

To the editor
Continuing evolution of DNA sequencing has transformed modern biology. Reduced sequencing costs coupled with novel sequencing based assays has led to rapid adoption of next generation sequencing (NGS) across diverse areas of life science research1-4. Sequencing has moved out of the genome centers into core facilities and individual labs where any investigator can access them for modest and progressively declining cost. While easy to generate in tremendous quantities, sequence data is still difficult to manage and analyze. Sophisticated informatics techniques and supporting infrastructure are needed to make sense of even conceptually simple sequencing experiments — let alone the more complex analysis techniques being developed. The most pressing challenge facing the sequencing community today is providing the informatics infrastructure and accessible analysis methods needed to make it possible for all investigators to realize the power of high-throughput sequencing to advance their research.
A possible solution to this infrastructure challenge comes in the form of “cloud computing”, a model where computation and storage exist as virtual resources, accessed via the Internet, which can be dynamically allocated and released as needed5. Where previously acquisition of large amounts of computing power required significant initial and ongoing costs, the cloud model radically alters this by allowing computing resources and services to be acquired and paid for on demand. Importantly, cloud resources can provide storage and computation at far less cost than dedicated resources for certain use cases. For several specific applications, effective use of cloud resources has already been demonstrated6-8. In general however, cloud resources are not provided in a form that can be immediately used by a researcher without informatics expertise. Several commercial vendors provide cloud-based sequence analysis services through the web that hide all complexity of the underlying infrastructure. Yet these contain limited sets of analysis tools, and because they are proprietary solutions users must give up some control over their own data and risk vendor lock-in. All “battle-tested” NGS analysis practices (such as analysis of human variation exemplified by the 1000 Genome Consortium publication) are open-source.
One popular open-source platform that has made substantial progress toward making complex analysis available to researchers is Galaxy9, 10. Galaxy enables users to perform analysis using nothing more than a web-browser. The environment automatically and transparently tracks every detail of the analysis, allows the construction of complex workflows, and allows the results to be documented, shared, and published with complete provenance, guaranteeing transparency and reproducibility. Importantly, Galaxy is an extensible platform; nearly any software tool can easily be integrated into Galaxy, and there is an active community of developers ensuring the latest tools are wrapped and made available through the Galaxy Tool Shed (http://usegalaxy.org/community). Galaxy is provided as a free public service (http://usegalaxy.org) with which thousands of users perform hundreds of thousands of analyses each month. However, this free public resource cannot meet increasing demand without implementing limits on data transfer and compute usage, resulting in delays that users may find unacceptable. Fortunately the Galaxy platform is easily deployed on local resources, and many groups working with large-scale sequence data now run their own Galaxy instances. However, this still requires local compute resources and informatics knowledge.
To bring the virtually unlimited resources of cloud computing into the hands of biomedical researchers we have developed Galaxy Cloud. It allows anyone to run a private Galaxy installation on the Cloud exactly replicating functionality of the main site (http://usegalaxy.org) but without the need to share compute resources with other users (Fig. 1). Unlike software service solutions, with Galaxy Cloud the user can customize their deployment as well as retain complete control over their instance and associated data; the analysis can also be moved to other cloud providers or local resources, avoiding concerns about vendor lock-in.
Figure 1.
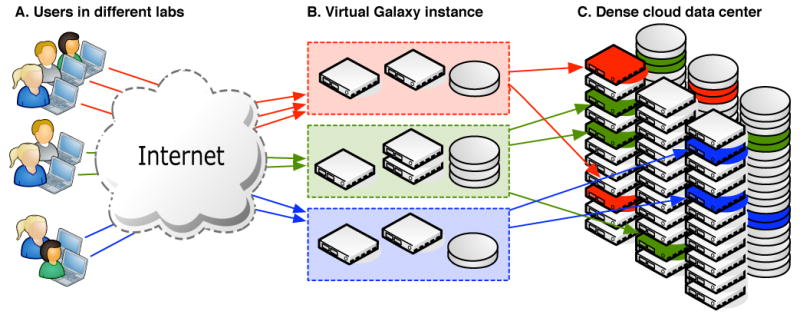
Overview of Galaxy instances running on cloud resources: (A) Users in different labs access a dedicated Galaxy instance over the internet with nothing more than a web browser, (B) these Galaxy instances appear to the users to be dedicated infrastructure with apparently infinite compute and storage resources, but are in fact virtual resources (C) which Galaxy's autoscaling acquires and releases on demand in response to changing workloads.
Currently we provide a public Galaxy Cloud deployment on the popular Amazon Web Services (AWS) cloud, however it is compatible with Eucalyptus and other clouds. If starting for the first time, the instance is configured by the user (e.g. by specifying the amount of initial storage allocated; exact step-by-step instructions are provided at http://usegalaxy.org/cloud). Once configured, users can then access their Galaxy, which will function exactly like the Galaxy public site. Every analysis tool that is available through the public Galaxy instance is installed an available for immediate use, as well as all the necessary supporting data (e.g. genome sequences, alignments, indexes). In addition, a number of tools that are too computationally intensive to provide on the public Galaxy are also included. This ready-to-use environment is combined with the ability to allocate practically unlimited computing power on demand thanks to use of cloud computing. When the user has finished analysis and the instance is no longer needed, all compute resources can be released, while the users data and instance state are preserved to be used later.
Galaxy Cloud's deployment is achieved by coupling the Galaxy framework to CloudMan11, which automates management of the underlying infrastructure cloud resources (see Supplemental Notes, Supplemental Figure 1). CloudMan handles all aspects of infrastructure management, including resource acquisition, configuration, and data persistence, necessary to support the Galaxy application. In the above scenario, CloudMan has allocated dedicated storage for the user's own data, initialized the Galaxy database, as well as composed additional data volumes containing the tools and secondary data they require. As with any instance of Galaxy, additional tools and data can easily be added by the owner of the instance and shared with others.
As a case study into the use of Galaxy Cloud, we consider the problem of identifying heteroplasmic sites — variation among the multiple copies of the mitochondrial genome (mtDNA) within a cell or individual. Mutations in mtDNA have been implicated in hundreds of diseases, and in many cases the disease causing variants can be heteroplasmic, with manifestation dependent on the relative proportion of variants12, 13. Further, this task emphasizes many of the key motivations for Galaxy Cloud. 1) It involves the use of clinical samples, which often involve strict privacy concerns and should not be analyzed on a public site, but can be analyzed on a secure public or private cloud resource. 2) It is both a data intensive problem and one with compute needs that vary over the course of the analysis. 3) It requires different methodology than the related problem of SNP calling in diploid genomes, showing the power of Galaxy's workflow system to construct solutions to non-standard tasks. 4) There is currently has no commonly accepted approach, which has led to questions about the validity of published heteroplasmic sites, emphasizing the need for a system that makes analysis completely transparent and reproducible.
Using mtDNA sequence data from nine individuals across three families14, we developed Galaxy workflows to perform the identification of heteroplasmic sites. These workflows map the sequencing reads, separate them by strand, transform datasets from read-centric to genome-centric form and perform a number of filtering and thresholding steps before merging the branches and generating a list of sites that contain allelic variants above a certain frequency supported by high quality reads on both strands. Running the workflows identified four heteroplasmic sites in two of the three examined families.
This analysis was computed entirely using Galaxy Cloud on AWS, and can be replicated exactly by importing the datasets and workflow available at http://s3.amazonaws.com/heteroplasmy/index.html. For complete description and explanation of the acquired data as well as how to use, import, and modify workflows used for the described heteroplasmy study see the Galaxy Page9 at http://usegalaxy.org/heteroplasmy/.
To perform the analysis, we uploaded 45GB of sequence datasets to S3, which took 9 hours at an average transfer rate of 1.5MB/sec and cost $5. During the execution of the analysis workflow, the cluster size was managed by CloudMan's auto-scaling feature and the cluster size varied between 1 and 16 nodes. It took approximately 6 hours and cost $20 to complete the workflow. With auto-scaling disabled, for fixed cluster sizes of 5 and 20 nodes the runtime was 9 hours at a cost of $20 and 6 hours at a cost of $50 respectively. By adapting the compute resource as the workflows demands change, auto-scaling is able to provide both the smallest total runtime and cost. Once the workflow is executed, the obtained results can be further analyzed directly on the cloud, downloaded, or left on the cloud for future reference. Overall, a complete analysis utilizing a compute cluster and a variety of open source NGS tools was performed within 15 hours for a cost of $25 using nothing but a web browser.
Cloud computing resources may not be as cost-effective for all usage scenarios. The workflow was already developed, which made it straightforward to execute in entirety. The interactive analysis and trial-and-error involved in building and refining the workflows is less cost-effective, though auto-scaling helps avoid excessive waste. This particular workflow has steps that could be executed in parallel, which allowed it to take advantage of cloud elasticity. Cloud instances of Galaxy will be limited by the resources available from a given cloud provider. For example, the largest memory instances currently provided by AWS are not sufficient to run certain de-novo assemblers. However, these are limitations of the provider used, not the cloud model. An advantage of the virtualization-based cloud model is the ability to move to a different cloud provider or to local resources. Cloud computing offers a new avenue for accessing computational infrastructure and Galaxy Cloud helps harness the potential in a very general way, but may not be appropriate or cost-effective for some workloads.
As NGS becomes an indispensible tool for biomedical research, it is crucial to provide analysis solutions that are usable for biomedical researchers and cost effective. Galaxy Cloud addresses this by combining the accessible Galaxy interface with automated management of cloud computing resources. Unlike purpose built solutions, Galaxy allows users both to use existing tested best practices in the form of workflows, or construct their own analyses for novel tasks. Galaxy Cloud instance are owned and controlled entirely by the user who created them, and can be used effectively in secure private clouds. Thus Galaxy Cloud provides a solution that retains user control and privacy, makes complex analysis accessible, and enables the use of practically limitless on-demand compute resources.
Supplementary Material
Acknowledgments
The authors are grateful to Jessica Beiler for coordinating sample collection, to clinical nurses from Penn State College of Medicine's Pediatric Clinical Research Office for collecting the samples and to volunteers for donating the samples. Efforts of the Galaxy Team (Enis Afgan, Dannon Baker, Dan Blankenberg, Nate Coraor, Jeremy Goecks, Greg Von Kuster, Ross Lazarus, Kanwei Li, Kelly Vincent) were instrumental for making this work happen. This work was funded by NIH grants HG005133 and HG005542 to JT and AN, NSF grant DBI 0543285 and NIH grant HG004909 to AN and JT, and NIH grant GM07226405S2 to KDM. Additional funding is provided, in part, under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
References
- 1.Schatz MC, Langmead B, Salzberg SL. Cloud computing and the DNA data race. Nat Biotechnol. 2010;28:691–693. doi: 10.1038/nbt0710-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepke S, Wold B, Mortazavi A. Computation for ChIP-seq and RNA-seq studies. Nature methods. 2009;6:S22–S32. doi: 10.1038/nmeth.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein L. The case for cloud computing in genome informatics. Genome biology. 2010;11:207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009;10:R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schatz MC. CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics. 2009;25:1363–1369. doi: 10.1093/bioinformatics/btp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Afgan E, et al. In: Guide to e-Science: Next Generation Scientific Research and Discovery. Yang K, editor. Springer; p. 35. in press. [Google Scholar]
- 11.Afgan E, et al. Galaxy CloudMan: delivering cloud compute clusters. BMC Bioinformatics. 2010;11(12):S4. doi: 10.1186/1471-2105-11-S12-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiMauro S. The many faces of mitochondrial diseases. Mitochondrion. 2004;4:799–807. doi: 10.1016/j.mito.2004.07.032. [DOI] [PubMed] [Google Scholar]
- 13.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goto H, et al. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome biology. 2011 doi: 10.1186/gb-2011-12-6-r59. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.