

Abstract
Ras proteins are monomeric GTPases that act as binary molecular switches to regulate a wide range of cellular processes. The exchange of GTP for GDP on Ras is regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs), which regulate the activation state of Ras without covalently modifying it. In contrast, post-translational modifications (PTMs) of Ras proteins direct them to various cellular membranes and, in some cases, modulate GTP–GDP exchange. Important Ras PTMs include the constitutive and irreversible remodelling of its C-terminal CAAX motif by farnesylation, proteolysis and methylation, reversible palmitoylation, and conditional modifications including phosphorylation, peptidyl-proly isomerisation, mono- and di-ubiquitination, nitrosylation, ADP ribosylation and glucosylation.
The intense interest in the actions and regulation of Ras among cancer researchers and cell biologists can be traced to the recognition of ras genes as the transforming principle of tumor viruses1 and the identification of hras as the first oncogene isolated from a human tumor2–4. Recent analyses of cancer genomes have reconfirmed the central role of Ras as a driver of oncogenesis in several human tumors5. In addition, germline mutations in ras genes have recently been recognized as the underlying cause of three developmental disorders, Costello syndrome6, Noonan syndrome 7 and Cranio-Facio-Cutaneous8 syndrome, providing further links between ras mutation and disease.
To cell biologists, Ras serves as the paradigm of a monomeric GTPase switch, a protein that exists in two states depending on the guanine nucleotide that it binds. As a binary switch Ras regulates the flow of information down several signaling pathways. Cell biologists have also devoted attention to Ras because it represents the archetypal CAAX protein. This class of protein terminates in a CAAX sequence, where C is cysteine, A is usually, but not always, an aliphatic amino acid and X is any amino acid. The CAAX sequence directs the post-translational modification of the C-terminus of the protein with a polyisoprenoid lipid which, in the case of Ras, is a farnesyl moiety. This modification converts an otherwise globular, hydrophilic protein to one that associates with the cytoplasmic leaflet of cellular membranes, a process required for Ras activation and signaling.
The first two decades of research into the cell biology of Ras were marked by an exponential growth in the understanding of how the exchange of GTP for GDP on this protein is regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) (FIG. 1a). We now know that GEFs activate Ras by inducing the release of GDP and permitting GTP binding, whereas GAPs inactivate small GTPases like Ras by increasing their intrinsic rate of GTP hydrolysis to return the protein to the GDP-bound state. The effectors and downstream pathways regulated by Ras, as well as the enzymes that modify the C-terminal hypervariable region that targets Ras to membranes, were also characterized9. More recent insights into Ras biology have been gained from experiments carried out in vivo and in cultured cells. For example, the generation of transgenic mice has confirmed what cell biologists and cancer geneticists already suspected, that is, that there are significant biological differences between Ras isoforms10. Furthermore, although Ras was initially thought to be expressed on, and to signal exclusively from, the plasma membrane, live cell imaging of fluorescently labeled Ras proteins has revealed that Ras traffics between various subcellular compartments, including the Golgi apparatus and endosomes, and it is capable of signaling from multiple locations 11–14. It has become clear that post-translational modifications (PTMs) control the localization of Ras proteins (BOX 1). These include both the modifications of the C-terminal hypervariable regions of Ras, which have been studied for two decades, and the more recently appreciated Ras modifications, such as phosphorylation, nitrosylation, ubiquitination and peptidyl-prolyl isomerization.
Figure 1. Ras Signaling.

(A) The GDP/GTP cycle of Ras is shown. Inactive, GDP-bound Ras is activated by a GEF that induces the release of GDP and thereby permits GTP to bind. GTP binding induces a marked conformational change in Ras that allows it to bind effectors via their Ras binding domains (RBD). The “on” state of Ras is limited by its slow intrinsic GTPase activity, which is accelerated up to 105 fold by the binding of a GAP, and allows Ras to return to its inactive, GDP-bound state. (B) The Ras/Raf-1/Erk pathway. This pathway is engaged by receptor tyrosine kinases (RTKs), which are activated upon growth factor binding. The adaptor protein GRB2 binds to activated (that is, phosphorylated) RTKs. GRB2 also binds the GEF son of sevenless (SOS) and brings it to the membrane where it can activate Ras. Ras initiates downstream signaling by bringing RAF1 to the membrane and activating its kinase activity. This is the best characterized Ras regulated pathway and it is frequently dysregulated in cancer. (C) Multiplex regulation of, and signaling from, Ras. The various families of GEFs, GAPs and effectors that have been reported to regulate Ras or transmit signals from Ras-GTP, are shown.
Box 1 . Post-Translational Modifications of Ras.
Post-translational modification (PTM) of proteins is exceedingly common, according to Princeton’s proteome-wide PTM statistics curator (http://selene.princeton.edu/PTMCuration/). The purpose of PTM is to extend the complexity and function of proteins, which are constructed from only twenty amino acids. PTMs play numerous roles ranging from allowing proper folding and localization, to signaling for protein senescence and degradation. Readily reversible PTMs such as phosphorylation are used extensively in signaling pathways as the molecular currency that transmits information. It is therefore not surprising that a signaling molecule as important as Ras can be post-translationally modified in multiple ways, including by farnesylation, palmitoylation, methylation, peptidyl-prolyl isomerization, phosphorylation, nitrosylation and mono- and di-ubiquitination as well as by proteolytic removal of a C-terminal pro-peptide (Fig. 6).
The mechanism and function of each Ras PTM has been fairly well characterized. Farnesylpyrophosphate is a lipid intermediate in the cholesterol biosynthetic pathway, and it is added to the CAAX cysteine of Ras44 by farnesyltransferase45 via a stable thioether linkage. Palmitate, an abundant saturated (acyl) fatty acid, which is used to modify many proteins, is added to one or two cysteines immediately upstream of the Ras CAAX sequence58 by one or more protein acyltransferases (PATs)61 via a labile thioester bond. Reversal of this modification is catalyzed by one or more thioesterases such as acyl protein thioesterase 1 (APT1)76. The AAX amino acids of the CAAX sequence are substrates for an endoprotease designated Ras converting enzyme 1 (Rce1)50. Following Rce1-mediated proteolysis, the α-carboxyl group of the now C-terminal prenylcysteine of Ras is methylesterified by isoprenylcysteine carboxylmethyltransferase (Icmt)51. The G-P peptidyl-prolyl bond of H-Ras at position 178–179 undergoes cis-trans isomerization catalyzed by FKBP1278. Protein kinase C (PKC) phosphorylates K-Ras4B on serine 18181. Cysteine 118, which is conserved in all Ras isoforms, functions as a redox indicator that can be nitrosylated90. Finally, H-Ras and K-Ras can be mono- and di-ubiquitinated on several lysines86, 87 by the E3 ligase Rabex-588. In addition to these endogenous PTMs, two pathogenic bacteria, Pseudomonas aeruginosa and Clostridium sordelli, produce toxins that can ADP-ribosylate95 or glucosylated96 Ras.
The interaction of Ras with its myriad of GEFs, GAPs and effectors (FIG. 1c) has been the subject of numerous reviews15 and will not be discussed in detail here. Rather, this review will focus on the PTMs that are largely responsible for the trafficking and localization of Ras proteins. These PTMs present targets for the development of small molecule inhibitors that might limit Ras activity and thereby have utility as drugs for a wide range of disorders characterized by dysregulated signaling, including cancer.
Ras as a molecular switch
There are three Ras genes in mammalian genomes: hras, nras, and kras16. Because the transcript from the kras locus can be alternatively spliced, the three genes give rise to four protein isoforms: N-Ras, H-Ras, K-Ras4A and K-Ras4B. These proteins are >90% identical in the first 168–169 amino acids (G-domain) but differ in the C-terminal 20 amino acids that are known as the hypervariable region (HVR) (FIG. 2a).
Figure 2. Post-translational modification of the C-terminal membrane-targeting region of Ras.

(A) The hypervariable regions (HVR) of the four Ras isoforms are shown. These sequences contain all of the information required to target the different Ras proteins to various subcellular membrane compartments as demonstrated by the fact that they can be used in isolation to target unrelated proteins in the same way. The CAAX motif is often considered the “first signal” because a “second signal” immediately upstream of that sequence is also required for plasma membrane targeting. For N-Ras, H-Ras and K-Ras4A this consists of cysteines that are palmitoylated. For K-Ras4B (shown separately) the second signal consists of a polybasic region with a net charge of +8. The cysteines of the CAAX motifs that are farnesylated are shown in yellow. Cysteines in the second signal region that are modified by palmitate are shown in green. The lysines of the polybasic region of K-Ras4B are shown in red and serine 181, which is the principal site of phosphorylation, is show in blue. (B) CAAX processing is catalyzed by three enzymes that work sequentially: farnesyltransferase (FTase), Ras converting enzyme 1 (Rce1) and isoprenylcysteine carboxyl methyltransferase (Icmt). FTase is a cytosolic enzyme that catalyzes the first and rate-limiting reaction in the sequence. Rce1 is an ER-localized endoprotease that removes the AAX amino acids rendering the farnesylcysteine the new C-terminus. Icmt, also localized in ER membranes, methylesterifies the α-carboxyl group of the farnesylcysteine. S-adenosylmethionine (AdoMet) is used as the methyl donor in this reaction generating S-adenosylhomocysteine (AdoHcy) as a product. The end result of these modifications is to convert the C-terminus of Ras proteins from a hydrophilic to a hydrophobic domain: lapidated peptide with the charge of the C-terminal carboxylate negated by methylation. Whereas the reacions catalyzed by FTase and Rce1 are irreversible, prenylcysteine carboxyl methylation catalyzed by Icmt is readily reversible at physiologic pH, although a specific esterase that catalyzes the reverse reaction has not been identified.
Each Ras protein is a 21 kD guanine nucleotide binding protein with intrinsic GTPase activity and is therefore designated a small or monomeric GTPase, distinguishing Ras proteins from the α subunits of heterotrimeric G proteins. The conformation of one surface of Ras proteins, consisting of the switch I and switch II regions, changes radically when GTP is substituted for GDP in the guanine nucleotide binding pocket17. This is the physical basis for the molecular switch that is at the core of Ras as a regulatory machine. Ras proteins transduce signals by interacting with effectors only when in the GTP-bound conformation. Thus, on/off signaling through Ras is ultimately determined by the factors that initiate GTP–GDP exchange and those that affect its GTPase activity.
Ras proteins are activated by GEFs, eight of which are encoded in mammalian genomes15. These include two isoforms each of the Son of Sevenless (SOS) proteins and Ras Guanyl-nucleotide Releasing Factors (RasGRF), and four isoforms of Ras Guanine-nucleotide Releasing Protein (RasGRP) (FIG. 1c). Each GEF contains a Cdc25 homology region, which is the catalytic domain that stimulates the release of GDP. GDP release promotes GTP-binding because GTP is ten times more abundant than GDP in the cytosol. SOS is the best-characterized Ras GEF owing to the fact that its role in the Ras–MAPK pathway is firmly established. SOS binds constitutively to the adaptor protein Grb2 and this complex is brought, via the SH2 domain of the adaptor, to protein tyrosine kinase receptors (PTKRs) at the plasma membrane that have been activated by the phosphorylation of specific tyrosines in their cytosolic domains (FIG. 1b). The translocation of the Grb2–SOS complex from the cytosol to the membrane-associated PTKR is aided by the Plextrin and histone homology domains of SOS, both of which bind to negatively charged phospholipids18, 19.
The intrinsic GTPase activity of Ras is very weak (kcat≈2 × 10−4 s−1)20. The rate of catalysis can be increased by a factor of up to 105 by the action of GAPs, and it is this class of protein that negatively regulates Ras and limits signaling. Mammalian genomes encode seven Ras GAPs, the best studied of which are p120 Ras GAP (aka RASA1) and neurofibromin15. The others are synGAP, GAP1m (aka RASA2), GAP1IP4BP (aka RASA3) and two calcium regulated GAPs, CAPRI (aka RASA4) and RASAL21. Ras GAPs function by stabilizing the transition state of the nucleophilic attack of water on GTP. They accomplish this by inserting a so called “arginine finger” into the active site of the GTPase22. Oncogenic mutations of Ras, such as the prevalent G12V mutation, prevent GAP proteins from increasing the catalytic rate of the GTPase and thereby lock Ras in the GTP-bound state23.
Molecular and cellular effects of Ras
Ras regulates many cellular functions, including gene expression, proliferation, survival, differentiation, cell cycle entry, and cytoskeletal dynamics. Dysregulation of these cellular functions are hallmarks of cancer24. The biological effects of Ras proteins are specified by the pathways that they regulate, which, in turn, are determined by the effectors with which they interact. Genetic studies in D. melanogaster and C. elegans, which were validated by concurrent biochemical studies, established Raf-1 as the first Ras effector25–30. Raf-1 is the first kinase in the mitogen activated protein kinase (MAPK) cascade, which proceeds through the activation of MEK and Erk (FIG. 1b). Numerous studies have implicated this pathway in biological processes as diverse as the negative selection of T lymphocytes in the thymus31 and the proliferation of epithelial cells10. The central role of the Ras/MAPK pathway in many cancers is substantiated by the fact that multiple nodes in this cascade have been identified as naturally occurring oncogenes, including mutant forms of EGFR, Ras, Raf, and Ets.
Besides Raf-1, at least six other families of proteins have been shown to interact with Ras in a GTP-dependent fashion and are therefore also considered effectors 9 (FIG. 1c). Of these, phosphatidylinositol 3-kinase (PI3K) and a family of Ral GEFs that includes RalGDS have been the most extensively studied. The ability of these effectors to contribute to oncogenesis is established, in part, by the fact that mutant forms of these proteins are themselves oncogenes32, 33. In addition, their ability to promote oncogenesis has been established in animal models. For example, mice deficient in RalGDS are protected from Ras driven skin cancer34. Knock-in mice homozygous for a PI3K p110α allele lacking a Ras binding domain were protected from lung tumors induced by oncogenic K-Ras4B 35. Another important Ras effector is Tiam1, a Rac GEF that links Ras signaling to Rac36, which is a Rho family GTPase that regulates the actin cytoskeleton and activates p21 activated protein kinases (PAKs) and c-Jun amino-terminal kinase (JNK). Finally, PLCΣ is a Ras effector that enables Ras to directly stimulate the production of the second messengers diacylglycerol and calcium 37. Although Tiam1 and PLCΣ are not proto-oncogenes, silencing either of these effectors ameliorates H-Ras driven oncogenesis in a murine skin tumor model 38, 39.
From this brief overview it should be apparent that Ras is a prolific signaling molecule that is involved both in normal cellular homeostasis and in pathologic conditions. Accordingly, the PTMs that regulate Ras proteins have great significance in both normal physiology and in disease.
Regulation of Ras by constitutive PTMs
In addition to GTP binding, Ras proteins must associate with cellular membranes in order to transduce signals. Membrane association constrains Ras in two dimensions and thereby greatly facilitates its interaction with GEFs and GAPs. Indeed, Ras GEFs are primarily regulated through their translocation to membranes via domains that bind membrane proteins (e.g. SH2) or membrane phospholipids (e.g. PH). Activated Ras, in turn, recruits its effectors to membranes. In the case of Raf-1, the membrane itself participates in the activation of this kinase via a poorly understood mechanism40. Thus Ras is not only an allosteric regulator of the Raf-1 kinase but also a membrane tether for the protein41, 42.
Nascent Ras is a globular, hydrophilic protein and its association with cellular membranes is mediated by a series of PTMs, some of which are constitutive and occur immediately after translation, and some of which are conditional. Constitutive PTMs will be discussed here, and conditional PTMs will be discussed in the next section.
CAAX processing: prenylation, proteolysis and methylation
Ras is a member of a large class of proteins known as CAAX proteins. The CAAX sequence is modified by three enzymes that work sequentially43. First, unmodified CAAX sequences serve as substrates for prenylation by one of two cytosolic prenyltransferases44, 45. If the amino acid in the X position is leucine46, as is the case for most Rho family GTPases and γ subunits of heterotrimeric G proteins, then geranylgeranyltransferase type I (GGTase I) adds a 20-carbon polyisoprene lipid to the CAAX cysteine via a stable thioether bond. Geranylgeranylation of Rho proteins not only allows them to associate with membranes, upon which they exert their biological effects, but it also promotes their association with a cytosolic chaperone, RhoGDI. Because RhoGDI possesses a hydrophobic pocket into which the geranylgeranyl lipid is sequestered it can regulate the trafficking of Rho proteins on and off membranes47. If the amino acid in the X position of CAAX is not leucine46, as is the case for all Ras proteins, then farnesyltransferase (FTase) modifies the CAAX cysteine with a 15-carbon farnesyl lipid (FIG. 2b). Farnesylation affords Ras proteins relatively weak affinity for cellular membranes48. Several farnesyl binding proteins analogous to RhoGDI, such as PDE™49, have been described that may facilitate the trafficking of farnesylated proteins.
Proteins such as Ras that are modified only with a farnesyl lipid accumulate on the cytoplasmic face of the endoplasmic reticulum11, where they encounter the next CAAX processing enzyme, Ras converting enzyme type 1 (Rce1). Ras prenylation is a prerequisite for the action of Rce1, not only because it is required for the co-localization of Ras and Rce1 on the ER, but also because of the substrate specificity of this protease. Rce1 is an endoprotease that removes the AAX amino acids so that the farnesylcysteine is the new C-terminus50. Ras is then modifed by another ER resident enzyme, known as isoprenylcysteine carboxylmethyltransferase (Icmt), which catalyzes the methyl esterification of the α-carboxyl group of the farnesylcysteine51. Of these three modifications, only the one catalyzed by Icmt is reversible (FIG. 2b). The end result of these three modifications is the remodeling of the C-terminus of Ras proteins from a hydrophilic region to a hydrophobic one that is capable of insertion into cellular membranes, which is a requirement for Ras biological activity.
There is no evidence that CAAX processing is regulated with respect to cell activation, metabolic status or cell cycle, suggesting that it is a housekeeping process. One caveat is the recent description of the binding of a splice variant of smgGDS, a GEF that is active against a wide range of small GTPases, to unprenylated Ras and thereby retarding its prenylation, suggesting a mode of regulation52. Two classes of drugs can block prenylation of Ras and other proteins: statins that inhibit HMG-CoA and therefore limit the availability of farnesyl pyrophoshate, which is an intermediate in the cholesterol biosynthetic pathway initiated by HMG-CoA reductase, and farnesyltransferase inhibitors (FTIs) that directly inhibit farnesylation. A pool of unprocessed, endogenous Ras in the absence of prenylation-inhibiting drugs has not been described. Ras processed by the three sequential PTMs of the CAAX sequence can be distinguished from unprocessed Ras by SDS-PAGE because the processed form has a slightly faster electrophoretic mobility. The unprocessed form of endogenous Ras can be observed in SDS-PAGE only after treatment with statins or FTIs53, 54. Concordant with these results, whereas endogenous Ras is readily detected in the cytosolic fraction of cells treated with statin or FTI, the vast majority of Ras is in the membrane fraction of untreated cells (M. Zhou and M.R.P, unpublished observation). These observations argue strongly against a pool of unprocessed Ras that is awaiting processing, and suggest that CAAX processing follows translation immediately in an efficient and constitutive manner. Nevertheless, the CAAX processing pathway can be saturated since, when Ras is overexpressed, a significant pool is unprocessed based on the results of electrophoretic mobility and subcellular fractionation (M. Zhou and M.R.P, unpublished observation).
Although CAAX processing is necessary for the delivery of Ras proteins to, and stable association with, the plasma membrane, it is not sufficient for these events. Elements within the HVR that are upstream of the CAAX sequence55 are also required. These elements have been called “second signals” for plasma membrane targeting. There are two types of second signal: one consists of cysteines that serve as acceptor sites for palmitoylation and the other consists of polybasic regions rich in lysines. N-Ras, H-Ras and K-Ras4A have the former second signal and K-Ras4B has the latter (FIG 2a).
Palmitoylation as a second signal
The covalent attachment of the acyl chain of a fatty acid to a protein is known as protein acylation56. Although acyl chains of a variety of lengths have been shown to be incorporated into proteins, the 14-carbon myristoyl chain and the 16-carbon palmitoyl chain are the most commonly utilized. Whereas myristoylation is largely restricted to the N-termini of proteins, palmitoylation of cysteine residues occurs throughout the polypeptide chain. The palmitoylation of Ras was first described 25 years ago57, and it was later appreciated that this PTM is isoform specific58. Whereas H-Ras has two cysteines that can accept palmitate (residues Cys181 and Cys184), N-Ras only has one (residue Cys181). Although the best studied splice variant of K-Ras, K-Ras4B, does not undergo palmitoylation, the other splice form, K-Ras4A, is palmitoylated at Cys18059. Palmitate is linked to Ras via a labile thioester bond and the modification is readily reversible60.
A palmitoyl acyltransferase (PAT) capable of modifying Ras was recently identified as the DHHC9–GPC16 complex61, which is an ortholog of the yeast Erf4–Erf2 complex62. DHHC9–GPC16 is a member of a family of 25 PATs that all contain a DHHC motif but differ in their subcellular localization and substrate specificity63. Whereas all members of this family have a DHHC motif, most do not have binding partners analogous to GPC16. Knockdown studies suggest that DHHC9–GPC16 is not the only PAT with activity toward Ras64. DHHC9 and other DHHC motif-containing members of this family of enzymes are transmembrane proteins. Thus, CAAX processing of Ras, which affords the protein some affinity for membranes, would appear to be a prerequisite for palmitoylation. However, Ras mutants in S. cerevisiae that lack the CAAX cysteine can still be palmitoylated, demonstrating that prenylation is not absolutely required for palmitoylation, at least in yeast65.
Palmitoylation is required for the trafficking of N-Ras and H-Ras from the endomembrane system to the plasma membrane11, 66 (FIG 3). Palmitoylation of Ras takes place on the cytosolic face of the Golgi apparatus where DHHC9–GPC16 resides. Whereas farnesylated Ras has modest affinity for membranes, Ras that is both farnesylated and palmitoylated has more than 100 fold higher affinity67, 68 and therefore palmitoylation of Ras at the Golgi serves as an affinity trap for the protein. The high affinity binding to membranes of dually lipidated proteins promotes their subcellular trafficking via vesicular transport.
Figure 3. Ras trafficking.

Ras is synthesized on cytosolic free polysomes as a globular hydrophilic protein. Nascent Ras encounters farnesyltransferase in the cytosol (1) and, after farnesylation, it gains affinity for, and is transported to, membranes of the endoplasmic reticulum (ER) (2) where it encounters the subsequent CAAX processing enzymes Rce1 (3) and Icmt (4). Following CAAX processing, K-Ras4B deviates from the path of the palmitoylated Ras isoforms and proceeds directly to the plasma membrane (5) via a poorly understood pathway that may involve cytosolic chaperones. N-Ras and H-Ras proceed to the cytosolic face of the Golgi apparatus where they are palmitoylated by DHHC9–GCP16 and thereby trapped in that membrane compartment (6). From the Golgi they traffic via vesicles to the plasma membrane (7). Upon phosphorylation of serine 181, K-Ras4B can be discharged from the plasma membrane and travel back to the endomembrane system (5). N-Ras and H-Ras are discharged from the membrane by depalmitoylation, and move by retrograde transport back to the Golgi for another round of palmitoylation (8).
The polybasic region as a second signal for K-Ras4B
K-Ras4B is unique among Ras proteins in that it cannot be palmitoylated. Nevertheless, as is the case for all CAAX proteins, it still requires a second signal for trafficking to the plasma membrane55. The K-Ras4B second signal consists of a lysine-rich domain in the HVR that has a net positive charge of 8. Thus, although no PTM is required to create this second signal, the motif is considered here because it complements farnesylation and can itself be modified by a PTM (see below). The polybasic region forms an electrostatic interaction with the negatively charged headgroups of the inner leaflet of the plasma membrane. Neither this electrostatic interaction alone nor the insertion of the farnesyl group on Ras into the phospholipid bilayer provides sufficient affinity for stable membrane association, but the two interactions together provide sufficient affinity for relatively stable membrane association. Although the steady-state distribution of K-Ras4B appears to be predominantly at the plasma membrane, molecular trapping studies in live cells have revealed that the interaction of K-Ras4B with the plasma membrane is dynamic69.
Palmitoylation–depalmitoylation cycling
Unlike farnesylation, palmitoylation is readily reversible under physiologic conditions. Indeed, it was shown more than 25 years ago that the half-life of palmitate on Ras was considerably shorter than the half-life of the protein60. More recently it was found that N-Ras and H-Ras undergo an acylation–deacylation cycle that is linked to Ras trafficking to and from the Golgi apparatus70, 71(FIG 3). Anterograde trafficking from the Golgi to the plasma membrane requires palmitoylation and proceeds via vesicular transport11, 66. The localization of DHHC9–GPC16 on the Golgi and the fact that dually lipidated proteins are affinity trapped by membranes supports this model. Photobleaching70, 71 and photoactivation70 studies have revealed that retrograde trafficking of Ras from the plasma membrane to the Golgi requires depalmitoylation and is too rapid to occur via vesicular trafficking. These observations support the current model in which N-Ras and H-Ras are palmitoylated on the Golgi apparatus and thereby affinity trapped in a membrane compartment, transported to the plasma membrane on vesicles and, after a certain period of time, depalmitoylated there and released back into the cytosol (FIG 4). From the cytosol the Ras proteins can diffuse back to the Golgi for another round of palmitoylation and a return to the plasma membrane. Evidence that Ras signaling from the Golgi apparatus differs from Ras signaling from the plasma membrane in terms of relative downstream pathway utilization12, 13, 72–74 and, in the case of T lymphocytes, in biological outcome31, supports the idea that the acylation/deacylation cycle is a way to modulate signaling.
Figure 4. The acylation/deacylation cycle of H-Ras.

During acylation, the acyl chain of a fatty acid is covalently attached to a cysteine residue of a protein via a labile thioester linkage. The most common fatty acid utilized for this purpose is palmitate, which contributes its 16 carbon saturated acyl chain to the protein. The enzymes that catalyze this lipidation reaction are known as palmitoyl-acyltransferases (PATs). H-Ras is palmitoylated on the Golgi apparatus by the PAT DHHC9-GCP16 (1) and sent to the plasma membrane via vesicular transport (2). Once on the membrane H-Ras is susceptible to depalmitoylation by a thioesterase such as acyl protein thioesterase 1 (APT1). Palmitoylated H-Ras binds to FKBP12, which catalyzes cis-trans isomerization of the peptidyl-prolyl bond immediately adjacent to the palmitoylated cysteine. This isomerization constitutes a molecular timer that promotes depalmitoylation, which allows H-Ras to leave the plasma membrane (4) and diffuse back to the Golgi (5) for another round of acylation. FK506, rapamycin, cycloheximide (CHX) and other drugs that inhibit the prolylisomerase activity of FKBP12 augment H-Ras palmitoylation by inhibiting depalmitoylation.
There is no evidence that palmitoylation of Ras by DHHC9–GPC16 or any other PAT is regulated, suggesting that, like farnesylation, palmitoylation is an immediately post-translational “housekeeping” modification, although this is an under-investigated area. In contrast, there is evidence that depalmitoylation is regulated by GTP-loading of H-Ras75. Thus, elucidation of the mode of regulation of depalmitoylation rests, to some extent, on identifying the mechanism of palmitate removal. The thioester linkage between palmitate and its substrates is quite labile, and one school of thought holds that depalmitoylation may not be enzymatic. Another school holds that there are one or more Ras specific palmitoyl thioesterases that catalyze the hydrolysis reaction. One candidate is acyl protein thioesterase 1 (APT1)76. However, this protein has many other substrates and is found in the cytosol, raising the question of how it could have access to the palmitate modifications of membrane-bound Ras77. A recent, highly innovative study used semisynthetic Ras proteins with cleavable or non-cleavable acyl modifications and came to the conclusion that, although Ras could only be palmitoylated on the Golgi, depalmitoylation can occur anywhere in the cell64. This suggests a model whereby the entropy that would otherwise distribute Ras over all intracellular membranes is overcome by the restricted localization of palmitoylation on the Golgi and unidirectional vectorial vesicular transport 64.
Regulation of Ras by conditional PTMs
As discussed above, lipidation of the C-terminal membrane targeting domain of Ras with a farnesyl and palmitoyl lipid are constitutive modifications that follow translation rapidly and efficiently. Ras proteins can also be modified in several other ways that are conditional upon cell activation, redox state or microbial pathogenesis. The conditions upon which these modifications occur, and their physiologic relevance, are only beginning to be elucidated. These modifications are discussed below.
Peptidyl-prolyl isomerization
A recent study has implicated FKBP12, a cis-trans prolyl isomerase, in the regulation of Ras depalmitoylation78 (FIG 4). The extent of H-Ras palmitoylation was shown to depend on the presence or absence of a proline at position 179 in the HVR. FK506 and other chemical inhibitors of the prolyl isomerase activity of FKBP12, including cycloheximide and rapamycin, inhibited H-Ras depalmitoylation and this effect was recapitulated by silencing FKBP12. In addition, H-Ras bound to FKBP12 in a palmitoylation-dependent fashion. Together these observations suggest that isomerization of the Gly–Pro peptidyl-prolyl bond at position 178–179 of H-Ras regulates depalmitoylation and thereby constitutes a molecular timer for acylation. Cis-trans isomerization about this bond, which accelerates Ras depalmitoylation, is catalyzed by FKBP12. Thus, peptidyl-prolyl isomerization is the most recently recognized Ras PTM. Although originally thought to only play a role in the proper folding of nascent proteins, cis-trans isomerization of peptidyl-prolyl bonds is increasingly recognized as a mechanism for signaling, particularly when a molecular timer is required79.
Phosphorylation and the farnesyl-electrostatic switch
K-Ras4B was first shown to be phosphorylated by protein kinase C (PKC) at its C-terminus in 1987, although no biochemical effect of this modification was reported80. Recently, the PKC phosphorylation site was mapped to serine 181, which is positioned within the polybasic region81. Phosphorylation is stimulated by calcium ionophore, implicating a conventional PKC as the relevant kinase, although which member(s) of this class of kinase are involved in regulating K-Ras4B has not been determined. The physiologic stimulus that induces K-Ras4B phosphorylation also remains to be elucidated. Phosphorylation of serine 181 reduces the net charge of the polybasic region, causing K-Ras4B to lose affinity for the plasma membrane and to accumulate on endomembranes. This mechanism is reminiscent of the myristoyl-electrostatic switch that regulates the membrane association of the myristoylated alanine-rich C-kinase substrate (MARCKS) protein82, and therefore it has been called the farnesyl-electrostatic switch (FIG 5).
Figure 5. The farnesyl-electrostatic switch of K-Ras4B.

The MARKS protein depicted above K-Ras4B is known to associate conditionally with the plasma membrane by virtue of a myristoylated N-terminus and a nearby polybasic region. Protein kinase C (PKC)-induced phosphorylation of serines (shown in magenta) within the polybasic region partially neutralizes its positive charge and allows MARCKS to fall of the membrane in a process known as a myristoyl-elecrostatic switch. The C-terminal farnesyl modification of K-Ras4B and the nearby polybasic region are similarly regulated by a farnesyl-electrostatic switch that is activated by PKC-mediated phosphorylation of serine 181. Serine 171 is also a phosphate acceptor that may contribute but is not required for the operation of the farnesyl-electrostatic switch, which is primarily regulated by phosphorylation of serine 181. Specifically, phosphorylation of serine 181 in K-Ras4B promotes the dissociation of Ras from membranes. The inset shows that GFP-K-Ras4B dissociates from the membrane and is internalized in live Jurkat T cells upon exposure to the PKC agonist phorbol myristate acetate (PMA).
K-Ras4B is not the only small GTPase that has a farnesyl-electrostatic switch; the Rho family GTPase Rnd3 is also regulated in this fashion83. Furthermore, RalA, a Ras family GTPase, is phosphorylated on serine 194 in its HVR by Aurora A kinase and this causes it to translocate to mitochondria where it regulates mitochondrial fission 84. Interestingly, when K-Ras4B has an activating mutation, relocation to endomembranes as a consequence of the farnesyl-electrostatic switch is associated with cell death81. The mechanism for the cell toxicity is unclear, but, paradoxically, the anti-apoptotic protein Bcl-XL is required81. The farnesyl-electrostatic switch of oncogenic K-Ras4B might be exploited in the development of anti-cancer drugs. Dissociation of K-Ras4B from the plasma membrane was also observed in neurons stimulated with glutamate, and a mechanism involving Ca++/calmodulin binding to the K-Ras polybasic region was proposed85. Because calmodulin binds to the polybasic region of K-Ras4B and other proteins through a calcium-regulated electrostatic interaction, phosphorylation of serine 181 would be expected to diminish calmodulin binding. Thus, under conditions in which the farnesyl-electrostatic switch is engaged by the phosphorylation of K-Ras4B on serine 181, calmodulin is unlikely to play a role in causing the dissociation of K-Ras4B from the plasma membrane.
Ubiquitination
H-Ras,N-Ras and K-Ras4B were recently shown to be substrates for mono- and diubiquitination86, 87. The conditions under which Ras becomes ubiquitinated have not been determined. Nor have all of the ubiquitin acceptor sites been mapped, although, based on mutagenesis studies, several surface exposed lysines are candidates86. Mass spectrometry of affinity purified, dual epitope-tagged H-Ras from cells expressing tagged ubiquitin revealed mono- and di-ubiquitination on lysines 117, 147 and 17087. The E3 ligase responsible for these modifications was identified as Rabex-588. Furthermore, the Ras effector RIN1 is required for Rabex-5-dependent Ras ubiquitination88. RIN1, through its function as a Rab5 GEF, is thought to promote the Rab5-GTP-dependent recruitment of Rabex-5 to endosomal sites where H-Ras ubiquitination takes place. Whereas the presence of H-Ras that could not be ubiquitinated was rare on endosomes, H-Ras that was artificially and constitutively ubiquitinated was enriched on endosomes. Thus, ubiquitination regulates the trafficking of H-Ras to and from endosomes. Importantly, the ubiquitin-mediated endosomal restriction of H-Ras was also associated with a reduction in MAPK signaling, demonstrating that ubiquitination is yet another PTM that regulates Ras compartmentalization and the spatial control of its signaling output. K-Ras4B was recently found to be monoubiquitinated on lysines 104 and 14787. This modification led to enhanced GTP loading and, in the setting of an oncogenic V12 mutation, increased the affinity of K-Ras4B for the downstream effectors Raf-1 and PI3K.
S-Nitrosylation
Cysteine 118 is highly conserved among Ras isoforms and orthologs and it is the most surface exposed cysteine on these GTPases. This residue was shown to be a redox switch more than fifteen years ago89, and subsequent studies revealed that cysteine 118 could be nitrosylated when exposed to nitric oxide (NO)90. The mechanism of nitrosylation was later shown to be a direct interaction of the thiol of cysteine 118 with either a nitrogen dioxide radical (•NO2) formed when NO interacts with O2, or with a glutathionyl radical (GS•)91. S-nitrosylation does not alter the structure of Ras but leads to enhanced guanine nucleotide exchange92, 93, which promotes more efficient Ras activation. This has led to the speculation that Ras signaling can be modulated by redox reactions, although the physiologic relevance of Ras S-nitrosylation remains to be determined.
Bacterial toxins and exoenzymes
The Ras superfamily of small GTPases is a favorite target of bacterial virulence factors that evolved as enzymes that modify eukaryotic signaling molecules. Most such toxins and exoenzymes target the Rho family of small GTPases that regulate the actin cytoskeleton94. However, there are also bacterial enzymes that are active against Ras. Exoenzyme S (ExoS) of Pseudomonas aeruginosa is an ADP-ribosyl transferase that modifies Ras. ExoS adds ADP-ribose to arginines 41 and 128 of Ras, leading to inefficient guanine nucleotide exchange95. Lethal toxin (LT) of Clostridium sordelli is a monoglucosyltranferase that uses UDP-glucose as a substrate and glucosylates Ras on threonine 35. Toxin B of Clostridium difficile has a similar activity towards Rho proteins96. Glucosylation of Ras inhibits signaling to MAPK97.
Whereas nascent Ras requires stoichiometric CAAX processing for maturation as a membrane protein, it is now clear that the mature protein can be modified in ways that modulate subcellular trafficking and signaling (FIG 6). Although these modifications are conditional in the sense that pools of Ras exist in both the modified and unmodified forms, the conditions in which these PTMs are stimulated, the stoichiometry of the modifications and their biological significance remain to be elucidated.
Figure 6. Post-translational modifications of Ras.
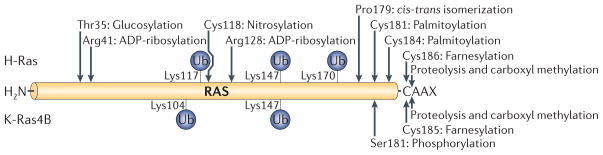
All PTMs reported for H-Ras (top) and K-Ras4B (bottom) are shown along the backbone of Ras. Sites of mono- and di-ubiquitination are indicated with blue spheres (Ub). Glucosylation and ADP-ribosylation only occur in cells intoxicated with bacterial virulence factors. The other PTMs are intrinsic to all eukaryotic cells. All of these PTMs have consequences both for Ras trafficking and signaling.
Consequences of Ras PTMs
The primary function of most of the PTMs of Ras is to deliver the molecule to the right place within the cell. In this section we will describe how the PTMs discussed above influence Ras trafficking to the plasma membrane and its partitioning into membrane microdomains.
Ras trafficking
CAAX processing and palmitoylation of Ras were originally thought to be a way to convert a globular hydrophilic protein into one with a hydrophobic C-terminus that has generalized affinity for cell membranes. The discovery that two of the CAAX processing enzymes, Icmt and Rce1, are polytopic membrane proteins restricted to the ER revealed that Ras trafficking is more complex than originally believed51, 98, 99.
Our current understanding of Ras trafficking begins in the cytosol where nascent Ras is synthesized on free polysomes (FIG. 3). FTase, the first and rate limiting, of the CAAX processing enzymes, is cytosolic 100. Once farnesylated, Ras gains modest affinity for membranes, particularly those of the ER. How farnesylated Ras is transferred from the cytosolic FTase to membranes is a matter of speculation. Are there chaperones involved? If so, does a single chaperone or family of chaperones both deliver farnesylated Ras to Icmt and Rce1 at the ER and also retrieve the fully CAAX processed product? The existence of and physiologic role of RhoGDI, a geranylgeranyl-binding chaperone for Rho family GTPases47, has led Ras biologists to speculate that an analogous chaperone may exist for farnesylated proteins. Several proteins have been identified in yeast-two hybrid screens as farnesyl-binding proteins, including PDE6δ101, PRA1102 and Galectin103. Among these proteins, PDE6δ is particularly intriguing because its farnesyl binding domain bears structural similarity to RhoGDI and because it was recently reported that its capacity to bind Ras and other farnesylated proteins is regulated by its interaction with the non-prenylated small GTPases Arl1 and Arl2 49. Moreover, a recent study reported that PDE6δ is required for proper trafficking of Ras and other farnesylated proteins as well as for efficient Ras signaling. Another intriguing possibility is that FTase itself serves as the chaperone, since it was found to have an unusual mode of catalysis; it has the ability to retain its product in a secondary exit surface near the active site100.
Whether or not chaperones plays a role in Ras trafficking, the first membrane compartment visited by nascent Ras is the ER. The basis for the particular affinity of Ras for ER membranes is not universally accepted nor understood, but it may relate to the biophysical properties of this compartment and the branched, unsaturated nature of the polyisoprene lipid that is added by farnesylation. Some investigators have argued that GFP-tagged prenylated proteins appear on the ER simply because the ER constitutes by far the greatest reservoir of membrane in the cell64. However, without a second signal, GFP-tagged Ras and other CAAX proteins appear neither on the plasma membrane nor on other abundant membrane-bound organelles, such as mitochondria11, calling into question the notion of non-specific membrane association. A particular affinity of Ras for the ER makes teleological sense since it is here that the next two enzymes in the CAAX processing cascade (Rce1 and Icmt) reside. Indeed, it is conceivable that Rce1 itself helps attract proteins with farnesylated CAAX sequences to the ER and thereby accounts for the observed membrane localization.
Once CAAX processing is complete, Ras isoforms diverge in their subsequent trafficking43. Palmitoylated isoforms visit the cytoplasmic face of the Golgi where they are acylated and thereby affinity trapped in Golgi membranes from where they can be incorporated into transport vesicles and enter the secretory pathway. K-Ras4B cannot be trapped on the Golgi and proceeds to the plasma membrane either by passive diffusion or by an as yet uncharacterized delivery system that could involve cytosolic chaperones. Recent observations have revealed that the plasma membrane is not the end of the road for palmitoylated Ras or K-Ras4B. The former undergoes retrograde trafficking, which is mediated by the acylation cycle described above70, 71, and the latter undergoes retrograde trafficking from the plasma membrane to endomembranes upon phosphorylation and engagement of the farnesyl-electrostatic switch81.
Ras is also trafficked to endosomes by two routes, one from the plasma membrane and one from the Golgi. Ras that is associated with clathrin-coated regions of the plasma membrane is internalized into primary endosomes during clathrin-mediated endocytosis. As clathrin-coated membrane domains also contain the activated protein tyrosine kinase receptors that signal through Ras and Raf-1, all of these elements are present on these so-called ‘signaling endosomes’ and signaling can persist104. Indeed, in some contexts, endocytosis is absolutely required for Ras–MAPK signaling105. Recently, signaling endosomes were differentiated from their non-signaling counterparts by expression of the APPL1 adaptor protein106. APPL1 directly binds to Rab5, protein tyrosine kinase receptors and the phospholipid bilayer, thus serving as a scaffold for signaling complexes that include Ras. The association of APPL1 with endosome membranes was dependent on low PtdIns(3,4,5)P3 levels, suggesting that phosphoinositide remodeling interconverts signaling and non-signaling endosomes106. A second route to endosomes, which is taken by palmitoylated Ras, was recently described, in which N-Ras and H-Ras traffic from the Golgi to recycling endosomes and then on to the plasma membrane107.
There remains some disagreement as to whether K-Ras4B associates with, and signals from, endosomes like its palmitoylated cousins. Several groups reported that, whereas N-Ras and H-Ras were associated with endosomes, K-Ras4B was not86, 108, 109. These data are consistent with the observation that diubiquitination promotes the association of Ras with endosomes but, whereas all three Ras isoforms examined can be mono-ubiquitinated, only N-Ras and H-Ras are di-ubiquitinated86. More recently, K-Ras4B was reported to associate with early endosomes in a clathrin-dependent fashion and then to traffic to late endosomes, leaving H-Ras behind110. Moreover, K-Ras4B was found to reside on, signal from and undergo degradation in late endosomes, lysosomes and multivesicular bodies110. Clearly we have more to learn about Ras trafficking through the endosomal compartment.
Microdomain affinity and nanoclusters
Ras trafficking does not end at the plasma membrane because this organelle is not homogeneous but rather is composed of several types of microdomains111. Like its trafficking between organelles, the partitioning of Ras into membrane microdomains is regulated by PTMs. The best-characterized microdomain is a cholesterol-dependent, liquid-ordered domain often referred to as a “lipid raft.” Once thought to encompass relatively large patches of membrane analogous to the liquid-ordered domains observed in artificial bilayers112, cholesterol-dependent domains are now known to exist on a nanoscale, to be highly dynamic and to incorporate on the order of 10 or fewer signaling molecules such as Ras113, 114. In addition to lipid rafts, at least two cholesterol-independent microdomains have been detected, and these are also thought to be highly dynamic and on a nanoscale level111. Microdomains serve to bring together multiple signaling components. In addition, nanoclusters have been shown to convert analog Ras–MAPK signaling into a digital signal that improves fidelity115.
Palmitoylation appears to be one of the strongest determinants of protein association with lipid rafts. Therefore, it is not surprising that H-Ras, N-Ras, and K-Ras4B occupy distinct plasma membrane microdomains114. Inactive, GDP-bound H-Ras has been found to group in nanoclusters of ~6 molecules within ordered, cholesterol-rich microdomains. Interestingly, exchange of GDP for GTP causes H-Ras to lose affinity for the lipid raft domains and to migrate laterally into adjacent regions of disordered plasma membrane116, 117. Coincident with this migration is its interaction with a scaffolding protein, Galectin-1, which acts to enrich GTP-bound H-Ras in non-ordered microdomains118, 119. Surprisingly, although the final 10 amino acids of H-Ras, which encompasses the CAAX sequence and both palmitoylation sites, is sufficient to direct plasma membrane localization, it cannot fully recapitulate the dynamic behavior of full-length H-Ras with regard to plasma membrane nanoclusters and microdomains114, 120. Instead, additional elements within the HVR and even the G-domain help stabilize the membrane orientation and microdomain preference of full length H-Ras at the plasma membrane and thereby contribute to differential effector engagement120–122.
Interestingly, the GTP binding status of N-Ras also affects microdomain partitioning, but for this isoform it is the GTP-bound form that favors partition into cholesterol-dependent nanoclusters123, which in turn controls signal output. Thus, with regard to plasma membrane microdomains, mono- versus dipalmitoylation appears to differentially control localization. Moreover, whereas H-Ras that is mono-palmitoylated on cysteine 181 behaves like N-Ras, H-Ras that is mono-palmitoylated at cysteine 184 does not traffic efficiently to the plasma membrane123. Thus, it appears that the spacing between the prenyl and acyl modifications is also critical for proper trafficking and microdomain partitioning. The linker region comprising amino acids 170–179 within the H-Ras HVR has also been shown to specifically contribute to the association of H-Ras within non-raft microdomains120.
K-Ras4B partitions into cholesterol-independent microdomains that do not overlap with those into which GTP-bound H-Ras partitions114, 124. Interaction with the Galectin-3 scaffold enhances K-Ras4B nanocluster formation125. Phosphorylation of GTP-bound K-Ras4B on serine 181 reduces nanoclustering126. Thus, like palmitoylation, phosphorylation plays a role in nanocluster formation.
Generating a comprehensive model for how the C-terminus of Ras proteins interacts with phospholipid bilayers is confounded by the inability to produce crystals of Ras that include its C-terminus. All Ras structures have been solved from crystals of Ras proteins that lacked a C-terminus, which is thought to be disordered127. However, recent molecular dynamic simulations and NMR data collected from full-length farnesylated H-Ras in a 1,2-dimyristoylglycero-3-phosphocholine bilayer have provided computational models of membrane insertion, and revealed putative contributions of both the HVR and G-domains in this process128–130. In these models the GDP-bound conformation of H-Ras favors electrostatic interactions between basic residues within its HVR and the inner leaflet of the plasma membrane. In contrast, residues within the α-4 helix of the G-domain facilitate the membrane association of GTP-bound, active, H-Ras, control its orientation with respect to the membrane and thereby contribute directly to effector binding specificity131, 132. Moreover, in these models the depth of membrane insertion of palmitates was greater when H-Ras was GTP-loaded, perhaps explaining the activation-specific association of H-Ras with Galectin-1129.
Exploiting Ras PTMs for therapy
Oncogenic mutations of Ras block the ability of GAPs to accelerate GTP hydrolysis. Thus, the most direct approach to anti-Ras therapy would be to develop drugs that relieve this block. Unfortunately, efforts along these lines in academic laboratories have all failed. In addition to GTP loading, CAAX processing is required for oncogenic Ras to transform cells133. Accordingly, many have looked to Ras post-translational processing as an alternative approach to anti-Ras drug discovery134, 135. Two decades ago several pharmaceutical companies successfully targeted FTase and developed orally available FTase inhibitors (FTIs) that were surprisingly well tolerated136. This effort was among the first successful applications of rational drug design. Unfortunately, FTIs lacked efficacy in clinical trials designed to treat tumors driven by mutant N-Ras or K-Ras, the two Ras isoforms associated with human cancer. The lack of efficacy proved to result from alternative prenylation, whereby N-Ras and K-Ras, normally not substrates for geranylgeranylation, could be modified by geranylgeranyltransferase I (GGTase I) in the presence of FTI137. This monumental failure has dampened enthusiasm in the biopharmaceutical industry for targeting the Ras trafficking pathway. Nevertheless, the approach retains its scientific logic and remains among the most viable. Indeed, both GGTase I and Icmt inhibitors have shown promise in preclinical testing138, 139. Furthermore, as a protease, Rce1 is considered “drugable” and efforts at developing inhibitors against it have been reported140. Curiously, whereas genetic ablation of Icmt in mice has confirmed it to be a good target for anti-cancer drug discovery141, conditional knockout of Rce1 exacerbates K-Ras4B-driven myeloproliferative disease142, suggesting that this enzyme may not be a good target for anti-cancer drug discovery. The biological basis for the differences between targeted disruption of Icmt and Rce1 in tumor models remains elusive and likely reflects the fact that Ras is but one of many substrates for these enzymes.
Conclusions
The GTP binding, GTP hydrolysis and switching functions of Ras require no PTMs, although some PTMs affect exchange rates. However, PTMs are required for the proper trafficking and localization of Ras within the cell, which, in turn, are essential for Ras function. CAAX modification by FTase, Rce1 and Icmt remodels the C-terminus of the protein and creates a hydrophobic domain with affinity for membranes. Palmitoylation affinity traps N-Ras and H-Ras in Golgi membranes and initiates a cycle of traffic to and from the plasma membrane. Peptidyl-prolyl isomerization of H-Ras at proline 179 by FKBP12 regulates depalmitoylation and thereby acts as a molecular timer for plasma membrane association. Diubiquitination of N-Ras and H-Ras leads to their enrichment on endosomes. Phosphorylation of K-Ras4B in its polybasic region engages a farnesyl-electrostatic switch that repositions the protein on endomembranes, a process that is associated with cell death when K-Ras4B is activated. S-Nitrosylation, ADP-ribosylation and glucosylation are modifications that do not affect the localization of Ras but do affect guanine nucleotide exchange rates. Thus, the location and function of Ras is determined by post-translational modification making the enzymes that catalyze these modifications interesting targets for drug discovery.
Although much has been learned over the past two decades with regard to the PTM of Ras (FIG. 6), many questions remain. Will recently developed GGTIs alone or in common with FTI block Ras function without undue toxicity? What is the basis for the biological differences observed with Icmt versus Rce1 inhibition? Can substrates of these enzymes other than Ras explain the differences? Is there a Ras-specific cytosolic chaperone and could farnesyl-peptide mimetics be developed to block its activity? Is there a specific PAT and thioesterase pair that palmitoylates and depalmitoylates Ras, and would these serves as good drug targets? Does inhibition of cis-trans prolyisomerization by FK506 and rapamycin explain any of the activities of these widely used drugs? Further basic investigation into the network of PTMs that affect Ras trafficking and signaling will be required before these questions are answered.
Online Summary.
Ras proteins are molecular switches that regulate a wide variety of signaling pathways by engaging effectors on cellular membranes. They are themselves regulated by a variety of post-translational modifications.
Ras proteins associate with membranes by virtue of a series of constitutive post-translational modifications of their C-terminal CAAX sequence. These PTMs include prenylation, proteolysis and carboxyl methylation.
Membrane association and trafficking of all Ras isoforms other than K-Ras4B are also regulated by the reversible palmitoylation of cysteines in the C-terminal hypervariable regions of the proteins.
Cis-trans isomerization of a peptidyl-prolyl bond adjacent to a palmitate in H-Ras acts as a molecular timer that regulates depalmitoylation and retrograde trafficking.
Phosphorylation of K-Ras4B in its polybasic region allows this protein to dissociate from the plasma membrane through a mechanism known as the farnesyl-electrostatic switch.
Mono- and di-ubiquitination of H-Ras regulate its association with endosomes, and mono-ubiquitination of K-Ras4B enhances its activation.
S-nitrosylation of cysteine 118 of Ras promotes guanine nucleotide exchange.
Toxins produced by Pseudomonas aeruginosa and Clostridium sordelli ADP-ribosylate and monglucosylate Ras, respectively, leading to diminished signaling.
Glossary Terms
- G-domain
The first 169 amino acids of Ras proteins that fold into a globular, hydrophilic protein that contains a guanine-nucleotide (G) binding site
- heterotrimeric G protein
a member of the large subfamily of guanine nucleotide binding proteins that signal downstream of receptors that span the plasma membrane seven times. Composed of three subunits designated α, β and γ, where the α subunit binds nucleotide
- SH2 domain
the Src homology 2 domain is one of several types of domains found in numerous signaling molecules that bind to phospho-tyrosine in the context of adjacent amino acids
- cis-trans isomerization
transformation, usually by an enzyme, of a peptide bond, or more commonly a peptidyl-prolyl bond, from a cis to a trans conformation of vice versa
- myristoyl-electrostatic switch
a term used to describe the mechanism whereby the membrane association of N-myristoylated proteins like MARCKS is modulated by phosphorylation of serines in an adjacent polybasic region
- nitrosylation
modification of the sulfhydryl in a cysteine side chain of a protein with a nitrosyl group derived from nitric oxide
- polytopic membrane protein
a transmembrane protein that spans cellular membranes multiple times
- early endosomes
dynamic tubulovescicular organelles that form from the uncoating and fusing of clathrin-coated vesicles and represent the earliest element of the endocytic cycle
- late endosomes
larger, non-tubular organelles that mature from early endosomes, are partially acidified and fuse with primary lysosomes
- multivesicular bodies
late endosomes into which vesicles have budded off to form a cluster of smaller vesicles within the larger endosome
- Noonan syndrome
an autosomal dominant congenital disease that results in a variety of developmental defects including, but not limited to, dwarfism, pulmonary valve stenosis and learning disabilities. More than half of the cases are caused by mutations in the gene encoding SHP-2 and the others include gain-of-function mutations in the genes for K-Ras or SOS1 placing Noonan’s syndrome in the category of a Ras-opathy
- Cranio-Facio-Cutaneous syndrome
a rare genetic disorder characterized by distinctive facial appearance, congenital cardiac malformations and mental retardation. like Noonan Syndrome it is a Ras-opathy caused by gain of function mutations in the genes encoding K-Ras, Braf or Mek
- Costello syndrome
A rare genetic disorder similar to Noonan and Cranio-Facial-Cutaneous Syndromes that is caused by an activating mutation in the gene encoding H-Ras
Biographies
Ian Ahearn is an MD/PhD candidate at NYU School of Medicine. After graduating from Iona College he worked at the Harvard Center for Blood Research and later NYU School of Medicine before entering the NYU Medical Scientist Training Program and completing a PhD thesis in the Philips Lab. Dr. Ahearn plans for a career in translational research in academic dermatology.
Kevin Haigis is Assistant Professor of Pathology at Harvard Medical School and Assistant Pathologist at Massachesetts General Hospital. He received a BS from the University of New Hampshire and a PhD from the University of Wisconsin before completing post-doctoral studies at the Massachusetts Institute of Technology. Dr. Haigis uses transgenic mouse models to study signaling networks, including those that relate to the differential functions of Ras isoforms in the intestinal epithelium and colorectal cancer.
Dafna Bar-Sagi is Professor of Biochemistry and Vice Dean for Science at NYU School of Medicine. Dr. Bar-Sagi received her PhD from SUNY Stony Brook and completed a post-doctoral fellowship in cell biology at Cold Spring Harbor Laboratories. With her long-term focus on the mechanisms of cellular transformation, particularly those related to oncogenic Ras, Dr. Bar-Sagi is an internationally known leader in the field. Her current research focus includes the cellular mechanisms that drive pancreatic adenocarcinoma.
Mark Philips is Professor of Medicine, Cell Biology and Pharmacology at NYU School of Medicine. A graduate of Harvard College and Columbia College of Physicians & Surgeons, Dr. Philips received post-graduate training in rheumatology with Gerald Weissman and cell biology with David Sabatini. His research focus had been on the post-translational modification and subcellular trafficking of small GTPases, particularly Ras.
References
- 1.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 2.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297:474–8. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 3.Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A. 1982;79:3637–40. doi: 10.1073/pnas.79.11.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos E, Tronick SR, Aaronson SA, Pulciani S, Barbacid M. T24 human bladder carcinoma oncogene is an activated form of the normal human homologue of BALB- and Harvey-MSV transforming genes. Nature. 1982;298:343–7. doi: 10.1038/298343a0. [DOI] [PubMed] [Google Scholar]
- 5.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoki Y, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–40. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 7.Schubbert S, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–6. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 8.Niihori T, et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294–6. doi: 10.1038/ng1749. [DOI] [PubMed] [Google Scholar]
- 9.Cox AD, Der CJ. Ras history: The saga continues. Small Gtpases. 2011;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haigis KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–8. doi: 10.1038/ngXXXX. This study demonstrates significant biological differences in vivo between Ras isoforms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choy E, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. The involvement of the endomembrane in Ras processing was established in this report. [DOI] [PubMed] [Google Scholar]
- 12.Chiu VK, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343–50. doi: 10.1038/ncb783. Ras signaling from endomembranes was first reported in this study. [DOI] [PubMed] [Google Scholar]
- 13.Bivona TG, et al. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003;424:694–8. doi: 10.1038/nature01806. [DOI] [PubMed] [Google Scholar]
- 14.Casar B, et al. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol. 2009;29:1338–53. doi: 10.1128/MCB.01359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–57. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbacid M. ras Genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 17.Schlichting I, et al. Time-resolved X-ray crystallographic study of the conformational change in Ha-Ras p21 protein on GTP hydrolysis. Nature. 1990;345:309–15. doi: 10.1038/345309a0. [DOI] [PubMed] [Google Scholar]
- 18.Zhao C, Du G, Skowronek K, Frohman MA, Bar-Sagi D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol. 2007;9:706–12. doi: 10.1038/ncb1594. [DOI] [PubMed] [Google Scholar]
- 19.Yadav KK, Bar-Sagi D. Allosteric gating of Son of sevenless activity by the histone domain. Proc Natl Acad Sci U S A. 2010;107:3436–40. doi: 10.1073/pnas.0914315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung HH, Benson DR, Cornish VW, Schultz PG. Probing the role of loop 2 in Ras function with unnatural amino acids. Proc Natl Acad Sci U S A. 1993;90:10145–9. doi: 10.1073/pnas.90.21.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rojas J, Santos E. Ras-Gefs and Ras Gaps. RAS Family GTPases. 2006;4:15–43. [Google Scholar]
- 22.Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat Struct Biol. 1997;4:686–9. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 23.Scheffzek K, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic mutants. Science. 1997;277:5324. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Dickson B, Sprenger F, Morrison D, Hafen E. Raf functions downstream of Ras1 in the Sevenless signal transduction pathway. Nature. 1992;360:600–3. doi: 10.1038/360600a0. [DOI] [PubMed] [Google Scholar]
- 26.Han M, Golden A, Han Y, Sternberg PWC. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature. 1993;363:133–40. doi: 10.1038/363133a0. [DOI] [PubMed] [Google Scholar]
- 27.Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci U S A. 1993;90:6213–7. doi: 10.1073/pnas.90.13.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warne PH, Viciana PR, Downward J. Direct interaction of ras and the amino-terminal region of Raf-1 in vitro. Nature. 1993;364:352–355. doi: 10.1038/364352a0. [DOI] [PubMed] [Google Scholar]
- 29.Zhang XF, et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993;364:308–13. doi: 10.1038/364308a0. [DOI] [PubMed] [Google Scholar]
- 30.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of ras GTP with RAf-1 and mitogen-activated protein kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 31.Daniels MA, et al. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 2006;444:724–9. doi: 10.1038/nature05269. This study revealed distinct biological outcomes from Ras/MAPK signaling on different subcellular compartments. [DOI] [PubMed] [Google Scholar]
- 32.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 33.D’Adamo DR, Novick S, Kahn JM, Leonardi P, Pellicer A. rsc: a novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene. 1997;14:1295–305. doi: 10.1038/sj.onc.1200950. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez-Garcia A, et al. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219–26. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 35.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 36.Lambert JM, et al. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621–5. doi: 10.1038/ncb833. [DOI] [PubMed] [Google Scholar]
- 37.Kelley GG, Reks SE, Ondrako JM, Smrcka AV. Phospholipase C(epsilon): a novel Ras effector. EMBO J. 2001;20:743–54. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai Y, et al. Crucial role of phospholipase Cepsilon in chemical carcinogen-induced skin tumor development. Cancer Res. 2004;64:8808–10. doi: 10.1158/0008-5472.CAN-04-3143. [DOI] [PubMed] [Google Scholar]
- 39.Malliri A, et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867–71. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- 40.Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–9. doi: 10.1016/s0955-0674(97)80060-9. [DOI] [PubMed] [Google Scholar]
- 41.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 42.Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 43.Wright LP, Philips MR. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J Lipid Res. 2006;47:883–91. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:8323–8327. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seabra MC, Reiss Y, Casey PJ, Brown MS, Goldstein JL. Protein farnesyltransferase and geranylgeranyltransferase share a common alpha subunit. Cell. 1991;65:429–434. doi: 10.1016/0092-8674(91)90460-g. [DOI] [PubMed] [Google Scholar]
- 46.Reid TS, Terry KL, Casey PJ, Beese LS. Crystallographic analysis of CaaX prenyltransferases complexed with substrates defines rules of protein substrate selectivity. J Mol Biol. 2004;343:417–33. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 47.Hoffman GR, Nassar N, Cerione RA. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- 48.Silvius JR, l’Heureux F. Fluorimetric evaluation of the affinities of isoprenylated peptides for lipid bilayers. Biochemistry. 1994;33:3014–3022. doi: 10.1021/bi00176a034. [DOI] [PubMed] [Google Scholar]
- 49.Ismail SA, et al. Regulation of a GDI-like transport system for farnesylated cargo by Arl2/3-GTP. 2011 doi: 10.1038/nchembio.686. In press. [DOI] [PubMed] [Google Scholar]
- 50.Boyartchuk VL, Ashby MN, Rine J. Modulation of Ras and a-Factor Function by Carboxyl-Terminal Proteolysis. Science. 1997;275:1796–1800. doi: 10.1126/science.275.5307.1796. [DOI] [PubMed] [Google Scholar]
- 51.Dai Q, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. Journal of Biological Chemistry. 1998;273:15030–15034. doi: 10.1074/jbc.273.24.15030. [DOI] [PubMed] [Google Scholar]
- 52.Berg TJ, et al. Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J Biol Chem. 2010;285:35255–66. doi: 10.1074/jbc.M110.129916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kohl NE, et al. Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science. 1993;260:1934–1937. doi: 10.1126/science.8316833. [DOI] [PubMed] [Google Scholar]
- 54.Lerner EC, et al. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–6. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 55.Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to loacalize p21ras to the plasma membrane. Cell. 1990;63:133–139. doi: 10.1016/0092-8674(90)90294-o. Described the polybasic sequence of K-Ras as an alternative membrane targeting motif. [DOI] [PubMed] [Google Scholar]
- 56.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. [Review] [138 refs] Biochimica et Biophysica Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 57.Buss JE, Sefton BM. Direct identification of palmitic acid as the lipid attached to p21ras. Mol Cell Biol. 1986;6:116–22. doi: 10.1128/mcb.6.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–77. doi: 10.1016/0092-8674(89)90054-8. Demonstrated that Ras proteins are isoprenylated. [DOI] [PubMed] [Google Scholar]
- 59.Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121:421–7. doi: 10.1242/jcs.020107. [DOI] [PubMed] [Google Scholar]
- 60.Magee AI, Gutierrez L, McKay IA, Marshall CJ, Hall A. Dynamic fatty acylation of p21N-ras. Embo J. 1987;6:3353–7. doi: 10.1002/j.1460-2075.1987.tb02656.x. Showed that palmitoylation of Ras is reversible. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swarthout JT, et al. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005;280:31141–8. doi: 10.1074/jbc.M504113200. Characterized a palmitoyl acyl transferase (PAT) that modifies Ras. [DOI] [PubMed] [Google Scholar]
- 62.Lobo S, Greentree WK, Linder ME, Deschenes RJ. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2002;21:21. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- 63.Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47:1118–27. doi: 10.1194/jlr.R600007-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Rocks O, et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141:458–71. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 65.Mitchell DA, Farh L, Marshall TK, Deschenes RJ. A polybasic domain allows nonprenylated Ras proteins to function in Saccharomyces cerevisiae. J Biol Chem. 1994;269:21540–6. [PubMed] [Google Scholar]
- 66.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–87. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shahinian S, Silvius JR. Doubly-lipid-modified protein sequence motifs exhibit long-lived anchorage to lipid bilayer membranes. Biochemistry. 1995;34:3813–22. doi: 10.1021/bi00011a039. [DOI] [PubMed] [Google Scholar]
- 68.Schroeder H, et al. S-Acylation and plasma membrane targeting of the farnesylated carboxyl-terminal peptide of N-ras in mammalian fibroblasts. Biochemistry. 1997;36:13102–9. doi: 10.1021/bi9709497. [DOI] [PubMed] [Google Scholar]
- 69.Silvius JR, Bhagatji P, Leventis R, Terrone D. K-ras4B and Prenylated Proteins Lacking “Second Signals” Associate Dynamically with Cellular Membranes. Mol Biol Cell. 2006;17:192–202. doi: 10.1091/mbc.E05-05-0408. This study demonstrates that the association of K-Ras with the plasma membrane is reversible and highly dynamic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rocks O, et al. An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science. 2005;307:1746–52. doi: 10.1126/science.1105654. Along with Ref. 71 this study established that the palmitoylation/depalmitoylation of N-Ras and H-Ras mediates a cycle of transport between the Golgi and plasma membrane. [DOI] [PubMed] [Google Scholar]
- 71.Goodwin JS, et al. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J Cell Biol. 2005;170:261–72. doi: 10.1083/jcb.200502063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mor A, et al. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat Cell Biol. 2007;9:713–9. doi: 10.1038/ncb1592. [DOI] [PubMed] [Google Scholar]
- 73.Matallanas D, et al. Distinct Utilization of Effectors and Biological Outcomes Resulting from Site-Specific Ras Activation: Ras Functions in Lipid Rafts and Golgi Complex Are Dispensable for Proliferation and Transformation. Mol Cell Biol. 2006;26:100–116. doi: 10.1128/MCB.26.1.100-116.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mor A, Philips MR. Compartmentalized Ras/MAPK Signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 75.Baker TL, Zheng H, Walker J, Coloff JL, Buss JE. Distinct rates of palmitate turnover on membrane-bound cellular and oncogenic H-ras. J Biol Chem. 2003;278:19292–300. doi: 10.1074/jbc.M206956200. [DOI] [PubMed] [Google Scholar]
- 76.Dekker FJ, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–56. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 77.Zeidman R, Jackson CS, Magee AI. Protein acyl thioesterases (Review) Mol Membr Biol. 2009;26:32–41. doi: 10.1080/09687680802629329. [DOI] [PubMed] [Google Scholar]
- 78.Ahearn IM, et al. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol Cell. 2011;41:173–85. doi: 10.1016/j.molcel.2011.01.001. Shows that cis-trans peptidyl prolyl isomerization in the C-terminus of H-Ras regulates depalmitoylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–29. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 80.Ballester R, Furth ME, Rosen OM. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J Biol Chem. 1987;262:2688–95. [PubMed] [Google Scholar]
- 81.Bivona TG, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–93. doi: 10.1016/j.molcel.2006.01.012. Reports that phosphorylation of K-Ras at serine 181 regulates its subcellular localization and leads to cell death. [DOI] [PubMed] [Google Scholar]
- 82.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–6. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 83.Madigan JP, et al. Regulation of Rnd3 localization and function by protein kinase C alpha-mediated phosphorylation. Biochem J. 2009;424:153–61. doi: 10.1042/BJ20082377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kashatus DF, et al. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol. 2011;13:1108–1115. doi: 10.1038/ncb2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fivaz M, Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J Cell Biol. 2005;170:429–41. doi: 10.1083/jcb.200409157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell. 2006;21:679–87. doi: 10.1016/j.molcel.2006.02.011. Established mono- and di-ubiquitination of H-Ras and showed the effects of this PTM on the localization on endosomes. [DOI] [PubMed] [Google Scholar]
- 87.Sasaki AT, et al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci Signal. 2010;4:ra13. doi: 10.1126/scisignal.2001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu L, Lubkov V, Taylor LJ, Bar-Sagi D. Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr Biol. 2010;20:1372–7. doi: 10.1016/j.cub.2010.06.051. Rabex-5 was identified as the E3 ubiquitin ligase that modifies Ras in a reaction that requires the Ras effector RIN1, suggesting feedback between this modification and signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195–8. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 90.Lander HM, et al. Redox regulation of cell signalling. Nature. 1996;381:380–1. doi: 10.1038/381380a0. Mass spec analysis revealed cysteine 118 to be the site of redox regulation of Ras and that S-nitorsylation of this cysteine promotes guanine nucleotide exchange. [DOI] [PubMed] [Google Scholar]
- 91.Heo J, Campbell SL. Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry. 2004;43:2314–22. doi: 10.1021/bi035275g. [DOI] [PubMed] [Google Scholar]
- 92.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–20. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 93.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003;100:6376–81. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aktories K, Schmidt G, Just I. Rho GTPases as targets of bacterial protein toxins. Biol Chem. 2000;381:421–6. doi: 10.1515/BC.2000.054. [DOI] [PubMed] [Google Scholar]
- 95.Ganesan AK, Vincent TS, Olson JC, Barbieri JT. Pseudomonas aeruginosa exoenzyme S disrupts Ras-mediated signal transduction by inhibiting guanine nucleotide exchange factor-catalyzed nucleotide exchange. J Biol Chem. 1999;274:21823–9. doi: 10.1074/jbc.274.31.21823. [DOI] [PubMed] [Google Scholar]
- 96.Just I, Selzer J, Hofmann F, Green GA, Aktories K. Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem. 1996;271:10149–53. doi: 10.1074/jbc.271.17.10149. This is the first report of a bacterial toxin that inactivates Ras by post-translational modification. [DOI] [PubMed] [Google Scholar]
- 97.Herrmann C, Ahmadian MR, Hofmann F, Just I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J Biol Chem. 1998;273:16134–9. doi: 10.1074/jbc.273.26.16134. [DOI] [PubMed] [Google Scholar]
- 98.Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proceedings of the National Academy of Sciences (USA) 1998;95:11175–11180. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wright LP, et al. Topology of mammalian isoprenylcysteine carboxyl methyltransferase determined in live cells with a fluorescent probe. Mol Cell Biol. 2009;29:1826–33. doi: 10.1128/MCB.01719-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lane KT, Beese LS. Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J Lipid Res. 2006;47:681–99. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Nancy V, Callebaut I, El Marjou A, de Gunzburg J. The delta subunit of retinal rod cGMP phosphodiesterase regulates the membrane association of Ras and Rap GTPases. J Biol Chem. 2002;277:15076–84. doi: 10.1074/jbc.M109983200. [DOI] [PubMed] [Google Scholar]
- 102.Figueroa C, Taylor J, Vojtek AB. Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J Biol Chem. 2001;276:28219–25. doi: 10.1074/jbc.M101763200. [DOI] [PubMed] [Google Scholar]
- 103.Paz A, Haklai R, Elad-Sfadia G, Ballan E, Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001;20:7486–93. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 104.Fehrenbacher N, Bar-Sagi D, Philips M. Ras/MAPK signaling from endomembranes. Mol Oncol. 2009;3:297–307. doi: 10.1016/j.molonc.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Di Fiore PP, De Camilli P. Endocytosis and signaling. an inseparable partnership. Cell. 2001;106:1–4. doi: 10.1016/s0092-8674(01)00428-7. [DOI] [PubMed] [Google Scholar]
- 106.Zoncu R, et al. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–21. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Misaki R, et al. Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J Cell Biol. 2010;191:23–9. doi: 10.1083/jcb.200911143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Omerovic J, Hammond DE, Clague MJ, Prior IA. Ras isoform abundance and signalling in human cancer cell lines. Oncogene. 2008;27:2754–62. doi: 10.1038/sj.onc.1210925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roy S, Wyse B, Hancock JF. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol Cell Biol. 2002;22:5128–40. doi: 10.1128/MCB.22.14.5128-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lu A, et al. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol. 2009;184:863–79. doi: 10.1083/jcb.200807186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Henis YI, Hancock JF, Prior IA. Ras acylation, compartmentalization and signaling nanoclusters (Review) Mol Membr Biol. 2009;26:80–92. doi: 10.1080/09687680802649582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Edidin M. The state of lipid rafts: from model membranes to cells. Annu Rev Biophys Biomol Struct. 2003;32:257–83. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 113.Sharma P, et al. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 2004;116:577–89. doi: 10.1016/s0092-8674(04)00167-9. [DOI] [PubMed] [Google Scholar]
- 114.Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–70. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tian T, et al. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–14. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 116.Plowman SJ, Muncke C, Parton RG, Hancock JF. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A. 2005;102:15500–5. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Prior IA, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–75. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 118.Belanis L, Plowman SJ, Rotblat B, Hancock JF, Kloog Y. Galectin-1 is a novel structural component and a major regulator of h-ras nanoclusters. Mol Biol Cell. 2008;19:1404–14. doi: 10.1091/mbc.E07-10-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Elad-Sfadia G, Haklai R, Ballan E, Gabius HJ, Kloog Y. Galectin-1 augments Ras activation and diverts Ras signals to Raf-1 at the expense of phosphoinositide 3-kinase. J Biol Chem. 2002;277:37169–75. doi: 10.1074/jbc.M205698200. [DOI] [PubMed] [Google Scholar]
- 120.Rotblat B, et al. Three separable domains regulate GTP-dependent association of H-ras with the plasma membrane. Mol Cell Biol. 2004;24:6799–810. doi: 10.1128/MCB.24.15.6799-6810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prior IA, Hancock JF. Compartmentalization of Ras proteins. J Cell Sci. 2001;114:1603–8. doi: 10.1242/jcs.114.9.1603. [DOI] [PubMed] [Google Scholar]
- 122.Jaumot M, Yan J, Clyde-Smith J, Sluimer J, Hancock JF. The linker domain of the Ha-Ras hypervariable region regulates interactions with exchange factors, Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 2002;277:272–8. doi: 10.1074/jbc.M108423200. [DOI] [PubMed] [Google Scholar]
- 123.Roy S, et al. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol. 2005;25:6722–33. doi: 10.1128/MCB.25.15.6722-6733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Niv H, Gutman O, Kloog Y, Henis YI. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J Cell Biol. 2002;157:865–72. doi: 10.1083/jcb.200202009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shalom-Feuerstein R, Cooks T, Raz A, Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005;65:7292–300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]
- 126.Plowman SJ, Ariotti N, Goodall A, Parton RG, Hancock JF. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol Cell Biol. 2008;28:4377–85. doi: 10.1128/MCB.00050-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 128.Gorfe AA, Babakhani A, McCammon JA. Free energy profile of H-ras membrane anchor upon membrane insertion. Angew Chem Int Ed Engl. 2007;46:8234–7. doi: 10.1002/anie.200702379. Computational modeling of insertion of the lipids that modify H-Ras into a membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, McCammon JA. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 2007;50:674–84. doi: 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- 130.Thapar R, Williams JG, Campbell SL. NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J Mol Biol. 2004;343:1391–408. doi: 10.1016/j.jmb.2004.08.106. [DOI] [PubMed] [Google Scholar]
- 131.Abankwa D, et al. A novel switch region regulates H-ras membrane orientation and signal output. EMBO J. 2008;27:727–35. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abankwa D, Gorfe AA, Inder K, Hancock JF. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci U S A. 2010;107:1130–5. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Willumsen BM, Cox AD, Solski PA, Der CJ, Buss JE. Novel determinants of H-Ras plasma membrane localization and transformation. Oncogene. 1996;13:1901–9. [PubMed] [Google Scholar]
- 134.Philips MR, Cox AD. Geranylgeranyltransferase I as a target for anti-cancer drugs. J Clin Invest. 2007;117:1223–5. doi: 10.1172/JCI32108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Winter-Vann AM, Casey PJ. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat Rev Cancer. 2005;5:405–12. doi: 10.1038/nrc1612. [DOI] [PubMed] [Google Scholar]
- 136.Basso AD, Kirschmeier P, Bishop WR. Thematic review series: lipid posttranslational modifications. farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 137.Whyte DB, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–64. doi: 10.1074/jbc.272.22.14459. This study showed that in the presense of an FTI, oncogenic Ras could be alternatively prenylated by geranylgeranyl transferase I. [DOI] [PubMed] [Google Scholar]
- 138.Kazi A, et al. Blockade of protein geranylgeranylation inhibits Cdk2-dependent p27Kip1 phosphorylation on Thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol Cell Biol. 2009;29:2254–63. doi: 10.1128/MCB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Winter-Vann AM, et al. A small-molecule inhibitor of isoprenylcysteine carboxyl methyltransferase with antitumor activity in cancer cells. Proc Natl Acad Sci U S A. 2005;102:4336–41. doi: 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schlitzer M, Winter-Vann A, Casey PJ. Non-peptidic, non-prenylic inhibitors of the prenyl protein-specific protease Rce1. Bioorg Med Chem Lett. 2001;11:425–7. doi: 10.1016/s0960-894x(00)00685-5. [DOI] [PubMed] [Google Scholar]
- 141.Bergo MO, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. Journal of Clinical Investigation. 2004;113:539–550. doi: 10.1172/JCI18829. This work showed that modification of K-Ras by Icmt is required for its transforming activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wahlstrom AM, et al. Rce1 deficiency accelerates the development of K-RAS-induced myeloproliferative disease. Blood. 2007;109:763–8. doi: 10.1182/blood-2006-05-024752. [DOI] [PMC free article] [PubMed] [Google Scholar]