrocyte activation were observed in Siat9+/−NPC1−/− mice, compared to NPC1−/− mice (Figs. 1D and 1E). Together, these data indicated that complete and heterozygotic deletion of GM3 synthase exerted distinct effects on the NP-C phenotype. Homozygotic deletion of GM3 synthase in NPC1−/− mice resulted in earlier and more severe neurodegeneration. However, heterozygotic deletion of GM3 synthase resulted in modest alleviation of motor symptoms and extended lifespan of NPC1−/− mice. These results suggest that gangliosides, especially GM3, are contributing factors in the pathogenesis of NP-C.
Heterozygotic deletion of GM3 synthase resulted in reduced severity of the dendritic phenotype of NP-C, whereas homozygotic deletion of GM3 synthase induced severe dendritic abnormalities
Purkinje neurons, which undergo prominent neurodegeneration in NP-C, were visualized by immunostaining with anti-calbindin antibody. At four weeks of age, similar Purkinje neuron density and dendritic trees were observed in cerebellar sections of WT, Siat9−/−, Siat9+/−NPC1−/−, and NPC1−/− mice, however, dystrophic changes of dendrites were observed in the cerebellar molecular layer of sections from Siat9−/−NPC1−/− mice (Supplementary Figs. 4A and 5A). With progression of the disease, at seven weeks of age, we observed loss of Purkinje neurons in both Siat9+/−NPC1−/− and NPC1−/− mice, however, cere -bellar sections from Siat9+/−NPC1−/− mice showed significantly more immunopositive dendrites in the molecular layer, compared with NPC1−/− mice (Fig. 2A). We next examined the effects of deletion of GM3 synthase on the dendritic morphology of cultured Purkinje neurons. A noticeable reduction in neuronal arbors and the diameter of the dendrites was observed in Siat9−/−NPC1−/− Purkinje neuron cultures (Fig. 2B). To quantify the abnormality, the main dendrite width was estimated from micrographs of anti-calbindin stained cells. A significant reduction in the mean main dendrite width was observed in cultured Purkinje neurons from Siat9−/−NPC1−/− mice, compared with NPC1−/− mice (Fig. 2C). A slight increase in the main dendrite widths was observed in Siat9+/−NPC1−/− Purkinje neurons, compared with NPC1−/− neurons; however, it did not reach significance (Fig. 2C). Siat9−/− Purkinje neurons were not noticeably different from WT Purkinje neurons (Supplementary Figs. 5B and 5C).
Fig. 2.

Heterozygotic deletion of GM3 synthase resulted in reduced severity of the dendritic phenotype of NP-C, whereas homozygotic deletion of GM3 synthase resulted in induction of severe dendritic abnormalities. (A) Cerebellar sections from seven-week-old Siat9+/−NPC1−/− and NPC1−/− mice were processed for immunostaining with anti-calbindin antibody for labeling of Purkinje neurons. A marked loss of Purkinje neurons was observed in both Siat9+/−NPC1−/− and NPC1−/− mice. No significant difference in Purkinje cell density was observed between the two groups, however, cerebellar sections from Siat9+/−NPC1−/− mice showed more calbindin-positive dendrites in the molecular layer, compared with NPC1−/− mice. Photos are representative of three slices per genotype. (B) Cerebellar Purkinje neuron cultures (10 DIV) from Siat9−/−NPC1−/−, Siat9+/−NPC1−/−, and NPC1−/− mice were immunostained with anti-calbindin antibody. (C) Quantitative image analysis of the main dendrite width at a distance of one cell-body diameter from the cell body. The mean of the main dendrite width was significantly reduced in Siat9−/−NPC1−/− Purkinje neurons, compared with NPC1−/− neurons (**p < 0.01). However, no significant differences were observed between Siat9+/−NPC1−/− and NPC1−/− Purkinje neurons, although the main dendrite widths of Siat9+/−NPC1−/− Purkinje neurons were slightly increased, compared with NPC1−/− neurons. (D) Sections of seven-week-old Siat9+/−NPC1−/− and NPC1−/− cerebral cortex were immunostained with anti-SMI-32 antibody for visualization of pyramidal neurons in the cerebral cortex. Apical dendrites of SMI-32-stained pyramidal cells were more pronounced in sections of Siat9+/−NPC1−/− mice, compared to NPC1−/− mice. Photos are representative of three slices per genotype. Scale bar, 50 μm. (E) Cerebral cortical neuron cultures (4 DIV) were immunostained with anti-MAP2 antibody. Scale bar, 50 μm. (F) Quantitative analysis of total dendrite length, max process length, and the number of dendritic branches are shown. Siat9−/−NPC1−/− cortical neurons showed significant decreases in total dendritic length and max process length, compared with Siat9+/−NPC1−/− and NPC1−/− cortical neurons. The number of dendritic branches was significantly reduced in Siat9−/−NPC1−/− and Siat9+/−NPC1−/− cortical neurons, compared with NPC1−/− neurons. Data are expressed as mean ± SEM of 80–90 cells measured per genotype from four embryos of different origins. Anova, Tukey’s HSD test. *p < 0.05, **p < 0.01 when compared with NPC1−/− mice.
For analysis of the neuronal morphology in the cerebrum, we performed immunohistochemistry using a monoclonal antibody against a non-phosphorylated epitope of the neurofilament heavy chain, SMI-32 (Fig. 2D). This antibody labels pyramidal neurons in layer V (Campbell and Morrison, 1989). Our results showed that the neuronal morphology in layer V of Siat9−/−NPC1−/− mouse brains was distinct from that in NPC1−/− mice (Supplementary Fig. 4B). Neurons in layer V of the NPC1−/− and Siat9+/−NPC1−/− cortex formed long, straight apical dendrites radial to the surface of the brain (arrowhead in Supplementary Fig. 4B). In contrast, neurons in the Siat9−/−NPC1−/− cortex did not form long apical dendrites and these dendrites displayed a different outgrowth pattern with multiple curved neurites (arrows in Supplementary Fig. 4B). Particularly notable, however, was that dendrites of SMI-32-stained pyramidal cells were more pronounced in the Siat9+/−NPC1−/− cortex, compared to the NPC1−/− cortex at seven weeks of age (Fig. 2D). Neurons in the Siat9−/− cortex were comparable to WT neurons (Supplementary Fig. 5D). We further examined dendrite formation in cultured cortical neurons. E 18 cortical neurons were cultured for four days, and immunostained using an antibody against MAP2. Compared to NPC1−/− neurons, significantly reduced neuritic outgrowth length was observed in cortical neurons from Siat9−/−NPC1−/− mice (23 %; Figs. 2E and 2F). In addition, Siat9−/−NPC1−/− cortical neurons showed decreased maximum dendritic length and branch numbers, 20% and 22%, respectively (Figs. 2E and 2F). Siat9+/−NPC1−/− cortical neurons showed no significant differences in neuritic outgrowth length and maximum dendritic length, however, branch numbers were decreased (18%), compared to NPC1−/− neurons (Figs. 2E and 2F). Siat9−/− single mutant showed no significant differences in any of these dendritic parameters, compared with WT control (Supplementary Figs. 5E and 5F). These results indicate that homozygotic deletion of GM3 synthase affects neuronal morphogenesis in NPC1−/− neurons, likely contributing to the worsened neurological phenotype, whereas heterozygotic deletion of GM3 synthase resulted in slightly reduced severity of the dendritic phenotype of NP-C.
Complete and heterozygotic deletion of GM3 synthase exerted opposite effects on demyelinating neuropathy in NPC1−/− mice
In NPC1−/− mice, hypomyelination occurs in the cerebral cortex and the corpus callosum, accounting for the tremor and ataxia observed in these animals (Takikita et al., 2004). As previously described, heterozygotic deletion of GM3 synthase resulted in reduced motor abnormalities in NP-C mice, while complete deletion exacerbated the tremor/ataxia phenotype, suggesting severe myelin deficiency. To examine the effects of GM3 synthase deletion on the demyelinating process in NP-C mice, we labeled brain sections with antibody against myelin basic protein (MBP) in order to visualize the myelin fibers (Figs. 3A–3C and Supplementary Figs. 6A–6C). At four weeks of age, slight demyelinating changes were observed in sections of cerebellum (Supplementary Fig. 6A), cerebral cortex (Supplementary Fig. 6B), and corpus callosum (Supplementary Fig. 6C) of Siat9+/−NPC1−/− and NPC1−/− mice. However, MBP immunoreactivity was almost completely absent in brain sections of Siat9−/−NPC1−/− double mutant mice (Supplementary Figs. 6A–6C). No significant differences were observed in the pattern of MBP-positive myelination in the Siat9−/− cortex, compared to the WT cortex (data not shown). In order to confirm and extend the above findings, using electron microscopy, we examined the corpus callosum in WT, Siat9−/−, Siat9−/−NPC1−/−, Siat9+/−NPC1−/−, and NPC1−/− mice at four weeks of age (Supplementary Fig. 6D). In Siat9−/−NPC1−/− mice, most of myelinated fibers showed hypomyelination and some also showed severe degenerative changes, including lysis of axoplasm and the myelin sheath, confirming the results obtained by MBP immunohistochemistry (Supplementary Fig. 6D). Of particular interest, in contrast to Siat9−/−NPC1−/− mice, at seven weeks of age, increased intensity of MBP staining was observed throughout the cerebellum (Fig. 3A), cerebral cortex (Fig. 3B), and corpus callosum (Fig. 3C) in Siat9+/−NPC1−/− mice, when compared to NPC1−/− mice. Western blotting of lysates from cerebellum (Fig. 3D) and cerebrum (Fig. 3E) showed decreased levels of MBP in the cerebellum and cerebral cortex of Siat9+/−NPC1−/− and NPC1−/− mice, compared to WT mice. However, slightly higher levels of expression of MBP were observed in the cerebellum and cerebral cortex of Siat9+/−NPC1−/− mice, compared with NPC1−/− mice (Figs. 3D and 3E). In addition, electron micrographs showed less progress of demyelination in Siat9+/−NPC1−/− mice, compared to NPC1−/− mice (Fig. 3F). Taken together, these data suggest the importance of gangliosides in maintenance of myelination, and that heterozygotic of GM3 synthase can result in delayed progression of demyelinating neuropathy in NP-C.
Fig. 3.
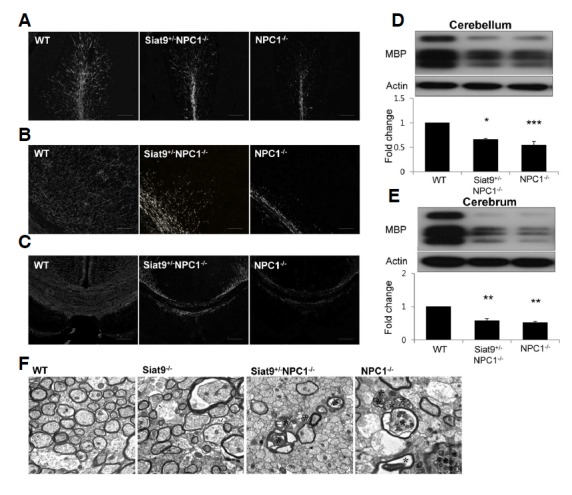
Heterozygotic deletion of GM3 synthase resulted in reduced demyelination in NPC1−/− mice. (A–C) Sections of mouse brain were immunostained with anti-MBP antibody for detection of myelin sheaths in cerebellar white matter (A) cerebral cortex (B) and corpus callosum (C) of seven-week-old WT, Siat9+/−NPC1−/−, and NPC1−/−mice. Scale bar, 100 μm. (D, E) Western blot analysis for MBP in cerebellum (D) and cerebrum (E) of seven-week-old WT, Siat9+/−NPC1−/−, and NPC1−/− mice (n = 3 per group). (F) Electron micrograph showing regions of the corpus callosum in WT, Siat9−/−, Siat9+/−NPC1−/−, and NPC1−/− mice at seven weeks of age. Asterisks in NPC1−/− and Siat9+/−NPC1−/− indicate degenerative change and hypomyelination. Many of the myelinated fibers showed hypomyelination in NPC1−/−mice, however, Siat9+/−NPC1−/− mice exhibited less demyelinating change. Scale bar, 500 nm.
Homozygotic deletion of GM3 synthase in NPC1−/− oligodendrocytes leads to defective differentiation of oligodendrocytes, which can be rescued by treatment with exogenous GM3
To determine whether hypomyelination in the Siat9−/−NPC1−/− mouse was caused by a defect in oligodendrocyte precursor cell development or by an inability of oligodendrocytes to produce myelin, we used primary cultures of oligodendrocytes. This cell culture system contains cells that are formed from precursor cells with multiple branches to become myelin-membrane sheet producing cells. Oligodendrocyte cultures from WT, Siat9−/−, Siat9−/−NPC1−/−, Siat9+/−NPC1−/−, and NPC1−/− mice were analyzed using antibodies against MBP for identification of differentiated cells (Supplementary Fig. 6E). A lower number of MBP+ cells with a reduced area of myelin-like membrane sheets was observed in oligodendrocytes obtained from Siat9−/−NPC1−/− mice, compared with cells derived from other genotype litter-mates (Supplementary Fig. 6F), whereas Siat9+/−NPC1−/− oligodendrocytes were not significantly different from NPC1−/− oligodendrocytes (Supplementary Fig. 6F). To further investigate the role of specific gangliosides during oligodendrocyte development, we added exogenous gangliosides to cultured, differentiated Siat9−/−NPC1−/− oligodendrocytes and characterized changes in the capacity of these cells to elaborate myelin-like membrane sheets in vitro. Mature oligodendrocytes were labeled with MBP antibody in order to visualize extended myelin-like membrane sheets (Fig. 4A). Oligodendrocytes grown in the presence of 1 μM GM3 ganglioside exhibited a significant increase in the area of MBP-positive sheets (Fig. 4B). These effects on the developing oligodendrocytes were also obtained by addition of GM2, but did not reach the level of the GM3 effect. Taken together, these data suggest that the GM3 ganglioside plays a critical role during oligodendrocyte development in NP-C.
Fig. 4.
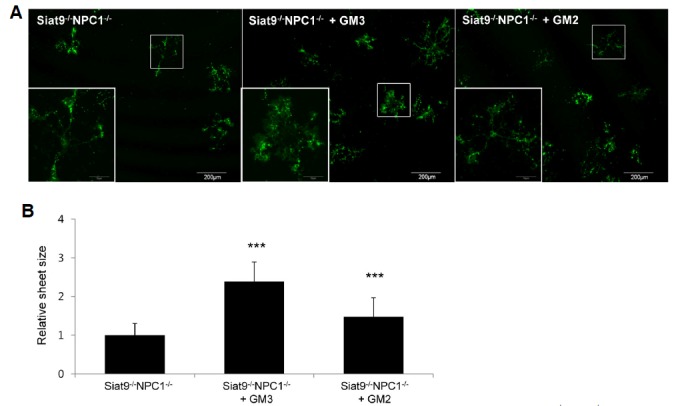
Treatment with exogenous ganglioside GM3 promoted spreading of myelin-membrane sheets of Siat9−/−NPC1−/− oligodendrocytes. (A) Differentiation of oligodendrocyte precursor cells occurred in the absence or in the presence of 1 μM GM3 or GM2 for two weeks. The cells were then stained for MBP and analyzed for changes in the size of myelin membrane sheets. Higher magnification of the boxed areas showed the myelin membrane sheets of a single oligodendrocyte. (B) The relative sheet size of primary oligodendrocytes was quantified. Treatment of oligodendrocytes with exogenous ganglioside 1 μM GM3 resulted in the greatest increase in area of MBP positive sheets, compared with the control. Data are expressed as mean ± SEM of 40 cells measured per genotype from four embryos of different origins. Anova, Tukey’s HSD test. ***p < 0.001 when compared with Siat9−/−NPC1−/− oligodendrocytes.
Heterozygotic deletion of GM3 synthase resulted in reduced accumulation of cholesterol and modulated sphingolipid metabolism
Together with ganglioside, accumulation of cholesterol and sphingolipids (sphingomyelin and sphingosine) occurs in cells with a defective NPC1 gene (Vanier, 2010). These lipids have high affinity toward each other (Simons and Ikonen, 1997) and the mass and/or synthesis are coordinately regulated (Ridgway, 2000). Therefore, we analyzed the effect of deletion of GM3 synthase on accumulation of these lipids. At four weeks of age, the levels of unesterified cholesterol in cerebellum (Fig. 5A) and cerebrum (Fig. 5B) of NPC1−/− mice were significantly increased, compared with those of WT mice, however, significantly lower levels of cholesterol were observed in cerebellum of Siat9+/−NPC1−/− mice, compared with NPC1−/− mice. We next examined the question of whether heterozygotic deletion of GM3 synthase also affects sphingolipids and their related enzymes. Sphingomyelin, acidic sphingomyelinase (ASM) activity, acid ceramidase (AC) activity, ceramide, sphingosine, and sphingosine-1-phosphate (S1P) were determined in brains of WT, Siat9+/−NPC1−/−, NPC1−/−, and Siat9−/− mice at seven weeks of age (Fig. 5, and Supplementary Fig. 7). As previously reported (Vanier, 2010), increased levels of sphingomyelin were observed in NPC1−/− mice, compared to WT mice, and these levels were decreased by heterozygotic deletion of GM3 synthase (Figs. 5C and 5D). ASM, which is responsible for lysosomal hydrolysis of sphingomyelin, is partially reduced in NP-C cells (Tamura et al., 2006). Likewise, ASM showed decreased activity in cerebellum of NPC1−/− mice, compared to WT mice, while, in cerebellum of Siat9+/−NPC1−/− mice, the levels were slightly increased (Fig. 5E). However, we did not observe any significant change of ASM in cerebrum. Activity of AC, which catalyzes hydrolysis of ceramide, was increased in cerebrum of Siat9+/− NPC1−/− mice, compared to NPC1−/− mice (Supplementary Fig. 7A), however, no statistical difference in the levels of ceremide was observed between groups (Supplementary Fig. 7B). Increased levels of sphingosine were observed in NPC1−/− mice, compared to WT mice, and these levels were decreased by partial deletion of GM3 synthase, however, this difference did not reach statistical significance (Supplementary Fig. 7C). Decreased levels of S1P were observed in cerebellum of NPC1−/− mice, compared to WT mice, and these levels were increased by partial deletion of GM3 synthase, however, this difference did not reach statistical significance (Supplementary Fig. 7D). These results indicate that heterozygotic deletion of GM3 synthase affects accumulation of cholesterol and sphingolipid metabolism, likely contributing to the milder neurological phenotype of NP-C.
Fig. 5.
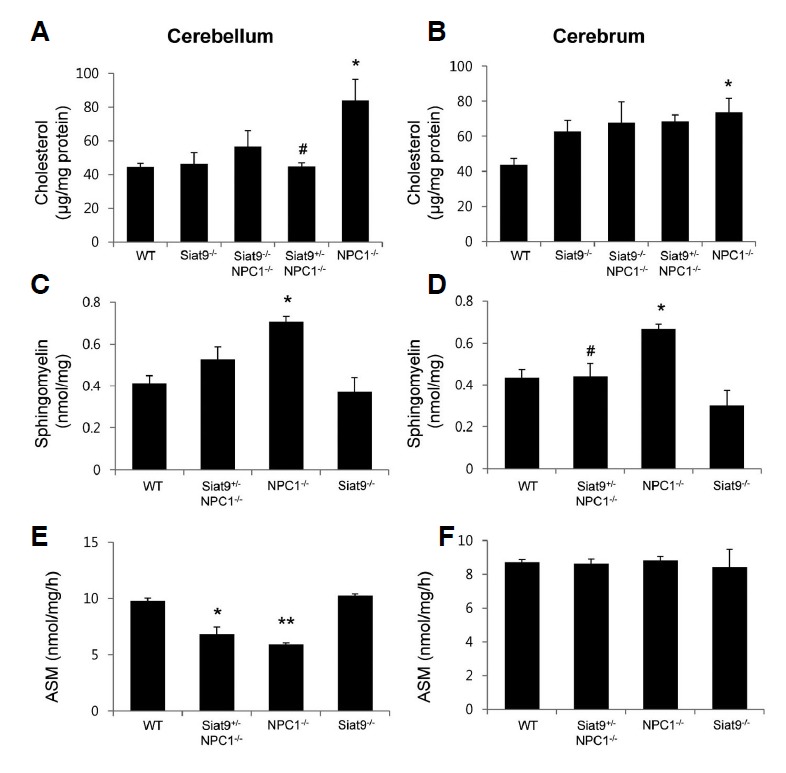
Heterozygotic deletion of GM3 synthase resulted in reduced accumulation of cholesterol and sphingomyelin. Cholesterol was extracted from the brains of WT, Siat9−/−, Siat9−/−NPC1−/−, NPC1−/−, and Siat9+/−NPC1−/− mice (n = 5) at four weeks of age, and their levels per mg of protein were determined. Significantly increased levels of unesterified cholesterol were observed in cerebellum (A) and cerebrum (B) of NPC1−/− mice, compared with WT mice. However, the levels of cholesterol were significantly lower in cerebellum of Siat9+/−NPC1−/− mice, compared with NPC1−/− mice. Sphingomyelin (C, D) and ASM activities (E, F) were determined using UPLC in brains of WT, Siat9+/−NPC1−/−, NPC1−/−, and Siat9−/− mice (n = 5) at seven weeks of age. (C, D) Increased levels of sphingomyelin were observed in NPC1−/− mice, compared to WT mice, and these levels were decreased by partial deletion of GM3 synthase. (E, F) Decreased activity of ASM was observed in cerebellum of NPC1−/− mice, compared to WT mice. Activity of ASM was slightly increased in cerebellum of Siat9+/−NPC1−/− mice; however, this difference did not reach statistical significance. Data are expressed as mean ± SEM. Anova, Tukey’s HSD test. *p < 0.05, **p < 0.001 when compared with WT mice. # p < 0.05 when compared with NPC1−/− mice.
DISCUSSION
Significant alterations of GSLs, especially gangliosides GM2 and GM3, occur in brain tissues from NP-C patients (Vanier, 2010). Therefore, ganglioside accumulation has been suspected of contributing to neuronal degeneration and dysfunction in NP-C. Indeed, GSLs play an important role in the etiology of several neurodegenerative diseases. For example, some GSLs are potent antigens that may underlie certain autoimmune neurodegenerative diseases (Ariga et al., 2001). Alzheimer’s disease may arise as a result of aggregation of amyloid-β-proteins initiated by GM1 (Hayashi et al., 2004). Several lysosomal storage diseases are also caused by defects of GSL catabolism (Futerman and van Meer, 2004). In these disorders, defects in lysosomal glycosidases result in accumulation of undegraded GSL substrates in lysosomes, leading to development of neurological disease. Therefore, inhibitors of GSL synthesis may improve the clinical phenotype in such neurodegenerative diseases. Several studies have reported that miglustat, an inhibitor that can diminish GSL biosynthesis, provides a therapeutic benefit in NP-C (Lachmann et al., 2004; Stein et al., 2012; Zervas et al., 2001b). Case reports have also suggested that it may slow the course of NP-C in humans (Di Rocco et al., 2012; Karimzadeh et al., 2013; Skorpen et al., 2012). However, the contribution of GSL accumulation to the pathogenesis of these disorders has remained poorly defined.
The objective of this study was to investigate the role of gangliosides, particularly GM3, in the neuropathology of NP-C. In the first phase of the study, we generated NPC1−/− mice models with complete or partial genetic deletion of the GM3 synthase gene and evaluated the effect of the genetic deletion on the neuropathology of NP-C. Of particular interest, complete and partial deletion of the GM3 synthase gene exerted distinct effects on the NP-C phenotype.
One overall conclusion of this study is that complete deletion of the GM3 synthase gene results in exacerbation of the phenotypes of NP-C mice. One of the most striking features of these mice was the dendritic structural abnormalities. In vivo and in vitro studies have demonstrated that a lack of a- and b-series gangliosides results in impaired dendritogenesis in NPC1−/− neurons. Indeed, the functional roles of GSLs, including gangliosides, in the developing nervous system have been analyzed at the cellular level. Gangliosides function in glycosphingolipid-enriched microdomains (lipid rafts) to promote dendritic initiation through modulation of specific membrane proteins and/or their associated second messenger cascades (Walkley et al., 2000). Among the gangliosides, GM3, GM2, and GD3 are especially predominant in embryonic brains, and play a pivotal role in regulation of dendritogenesis (Lehmann et al., 2003; Walkley et al., 1998; Yu et al., 2009). Despite the important roles of these gangliosides in dendrite development, no dendritic structural abnormalities were observed in neurons prepared from Siat9−/− mice. These data suggest that the functions of a- and b-series gangliosides may be compensated for by the remaining GSLs during brain development under normal conditions. In contrast, Siat9−/−NPC1−/− neurons showed abnormal dendritogenesis, suggesting that complete deletions of a- and b-series gangliosides is detrimental under NPC1-deficiency condition. The results of this study also showed severe hypomyelination in Siat9−/−NPC1−/− double mutant mice. In addition, cultured oligodendrocytes derived from these mice showed markedly impaired myelin-like membrane process extension, and this defective oligodendrocyte development was effectively prevented by treatment with GM3. These results suggest that hypomyelination in double mutant mice corresponds to abnormal oligodendrocyte development, and that GM3 plays a dominant role in this process. Under normal conditions, however, the elimination of gangliosides from oligodendrocytes is not detrimental to myelination. In fact, oligodendrocytes derived from Siat9−/− mice did not show any striking defects in oligodendrocyte development and myelination. These results support Saadat’s (2010) finding that a line of mice in which UDP-glucose:ceramide glucosyltransferase (Ugcg) was selectively disrupted in oligodendrocytes did not show myelin or oligodendroglial abnormalities (Saadat et al., 2010). Taken together, these data demonstrate that NPC1 deficiency sensitizes mice to the stresses of GM3 deletion during neural development. Thus, the precise amount and appropriate timing of expression of GM3 play a vital role in development of the nervous system under disease conditions, particularly NP-C.
One additional important finding in our study was the degree of pathological change in NPC1−/− mice with partial deletion of the GM3 synthase gene (Siat9+/−NPC1−/−). Reports analyzing the ganglioside composition of partial GM3 synthase deletion (Siat9+/−) in mouse brain (Yoshikawa et al., 2009) and skin fibroblasts (Shevchuk et al., 2007) identified similar ganglioside expression profiles, although the ratios differed. Likewise, similar expression profiles of complex gangliosides were observed in both cerebellum and cerebrum of NPC1−/− and Siat9+/−NPC1−/− mice. However, Siat9+/−NPC1−/− mice exhibited several different pathological features. Analysis of Siat9+/−NPC1−/− mice showed milder demyelinating neuropathy, suggesting that accumulation of ganglioside contributes, at least in part, to this neuropathy. In addition, we demonstrated here that cerebellar cholesterol concentrations were altered by partial deletion of the GM3 synthase gene. Previous studies have explored the effects of ganglioside synthase deletion on tissue cholesterol concentrations (Gondre-Lewis et al., 2003; Li et al., 2008). In 2003, ganglioside and cholesterol expression in neurons of mice doubly deficient in both NPC1 and the GSL synthetic enzyme, GM2/GD2 synthase, was examined (Gondre-Lewis et al., 2003). As expected, neurons in double-deficient mice lacked intra-neuronal GM2 accumulation, however, remarkably, some also exhibited absent or dramatic reduction in the levels of free cholesterol (Gondre-Lewis et al., 2003). These findings provide a compelling argument that cholesterol sequestration in NPC1-deficient neurons is ganglioside dependent. However, in a recent study, in animals lacking either GM2/GD2 or GM3 synthase, tissue cholesterol concentrations and synthesis rates were not different from those of control animals (Li et al., 2008). Likewise, in this study, we have shown that the levels of cholesterol observed in Siat9−/− mice did not differ significantly from those of WT mice. However, the levels of cholesterol in cerebellum of NP-C mice were significantly decreased by partial depletion of GM3 synthase. Although the exact mechanism underlying the different effects of partial deletion of the Siat9 gene is not fully understood, our results suggest that the activity of GM3 synthesis is related to cellular cholesterol metabolism in the NP-C brain. Another interesting finding is that partial depletion of GM3 synthase affects sphingolipid metabolism. Sphingomyelin is the major accumulating storage substrate in NP-C, and NPC1-deficient cells have much lower levels of ASM activity (Rosenbaum and Maxfield, 2011). Restoration of ASM activity levels reverted cholesterol accumulation in NPC1 mutant cells, and restored normal protein trafficking (Rosenbaum and Maxfield, 2011). In this study, we found that accumulation of sphingomyelin is decreesed and ASM activity and the levels of S1P, a key antiapoptotic molecule, were increased by partial depletion of GM3 synthase in NP-C brain. These data suggest that the previously descrybed effects of partial depletion of GM3 synthase on NP-C neuropathy may be due to modulation of these sphingolipid metabolisms.
In conclusion, these results indicate that partial reduction of GM3 synthesis could have an ameliorating effect on NP-C, while complete deletion has deleterious effects on neuronal morphogenesis and myelination during neural development. Therefore, recognition of the overall role of gangliosides in neural development and NP-C pathogenesis is important, and essential to a full understanding, in order to develop optimized substrate reduction therapy with reduced adverse effects. In addition, our data raise the possibility that allelic differences in genes affecting ganglioside metabolism may modify the NP-C phenotype. At the current time, no correlation between specific NPC1 mutations and disease age of onset or rate of progression has been established (Runz et al., 2008). These clinical observations suggest the existence of genetic modifiers (Vanier et al., 1996), and the work presented here identifies Siat9 as a candidate modifier, a possibility that will be explored in future studies.
Supplementary information
Acknowledgments
This work was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health & Welfare, Republic of Korea (A120367). Additional support for this work came from the World Class University program (R32-10064) and the Bio & Medical Technology Development Program (2011-0019356) of the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT & Future Planning, Republic of Korea.
Note:
Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Ariga T., Miyatake T., Yu R.K. Recent studies on the roles of antiglycosphingolipids in the pathogenesis of neurological disorders. J. Neurosci. Res. 2001;65:363–370. doi: 10.1002/jnr.1162. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N., Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008;94:391–396. doi: 10.1016/j.ymgme.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Campbell M.J., Morrison J.H. Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J. Comp. Neurol. 1989;282:191–205. doi: 10.1002/cne.902820204. [DOI] [PubMed] [Google Scholar]
- Chen Y., Balasubramaniyan V., Peng J., Hurlock E.C., Tallquist M., Li J., Lu Q.R. Isolation and culture of rat and mouse oligodendrocyte precursor cells. Nat. Protoc. 2007;2:1044–1051. doi: 10.1038/nprot.2007.149. [DOI] [PubMed] [Google Scholar]
- Di Rocco M., Dardis A., Madeo A., Barone R., Fiumara A. Early miglustat therapy in infantile Niemann-Pick disease type C. Pediatr. Neurol. 2012;47:40–43. doi: 10.1016/j.pediatrneurol.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Fishman P.H., Brady R.O. Biosynthesis and function of gangliosides. Science. 1976;194:906–915. doi: 10.1126/science.185697. [DOI] [PubMed] [Google Scholar]
- Futerman A.H., van Meer G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell. Biol. 2004;5:554–565.20. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- German D.C., Liang C.L., Song T., Yazdani U., Xie C., Dietschy J.M. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–450. doi: 10.1016/s0306-4522(01)00517-6. [DOI] [PubMed] [Google Scholar]
- Gondre-Lewis M.C., McGlynn R., Walkley S.U. Cholesterol accumulation in NPC1-deficient neurons is ganglioside dependent. Curr. Biol. 2003;13:1324–1329. doi: 10.1016/s0960-9822(03)00531-1. [DOI] [PubMed] [Google Scholar]
- Hayashi H., Kimura N., Yamaguchi H., Hasegawa K., Yokoseki T., Shibata M., Yamamoto N., Michikawa M., Yoshikawa Y., Terao K., et al. A seed for Alzheimer amyloid in the brain. J. Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X., Dagan A., Gatt S., Schuchman E.H. Simultaneous quantitative analysis of ceramide and sphingosine in mouse blood by naphthalene-2,3-dicarboxyaldehyde derivatization after hydrolysis with ceramidase. Anal. Biochem. 2005;340:113–122. doi: 10.1016/j.ab.2005.01.058. [DOI] [PubMed] [Google Scholar]
- Karimzadeh P., Tonekaboni S.H., Ashrafi M.R., Shafeghati Y., Rezayi A., Salehpour S., Ghofrani M., Taghdiri M.M., Rahmanifar A., Zaman T., et al. Effects of miglustat on stabilization of neurological disorder in niemann-pick disease type C: iranian pediatric case series. J. Child Neuol. 2013;28:1599–1606. doi: 10.1177/0883073812464526. [DOI] [PubMed] [Google Scholar]
- Lachmann R.H., te Vruchte D., Lloyd-Evans E., Reinkensmeier G., Sillence D.J., Fernandez-Guillen L., Dwek R.A., Butters T.D., Cox T.M., Platt F.M. Treatment with miglustat reverses the lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol. Dis. 2004;16:654–658. doi: 10.1016/j.nbd.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Lee H., Lee J.K., Min W.K., Bae J.H., He X., Schuchman E.H., Bae J.S., Jin H.K. Bone marrow-derived mesenchymal stem cells prevent the loss of Niemann-Pick type C mouse Purkinje neurons by correcting sphingolipid metabolism and increasing sphingosine-1-phosphate. Stem Cells. 2010;28:821–831. doi: 10.1002/stem.401. [DOI] [PubMed] [Google Scholar]
- Lehmann F., Wegerhoff R., Rosenberg A., Schauer R., Kohla G. Early variations of the disialoganglioside GD3 in chicken embryonic brain support its role in cell migration. Biochimie. 2003;85:449–454. doi: 10.1016/s0300-9084(03)00077-4. [DOI] [PubMed] [Google Scholar]
- Liu B., Turley S.D., Burns D.K., Miller A.M., Repa J.J., Dietschy J.M. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. USA. 2009;106:2377–2382. doi: 10.1073/pnas.0810895106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus S.K., Morris J.A., Carstea E.D., Gu J.Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M.A., Tagle D.A., et al. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Paciorkowski A.R., Westwell M., Ounpuu S., Bell K., Kagan J., Mazzarella C., Greenstein R.M. Motion analysis of a child with Niemann-Pick disease type C treated with miglustat. Movement Disord. 2008;23:124–128. doi: 10.1002/mds.21779. [DOI] [PubMed] [Google Scholar]
- Ridgway N.D. Interactions between metabolism and intra-cellular distribution of cholesterol and sphingomyelin. Biochim. Biophys. Acta. 2000;1484:129–141. doi: 10.1016/s1388-1981(00)00006-8. [DOI] [PubMed] [Google Scholar]
- Rosenbaum A.I., Maxfield F.R. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J. Neurochem. 2011;116:789–795. doi: 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H., Dolle D., Schlitter A.M., Zschocke J. NPC-db, a Niemann-Pick type C disease gene variation database. Hum. Mutat. 2008;29:345–350. doi: 10.1002/humu.20636. [DOI] [PubMed] [Google Scholar]
- Saadat L., Dupree J.L., Kilkus J., Han X., Traka M., Proia R.L., Dawson G., Popko B. Absence of oligodendroglial glucosylceramide synthesis does not result in CNS myelin abnormalities or alter the dysmyelinating phenotype of CGT-deficient mice. Glia. 2010;58:391–398. doi: 10.1002/glia.20930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchuk N.A., Hathout Y., Epifano O., Su Y., Liu Y., Sutherland M., Ladisch S. Alteration of ganglioside synthesis by GM3 synthase knockout in murine embryonic fibroblasts. Biochim. Biophys. Acta. 2007;1771:1226–1234. doi: 10.1016/j.bbalip.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Skorpen J., Helland I.B., Tennoe B. Use of miglustat in a child with late-infantile-onset Niemann-Pick disease type C and frequent seizures: a case report. J. Med. Case Rep. 2012;6:383. doi: 10.1186/1752-1947-6-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnino S., Mauri L., Chigorno V., Mauri L., Chigorno V., Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology. 2007;17:1R–13R. doi: 10.1093/glycob/cwl052. [DOI] [PubMed] [Google Scholar]
- Stein V.M., Crooks A., Ding W., Prociuk M., O’Donnell P., Bryan C., Sikora T., Dingemanse J., Vanier M.T., Walkley S.U., et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J. Neuropathol. Exp. Neurol. 2012;71:434–448. doi: 10.1097/NEN.0b013e31825414a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikita S., Fukuda T., Mohri I., Yagi T., Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J. Neuropath. Exp. Neur. 2004;63:660–673. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- Tamura H., Takahashi T., Ban N., Torisu H., Ninomiya H., Takada G., Inagaki N. Niemann-Pick type C disease: novel NPC1 mutations and characterization of the concomitant acid sphingomyelinase deficiency. Mol. Genet. Metab. 2006;87:113–121. doi: 10.1016/j.ymgme.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Vanier M.T. Niemann-Pick disease type C. Orphanet. J. Rare. Dis. 2010;5:16. doi: 10.1186/1750-1172-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier M.T., Duthel S., Rodriguez-Lafrasse C., Pentchev P., Carstea E.D. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 1996;58:118–125. [PMC free article] [PubMed] [Google Scholar]
- Walkley S.U., Siegel D.A., Dobrenis K., Zervas M. GM2 ganglioside as a regulator of pyramidal neuron dendritogenesis. Ann. N.Y. Acad. Sci. 1998;845:188–199. doi: 10.1111/j.1749-6632.1998.tb09671.x. [DOI] [PubMed] [Google Scholar]
- Walkley S.U., Zervas M., Wiseman S. Gangliosides as modulators of dendritogenesis in normal and storage disease-affected pyramidal neurons. Cereb. Cortex. 2000;10:1028–1037. doi: 10.1093/cercor/10.10.1028. [DOI] [PubMed] [Google Scholar]
- Xu Y.H., Barnes S., Sun Y., Grabowski G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Knowles J.K., Lu Q., Zhang H., Arancio O., Moore L.A., Chang T., Wang Q., Andreasson K., Rajadas J., et al. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One. 2008;3:e3604. doi: 10.1371/journal.pone.0003604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T., Hashiramoto A., Haluzik M., Mizukami H., Beck S., Norton A., Kono M., Tsuji S., Daniotti J.L., Werth N., et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA. 2003;100:3445–3449. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M., Go S., Takasaki K., Kakazu Y., Ohashi M., Nagafuku M., Kabayama K., Sekimoto J., Suzuki S., Takaiwa K., et al. Mice lacking ganglioside GM3 synthase exhibit complete hearing loss due to selective degeneration of the organ of Corti. Proc. Natl. Acad. Sci. USA. 2009;106:9483–9488. doi: 10.1073/pnas.0903279106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R.K., Nakatani Y., Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009;50(Suppl):S440–445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervas M., Dobrenis K., Walkley S.U. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropath. Exp. Neur. 2001a;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- Zervas M., Somers K.L., Thrall M.A., Walkley S.U. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 2001b;11:1283–1287. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.