

Abstract
Oral cancer is one of the most common cancers and it constitutes a major health problem particularly in developing countries. Oral squamous cell carcinoma (OSCC) represents the most frequent of all oral neoplasms. Several risk factors have been well characterized to be associated with OSCC with substantial evidences. The etiology of OSCC is complex and involves many factors. The most clearly defined potential factors are smoking and alcohol, which substantially increase the risk of OSCC. However, despite this clear association, a substantial proportion of patients develop OSCC without exposure to them, emphasizing the role of other risk factors such as genetic susceptibility and oncogenic viruses. Some viruses are strongly associated with OSCC while the association of others is less frequent and may depend on cofactors for their carcinogenic effects. Therefore, the exact role of viruses must be evaluated with care in order to improve the diagnosis and treatment of OSCC. Although a viral association within a subset of OSCC has been shown, the molecular and histopathological characteristics of these tumors have yet to be clearly defined.
1. Introduction
The significant role of viruses in cancer was acknowledged finally in the second half of the past century after various rodent tumorigenic viruses were discovered, and evidence had accumulated supporting an association between viruses and human cancer. Indeed, the Nobel Prize was awarded to Rous in 1966 in recognition of his seminal discovery of tumor-inducing viruses. In addition, almost at the same time, a Special Virus Cancer Program (VCP) was launched by the US Congress in 1964 providing enormous funds for intensive research into the supposed role of viruses in human cancer. This program, criticized by some investigators as being a political moonshot-style plan, failed to identify candidate human cancer-causing viruses yet generated fundamental information about the molecular biology and mechanisms underlying, in particular, virus-related animal cancer and cancer in general [1].
The strong cohort effect that accounted for the increased incidence of head and neck cancers after 1915 indicates that oral cancer is a disease largely attributable to behaviors that expose an individual to environmental carcinogens. The majority of oral cancers in individuals above and below the age of 45 can be attributed to the combined effects of alcohol and tobacco smoking. Other risk factors for oral cancers include diet, Body Mass Index, oral hygiene, and viral infections [2]. The most commonly implicated viruses in oral cancer transformation have been the human papillomavirus (HPV) [3, 4], herpes group viruses [5], adenoviruses [6], and the hepatitis C viruses [7, 8].
Of these, HPV and herpes have been the most thoroughly studied and are now considered to be the most likely “synergistic viruses” involved in human oral cancer. The herpes viruses most often linked to oral cancer are the Epstein-Barr virus (EBV), human herpes virus- (HHV-) 8, and cytomegalovirus (CMV) [9].
This review will attempt to focus on the approaches taken to discover human cancer viruses and promising methods for detecting new viruses. We also discuss how causal association is established and possible cofactors that influence development of virus-associated cancers.
2. Criteria for Defining Viral Carcinogenesis
Even though human oncogenic viruses belong to different virus families and utilize diverse strategies to contribute to cancer development, they share many common features. One key feature is their ability to infect but not kill their host cell. In contrast to many other viruses that cause disease, oncogenic viruses have the tendency to establish long-term persistent infections [10]. Consequently, they have evolved strategies for evading the host immune response, which would otherwise clear the virus during these persistent infections. Additional cofactors, such as host immunity and chronic inflammation, as well as additional host cellular mutations, also play an important role in the transformation process [11]. Different guidelines have been proposed to aid in establishing a causal relationship between viruses and human cancers [12–15].
Evans and Mueller Guidelines [13].
Epidemiologic Guidelines
Geographic distribution of viral infection corresponds with that of tumor, adjusting for the presence of known cofactors.
Viral markers are higher in case subjects than in matched control subjects.
Viral markers precede tumor development, with a higher incidence of tumors in persons with markers than those without.
Tumor incidence is decreased by viral infection prevention.
Virologic Guidelines
Virus can transform cells in vitro.
Viral genome is present in tumor cells but not in normal cells.
Virus induces the tumor in an experimental animal.
Hill Criteria for Causality [14, 15]
Strength of association (how often is the virus associated with the tumor?).
Consistency (has the association been observed repeatedly?).
Specificity of association (is the virus uniquely associated with the tumor?).
Temporal relationship (does virus infection precede tumorigenesis?).
Biologic gradient (is there a dose response with viral load?).
Biologic plausibility (is it biologically plausible that the virus could cause the tumor?).
Coherence (does the association make sense with what is known about the tumor?).
Experimental evidence (is there supporting laboratory data?).
3. Human Papillomavirus and OSCC
The human papillomavirus (HPV) family consists of more than 200 genotypes, classified in accordance with the ability to infect and transform epithelial cells. HPVs are DNA viruses that specifically target the basal cells of the epithelial mucosa [16]. Genotypes, such as HPV1, infect epidermal cells, whereas HPVs 6, 11, 16, and 18 infect epithelial cells of the oral cavity and other mucosal surfaces [17].
HPV belongs to the family Papovaviridae. These are small nonenveloped icosahedral viruses with an 8 kbp long double-stranded circular DNA genome. The papilloma virus genome comprises early and late genes that encode early proteins E1–E7 and late proteins L1-L2. The early proteins are nonstructural proteins involved in replication and transcription of the genome (E1–E5) or in host cell tumoral transformation (E6 and E7), whereas L1 and L2 are the structural capsid proteins of the virion. The late genes (L1-L2) code for the structural proteins of viral capsid and are activated during the final stages of the viral cycle. Up to six early genes and two late genes can be detected in HPV [18–23] (Table 1).
Table 1.
Overview of HPV gene products (E) early and (L) late.
| Gene product | Description |
|---|---|
| E1 | Helicase function; essential for viral replication and control of gene transcription. |
| E2 | Viral transcription factor; essential for viral replication and control of gene transcription. |
| E4 | Interaction with cytoskeleton proteins; viral assembly. |
| E5 | Growth stimulation by interaction with growth factor receptors; downregulation surface HLA class I molecules. |
| E6 | Cell immortalization; p-53 degradation; telomerase activation; antiapoptotic effect; induction of genomic instability. |
| E7 | Cell immortalization; interaction with pRb and pRb-associated pocket proteins; transactivation of E2F dependent promoters; induction of genomic instability. |
| L1 | Major capsid proteins. |
| L2 | Minor capsid protein; role in recruiting viral genomes for encapsidation; involvement in nuclear transport of viral DNA. |
The ability of HPV to transform epithelial cells is divided into high-risk and low-risk types. Low-risk types are associated with development of benign lesions such as warts, while infections with high-risk types may progress to malignant lesions [17].
Both high-risk and low-risk types of HPV can cause the growth of abnormal cells, but only the high-risk types lead to cancer because only the E7 protein encoded by high-risk HPVs can immortalize human epithelial cells. Sexually transmitted, high-risk HPVs include types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, and 73 [24]. It is important to note, however, that in the genital tract the large majority of high-risk HPV infections regress on their own and do not cause cancer [25].
The HPV involvement in oral carcinogenesis was supported on the basis of the following evidences:
4. Genital HPV Infection and Risk of OSCC
Several studies examined the incidence of second cancers after an initial diagnosis of anogenital cancers [34, 35] and have showed that there is an increased risk of head and neck cancer as well as other HPV-associated anogenital cancers in the two major HPV 16 oncogenes E6- and E7-positive patients. This association between HPV-associated anogenital cancers and head and neck cancer was further strengthened by two larger studies [36, 37]. Additionally, the presence of antibodies to HPV E6 and E7 proteins was found to be more associated with tumors of the oropharynx than with these of the oral cavity.
A recent study has shown that besides the classical horizontal transmission during the sexual life, a vertical transmission occurs in approximately 20% of case HPV-positive people. In these individuals, HPV-DNA is detected in amniotic fluid, foetal membranes, blood, and placental trophoblastic cells, all suggesting HPV infection in utero, that is, prenatal transmission [38].
4.1. Human Papillomavirus- (HPV-) Induced Carcinogenesis
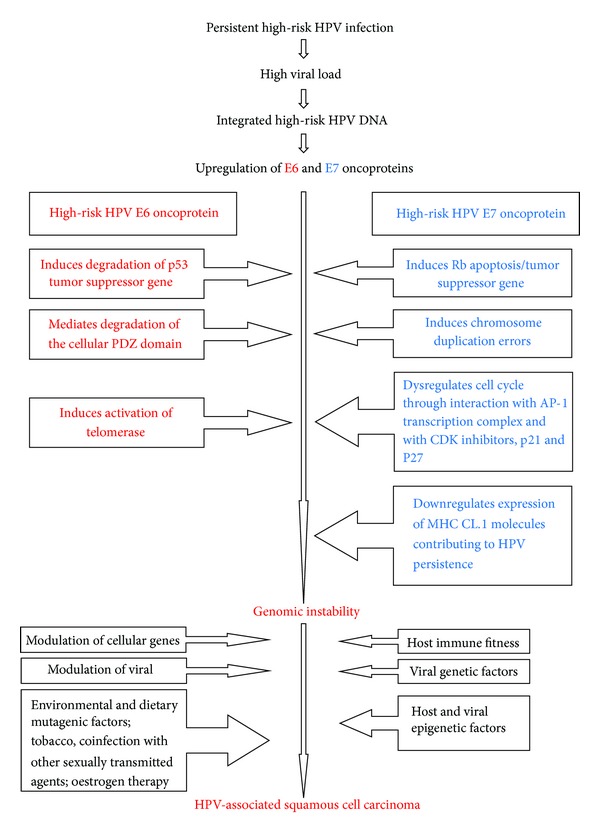
E6 and E7 oncoproteins can inactivate the genetic mechanisms that control both the cell cycle and apoptosis. The hallmark of high-risk HPV E6 oncogenic activity is degradation of the p53 tumour-suppressor gene [39, 40]. The functions of p53 in the cell cycle include controlling the G1 transition to the S phase of the cell cycle at the G1 checkpoint by inducing expression of cyclin inhibitors p16, p21, and p27 that block the activities of cyclin-CDKs (cyclin-dependent kinases) complexes, thus mediating arrest of the cell cycle by blocking the progression of the cell cycle at the G1/S transition. p53 activities mediate cell proliferation in response to mitogenic stimulation; mediate arrest of the cell cycle growth at the G1 checkpoint following DNA damage, hence permitting repair of the damaged DNA before the cell enters the DNA synthesis phase; and mediate induction of apoptosis of cells in which the DNA damage is beyond repair. Therefore, inactivation, degradation, or mutation of the p53 gene may dysregulate its functions resulting in increased cell proliferation, in accumulation of damaged DNA, in growth of cells harbouring DNA errors, and in prolonged cell survival. However, loss of p53 function alone is not sufficient for the development of cancer, and other cytogenetic alterations are required for complete malignant transformation [41].
In addition to these properties, E6 oncoprotein of high-risk HPV types can also mediate cell proliferation through the PDZ-ligand domain. The PDZ domain is located at areas of cell-to-cell contact, such as tight junctions of epithelial cells, and is associated with signal transduction pathways. The binding of high-risk HPV E6 oncoprotein to the PDZ family of proteins may result in degradation of the PDZ domain leading to dysregulation of organization, differentiation, and the chromosomal integrity of HPV infected epithelial cells [41]. This may contribute to morphological transformation of keratinocytes infected with high-risk HPV and to induction of epithelial hyperplasia. Telomerase is an enzyme that adds hexanucleotide repeats onto the end of the chromosome telomere. Telomerase activity is usually restricted to embryonic cells and is absent in normal somatic cells. When telomerase is absent, there is progressive shortening of telomeres as the cells repetitively divide, ultimately resulting in senescence of these cells [42].
HPV-induced activation of telomerase prevents the shortening of telomeres resulting in prolongation of the life-span of HPV-infected cells. High-risk HPV E7 oncoprotein has the capacity to initiate DNA synthesis in differentiated epithelial cells mainly by binding and inactivating the Rb apoptosis/tumour suppressor gene. The Rb family of proteins plays an essential role in controlling the cell cycle by governing the checkpoint between the G1 and the S phase. Hypophosphorylated Rb binds to E2F transcription factor forming a Rb-E2F complex, making E2F unavailable for transcription of genes associated with DNA synthesis. Upon phosphorylation of Rb by cyclin-CDK complexes, E2F is released from the Rb-E2F transcription repressor complex, and it induces transcription of the S-phase genes. E7 oncoprotein of high-risk HPV types functionally inactivates the Rb family of proteins resulting in overexpression of E2F transcription factor with upregulation of cell cycle genes resulting in DNA replication, in the transition of the cell from the G1 to the S phase, and in increased cell proliferation [43–45].
E7 oncoprotein can also interact with other cellular factors that control the cell cycle including histone deacetylases, AP-1 transcription complex, and CDK inhibitors, p21 and p27. Furthermore, E7 of high-risk HPV 16 and HPV 18 can decrease the expression of major histocompatibility complex (MHC) class I molecules, thus interfering with MHC class I antigen presentation, resulting in downregulation of cellular immune responses, allowing HPV to persist in infected epithelial cells [46].
In addition to these properties, high-risk HPV E7 oncoprotein can induce chromosome duplication errors leading to dysregulation of mitotic spindle formation and function, contributing to the genomic instability of the cell. The separate pathological effects of high-risk HPV E6 and E7 on the cell cycle complement each other, and together E6 and E7 mediate the HPV-associated epithelial cell transformation and promote cellular genomic instability that predisposes the infected cells to full malignant transformation. High-risk HPV E7 activates the DNA synthesis and cell replication mechanisms that are normally inactive in matured epithelial cells, thus initiating pathological cell growth. By inducing cell survival and delayed apoptosis of cells with DNA damage, E6 allows E7 to exert and sustain its pathological effect [47].
Typically, infected epithelial cells of HPV-associated benign lesions harbor low-risk HPV episomally in the nuclei. In HPV-associated malignancies, high-risk HPV DNA may either be integrated within the cellular genome or it may be maintained as an episome in the nuclei of the malignant cells. It is unclear how the HPV genome, whether episomal within the nucleus or integrated into the nuclear cellular genome, brings about the same end result of malignancy. The integration of HPV DNA favours the inactivation of tumour suppressor genes, p53 and Rb, contributing to increased cellular chromosomal instability and prolonging the life-span of the cell, which are essential steps in the multistep process of HPV-associated carcinogenesis. It is probable that following the initial HPV-induced cellular transformation, additional interactions with chemical carcinogens will provide the necessary additional impetus for the development of frank malignancy [47] (Figure 1).
Figure 1.
The HPV involvement in oral and oropharyngeal carcinogenesis was first proposed in 1983 by Syrjänen et al. [121] (Table 2). Their results showed that 40% of the laryngeal and oral cancers contained histological and morphological similarities with HPV-infected lesions, and 50% of the samples demonstrated HPV structural proteins by immunohistochemistry [122]. Since then, several studies have focused on HPV detection in oral cancer but results have been conflicting [65, 123, 124]. It was found that the prevalence of HPV detection varies broadly, depending on the population, on the location of the cancerous lesion, on type of specimen, and on detection method. By contrast, HPV was more frequently detected in OSCCs of the oropharynx and tonsil than at other head and neck sites [125–127]. However, in a systematic review that was performed by Syrjänen et al. [128] it was suggested that a potentially important causal association between HPV (specifically HPV 16) and OSCC exists. In a recent study the overall HPV prevalence was 10.5% in oral cavity carcinomas and was higher in female than in male cases. Ninety-five percent of HPV-positive cases were infected by a single HPV type. HPV 16 was the most prevalent type and was found in 95.5% of HPV-positive oral cavity carcinoma cases [129].
Table 2.
Studies on HPV in oral squamous cell carcinoma.
| Study/year | Geographic location | Tumour site(s) | No. of cases | HPV +ve N (%) |
Genotype(s) | DNA method |
|---|---|---|---|---|---|---|
| Loning et al., 1985 [48] | Germany | Oscc | 6 | 50% | 16, 11 | SB |
| Maitland et al., 1987 [49] | United Kingdom | Oscc | 15 | 46.7% | 16 | SB |
| Gassenmaier and Hornstein, 1988 [50] | Germany | Oscc | 68 | 23.5% | 16 | ISH |
| Syrjänen et al., 1988 [51] | Finland | Oscc | 51 | 11.8% | 16, 18 | ISH |
| Chang et al., 1990 [52] | Finland | Oscc | 40 | 2.5; 27.5% | 16 | ISH; PCR |
| Greer et al., 1990 [53] | USA | Oscc | 100 | 6% | n.d | ISH |
| Kashima et al., 1990 [54] | USA | Oscc | 26 | 19.2% | SB | |
| Young and Min et al., 1991 [55] | USA | Oscc | 27 | 0% | 6, 11, 16, 18, 31, 33, 35 | PCR |
| Zeuss et al., 1991 [56] | Spain | Oscc | 15 | 0% | 6, 11 | ISH |
| Holladay and Gerald, 1993 [57] | USA | Oscc | 39 | 17.9% | 16 | PCR |
| Cox et al., 1993 [58] | UK | Oscc | 8 | 50% | 16 | SB |
| Brandwein et al., 1994 [59] | USA | Oscc | 64 | 25% | 16 | PCR |
| González-Moles et al., 1994 [60] | Spain | Oscc | 27 | 37% | 6, 11 | ISH |
| Ostwald et al., 1994 [61] | Germany | Oscc | 26 | 61.5% | 16, 18 | PCR |
| Balaram et al., 1995 [62] | India | Oscc | 91 | 73.6% | 6, 11, 16, 18 | PCR |
| Shindoh et al., 1995 [63] | Japan | Oscc | 77 | 31.2% | 16 | PCR |
| Van Rensburg et al., 1995 [64] | South Africa | Oscc | 66 | 1.5% | 18 | ISH |
| Cruz et al., 1996 [65] | The Netherlands | Oscc | 35 | 54.3% | 16 | PCR |
| Mao et al., 1996 [66] | USA | Oscc | 64 | 31% | n.d. | PCR |
| Van Rensburg et al., 1996 [67] | South Africa | Oscc | 146 | 1.4% | 11, 16 | PCR |
| Chiba et al., 1996 [68] | Japan | Oscc | 38 | 21% | 16, 18, 33 | PCR |
| Wen et al., 1997 [69] | Japan | Oscc | 45 | 31.1% | 16, 18 | PCR |
| Gopalakrishnan et al., 1997 [70] | USA | Oscc | 10 | 30% | 16 | PCR |
| Ibrahim et al., 1998 [71] | Sudan | Oscc | 88 | 0; 0% | n.d. | ISH; PCR |
| Koh et al., 1998 [72] | Korea | Oscc | 42 | 52% | n.d. | PCR |
| Premoli-De-Percoco et al., 1998 [73] | Venezuela | Oscc | 50 | 70% | 16, 18 | ISH |
| D'Costa et al., 1998 [74] | India | Oscc | 100 | 15% | 16, 18 | PCR |
| Schwartz et al., 1998 [33] | USA | Oscc | 193 | 21.2% | 16 | PCR |
| Elamin et al., 1998 [75] | UK | Oscc | 28 | 50% | 6, 16 | PCR |
| Aggelopoulou et al., 1999 [76] | Greece | Oscc | 81 | 49% | n.d | PCR |
| Pillai et al., 1999 [77] | India | Oscc | 61 | 16.8% | 16 | PCR |
| Pintos et al., 1999 [78] | Canada | Oral cavity | 29 | 10% | n/s | PCR |
| Badaracco et al., 2000 [79] | Italy | Oscc | 66 | 36.4% | 16, 6 | PCR |
| Bouda et al., 2000 [80] | Greece | Oscc | 19 | 94.7% | 16, 18, 33 | PCR |
| Cao et al., 2000 [81] | China | Oscc | 40 | 72.5% | 16, 18 | PCR |
| Patima Cao et al., 2000 [82] | China | Oscc | 73 | 74% | 16, 18 | PCR |
| Sand et al., 2000 [83] | Sweden | Oscc | 24 | 12.5% | 16, 18, 6, 11 | PCR |
| Tsuhako et al., 2000 [84] | Japan | Oscc | 83 | 56.9% | n.d. | PCR |
| Gillison et al., 2000 [30] | USA | Oscc | 84 | 11.9% | 16 | PCR & ISH & SB |
| Bouda et al., 2000 [80] | Greece | Oscc | 19 | 89.5% | 16, 18 | PCR |
| Premoli-De-Percoco and Ramirez 2001 [85] | Venezuela | Oscc | 50 | 60% | 16, 18 | PCR |
| Schwartz et al., 2001 [86] | USA | Oscc | 254 | 24.4% | 16 | PCR |
| Chen et al., 2002 [87] | Taiwan | Oscc | 29 | 82.7% | 16, 18, 6 | PCR |
| Kojima et al., 2002 [88] | Japan | Oscc | 53 | 66% | 38 | PCR |
| Nagpal et al., 2002 [89] | India | Oscc | 110 | 33.6% | 16, 18 | PCR |
| Chang et al., 2003 [90] | Japan | Oscc | 103 | 49.5% | HR | PCR |
| Fregonesi et al., 2003 [91] | Brazil | Oscc | 46 | 39% | 16, 18, 6, 11 | ISH |
| Kansky et al., 2003 [92] | Slovenia | Oscc | 62 | 95% | 16, 33, 58, 11, 31, 68 | PCR |
| Ritchie et al., 2003 [93] | USA | Oscc | 141 | 15% | 16, 33 | PCR |
| Sugiyama et al., 2003 [94] | Japan | Oscc | 86 | 34.8% | 16 | PCR |
| Herrero et al., 2003 [95] | France | Oscc | 58 | 5.17% | 16 | PCR |
| Ostwald et al., 2003 [96] | Germany | Oscc | 118 | 43.2% | 6, 11, 16, 18 | PCR/SB |
| Correnti et al., 2004 [97] | Venezuela | Oscc | 16 | 50% | HR | PCR |
| Dahlgren et al., 2004 [98] | Sweden | Oscc | 110 | 10.9% | 16 | PCR |
| Smith et al., 2004 [99] | USA | Oscc | 106 | 9.4% | 16, 33 | PCR |
| Zhang et al., 2004 [100] | Japan | Oscc | 73 | 34% | 16, 18 | PCR |
| Yang et al., 2004 [101] | Taiwan | Oscc | 37 | 10.8% | 16, 18 | PCR |
| Koppikar et al., 2005 [102] | India | Oscc | 101 | 31.7% | 16, 18 | PCR |
| Lo Muzio et al., 2005 [103] | Italy | Oscc | 18 | 50% | n.d. | PCR |
| Boy et al., 2006 [104] | South Africa | Oscc | 59 | 11% | 18 | PCR; ISH |
| El-Mofty and Patil 2006 [105] | USA | Oscc | 94 | 30% | 16, 31, 33 | PCR |
| Nemes et al., 2006 [106] | Hungary | Oscc | 79 | 41.7% | 16 | PCR |
| Rivero and Nunes 2006 [107] | Brazil | Oscc | 23 | 0% | n.d. | PCR |
| Campisi et al., 2006 [108] | Italy | Oscc | 63 | 38.1% | n.d | PCR |
| Furrer et al., 2006 [109] | Argentina | Oscc | 14 | 42.8% | 16, 18 | PCR/SB |
| Koyama et al., 2007 [110] | Japan | Oscc | 20 | 100% | 18, 22, 16, 70 | PCR |
| Sugiyama et al., 2007 [111] | Japan | Oscc | 66 | 100% | 16 | PCR |
| Luo et al., 2007 [112] | Taiwan | Oscc | 51 | 73% | — | PCR |
| Khovidhunkit et al., 2008 [113] | Thailand | Oscc | 65 | 1.54% | 16, 18 | PCR |
| Szarka et al., 2009 [114] | Hungary | Oscc | 65 | 47.7% | 16, 18 | PCR |
| Zhao et al., 2009 [115] | China | Oscc | 52 | 40.4% | 16, 18, 11, 6 | PCR |
| Attner et al., 2010 [116] | Sweden | Oscc | 87 | 68 (78)% | 16, 33 | PCR |
| Elango et al., 2011 [117] | India | Oscc | 60 | 48% | 16 | PCR/IHC |
| Kristoffersen et al., 2012 [118] | Norway | Oscc | 50 | 30% | 6, 11, 16 | PCR |
| Chen et al., 2012 [119] | Taiwan | Oscc | 64 | 39% | 16 | ISH/IHC |
| Lee et al., 2012 [120] | Taiwan | Oscc | 173 | 22% | 16, 18 | PCR |
PCR: polymerase chain reaction; SB: southern blotting; ISH: in situ hybridization; IHC: immunohistochemistry.
Furthermore, other studies have proved the existence of a synergistic effect between HPV and alcohol. The risk of head and neck cancer was statistically significantly increased in heavy alcohol users detected with the virus, compared to that of HPV-negative cancer drinkers. Therefore, it has been proposed that alcohol can biologically modify mucosal tissue, by increasing its permeability to viral infection or by influencing the immune response to HPV [99]. It is believed that one of the major events of HPV-induced carcinogenesis is the integration of the HPV genome into a host chromosome. HPV genome integration often occurs near fragile sites of the human genome, but there are no apparent sites for integration and no evidence for insertional mutagenesis [130].
Slightly modifying Koch's postulates, in order to establish a relationship between a causative virus and a disease, four criteria are needed: (1) viral genome ought to be present in tumor lesions or in tumor cells; (2) virus must be isolated from a pathologic lesion and grown in culture; (3) cultured virus should cause disease when inoculated into a healthy organism; and (4) virus must be reisolated from the inoculated host and identified as being identical to the original specific causative factor. However, the use of Koch's postulates to establish disease causation does not fully apply to these phenomena, since the etiology of cancer is multifactorial [131]. On the contrary, other authors suggest that the incidence of HPV infection in the oral cavities of healthy population is very low and therefore other risk factors are most likely responsible to promote oral carcinogenesis [132]. Hence, we have tried to collect the studies done on HPV in oral cancer.
5. HPV Testing
HPV testing is critical for the estimation of HPV prevalence in various oral diseases. HPV testing is usually based on PCR method. General or consensus primers targeting L1 gene are most frequently used for HPV detection because they are able to identify several HPV genotypes at the same time. Sampling techniques together with widely divergent PCR methods in different studies explain most of the variability in HPV prevalence among OSCC and control samples [133]. In situ hybridization and in situ oncogenic protein staining techniques have also increased sensitivity and specificity and are used for HPV testing. These techniques have allowed for not only the detection of HPV in cytological smears or histopathological immunesections but also the determination of the topographic site of the infection [134]. According to recent studies, HPV-positive squamous cell carcinomas have intact p16 gene and wild type p53 compared to HPV-negative ones [135]. Other authors have noted that a distinctive mark of the presence of HPV in oral cancer could be found in p16 nuclear or cytoplasmic overexpression [136, 137]. However, one goal of the scientific research is to find new biological markers able to identify the set(s) of genes involved in oral carcinogenesis.
6. HPV Serology
The immune response to HPV infection involves both the cell-mediated and humoral responses. However, serological evidence is circumstantial since it provides only data on prior exposure to HPV. Since not all patients with HPV-associated cancers have detectable HPV antibodies, serum antibody determination may be a limited biomarker for HPV infection and carcinogenesis. Serum antibodies to HPV capsid proteins (virus-like particles) are thought to be a marker of lifetime HPV infection [138, 139]. Antibodies against HPV E6 and E7 proteins are associated with increased risk of HPV-associated cancer [140, 141] but are rather linked more with tumors from the oropharynx than from the oral cavity. The use of HPV viral load in oral biopsies in conjunction with serological markers may serve to identify a subset of HPV-associated oral cancers in which HPV is biologically active.
7. Herpes Simplex Virus (HSV)
HSV is a double-stranded DNA virus that is enveloped. When it infects cells the series of events that takes place is very complex but can be analyzed logically since it follows a coordinated series of steps. The virus exists in two closely related forms, known as HSV 1 and HSV 2. HSV 1 causes mainly oral and ocular infections, while HSV 2 causes mainly genital infections. However, the overall behavior and structure of the two viruses are very similar. Each virus causes a severe primary infection followed by a latent infection that may be followed by recurrent infections. Cells that are infected by HSV are killed. Gene maps of HSV 1 and HSV 2 have been developed and show that the various functions of the two viruses are encoded by identical regions of the genome of each virus [142].
The only viral function that is not in a related location between the two types is the ability to transform cells, and the reason is unknown. The transforming region of HSV 1 (minimum transforming region 1, mtr-1) is located in the left third of the genome, while the equivalent two functions of HSV 2 (mtr-2 and mtr-3) are close to the center of the genome [143].
7.1. Transformation by HSV
Induction of cellular proteins has been studied as a possible mechanism for transformation by HSV. It is known that infection by HSV 1 induces the expression of “stress” or “heat shock” proteins. The mechanism of induction is not known but does depend on the expression of the immediate-early family of HSV proteins. Since HSV might transform cells by stimulating the expression of cellular proteins, some workers have started to study such proteins by isolating cDNA from cells that were transformed by HSV 2 [142–144].
However, the function of these cloned fragments is still not known. Host cell shutoff is recognized as being an important event during infection of cells by HSV. The infected cell ceases to synthesize cellular proteins, and cell RNA is degraded very quickly. Recently, it was found that the gene of HSV that mediates shutoff is located in the same region of the genome as the mtr-2 region of HSV 2, which mediates cell transformation [145]. This has raised the possibility that the mechanism of transformation might be related to the mechanism of shutoff. Shutoff is known to be a multistep process, depending on more than one viral activity [146].
The first phase of host cell shutoff is due to some activity by the virus particle itself. It happens very soon after infection and occurs even if virus gene expression is prevented by Actinomycin D or by irradiation of the virus [147].
The second phase of the host cell shutoff does require the expression of virus genes and eliminates any remaining host protein synthesis. It seems likely that these phenomena will be studied quite closely in the future to find which of them is most likely to be involved in cell transformation. Other activities of HSV that might be related to cell transformation include the fact that the virus can stimulate the replication of other viruses. This can also occur when cells are exposed to chemical carcinogens [148].
7.1.1. Induction of Cellular Proteins
Infection by HSV induces the expression of stress or heat shock proteins. According to Steele and Shillitoe, the exact mechanism is not known but it depends on the expression of the immediate early family of HSV proteins. It was, therefore, concluded that HSV might transform cells by stimulating the expression of cellular proteins [149].
7.1.2. Host Cell Shutoff Process
Infected cell ceases to synthesize cellular proteins and cell RNA would be very quickly degraded. A mediating gene was located in the same region of the genome as the mtr-2 region of HSV 2 that mediates cell transformation [145]. Steele and Shillitoe raised the possibility that the mechanism of cell transformation might be related to the mechanism of shutoff [149].
7.1.3. Stimulation of Other Viruses by HSV
This comes from the idea that at least in cervical carcinogenesis, HSV and HPV may act as cocarcinogens, with HSV as an initiator and HPV as a promoter. In oral lesions, however, Scully et al. in 1993 showed that very few premalignant or carcinoma specimens appeared to have both HPV 16 and HSV 1 DNA sequences. They, therefore, concluded that there is no evidence that these potentially oncogenic DNA viruses do play a synergistic role in oral cancer development, but the possibility cannot be discounted [150].
7.1.4. Chromosomes as Targets
Stich et al. [151], Mincheva et al. [152], and Peat and Stanley [153] suggested that when cells are infected by HSV there is chromosomal damage which is at first restricted to a site on chromosome 1q and to some extent on chromosomes 3, 9, and 16. Based on these studies, Steele and Shillitoe gave a plausible explanation for the possibility that HSV has specific chromosomal targets for rearrangement. Damage to a particular chromosomal site might be another possible mechanism of cell transformation by HSV [149].
El Sissy [154] attempted to search for possible presence of HSY-2 protein in 21 lesional tissues of oral squamous cell carcinoma, as well as in normal oral mucosa, using immunohistochemical peroxidase-antiperoxidase (PAP) technique. Specimens of normal oral mucosa revealed frequent positive staining for polyclonal HSY-2 protein marker, thus indicating that HSY-2 is not an uncommon inhabitant of the oral mucosa. Only highly differentiated grades of oral squamous cell carcinoma showed positive staining for HSY-2, while less differentiated carcinomas failed to reveal any immunoreactivity. This raised the suggestion of a possible causative role played by HSY-2 in the establishment of the neoplastic process in oral squamous cell carcinoma through a “hit and run” mechanism. The present results point to HSY-2 as a promoter or an initiator in early neoplastic changes in well-differentiated oral squamous cell carcinoma that becomes denatured and consequently not evident in less highly differentiated tumours.
Jalouli et al. [155], using PCR/DNA sequencing, investigated the prevalence of human papillomavirus (HPV), herpes simplex virus (HSV), and Epstein-Barr virus (EBV) DNA in brush biopsies obtained from 150 users of Sudanese snuff (toombak) and 25 nonusers of toombak in formalin-fixed paraffin-embedded tissue samples obtained from 31 patients with oral dysplasias (25 toombak users and 6 nonusers) and from 217 patients with oral cancers (145 toombak users and 72 nonusers). In the brush tissue samples from toombak users, HPV was detected in 60 (40%), HSV in 44 (29%), and EBV in 97 (65%) of the samples. The corresponding figures for the 25 samples from non-users were 17 (68%) positive for HPV, 6 (24%) positive for HSV and 21 (84%) for EBV. The formalin-fixed samples with oral dysplasias were all negative for HPV. In the 145 oral cancer samples from toombak users, HPV was detected in 39 (27%), HSV in 15 (10%), and EBV in 53 (37%) of the samples. The corresponding figures for the samples from nonusers were 15 (21%) positive for HPV, 5 (7%) for HSV and 16 (22%) for EBV. These findings illustrate that prevalence of HSV, HPV, and EBV infections is common and may influence oral health and cancer development.
Delavarian et al. [156] investigated the presence of viruses in oral squamous cell carcinoma (OSCC) in young patients (20–40 years old) attending Mashhad Dental Faculty from 1996 to 2009 for the first time in Iranian population. Twenty-one formalin-fixed, paraffin-embedded sections of patients under 40 years with clinical diagnosis of OSCC, who had been referred to Mashhad Dental Faculty from 1996 and 2009, were evaluated for DNA extraction. All specimens were tested for presence of human papillomavirus, Epstein-Barr virus, Herpes simplex virus type 1, and cytomegalovirus virus. From 21 specimens, viruses were detected only in three cases. Two samples were positive for EBV and the third one was coinfected with EBV and HSV 1. All specimens were negative for HPV and CMV. Authors concluded that viruses had no important role in OSCC in young patients. Further researches are needed to clarify this role and to identify other possible risk factors.
7.1.5. Epidemiological Studies
The herpes simplex viruses (HSVs), types 1 and 2, have been investigated in the past for possible associations with human cancers. Levels of antibody to HSV 1 have been reported to be higher in patients with oral cancer than in control subjects [157]. These antibodies were largely of the IgA and IgM classes, and patients with the highest levels of anti-HSV IgM had a shorter survival than other patients [158]. It has been reported that a combination of HSV seropositivity and a history of cigarette smoking is associated with a higher risk of oral cancer than would be expected from a purely additive effect [159]. Early studies attempted to find if DNA of HSV could be detected in oral cancers, and preliminary reports did indicate the presence of both viral DNA and RNA [58, 160]. However, the nature of the HSV genome makes it difficult to produce specific probes, and the possible sequences that were detected have not been identified [58].
7.2. Molecular Mechanisms of Carcinogenesis
The explanation for the association between oral cancer and HSV could well be that of a confounding variable, except for the fact that HSV can transform some animal cells to a malignant phenotype in vitro [161]. Indeed HSV has shown cocarcinogenic activity in combination with chemicals in vivo [162]. Unfortunately, the association is difficult to study, because cells that are transformed by HSV do not express specific virus antigens or retain any specific genes of the virus [163]. Instead it seems likely that the transformation is due to the virus acting as a mutagen, and a region of the viral genome has been isolated which raises the mutation frequency in cultured cells [164]. This results in some but not all of the features of malignancy [165]. Neither the mutations nor the phenotypic changes are sufficiently specific to act as markers by which a herpes-induced malignancy could be diagnosed [166]. Recent years have seen little or no progress in the study of HSV and its malignant potential.
8. Epstein-Barr Virus Infection (EBV)
EBV was named after Tony Epstein and Yvonne Barr who were the two scientists who first isolated and described the virus in 1964 from lymphoma samples collected by Denis Burkitt. The EBV genome is a double-stranded DNA molecular of approximately 172,000 base pairs; as in other herpes viruses, the molecule is divided into unique, internal repeat, and terminal repeat domains [167]. The genome encodes approximately 80 proteins. The function of many of the genes involved in viral replication has been inferred from their homology to herpes simplex virus genes; however, genes expressed during latent infection of B cells do not have recognized counterparts in other human herpes viruses.
All phases of the EBV life cycle are associated with human disease. In immunocompromised individuals, infected cells increase in number and eventually B cell growth control pathways are activated, inducing transformation and leading to malignancies such as nasopharyngeal carcinoma (NPC), Burkitt's lymphoma (BL), posttransplant lymphomas, and gastric carcinomas [168]. EBV encodes several viral proteins that have transforming potential, including EBV latent membrane proteins 1 and 2 (LMP1 and LMP2) and EBV nuclear antigens 2 and 3 (EBNA2 and EBNA3). LMP1 can transform a variety of cell types, including rodent fibroblasts [169], and is essential for the ability of EBV to immortalize B cells [170]. The multiple transmembrane-spanning domains and the carboxyl terminus of LMP1 can interact with several tumor necrosis factor receptor associated factors (TRAFs) [171, 172]; this interaction results in high levels of activity of NF-jB, Jun, and p38 in LMP1-expressing epithelial and B cells [173–175].
LMP1 also upregulates the expression of numerous antiapoptotic and adhesion genes and activates the expression of IRF-7 [176], matrix metalloproteinase-9 (MMP-9), and fibroblast growth factor-2 (FGF-2) [177]. A second viral membrane protein, LMP2, is dispensable for transformation of naïve B cells but is required for transformation of postgerminal center B cells. LMP2 interacts with Lyn and Syk to mimic B cell receptor (BCR) signaling, including activation of the PI3 K/AKT survival pathway [178]. These pathways are constitutively active and drive proliferation through a normal cell cycle cascade.
Horiuchi et al. [179] carried out a study to determine the presence of EBV in various squamous cell proliferative lesions in the oral cavity. They made use of PCR and in situ hybridization for detecting the presence of EBV DNA and EBV encoded small messenger RNA. Nearly 60% of SCC was EBV genome positive, but none of papilloma demonstrated EBV genome. On the other hand, oral hairy leukoplakia lesion seen in patients with AIDS has been proved to be EBV associated. Horiuchi et al. concluded that EBV virus infection of oral squamous epithelium may be carcinogenic or, alternatively, the virus may merely exist in epithelial cells of squamous cell carcinoma, carcinoma in situ, and leukoplakia.
González-Moles et al. [180] showed a positive correlation between different grades of OSCC and EBV DNA positivity and also showed that percentage positivity of EBV DNA increases from well-differentiated OSCC to poorly differentiated OSCC. In a study by Higa et al. [181], fifty-four patients with oral squamous cell carcinoma reported from 1997 to 1999 in Okinawa were compared with 21 and 20 patients from Kitakyushu and Kumamoto in Kyushu, mainland Japan, respectively. 51 diagnoses were confirmed by conventional histological examination of paraffin wax sections. EBV was detected by nonisotopic in situ hybridization and PCR (Bam HI-F, EBV nuclear antigen 2 (EBNA2), and latent membrane protein 1 (LMP-1) regions). Sequence analysis of the PCR products was also carried out. In Okinawa, 25 patients were found to be infected with EBV type A by analyzing the 3-sequence divergence of the EBNA2 genes. Six patients were positive for EBV type B and 8 for both types A and B. Therefore, type A virus infection was demonstrated in 33 of 54 patients and type B in 14 of 54. In total, 39 of 54 patients were infected with EBV. Authors concluded that, in Okinawa, EBV infection was frequently demonstrated in oral squamous cell carcinoma (P < 0.001). However, in mainland Japan, there was no significant correlation between EBV and oral squamous cell carcinoma [181].
A study by Li et al. [182] to detect EBV infection and gene expression in oral cancer from patients in Taiwan by microarray analysis revealed that the majority of the specimens (82.5%) were EBV positive that probably expressed coincidently the genes for EBNAs, LMP2A, and 2B and certain structural proteins. Importantly, the genes fabricated at the spots 61 (BBRF1, BBRF2, and BBRF3) and 68 (BDLF4 and BDRF1) on EBV-chip were actively expressed in a significantly greater number of OSCC exhibiting exophytic morphology or ulceration than those tissues with deep invasive lesions.
In a study conducted by Sand et al. [183] examining 29 patients with OSCC, 23 with OLP, and 67 with clinically healthy oral mucosa, a nested polymerase chain reaction method for EBV DNA analysis was used. The overall EBV prevalence in patients with oral disease was 32.1%. Of the OSCC patients, 37.9% were EBV positive, and, of the OLP patients, 26.1% were EBV positive. Both percentages were statistically significant compared with that of control patients (7.3%). The difference in EBV prevalence between the smoking control group and the nonsmoking control group was without significance. Increased age did not enhance EBV prevalence. The authors were of the opinion that EBV is present in oral diseases such as OSCC and OLP. Smoking, alcohol use, or age does not seem to be a risk factor for EBV infection.
Jiang et al. [184] isolated epithelial cells by laser capture microdissection and levels of CD21, CK19, and EBV RNA were measured by quantitative reverse transcriptase PCR in oral dysplasia and squamous cell carcinoma. Results showed that expression of CD21 increased in frequency and intensity as oral epithelial cells become more dysplastic and that expression correlates with an increase in infection by EBV. Tumors or dysplastic lesions that carry EBV also generally express higher levels of CK19 than those that do not. Hence, the findings suggest that dysplasia may make cells more susceptible to infection by EBV and that infection by the virus may alter the phenotype of the infected cell in a manner which could affect prognosis.
9. Hepatitis C Virus
The worldwide prevalence of hepatitis C virus (HCV) is estimated to be around 3%, representing approximately 170 million infected individuals. Morbidity associated with HCV infection is due to not only the sequelae of chronic liver disease but also a variety of extra hepatic manifestations, including that involving the oral cavity [185].
Hepatitis C virus (HCV) is an enveloped, single-stranded positive-sense RNA virus that was isolated in 1989 from a chimpanzee chronically infected by contamination with a human factor VIII concentrate (Choo et al.) [186].
There are six HCV genotypes and more than 100 subtypes. Its envelope contains two glycoproteins, E1 and E2, which form heterodimers at the surface of the virion. The genomic RNA is translated into a viral polyprotein which is cleaved by cellular proteases to generate the capsid protein (C), the two glycoproteins E1 and E2, a small protein the role of which is unclear (p7), viral proteases NS2 and NS3, and nonstructural proteins NS4A and 4B and NS5A and 5B, which are required for viral RNA replication [187].
Although details of HCV replication are not known, it is thought to take place in the cytoplasm, where, among other products, negative-sense viral RNA (vRNA), replicative double-stranded forms, and nonstructural proteins are synthesized. Their presence can thus be used as evidence that virus multiplication, as opposed to passive transportation, is occurring (Negro et al.) [188]. Furthermore, sequence variants forming a quasispecies may circulate within an individual, possibly as a consequence of ongoing immune surveillance and viral mutations (Toyoda et al.) [189].
Johnson et al. suggested a possible involvement of HCV in diseases outside the liver and concluded that since oral cavity is frequently exposed to HCV viruses, this in turn increases the risk of genetic instability in these cells [185]. Nagao et al. studied a group of 100 patients including 88 SCC. Anti-HCV antibodies were detected in sera of 25 patients [190]. The exact mechanism is unclear. Gandolfo et al. found very high prevalence of anti-HCV antibodies in patients with oral lichen planus (OLP) [191]. Nagao et al. suggested that, since OLP is also histologically a disease of squamous cells, the squamous cells of oral region are continuously exposed to HCV from saliva as well as from serum in HCV-positive patients and that this may be involved in the development of SCC and OLP in these patients [190].
10. Conclusion
Oral cancer is an important cause of morbidity and mortality, especially in developing countries, and its prevalence may rise in the foreseeable future. The studies of virus-associated head and neck cancers have provided many critical insights into key mechanisms of carcinogenesis. The human tumor virus oncogenes play central roles in viral life cycles and their oncogenic potential is a manifestation of these activities.
Some viruses, most notably the high-risk HPVs, play essential roles in the initiation as well as progression of cancers and continued expression of their viral transforming activities is necessary for the maintenance of the transformed phenotype. Recent reviews establish the increased evidence of HPV-related oral cavity cancer in man.
Standardization of the methods for sample collection and analysis is mandatory to obtain reliable data and to compare the results obtained in different studies in the presence of HPV in variable proportions in oral squamous cell carcinoma tissues. Some tumors are associated with papillomaviruses and some with viruses of the herpes family; however, the exact role of these viruses must still be evaluated carefully. These viruses may provide targets for therapy and for diagnostic tests and may widen our understanding of the mechanisms by which the tumors develop.
Vaccines are designed strictly for prevention of such viral infections which may be involved in cancers of the head and neck. Few studies found sufficient evidence to support a global immunization program against HPV, irrespective of gender and geography, to help to achieve a reduction in HPV-related malignant diseases in the future. Clearly, large, prospective randomized trials are needed to document the clinical usefulness of these vaccines against oral cancer.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Javier RT, Butel JS. The history of tumor virology. Cancer Research. 2008;68(19):7693–7706. doi: 10.1158/0008-5472.CAN-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McLaughlin-Drubin ME, Munger K. Viruses associated with human cancer. Biochimica et Biophysica Acta. 2008;1782(3):127–150. doi: 10.1016/j.bbadis.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gondivkar SM, Parikh RV, Gadbail AR, et al. Involvement of viral factors with head and neck cancers. Oral Oncology. 2012;48(3):195–199. doi: 10.1016/j.oraloncology.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Greer RO, Jr., Douglas JM, Jr., Breese P, Crosby LK. Evaluation of oral and laryngeal specimens for human papillomavirus (HPV) DNA by dot blot hybridization. Journal of Oral Pathology and Medicine. 1990;19(1):35–38. doi: 10.1111/j.1600-0714.1990.tb00778.x. [DOI] [PubMed] [Google Scholar]
- 5.Greer RO, Shroyer K, Crosby L. NIH Publication No. 92-3461. Bethesda, Md, USA: US Department of Health and Human Services, Public Health Service, National Institutes of Health; 1992. Identification of human papillomavirus DNA in smokeless tobacco keratoses and premalignant and malignant oral lesions by PCR amplification with consensus sequence primers. [Google Scholar]
- 6.Park NH, Byung MM, Sheng LL. NIH Publication No. 92-3461. Bethesda, Md, USA: US Department of Health and Human Services, Public Health Service, National Institutes of Health; 1992. Role of viruses in oral carcinogenesis. [Google Scholar]
- 7.Johnson NW. Risk Markers for Oral Disease, Oral Cancer Detection of Patients and Lesions at Risk. Cambridge, UK: Cambridge University Press; 1991. [Google Scholar]
- 8.Nagao Y, Sata M, Tanikawa K, Itoh K, Kameyama T. High prevalence of hepatitis C virus antibody and RNA in patients with oral cancer. Journal of Oral Pathology and Medicine. 1995;24(8):354–360. doi: 10.1111/j.1600-0714.1995.tb01199.x. [DOI] [PubMed] [Google Scholar]
- 9.Porter SR, Lodi G, Chandler K, Kumar N. Development of squamous cell carcinoma in hepatitis C virus-associated lichen planus. Oral Oncology. 1997;33(1):58–59. doi: 10.1016/s0964-1955(96)00041-3. [DOI] [PubMed] [Google Scholar]
- 10.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 11.Cohen SM. Microbes and MAlignancy: Infection as a Cause of Human Cancers. Oxford, UK: Oxford University Press; 1999. pp. 89–106. J. Parsonnet (Ed.) [Google Scholar]
- 12.Koch R. Vernhandlungen des X Internationalen Medicinischen Congresses; 1890; Berlin. pp. 35–47. [Google Scholar]
- 13.Evans AS, Mueller NE. Viruses and cancer. Causal associations. Annals of Epidemiology. 1990;1(1):71–92. doi: 10.1016/1047-2797(90)90020-s. [DOI] [PubMed] [Google Scholar]
- 14.HILL AB. The environment and disease: association or causation? Proceedings of the Royal Society of Medicine. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill AB, Hill ID. Bradford Hill's Principles of Medical Statistics. 12th edition. London, UK: Edward Arnold; 1991. [Google Scholar]
- 16.Zur Hausen HZ, De Villiers E-M. Human papillomaviruses. Annual Review of Microbiology. 1994;48:427–447. doi: 10.1146/annurev.mi.48.100194.002235. [DOI] [PubMed] [Google Scholar]
- 17.Ragin CCR, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. Journal of Dental Research. 2007;86(2):104–114. doi: 10.1177/154405910708600202. [DOI] [PubMed] [Google Scholar]
- 18.Yabe Y, Sadakane H, Isono H. Connection between capsomeres in human papilloma virus. Virology. 1979;96(2):547–552. doi: 10.1016/0042-6822(79)90110-7. [DOI] [PubMed] [Google Scholar]
- 19.Gissmann L, Pfister H, Zur Hausen H. Human papilloma viruses (HPV): characterization of four different isolates. Virology. 1977;76(2):569–580. doi: 10.1016/0042-6822(77)90239-2. [DOI] [PubMed] [Google Scholar]
- 20.Danos O, Katinka M, Yaniv M. Human papillomavirus 1a complete DNA sequence: a novel type of genome organization among papovaviridae. The EMBO Journal. 1982;1(2):231–236. doi: 10.1002/j.1460-2075.1982.tb01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz E, Dürst M, Demankowski C, et al. DNA sequence and genome organization of genital human papillomavirus type 6b. The EMBO Journal. 1983;2(12):2341–2348. doi: 10.1002/j.1460-2075.1983.tb01744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kremsdorf D, Favre M, Jablonska S. Molecular cloning and characterization of the genomes of nine newly recognized human papillomavirus types associated with epidermodysplasia verruciformis. Journal of Virology. 1984;52(3):1013–1018. doi: 10.1128/jvi.52.3.1013-1018.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seedorf K, Krammer G, Durst M. Human papillomavirus type 16 DNA sequence. Virology. 1985;145(1):181–185. doi: 10.1016/0042-6822(85)90214-4. [DOI] [PubMed] [Google Scholar]
- 24.Munoz N, Bosch FX, Castellsague X. Against which human papillomavirus types shall we vaccinate and screen? The international perspective. International Journal of Cancer. 2004;111:278–285. doi: 10.1002/ijc.20244. [DOI] [PubMed] [Google Scholar]
- 25.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. The Lancet. 2007;370(9590):890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 26.Zur Hausen H, De Villiers EM, Gissmann L. Papillomavirus infections and human genital cancer. Gynecologic Oncology. 1981;12(2):S124–S128. doi: 10.1016/0090-8258(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 27.Bosch FX, Lorincz A, Muñoz N, Meijer CJLM, Shah KV. The causal relation between human papillomavirus and cervical cancer. Journal of Clinical Pathology. 2002;55(4):244–265. doi: 10.1136/jcp.55.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson IOC, van der Bijl P, van Wyk CW, van Eyk AD. A comparative light-microscopic, electron-microscopic and chemical study of human vaginal and buccal epithelium. Archives of Oral Biology. 2001;46(12):1091–1098. doi: 10.1016/s0003-9969(01)00082-6. [DOI] [PubMed] [Google Scholar]
- 29.Syrjänen KJ, Pyrhönen S, Syrjänen SM, Lamberg MA. Immunohistochemical demonstration of human Papilloma virus (HPV) antigens in oral squamous cell lesions. British Journal of Oral Surgery. 1983;21(2):147–153. doi: 10.1016/0007-117x(83)90060-4. [DOI] [PubMed] [Google Scholar]
- 30.Gillison ML, Koch WM, Capone RB, et al. Evidence for a causal association between human papilloma virus and a subset of head and neck cancers. Journal of the National Cancer Institute. 2000;92(9):709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- 31.Smith EM, Hoffman HT, Summersgill KS, Kirchner HL, Turek LP, Haugen TH. Human papillomavirus and risk of oral cancer. Laryngoscope. 1998;108(7):1098–1103. doi: 10.1097/00005537-199807000-00027. [DOI] [PubMed] [Google Scholar]
- 32.Miller CS, Johnstone BM. Human papillomavirus as a risk factor for oral squamous cell carcinoma: a meta-analysis, 1982–1997. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2001;91(6):622–635. doi: 10.1067/moe.2001.115392. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz SM, Daling JR, Doody DR, et al. Oral cancer risk in relation to sexual history and evidence of human papillomavirus infection. Journal of the National Cancer Institute. 1998;90(21):1626–1636. doi: 10.1093/jnci/90.21.1626. [DOI] [PubMed] [Google Scholar]
- 34.Bjorge T, Hennig EM, Skare GB, Soreide O, Thoresen SO. Second primary cancers in patients with carcinoma in situ of the uterine cervix. The Norwegian experience 1970–1992. International Journal of Cancer. 1995;62(1):29–33. doi: 10.1002/ijc.2910620108. [DOI] [PubMed] [Google Scholar]
- 35.Rabkin CS, Biggar RJ, Melbye M, Curtis RE. Second primary cancers following anal and cervical carcinoma: evidence of shared etiologic factors. American Journal of Epidemiology. 1992;136(1):54–58. doi: 10.1093/oxfordjournals.aje.a116420. [DOI] [PubMed] [Google Scholar]
- 36.Frisch M, Biggar RJ. Aetiological parallel between tonsillar and anogenital squamous-cell carcinomas. The Lancet. 1999;354(9188):1442–1443. doi: 10.1016/S0140-6736(99)92824-6. [DOI] [PubMed] [Google Scholar]
- 37.Hemminki K, Dong C, Frisch M. Tonsillar and other upper aerodigestive tract cancers among cervical cancer patients and their husbands. European Journal of Cancer Prevention. 2000;9(6):433–437. doi: 10.1097/00008469-200012000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Syrjänen S. Current concepts on human papillomavirus infections in children. APMIS. 2010;118(6-7):494–509. doi: 10.1111/j.1600-0463.2010.02620.x. [DOI] [PubMed] [Google Scholar]
- 39.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 40.Scheffner M, Whitaker NJ. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system. Seminars in Cancer Biology. 2003;13(1):59–67. doi: 10.1016/s1044-579x(02)00100-1. [DOI] [PubMed] [Google Scholar]
- 41.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 42.Reddel RR. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Letters. 2003;194(2):155–162. doi: 10.1016/s0304-3835(02)00702-4. [DOI] [PubMed] [Google Scholar]
- 43.Damania B. DNA tumor viruses and human cancer. Trends in Microbiology. 2007;15(1):38–44. doi: 10.1016/j.tim.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 44.Kim RH, Kang MK, Shin K-H, et al. Bmi-1 cooperates with human papillomavirus type 16 E6 to immortalize normal human oral keratinocytes. Experimental Cell Research. 2007;313(3):462–472. doi: 10.1016/j.yexcr.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 45.Liu X, Disbrow GL, Yuan H, Tomaić V, Schlegel R. Myc and human papillomavirus type 16 E7 genes cooperate to immortalize human keratinocytes. Journal of Virology. 2007;81(22):12689–12695. doi: 10.1128/JVI.00669-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chien W-M, Noya F, Benedict-Hamilton HM, Broker TR, Chow LT. Alternative fates of keratinocytes transduced by human papillomavirus type 18 E7 during squamous differentiation. Journal of Virology. 2002;76(6):2964–2972. doi: 10.1128/JVI.76.6.2964-2972.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang SG, Lee D, Kim J, Seo T, Choe J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. Journal of Biological Chemistry. 2002;277(4):2923–2930. doi: 10.1074/jbc.M109113200. [DOI] [PubMed] [Google Scholar]
- 48.Loning T, Ikenberg H, Becker J. Analysis of oral papillomas, leukoplakias, and invasive carcinomas for human papillomavirus type related DNA. Journal of Investigative Dermatology. 1985;84(5):417–420. doi: 10.1111/1523-1747.ep12265517. [DOI] [PubMed] [Google Scholar]
- 49.Maitland NJ, Cox MF, Lynas C, Prime SS, Meanwell CA, Scully C. Detection of human papillomavirus DNA in biopsies of human oral tissue. British Journal of Cancer. 1987;56(3):245–250. doi: 10.1038/bjc.1987.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gassenmaier A, Hornstein OP. Presence of human papillomavirus DNA in benign and precancerous oral leukoplakias and squamous cell carcinomas. Dermatologica. 1988;176(5):224–233. doi: 10.1159/000248710. [DOI] [PubMed] [Google Scholar]
- 51.Syrjänen SM, Syrjänen KJ, Happonen RP. Human papillomavirus (HPV) DNA sequences in oral precancerous lesions and squamous cell carcinoma demonstrated by in situ hybridization. Journal of Oral Pathology. 1988;17(6):273–278. doi: 10.1111/j.1600-0714.1988.tb01536.x. [DOI] [PubMed] [Google Scholar]
- 52.Chang F, Syrjanen S, Nuutinen J, Karja J, Syrjanen K. Detection of human papillomavirus (HPV) DNA in oral squamous cell carcinomas by in sity hybridization and polymerase chain reaction. Archives of Dermatological Research. 1990;282(8):493–497. doi: 10.1007/BF00371942. [DOI] [PubMed] [Google Scholar]
- 53.Greer RO, Jr., Eversole LR, Crosby LK. Detection of human papillomavirus-genomic DNA in oral epithelial dysplasias, oral smokeless tobacco-associated leukoplakias, and epithelial malignancies. Journal of Oral and Maxillofacial Surgery. 1990;48(11):1201–1205. doi: 10.1016/0278-2391(90)90538-d. [DOI] [PubMed] [Google Scholar]
- 54.Kashima HK, Levin LS, Kutcher M, De Villiers E-M, Kessis T, Shah K. Human papillomavirus in squamous cell carcinoma, leukoplakia, lichen planus, and clinically normal epithelium of the oral cavity. Annals of Otology, Rhinology and Laryngology. 1990;99(1):55–61. doi: 10.1177/000348949009900110. [DOI] [PubMed] [Google Scholar]
- 55.Young SK, Min KW. In situ DNA hybridization analysis of oral papillomas, leukoplakias, and carcinomas for human papillomavirus. Oral Surgery Oral Medicine and Oral Pathology. 1991;71(6):726–9. doi: 10.1016/0030-4220(91)90282-h. [DOI] [PubMed] [Google Scholar]
- 56.Zeuss MS, Miller CS, White DK. In situ hybridization analysis of human papillomavirus DNA in oral mucosal lesions. Oral Surgery Oral Medicine and Oral Pathology. 1991;71(6):714–720. doi: 10.1016/0030-4220(91)90280-p. [DOI] [PubMed] [Google Scholar]
- 57.Holladay EB, Gerald WL. Viral gene detection in oral neoplasms using the polymerase chain reaction. American Journal of Clinical Pathology. 1993;100(1):36–40. doi: 10.1093/ajcp/100.1.36. [DOI] [PubMed] [Google Scholar]
- 58.Cox M, Maitland N, Scully C. Human herpes simplex-1 and papillomavirus type 16 homologous DNA sequences in normal, potentially malignant and malignant oral mucosa. European Journal of Cancer B. 1993;29(3):215–219. doi: 10.1016/0964-1955(93)90025-a. [DOI] [PubMed] [Google Scholar]
- 59.Brandwein M, Zeitlin J, Nuovo GJ, et al. HPV detection using “hot start” polymerase chain reaction in patients with oral cancer: a clinicopathological study of 64 patients. Modern Pathology. 1994;7(7):720–727. [PubMed] [Google Scholar]
- 60.González-Moles MA, Ruiz-Avila I, González-Moles S, Martinez I, Ceballos A, Nogales F. Detection of HPV DNA by in situ hybridization in benign, premalignant and malignant lesions of the oral mucosa. Bulletin du Groupement International pour la Recherche Scientifique en Stomatologie & Odontologie. 1994;37(3-4):79–85. [PubMed] [Google Scholar]
- 61.Ostwald C, Muller P, Barten M, et al. Human papillomavirus DNA in oral squamous cell carcinomas and normal mucosa. Journal of Oral Pathology and Medicine. 1994;23(5):220–225. doi: 10.1111/j.1600-0714.1994.tb01117.x. [DOI] [PubMed] [Google Scholar]
- 62.Balaram P, Nalinakumari KR, Abraham E, et al. Human papillomaviruses in 91 oral cancers from indican betel quid chewers—High prevalence and multiplicity of infections. International Journal of Cancer. 1995;61(4):450–454. doi: 10.1002/ijc.2910610403. [DOI] [PubMed] [Google Scholar]
- 63.Shindoh M, Chiba I, Yasuda M, et al. Detection of human papillomavirus DNA sequences in oral squamous cell carcinomas and their relation to p53 and proliferating cell nuclear antigen expression. Cancer. 1995;76:1513–1521. doi: 10.1002/1097-0142(19951101)76:9<1513::aid-cncr2820760903>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 64.van Rensburg EJ, van Heerden WFP, Venter EH, Raubenheimer EJ. Detection of human papillomavirus DNA with in situ hybridisation in oval squamous carcinoma in a rural black population. South African Medical Journal. 1995;85(9):894–896. [PubMed] [Google Scholar]
- 65.Cruz IBF, Snijders PJF, Steenbergen RDM, et al. Age-dependence of human papillomavirus DNA presence in oral squamous cell carcinomas. European Journal of Cancer B. 1996;32(1):55–62. doi: 10.1016/0964-1955(95)00060-7. [DOI] [PubMed] [Google Scholar]
- 66.Mao EJ, Schwartz SM, Daling JR, et al. Human papilloma viruses and p53 mutations in normal pre-malignant and malignant oral epithelia. International Journal of Cancer. 1996;69:152–158. doi: 10.1002/(SICI)1097-0215(19960422)69:2<152::AID-IJC15>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 67.van Rensburg EJ, Engelbrecht S, van Heerden WFP, Raubennheimer EJ, Schoub BD. Human papillomavirus DNA in oral squamous cell carcinomas from an African population sample. Anticancer Research. 1996;16(2):969–973. [PubMed] [Google Scholar]
- 68.Chiba I, Shindoh M, Yasuda M, et al. Mutations in the p53 gene and human papillomavirus infection as significant prognostic factors in squamous cell carcinomas of the oral cavity. Oncogene. 1996;12(8):1663–1668. [PubMed] [Google Scholar]
- 69.Wen S, Tsuji T, Li X, Mizugaki Y, Hayatsu Y, Shinozaki F. Detection and analysis of human papillomavirus 16 and 18 homologous DNA sequences in oral lesions. Anticancer Research. 1997;17(1 A):307–311. [PubMed] [Google Scholar]
- 70.Gopalakrishnan R, Weghorst CM, Lehman TA, et al. Mutated and wild-type p53 expression and HPV integration in proliferative verrucous leukoplakia and oral squamous cell carcinoma. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 1997;83(4):471–477. doi: 10.1016/s1079-2104(97)90148-7. [DOI] [PubMed] [Google Scholar]
- 71.Ibrahim SO, Bertelsen B, Kalvenes MB, et al. Expression of keratin 13, 14 and 19 in oral squamous cell carcinomas from Sudanese snuff dippers: lack of association with human papillomavirus infection. APMIS. 1998;106(10):959–969. doi: 10.1111/j.1699-0463.1998.tb00246.x. [DOI] [PubMed] [Google Scholar]
- 72.Koh JY, Cho NP, Kong G, Lee JD, Yoon K. p53 mutations and human papillomavirus DNA in oral squamous cell carcinoma: correlation with apoptosis. British Journal of Cancer. 1998;78(3):354–359. doi: 10.1038/bjc.1998.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Premoli-De-Percoco G, Ramírez JL, Galindo I. Correlation between HPV types associated with oral squamous cell carcinoma and cervicovaginal cytology: an in situ hybridization study. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 1998;86(1):77–81. doi: 10.1016/s1079-2104(98)90153-6. [DOI] [PubMed] [Google Scholar]
- 74.D’Costa J, Saranath D, Dedhia P, Sanghvi V, Mehta AR. Detection of HPV-16 genome in human oral cancers and potentially malignant lesions from India. Oral Oncology. 1998;34(5):413–420. doi: 10.1016/s1368-8375(98)00028-1. [DOI] [PubMed] [Google Scholar]
- 75.Elamin F, Steingrimsdottir H, Wanakulasuriya S, Johnson N, Tavassoli M. Prevalence of human papillomavirus infection in premalignant and malignant lesions of the oral cavity in U.K. subjects: a novel method of detection. Oral Oncology. 1998;34(3):191–197. doi: 10.1016/s1368-8375(97)00081-x. [DOI] [PubMed] [Google Scholar]
- 76.Aggelopoulou EP, Skarlos D, Papadimitriou C, Kittas C, Troungos C. Human papilloma virus DNA detection in oral lesions in the Greek population. Anticancer Research. 1999;19(2B):1391–1395. [PubMed] [Google Scholar]
- 77.Pillai MR, Phanidhara A, Kesari AL, et al. Cellular manifestations of human papillomavirus infection in the oral mucosa. Journal of Surgical Oncology. 1999;71:10–15. doi: 10.1002/(sici)1096-9098(199905)71:1<10::aid-jso3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 78.Pintos J, Franco EL, Black MJ, Bergeron J, Arella M. Human papillomavirus and prognoses of patients with cancers of the upper aerodigestive tract. Cancer. 1999;85:1903–1909. doi: 10.1002/(sici)1097-0142(19990501)85:9<1903::aid-cncr4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 79.Badaracco G, Venuti A, Morello R, Muller A, Marcante ML. Human papillomavirus in head and neck carcinomas: prevalence, physical status and relationship with clinical/pathological parameters. Anticancer Research. 2000;20(2 B):1301–1305. [PubMed] [Google Scholar]
- 80.Bouda M, Gorgoulis VG, Kastrinakis NG, et al. ‘High risk’ HPV types are frequently detected in potentially malignant and malignant oral lesions, but not in normal oral mucosa. Modern Pathology. 2000;13(6):644–653. doi: 10.1038/modpathol.3880113. [DOI] [PubMed] [Google Scholar]
- 81.Cao J, Zhang ZY, Patima P, Zhang YX, Chen WT. Human papillomavirus infection and p53 alteration in oral squamous cell carcinoma. The Chinese Journal of Dental Research. 2000;3(3):44–49. [PubMed] [Google Scholar]
- 82.Patima Cao J, Chen WT, Zhang ZY. Detection of high risk human papillomavirus DNA in oral squamous cell carcinoma. Shanghai Kou Qiang Yi Xue. 2000;9:212–215. [PubMed] [Google Scholar]
- 83.Sand L, Jalouli J, Larsson P-A, Hirsch J-M. Human papilloma viruses in oral lesions. Anticancer Research. 2000;20(2B):1183–1188. [PubMed] [Google Scholar]
- 84.Tsuhako K, Nakazato I, Miyagi J, et al. Comparative study of oral squamous cell carcinoma in Okinawa, Southern Japan and Sapporo in Hokkaido, Northern Japan; with special reference to human papillomavirus and Epstein-Barr virus infection. Journal of Oral Pathology and Medicine. 2000;29(2):70–79. doi: 10.1034/j.1600-0714.2000.290204.x. [DOI] [PubMed] [Google Scholar]
- 85.Premoli-De-Percoco G, Ramirez JL. High risk human papillomavirus in oral squamous carcinoma: evidence of risk factors in a Venezuelan rural population. Preliminary report. Journal of Oral Pathology and Medicine. 2001;30(6):355–361. doi: 10.1034/j.1600-0714.2001.300605.x. [DOI] [PubMed] [Google Scholar]
- 86.Schwartz SR, Yueh B, McDougall JK, Daling JR, Schwartz SM. Human papillomavirus infection and survival in oral squamous cell cancer: a population-based study. Otolaryngology. 2001;125(1):1–9. doi: 10.1067/mhn.2001.116979. [DOI] [PubMed] [Google Scholar]
- 87.Chen PC-H, Kuo C, Pan C-C, Chou M-Y. Risk of oral cancer associated with human papillomavirus infection, betel quid chewing, and cigarette smoking in Taiwan—an integrated molecular and epidemiological study of 58 cases. Journal of Oral Pathology and Medicine. 2002;31(6):317–322. doi: 10.1034/j.1600-0714.2002.00129.x. [DOI] [PubMed] [Google Scholar]
- 88.Kojima A, Maeda H, Sugita Y, Tanaka S, Kameyama Y. Human papillomavirus type 38 infection in oral squamous cell carcinomas. Oral Oncology. 2002;38(6):591–596. doi: 10.1016/s1368-8375(01)00112-9. [DOI] [PubMed] [Google Scholar]
- 89.Nagpal JK, Patnaik S, Das BR. Prevalence of high-risk human papilloma virus types and its association with p53 codon 72 polymorphism in tobacco addicted oral squamous cell carcimona (OSCC) patients of Eastern India. International Journal of Cancer. 2002;97(5):649–653. doi: 10.1002/ijc.10112. [DOI] [PubMed] [Google Scholar]
- 90.Chang JY-F, Lin M-C, Chiang C-P. High-risk human papillomaviruses may have an important role in non-oral habits-associated oral squamous cell Carcinomas in Taiwan. American Journal of Clinical Pathology. 2003;120(6):909–916. doi: 10.1309/C5P6-NUQ2-NW6L-CTBP. [DOI] [PubMed] [Google Scholar]
- 91.Fregonesi PAG, Teresa DB, Duarte RA, Neto CB, De Oliveira MRB, Soares CP. p16INK4A immunohistochemical overexpression in premalignant and malignant oral lesions infected with human papillomavirus. Journal of Histochemistry and Cytochemistry. 2003;51(10):1291–1297. doi: 10.1177/002215540305101006. [DOI] [PubMed] [Google Scholar]
- 92.Kansky AA, Poljak M, Seme K, et al. Human papillomavirus DNA in oral squamous cell carcinomas and normal oral mucosa. Acta Virologica. 2003;47(1):11–16. [PubMed] [Google Scholar]
- 93.Ritchie JM, Smith EM, Summersgill KF, et al. Human papillomavirus infection as a prognostic factor in carcinomas of the oral cavity and oropharynx. International Journal of Cancer. 2003;104(3):336–344. doi: 10.1002/ijc.10960. [DOI] [PubMed] [Google Scholar]
- 94.Sugiyama M, Bhawal UK, Dohmen T, Ono S, Miyauchi M, Ishikawa T. Detection of human papillomavirus-16 and HPV-18 DNA in normal, dysplastic, and malignant oral epithelium. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2003;95(5):594–600. doi: 10.1067/moe.2003.36. [DOI] [PubMed] [Google Scholar]
- 95.Herrero R, Castellsagué X, Pawlita M, et al. Human papillomavirus and oral cancer: the international agency for research on cancer multicenter study. Journal of the National Cancer Institute. 2003;95(23):1772–1783. doi: 10.1093/jnci/djg107. [DOI] [PubMed] [Google Scholar]
- 96.Ostwald C, Rutsatz K, Schweder J, Schmidt W, Gundlach K, Barten M. Human papillomavirus 6/11, 16 and 18 in oral carcinomas and benign oral lesions. Medical Microbiology and Immunology. 2003;192(3):145–148. doi: 10.1007/s00430-002-0161-y. [DOI] [PubMed] [Google Scholar]
- 97.Correnti M, Rivera H, Cavazza ME. Detection of human papillomaviruses of high oncogenic potential in oral squamous cell carcinoma in a Venezuelan population. Oral Diseases. 2004;10(3):163–166. doi: 10.1046/j.1601-0825.2003.00989.x. [DOI] [PubMed] [Google Scholar]
- 98.Dahlgren L, Dahlstrand H, Lindquist D, et al. Human papillomavirus is more common in base of tongue than in mobile tongue cancer and is a favorable prognostic factor in base of tongue cancer patients. International Journal of Cancer. 2004;112(6):1015–1019. doi: 10.1002/ijc.20490. [DOI] [PubMed] [Google Scholar]
- 99.Smith EM, Ritche JM, Summersgill KF, et al. Human papillomavirus in oral exfoliated cells and risk of head and neck cancer. Journal of the National Cancer Institute. 2004;96(6):449–455. doi: 10.1093/jnci/djh074. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Z-Y, Sdek P, Cao J, Chen W-T. Human papillomavirus type 16 and 18 DNA in oral squamous cell carcinoma and normal mucosa. International Journal of Oral and Maxillofacial Surgery. 2004;33(1):71–74. doi: 10.1054/ijom.2002.0443. [DOI] [PubMed] [Google Scholar]
- 101.Yang Y-Y, Koh L-W, Tsai J-H, et al. Involvement of viral and chemical factors with oral cancer in Taiwan. Japanese Journal of Clinical Oncology. 2004;34(4):176–183. doi: 10.1093/jjco/hyh037. [DOI] [PubMed] [Google Scholar]
- 102.Koppikar P, De Villiers E-M, Mulherkar R. Identification of human papillomaviruses in tumors of the oral cavity in an Indian community. International Journal of Cancer. 2005;113(6):946–950. doi: 10.1002/ijc.20664. [DOI] [PubMed] [Google Scholar]
- 103.Lo Muzio L, D’Angelo M, Procaccini M, et al. Expression of cell cycle markers and human papillomavirus infection in oral squamous cell carcinoma: Use of fuzzy neural networks. International Journal of Cancer. 2005;115(5):717–723. doi: 10.1002/ijc.20940. [DOI] [PubMed] [Google Scholar]
- 104.Boy S, Rensburg EJV, Engelbrecht S, Dreyer L, van Heerden M, van Heerden W. HPV detection in primary intra-oral squamous cell carcinomas—commensal, aetiological agent or contamination? Journal of Oral Pathology and Medicine. 2006;35(2):86–90. doi: 10.1111/j.1600-0714.2006.00385.x. [DOI] [PubMed] [Google Scholar]
- 105.El-Mofty SK, Patil S. Human papillomavirus (HPV)-related oropharyngeal nonkeratinizing squamous cell carcinoma: characterization of a distinct phenotype. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontology. 2006;101(3):339–345. doi: 10.1016/j.tripleo.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 106.Nemes JA, Deli L, Nemes Z, Márton IJ. Expression of p16INK4A, p53, and Rb proteins are independent from the presence of human papillomavirus genes in oral squamous cell carcinoma. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontology. 2006;102(3):344–352. doi: 10.1016/j.tripleo.2005.10.069. [DOI] [PubMed] [Google Scholar]
- 107.Rivero ERC, Nunes FD. HPV in oral squamous cell carcinomas of a Brazilian population: amplification by PCR. Brazilian Oral Research. 2006;20(1):21–24. doi: 10.1590/s1806-83242006000100005. [DOI] [PubMed] [Google Scholar]
- 108.Campisi G, Giovannelli L, Calvino F, et al. HPV infection in relation to OSCC histological grading and TNM stage. Evaluation by traditional statistics and fuzzy logic model. Oral Oncology. 2006;42(6):638–645. doi: 10.1016/j.oraloncology.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 109.Furrer VE, Benitez MB, Furnes M, Lanfranchi HE, Modesti NM. Biopsy versus superficial scraping: detection of human papillomavirus 6, 11, 16, and 18 in potentially malignant and malignant oral lesions. Journal of Oral Pathology and Medicine. 2006;35(6):338–344. doi: 10.1111/j.1600-0714.2006.00423.x. [DOI] [PubMed] [Google Scholar]
- 110.Koyama K, Uobe K, Tanaka A. Highly sensitive detection of HPV-DNA in paraffin sections of human oral carcinomas. Journal of Oral Pathology and Medicine. 2007;36(1):18–24. doi: 10.1111/j.1600-0714.2006.00490.x. [DOI] [PubMed] [Google Scholar]
- 111.Sugiyama M, Bhawal UK, Kawamura M, et al. Human papillomavirus-16 in oral squamous cell carcinoma: clinical correlates and 5-year survival. British Journal of Oral and Maxillofacial Surgery. 2007;45(2):116–122. doi: 10.1016/j.bjoms.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 112.Luo C-W, Roan C-H, Liu C-J. Human papillomaviruses in oral squamous cell carcinoma and pre-cancerous lesions detected by PCR-based gene-chip array. International Journal of Oral and Maxillofacial Surgery. 2007;36(2):153–158. doi: 10.1016/j.ijom.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 113.Khovidhunkit S-OP, Buajeeb W, Sanguansin S, Poomsawat S, Weerapradist W. Detection of human papillomavirus in oral squamous cell carcinoma, leukoplakia and lichen planus in Thai patients. Asian Pacific Journal of Cancer Prevention. 2008;9(4):771–775. [PubMed] [Google Scholar]
- 114.Szarka K, Tar I, Feher E, et al. Progressive increase of human papillomavirus carriage rates in potentially malignant and malignant oral disorders with increasing malignant potential. Oral Microbiology and Immunology. 2009;24(4):314–318. doi: 10.1111/j.1399-302X.2009.00516.x. [DOI] [PubMed] [Google Scholar]
- 115.Zhao D, Xu Q-G, Chen X-M, Fan M-W. Human papillomavirus as an independent predictor in oral squamous cell cancer. International journal of oral science. 2009;1(3):119–125. doi: 10.4248/IJOS.09015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Attner P, Du J, Näsman A, et al. The role of human papillomavirus in the increased incidence of base of tongue cancer. International Journal of Cancer. 2010;126(12):2879–2884. doi: 10.1002/ijc.24994. [DOI] [PubMed] [Google Scholar]
- 117.Elango KJ, Suresh A, Erode EM, et al. Role of human papilloma virus in oral tongue squamous cell carcinoma. Asian Pacific Journal of Cancer Prevention. 2011;12(4):889–896. [PubMed] [Google Scholar]
- 118.Kristoffersen AK, Enersen M, Kverndokk E, et al. Human papillomavirus subtypes in oral lesions compared to healthy oral mucosa. Journal of Clinical Virology. 2012;53(4):364–366. doi: 10.1016/j.jcv.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 119.Chen S-F, Yu F-S, Chang Y-C, Fu E, Nieh S, Lin Y-S. Role of human papillomavirus infection in carcinogenesis of oral squamous cell carcinoma with evidences of prognostic association. Journal of Oral Pathology and Medicine. 2012;41(1):9–15. doi: 10.1111/j.1600-0714.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- 120.Lee L-A, Huang C-G, Liao C-T, et al. Human papillomavirus-16 infection in advanced oral cavity cancer patients is related to an increased risk of distant metastases and poor survival. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0040767.e40767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Syrjänen KJ, Pyrhönen S, Syrjänen SM, Lamberg MA. Immunohistochemical demonstration of Human papilloma virus (HPV) antigens in oral squamous cell lesions. British Journal of Oral Surgery. 1983;21(2):147–153. doi: 10.1016/0007-117x(83)90060-4. [DOI] [PubMed] [Google Scholar]
- 122.Syrjänen K, Syrjänen S, Lamberg M. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis. International Journal of Oral Surgery. 1983;12(6):418–424. doi: 10.1016/s0300-9785(83)80033-7. [DOI] [PubMed] [Google Scholar]
- 123.Miller CS, White DK. Human papillomavirus expression in oral mucosa, premalignant conditions, and squamous cell carcinoma a retrospective review of the literature. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 1996;82(1):57–68. doi: 10.1016/s1079-2104(96)80378-7. [DOI] [PubMed] [Google Scholar]
- 124.Syrjänen SM, Syrjänen KJ. HPV infections of the oral mucosa. In: Syrjänen KJ, Syrjänen SM, editors. Papillomavirus Infections in Human Pathology. New York, NY, USA: John Wiley & Sons; 2000. pp. 379–412. [Google Scholar]
- 125.Mellin H, Friesland S, Lewensohn R, Dalianis T, MunckWikland E. Human papillomavirus (HPV) DNA in tonsillar cancer: clinical correlates, risk of relapse, and survival. International Journal of Cancer. 2000;89:300–304. [PubMed] [Google Scholar]
- 126.D’Souza G, Kreimer AR, Viscidi R, et al. Case-control study of human papillomavirus and oropharyngeal cancer. The New England Journal of Medicine. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 127.Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England Journal of Medicine. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Syrjänen S, Lodi G, von Bültzingslöwen I, et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: a systematic review. Oral Diseases. 2011;17(1):58–72. doi: 10.1111/j.1601-0825.2011.01792.x. [DOI] [PubMed] [Google Scholar]
- 129.St Guily JL, Jacquard A-C, Prétet J-L, et al. Human papillomavirus genotype distribution in oropharynx and oral cavity cancer in France–the EDiTH VI study. Journal of Clinical Virology. 2011;51(2):100–104. doi: 10.1016/j.jcv.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 130.Ziegert C, Wentzensen N, Vinokurova S, et al. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene. 2003;22(25):3977–3984. doi: 10.1038/sj.onc.1206629. [DOI] [PubMed] [Google Scholar]
- 131.Haverkos HW. Viruses, chemicals and co-carcinogenesis. Oncogene. 2004;23(38):6492–6499. doi: 10.1038/sj.onc.1207822. [DOI] [PubMed] [Google Scholar]
- 132.Migaldi M, Pecorari M, Forbicini G, et al. Low prevalence of human papillomavirus infection in the healthy oral mucosa of a Northern Italian population. Journal of Oral Pathology and Medicine. 2012;41(1):16–20. doi: 10.1111/j.1600-0714.2011.01062.x. [DOI] [PubMed] [Google Scholar]
- 133.Pannone G, Santoro A, Papagerakis S, Lo Muzio L, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infectious Agents and Cancer. 2011;6(1, article 4) doi: 10.1186/1750-9378-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Burns JE, Baird MC, Clark LJ, et al. Gene mutations and increased levels of p53 protein in human squamous cell carcinomas and their cell lines. British Journal of Cancer. 1993;67(6):1274–1284. doi: 10.1038/bjc.1993.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gillespie MB, Rubinchik S, Hoel B, Sutkowski N. Human papillomavirus and oropharyngeal cancer: what you need to know in 2009. Current Treatment Options in Oncology. 2009;10(5-6):296–307. doi: 10.1007/s11864-009-0113-5. [DOI] [PubMed] [Google Scholar]
- 136.Goon PKC, Stanley MA, Ebmeyer J, et al. HPV & head and neck cancer: a descriptive update. Head & Neck Oncology. 2009;1, article 36 doi: 10.1186/1758-3284-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kirnbauer R, Hubbert NL, Wheeler CM, Becker TM, Lowy DR, Schiller JT. A virus-like particle enzyme-linked immunosorbent assay detects serum antibodies in a majority of women infected with human papillomavirus type 16. Journal of the National Cancer Institute. 1994;86(7):494–499. doi: 10.1093/jnci/86.7.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Carter JJ, Koutsky LA, Wipf GC, et al. The natural history of human papillomavirus type 16 capsid antibodies among a cohort of university women. Journal of Infectious Diseases. 1996;174(5):927–936. doi: 10.1093/infdis/174.5.927. [DOI] [PubMed] [Google Scholar]
- 139.Meschede W, Zumbach K, Braspenning J, et al. Antibodies against early proteins of human papillomaviruses as diagnostic markers for invasive cervical cancer. Journal of Clinical Microbiology. 1998;36(2):475–480. doi: 10.1128/jcm.36.2.475-480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stanley M. Antibody reactivity to HPV E6 and E7 oncoproteins and early diagnosis of invasive cervical cancer. American Journal of Obstetrics and Gynecology. 2003;188(1):3–4. doi: 10.1067/mob.2003.97. [DOI] [PubMed] [Google Scholar]
- 141.Markopoulos AK. Role of human papillomavirus in the pathogenesis of oral squamous cell carcinoma. The World Journal of Experimental Medicine. 2012;2(4):65–69. doi: 10.5493/wjem.v2.i4.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Filion M, Skup D, Suh M. Specific induction of cellular gene transcription in herpes simplex virus type -2-transformed cells. Journal of General Virology. 1988;69(8):2011–2019. doi: 10.1099/0022-1317-69-8-2011. [DOI] [PubMed] [Google Scholar]
- 143.La Thangue NB, Latchman DS. A cellular protein related to heat-shock protein 90 accumulates during herpes simplex virus infection and is overexpressed in transformed cells. Experimental Cell Research. 1988;178(1):169–179. doi: 10.1016/0014-4827(88)90388-6. [DOI] [PubMed] [Google Scholar]
- 144.Notarianni EL, Preston CM. Activation of cellular stress protein genes by herpes simplex virus temperature-sensitive mutants which overproduce immediate early polypeptides. Virology. 1982;123(1):113–122. doi: 10.1016/0042-6822(82)90299-9. [DOI] [PubMed] [Google Scholar]
- 145.Kwong AD, Kruper JA, Frenkel N. Herpes simplex virus virion host shutoff function. Journal of Virology. 1988;62(3):912–921. doi: 10.1128/jvi.62.3.912-921.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fenwick M. The effects of herpes viruses on cellular macromolecular synthesis. In: Fraenkel-Conrat H, Wagner RR, editors. Comprehensive Virology. Vol. 19. New York, NY, USA: Plenum; 1984. pp. 359–390. [Google Scholar]
- 147.Kwong AD, Frenkel N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(7):1926–1930. doi: 10.1073/pnas.84.7.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Read GS, Frenkel N. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of α (immediate early) viral polypeptides. Journal of Virology. 1983;46(2):498–512. doi: 10.1128/jvi.46.2.498-512.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Steele C, Shillitoe EJ. Viruses and oral cancer. Critical Reviews in Oral Biology and Medicine. 1991;2(2):153–175. doi: 10.1177/10454411910020020201. [DOI] [PubMed] [Google Scholar]
- 150.Boyd AL, Enquist L, Vande Woude GF, Hampar B. Activation of mouse retrovirus by herpes simplex virus type 1 cloned DNA fragments. Virology. 1980;103(1):228–231. doi: 10.1016/0042-6822(80)90140-3. [DOI] [PubMed] [Google Scholar]
- 151.Stich HF, Hsu TC, Rapp F. Viruses and mammalian chromosomes. I. Localization of chromosome aberrations after infection with herpes simplex virus. Virology. 1964;22(4):439–445. doi: 10.1016/0042-6822(64)90064-9. [DOI] [PubMed] [Google Scholar]
- 152.Mincheva A, Dundarov S, Bradvarova I. Effects of herpes simplex virus strains on human fibroblast and lymphocyte chromosomes and the localization of chromosomal aberrations. Acta Virologica. 1984;28(2):97–106. [PubMed] [Google Scholar]
- 153.Peat DS, Stanley MA. Chromosome damage induced by herpes simplex virus type 1 in early infection. Journal of General Virology. 1986;67(10):2273–2277. doi: 10.1099/0022-1317-67-10-2273. [DOI] [PubMed] [Google Scholar]
- 154.El Sissy AAI. Herpes simplex virus type 2 (HSV-2) proteins in oral squamous cell carcinoma—in situ detection. Cairo Dental Journal. 1997;13(2):265–270. [Google Scholar]
- 155.Jalouli J, Ibrahim SO, Sapkota D, et al. Presence of human papilloma virus, herpes simplex virus and Epstein-Barr virus DNA in oral biopsies from Sudanese patients with regard to toombak use. Journal of Oral Pathology and Medicine. 2010;39(8):599–604. doi: 10.1111/j.1600-0714.2010.00910.x. [DOI] [PubMed] [Google Scholar]
- 156.Delavarian Z, Pakfetrat A, Falaki F, Pazouki M. The role of viruses in oral squamous cell carcinoma in young patients in Khorasan (Northeast of Iran) Journal of Applied Sciences. 2010;10(11):981–985. [Google Scholar]
- 157.Shillitoe EJ, Greenspan D, Greenspan JS, Silverman S., Jr. Immunogloblin class of antibody to herpes simplex virus in patients with oral cancer. Cancer. 1983;51(1):65–71. doi: 10.1002/1097-0142(19830101)51:1<65::aid-cncr2820510116>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 158.Schantz SP, Shillitoe EJ, Brown B, Campbell B. Natural killer cell activity and head and neck cancer: a clinical assessment. Journal of the National Cancer Institute. 1986;77(4):869–875. [PubMed] [Google Scholar]
- 159.Starr JR, Daling JR, Fitzgibbons ED, et al. Serologic evidence of herpes simplex virus 1 infection and oropharyngeal cancer risk. Cancer Research. 2001;61(23):8459–8464. [PubMed] [Google Scholar]
- 160.Eglin RP, Scully C, Lehner T. Detection of RNA complementary to herpes simplex virus in human oral squamous cell carcinoma. The Lancet. 1983;2(8353):766–767. doi: 10.1016/s0140-6736(83)92299-7. [DOI] [PubMed] [Google Scholar]
- 161.Rapp F, Duff R. Transformation of hamster embryo fibroblasts by herpes simplex viruses type I and type II. Cancer Research. 1973;33(6):1527–1534. [PubMed] [Google Scholar]
- 162.Hirsch JM, Johansson SL, Vahlne A. Effect of snuff and herpes simplex virus-1 on rat oral mucosa: possible associations with the development of squamous cell carcinoma. Journal of oral pathology. 1984;13(1):52–62. doi: 10.1111/j.1600-0714.1984.tb01400.x. [DOI] [PubMed] [Google Scholar]
- 163.Galloway DA, McDougall JK. The oncogenic potential of herpes simplex viruses: evidence for a ’hit-and-run’ mechanism. Nature. 1983;302(5903):21–24. doi: 10.1038/302021a0. [DOI] [PubMed] [Google Scholar]
- 164.Shillitoe EJ, Zhang S, Wang G, Hwang CBC. Functions and proteins of herpes simplex virus type-1 that are involved in raising the mutation frequency of infected cells. Virus Research. 1993;27(3):239–251. doi: 10.1016/0168-1702(93)90036-m. [DOI] [PubMed] [Google Scholar]
- 165.Mikola H, Waris M, Tenovuo J. Inhibition of herpes simplex virus type 1, respiratory syncytial virus and echovirus type 11 by peroxidase-generated hypothiocyanite. Antiviral Research. 1995;26(2):161–171. doi: 10.1016/0166-3542(94)00073-h. [DOI] [PubMed] [Google Scholar]
- 166.Hwang CBC, Shillitoe EJ. DNA sequence of mutations induced in cells by herpes simplex virus type-1. Virology. 1990;178(1):180–188. doi: 10.1016/0042-6822(90)90392-5. [DOI] [PubMed] [Google Scholar]
- 167.Baer R, Bankier AT, Biggin MD. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310(5974):207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 168.Parkin DM. The global health burden of infection-associated cancers in the year 2002. International Journal of Cancer. 2006;118(12):3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 169.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43(3):831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 170.Rickinson A, Kieff E. In: Fields Virology. Knipe DM, Howley PM, editors. Philadelphia, Pa, USA: Lippincott Williams and Wilkins; 2001. pp. 2575–2627. [Google Scholar]
- 171.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80(3):389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 172.Kieff E, Rickinson A. In: Fields Virology. Knipe DM, Howley PM, editors. Philadelphia, Pa, USA: Lippincott Williams and Wilkins; 2001. pp. 2511–2573. [Google Scholar]
- 173.Miller WE, Cheshire JL, Baldwin AS, Jr., Raab-Traub N. The NPC derived C15 LMP1 protein confers enhanced activation of NF-κB and induction of the EGFR in epithelial cells. Oncogene. 1998;16(14):1869–1877. doi: 10.1038/sj.onc.1201696. [DOI] [PubMed] [Google Scholar]
- 174.Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) Oncogene. 1998;16(13):1731–1742. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- 175.Eliopoulos AG, Gallagher NJ, Blake SMS, Dawson CW, Young LS. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin- 6 and interleukin-8 production. Journal of Biological Chemistry. 1999;274(23):16085–16096. doi: 10.1074/jbc.274.23.16085. [DOI] [PubMed] [Google Scholar]
- 176.Zhang L, Pagano JS. Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. Journal of Virology. 2000;74(3):1061–1068. doi: 10.1128/jvi.74.3.1061-1068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wakisaka N, Murono S, Yoshizaki T, Furukawa M, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces and causes release of fibroblast growth factor-2. Cancer Research. 2002;62(21):6337–6344. [PubMed] [Google Scholar]
- 178.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9(3):405–411. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 179.Horiuchi K, Mishima K, Ichijima K, Sugimura M, Ishida T, Kirita T. Epstein-Barr virus in the proliferative diseases of squamous epithelium in the oral cavity. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and. 1995;79(1):57–63. doi: 10.1016/s1079-2104(05)80075-7. [DOI] [PubMed] [Google Scholar]
- 180.González-Moles M, Gutiérrez J, Ruiz I, Fernández JA, Rodriguez M, Aneiros J. Epstein-Barr virus and oral squamous cell carcinoma in patients without HIV infection: viral detection by polymerase chain reaction. Microbios. 1998;96(383):23–31. [PubMed] [Google Scholar]
- 181.Higa M, Kinio T, Kamiyama K, Iwamasa T, Hamada T, Iyama K. Epstein-Barr virus (EBV) subtype in EBV related oral squamous cell carcinoma in Okinawa, a subtropical island in southern Japan, compared with Kitakyushu and Kumamoto in mainland Japan. Journal of Clinical Pathology. 2002;55(6):414–423. doi: 10.1136/jcp.55.6.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Li C, Yen C-Y, Lu M-C, et al. Detection of EBV infection and gene expression in oral cancer from patients in taiwan by microarray analysis. Journal of Biomedicine and Biotechnology. 2009;2009:15 pages. doi: 10.1155/2009/904589.904589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sand LP, Jalouli J, Larsson P-A, Hirsch J-M. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 2002;93(5):586–592. doi: 10.1067/moe.2002.124462. [DOI] [PubMed] [Google Scholar]
- 184.Jiang R, Gu X, Moore-Medlin TN, Nathan C-A, Hutt-Fletcher LM. Oral dysplasia and squamous cell carcinoma: correlation between increased expression of CD21, Epstein-Barr virus and CK19. Oral Oncology. 2012;48(9):836–841. doi: 10.1016/j.oraloncology.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Johnson RJ, Gretch DR, Yamabe H, et al. Membranoproliferative glomerulonephritis associated with hepatitis C virus infection. The New England Journal of Medicine. 1993;328(7):465–470. doi: 10.1056/NEJM199302183280703. [DOI] [PubMed] [Google Scholar]
- 186.Choo Q-L, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 187.Carrozzo M, Quadri R, Latorre P, et al. Molecular evidence that the hepatitis C virus replicates in the oral mucosa. Journal of Hepatology. 2002;37(3):364–369. doi: 10.1016/s0168-8278(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 188.Negro F, Krawczynski K, Quadri R, et al. Detection of genomic- and minus-strand of hepatitis C virus RNA in the liver of chronic hepatitis C patients by strand-specific semiquantitative reverse-transcriptase polymerase chain reaction. Hepatology. 1999;29(2):536–542. doi: 10.1002/hep.510290223. [DOI] [PubMed] [Google Scholar]
- 189.Toyoda H, Fukuda Y, Nakano I, et al. Quasispecies nature of hepatitis C virus (HCV) in patients with chronic hepatitis C with mixed HCV subtypes. Journal of Medical Virology. 1998;54:80–85. [PubMed] [Google Scholar]
- 190.Nagao Y, Sata M, Itoh K, Tanikawa K, Kameyama T. Quantitative analysis of HCV RNA and genotype in patients with chronic hepatitis C accompanied by oral lichen planus. European Journal of Clinical Investigation. 1996;26(6):495–498. doi: 10.1046/j.1365-2362.1996.167314.x. [DOI] [PubMed] [Google Scholar]
- 191.Gandolfo S, Carbone M, Carrozzo M, Gallo V. Oral lichen planus and hepatitis C virus (HCV) infection: is there a relationship? A report of 10 cases. Journal of Oral Pathology & Medicine. 1994;23(3):119–122. doi: 10.1111/j.1600-0714.1994.tb01098.x. [DOI] [PubMed] [Google Scholar]