

Abstract
The allele E4 of apolipoprotein E (apoE4), the most prevalent genetic risk factor for Alzheimer's disease, is associated histopathologically with elevated levels of brain amyloid. This led to the suggestion that the pathological effects of apoE4 are mediated by cross-talk interactions with amyloid β peptide (Aβ), which accentuate the pathological effects of the amyloid cascade. The mechanisms underlying the Aβ-mediated pathological effects of apoE4 are unknown. We have shown recently that inhibition of the Aβ-degrading enzyme neprilysin in brains of wild-type apoE3 and apoE4 mice results in rapid and similar elevations in their total brain Aβ levels. However, the nucleation and aggregation of Aβ in these mice were markedly affected by the apoE genotype and were specifically enhanced in the apoE4 mice. We presently used the neprilysin inhibition paradigm to analyze the neuropathological and cognitive effects that are induced by apoE4 after activation of the amyloid cascade. This revealed that apoE4 stimulates isoform specifically the degeneration of hippocampal CA1 neurons and of entorhinal and septal neurons, which is accompanied by the accumulation of intracellular Aβ and apoE and with lysosomal activation. Furthermore, these neuropathological effects are associated isoform specifically with the occurrence of pronounced cognitive deficits in the ApoE4 mice. These findings provide the first in vivo evidence regarding the cellular mechanisms underlying the pathological cross talk between apoE4 and Aβ, as well as a novel model system of neurodegeneration in vivo that is uniquely suitable for studying the early stages of the amyloid cascade and the effects thereon of apoE4.
Keywords: apolipoprotein E4, β amyloid, neprilysin, neurodegeneration, CA1 neurons, entorhinal cortex, septum, lysosomes, learning and memory
Introduction
Alzheimer's disease (AD) is associated with several molecular hallmarks and genetic risk factors. These include amyloid β peptide (Aβ) and hyperphosphorylated tau, which are the major constituents of senile plaques and neurofibrilary tangles, respectively (Alzheimer et al., 1995; Goedert and Spillantini, 2006; Masters and Beyreuther, 2006), and the allele E4 of apolipoprotein E (apoE4), which is the most prevalent genetic risk factor for AD (Corder et al., 1993; Saunders et al., 1993; Roses, 2006). Histopathologically, apoE4 is associated with increased amyloid deposition, hippocampal atrophy, and neuronal loss, as well as with enhanced brain inflammation and impaired neuronal plasticity (Schmechel et al., 1993; Arendt et al., 1997; Mori et al., 2002).
The finding that brain amyloid levels are specifically elevated in apoE4-positive AD patients led to the suggestion that the pathological effects of apoE4 are mediated by cross-talk interactions with Aβ. Accordingly, in vitro studies revealed that apoE4 binds to Aβ isoform specifically (Strittmatter et al., 1993; LaDu et al., 1994), and that, unlike the AD benign allele apoE3, it does not protect against Aβ-induced toxicity (Puttfarcken et al., 1997; Jordan et al., 1998). In addition, amyloid β protein precursor (APP) × apoE double transgenic mice that express apoE4 have higher levels of Aβ deposits than the corresponding apoE3 × APP mice (Brendza et al., 2002; DeMattos, 2004; Holtzman, 2004).
The amyloid cascade is initiated by production of brain Aβ. This is followed by the formation of distinct Aβ oligomers that induce neuronal impairments and subsequent cell death (Hardy and Selkoe, 2002; Hardy, 2006). The steps of the amyloid cascade that are affected by apoE4 and the cellular and molecular mechanisms that underlie the resulting pathological effects are not known. Because the increased Aβ deposition in apoE4 × APP transgenic mice is apparent only in old mice (Holtzman et al., 2000; Fryer et al., 2005), and it does not seem to be associated with neuritic degeneration and neuronal loss (Holtzman et al., 2000; Hartman et al., 2002), there is a great need for models that reproduce the pathological interactions between apoE4 and Aβ in more detail. We have shown recently that intracerebroventricular infusion of thiorphan, which inhibits the Aβ-degrading enzyme neprilysin in wild-type mice, induces Aβ deposition and fibrilization, and that similar treatments in apoE3 and apoE4 transgenic mice result in marked and isoform-specific enhancement of the nucleation and aggregation of Aβ in apoE4 mice (Dolev and Michaelson, 2004, 2006). These effects are rapid and are apparent within 1 week after the initiation of treatment.
In the present study, we used the neprilysin inhibition paradigm to analyze the brain neuropathological processes and cognitive impairments induced by apoE4 at distinctive time intervals after activation of the amyloid cascade. Because apoE4 exacerbates the endosomal and lysosomal dysfunctions that occur in AD (Ji et al., 2002), we assessed the extent to which the pathological effects of apoE4 correlate spatially and temporally with the accumulation of intraneuronal Aβ and with the induction of lysosomal activation.
Materials and Methods
Transgenic mice.
APOE target replacement mice were created by gene targeting, as described previously (Sullivan et al., 1997). Construction of these mice differs from other apoE transgenic mice in that the human APOE gene was used to replace the mouse apoE gene. The two lines of apoE target replacement mice contain chimeric genes consisting of mouse 5′ regulatory sequences continuous with mouse exon 1 (noncoding), the 5′ half of mouse intron 1 continuous with the 3′ half of human intron 1, followed by human exons (and introns) 2–4 (Sullivan et al., 1997). The mice used were purchased from Taconic (Germantown, NY). Mice were back-crossed to C57BL/6J for eight generations and were homozygous for the APOE3 (3/3) or APOE4 (4/4) allele. The apoE genotype of the mice was confirmed by PCR analysis, as described previously (Levi et al., 2003). All experiments with mice were performed on age-matched male animals (3–4 months of age). All experiments were approved by the Tel-Aviv University Animal Care Committee, and every effort was made to reduce animal stress and to minimize animal usage.
Implantation of Alzet miniosmotic pumps.
Alzet (Palo Alto, CA) miniosmotic pumps (model 2004), which deliver their contents at a rate of 0.25 μl/h for up to 1 month, were each connected by means of a polyethylene catheter to a stainless steel cannula (Brain Infusion kit; Alzet) and loaded either with 0.5 mm thiorphan (Sigma, St. Louis, MO) in artificial CSF containing 1 mm ascorbic acid or with a similar solution without thiorphan. Mice were anesthetized by intraperitoneal injection of ketamine (120 mg/kg), their skulls were carefully exposed, and a small hole was drilled with a 25 gauge needle above the lateral ventricle (1 mm posterior and 1.5 mm lateral to the bregma). The tip of the brain-infusion cannula was inserted into the hole, and the cannula was glued to the skull (Luctite 454). To complete the procedure, the pump was inserted subcutaneously on the mouse's back, and the cut skin over the skull was sutured. An antibiotic (1% oxytetracycline) was added to the drinking water for 10 d.
Immunohistochemistry and immunofluorescence staining.
Mice were anesthetized with ketamine and xylazine and perfused transcardially with saline and then with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Their brains were removed, fixed overnight in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and then placed in 30% sucrose for 48 h. Frozen coronal sections (30 μm) were then cut on a sliding microtome and collected serially. The free-floating sections were immunostained as described previously (Matsumori et al., 2006), with the following primary antibodies (Abs): biotinylated anti-Aβ monoclonal Ab (mAb) 4G8 (1:200; Signet, Dedham, MA); rabbit anti-Aβ42 (1:500; Chemicon, Temecula, CA); rabbit anti-Aβ40 (1:500; Chemicon); goat anti-apoE (1:5000; Calbiochem, La Jolla, CA); biotinylated mouse anti-neuronal nuclei (anti-NeuN) (1:500; Chemicon); rabbit anti-microtubule-associated protein-2 (MAP-2) (1:1000; Chemicon); rabbit anti-synaptophysin (1:200, SYP H-93; Santa Cruz Biotechnology, Santa Cruz, CA); and rabbit anti-cathepsin D (1:500; Calbiochem). Accordingly, sections were washed with 10 mm PBS, pH 7.4, after which the primary antibody, diluted in PBS with 0.1% Triton X-100 (PBST) and with 2% of the appropriate serum, was applied overnight at 4°C. After having been rinsed in PBST, sections were incubated for 1 h at room temperature in the secondary antibody (Vector Laboratories, Burlingame, CA) and then diluted 1:200 in PBST that contained 2% of the appropriate serum (this step was omitted when biotinylated first Abs were used). After several additional rinses in PBST, sections were incubated for 0.5 h in avidin-biotin-horseradish peroxidase complex (ABC-Elite; Vector Laboratories) in PBST. After rinses in PBST, sections were placed for up to 10 min in diaminobenzidine chromagen solution (Vector Laboratories). The reaction was monitored visually and stopped by rinses of PBS. For the Aβ staining, sections were similarly treated, except that they were preincubated before adding the first antibody with 70% formic acid for 7 min. To minimize variability, sections from all animals were stained simultaneously. For hematoxylin staining (Mayer's Hematoxylin, #S3309; Dako Cytomation, Carpinteria, CA), free-floating sections were first mounted on slides and then stained according to the manufacturer instructions.
Immunofluorescence staining.
Coexpression of Aβ with MAP-2/apoE/cathepsin D and synaptophysin was evaluated by double and single immunostaining, using fluorescent chromogens. In brief, sections were first blocked by incubation with 0.1% Triton X-100 and 10% normal donkey serum in PBS for 1 h at room temperature. The primary antibodies were then dissolved in 0.1% Triton X-100 and 2% normal donkey serum in PBS, and finally incubated with the sections for 48 h at 4°C. Next, the bound primary antibodies were visualized by incubating the sections for 1 h at room temperature with Alexa-fluor 488/633-conjugated donkey anti-rabbit (1:1000; Invitrogen, Eugene, OR), Alexa-fluor 546-conjugated donkey anti-goat (1:1000; Invitrogen), or streptavidin-Alexa-fluor 488/633 (1:1000; Invitrogen), depending on the appropriate initial antibodies. The sections were then mounted on dry gelatin-coated slides, and fluorescence was visualized using a confocal scanning laser microscope (LSM 510; Zeiss, Oberkochen, Germany). Images (1024 × 1024 pixels) were obtained by a 63× water-immersion lens averaging eight scans per slice. All images were processed using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Image analysis.
The peroxidase-immunostained sections were viewed and photographed with a 40× objective and a Nikon DS-5M camera (Nikon Instech, Tokyo, Japan). The intensities of immunohistochemical staining as the percentage area stained were determined with the aid of the Image-Pro Plus system (version 5.1; Media Cybernetics, Silver Spring, MD). Other than making moderate adjustments for contrast and brightness, the images were not manipulated.
The image analysis was based on our previous studies indicating that the extracellular deposition of Aβ is maximal from bregma −1.5 to −3.5 (Dolev and Michaelson, 2004). Accordingly, the kinetic study, in which the levels of cellular Aβ and the neuronal pathology (NeuN) were assessed, focused on three serial sections from approximately −1.5 to −3.5 mm bregma, which were evenly spaced at 480 μm and of which CA1 domains were traced using a mouse brain atlas (Franklin and Paxinos, 1997). This revealed a maximal effect at 2 weeks and at bregma −2.5. The brain area specificity of the immunostaining was assessed using sections from mice that were treated with thiorphan for 2 weeks. Accordingly, images were taken at 40× from the CA1, dentate gyrus (DG), entorhinal, and visual cortex all at bregma −2.5 and from the medial septum and motor cortex at bregma +0.6, and their intensities of staining were then analyzed as described above. In addition, volumetric estimation of the levels of Aβ and NeuN staining of the entire hippocampal CA1 subfield was performed 2 weeks after the thiorphan treatment. Accordingly, the stained area of the CA1 subfield of each of the sections was calculated from every other frame at 40× and extrapolated for the entire slab, up to the next section. Summation of all the volumes of the positively stained slabs resulted in a total volumetric estimation of the entire stained CA1 subfield. This was calculated as (total CA1 area [μm2] × percentage of the CA1 area that was stained × 480 μm). Analysis of CA1 volume revealed no changes between mouse groups and no side effects of treatment.
Immunoblot analysis.
Total soluble Aβ and apoE of coronal brain slices (−1.5 to −3.5 mm from bregma) were extracted as described by Lesne et al. (2006). For determining apoE, samples were blotted and immunoreacted with goat anti-apoE (1:5000; Calbiochem). For Aβ analysis, samples were extracted with 6 m guanidine HCl, followed by immunoblot assays using mAb 4G8 (1:2000; Signet, Dedham, MA) as described previously (Dolev and Michaelson, 2004).
Behavioral experiments.
To prevent swimming-related complications resulting from the implanted Alzet pumps, the spatial navigation test was performed by a dry maze modification of the hole board test (Van der Zee et al., 1992), which monitors the ability of the mice to locate a small water-filled well in a circular arena.
Water deprivation.
Two days before implanting the Alzet pumps and throughout the entire experiment, the mice were subjected to a 23 h/day water deprivation regime, in which mice were able to drink ad libitum for 1 h every day. After the implantation of the Alzet pumps, the mice had 5 d to recover. For habituation, at day 6 after implantation of the Alzet pumps, the mice were placed in a circular arena (95 cm diameter, with 20 evenly separated wells; 1 cm depth, 0.5 cm diameter) in which all the wells were filled with 100 μl of water. Every mouse then underwent three runs per day for 2 d; each run consisted of 120 s. Mice were allowed to drink from all the wells they could locate during these runs. The arena was cleaned with 70% ethanol between every run. Learning the principle started after the habituation stage (day 8) by subjecting each mouse to five runs per day; each run lasted up to 120 s and only one well was filled with water. If the mouse found the water-filled well, then it was allowed to drink for 15 s, whereas if the mouse did not find the well, the mouse was brought to it after 120 s and allowed to remain there for 15 s. This was performed for 6 d (days 8–13 after the initiation of the thiorphan treatment). To elevate the level of complexity of the test, the location of the water-filled well was then changed to a new location on day 14, and the performance of the mice was then tested in this configuration for three more days. The trajectories of locomotion of the mice throughout the experiment were photographed and analyzed by EthoVision version 3.1 (Noldus Information Technology, Wageningen, The Netherlands), which was set up for the distance traveled (centimeters), the time required for the mice to locate the water-filled well (seconds), and their traveling velocity (centimeters/second).
Novel object recognition task.
This test was performed after the spatial navigation test. Briefly, the mice were first placed individually for 5 min in an empty arena (50 × 50 × 20 cm). This was followed on day 2 by a training session, during which one object was placed in the open field. The mice were then allowed to individually explore this object for 5 min, and the actual exploration time was then recorded. The ability of the mice to remember the object and to distinguish between novel and non-novel objects was then assessed by a retention test that was performed on day 3. Accordingly, each mouse was placed in the same box in which there was an object identical to the one used on day 2 and a different object placed in opposite corners of the box (10 cm from the corner). The mice were allowed to freely explore the arena for 5 min. The time spent exploring each of the objects was then recorded, and the extent to which the mice showed a preference for the new object, which is an indication that it remembered the old object, was then assessed by determining the ratio of the time spent at the new object to the total time spent at both objects. The objects used were ∼10–15 cm in height and were washed in alcohol between successive tests. Replacing the object used in the training session on day 2 with an identical object for the retention test on day 3 was done to control for possible olfactory effects.
Electron microscopy.
For electron microscopy studies, apoE3 and apoE4, sham- and thiorphan-treated transgenic mice were killed at 2 and 4 weeks after initiating the thiorphan treatment (n = 5 mice per group). Accordingly, the mice were anesthetized by intraperitoneal injection of ketamine and xylazine and perfused transcardially with PBS, followed by 4% paraformaldehyde and 0.2% glutaraldehyde in PBS. The heads were removed and kept refrigerated at 4°C for 1.5–2 h after perfusion, after which the brains were removed and postfixed with 4% paraformaldehyde in PBS. The next day, the brains were vibratomed at 50 μm, after which coronal sections from bregma −2.5 mm to bregma −2.70 mm, corresponding to images 51–53 in the study by Franklin and Paxinos (1997), were collected and maintained in their original sequence in PBS. The sections were osmicated, dehydrated in a series of graded ethanol, and then flat-embedded in Epon/Araldite resin. Two sections from the indicated bregma of each brain were glued on transparent cylinders made from Epon/Araldite. Each block was trimmed to include the CA1 layer of the hippocampus before ultrathin sectioning. The obtained 90 nm sections were mounted onto 200 mesh nickel grids, and every 10th section was examined using a JEOL (Akishima, Japan) 1200EX electron microscope. Accordingly, the number of sections per brain was six. The level of ultrastructural abnormalities in the form of flat vacuoles and membranes inclusions were assessed in distal and proximal dendrites and in the cell bodies. This was performed by scanning fields of 4 × 104 μm2 of coded sections and counting the number of dendrites and cell body profiles, which contained and which did not contain either vacuoles or membranous inclusions. Results shown are the percentage ±SD of the dentritic and cell body profiles in each of the mice groups that contained the ultrastructural abnormalities.
Statistical analysis.
All numerical analyses were performed using SPSS version 12. The two groups in each of the treatments (sham and thiorphan) were compared by two-way ANOVA (using a 2 × 2 design) and further analyzed, where appropriate, by Student's t test analysis. For the spatial navigation test, statistical analysis of repeated measurements was used (group × treatment × trial design).
Results
Intraneuronal accumulation of Aβ and ApoE
Inhibition of neprilysin by intracerebroventricular injection of thiorphan for 2 weeks triggered the formation of extracellular plaque-like deposits of Aβ, the levels of which were higher in the apoE4 than in the corresponding apoE3 mice (Fig. 1A). The size of the Aβ deposits was ∼10 μm, and, as observed previously with a different line of apoE4 transgenic mice (Dolev and Michaelson, 2004, 2006), they were present in the hippocampus as well as in the cortex and subcortical areas. Additional examination of the brain sections revealed enhanced Aβ staining of CA1 neurons of the thiorphan-treated apoE4 mice, which was more pronounced than that observed in either the corresponding apoE3 mice or the sham-treated apoE3 and apoE4 mice (Fig. 1A). Control experiments revealed that both the extracellular and CA1-associated Aβ staining were blocked by the addition of exogenous Aβ. Similar results were observed with an additional anti-Aβ antibody (rabbit anti-rodent; Chemicon). Furthermore, immunohistochemical experiments using AbR7334 (rabbit polyclonal), which is directed against the C-terminal of APP (Grimm et al., 2003), revealed no differences between the staining of CA1 and other hippocampal and cortical neurons of sham and naive apoE3 and apoE4 mice, and no changes after the thiorphan treatment (data not shown). This suggests that the increased immunostaining observed with the anti-Aβ mAb in CA1 neurons of the apoE4 mice, after the thiorphan treatment, is indeed a result of Aβ and not of APP- or other Aβ-containing fragments of APP. Double-labeling confocal microscopy, using the cytoplasmic marker MAP-2 (Fig. 1) and the neuronal nuclear marker NeuN (data not shown), confirmed the immunohistochemical findings and revealed that the CA1-associated Aβ of the apoE4 mice is neuronal and perinuclear (Fig. 1B).
Figure 1.
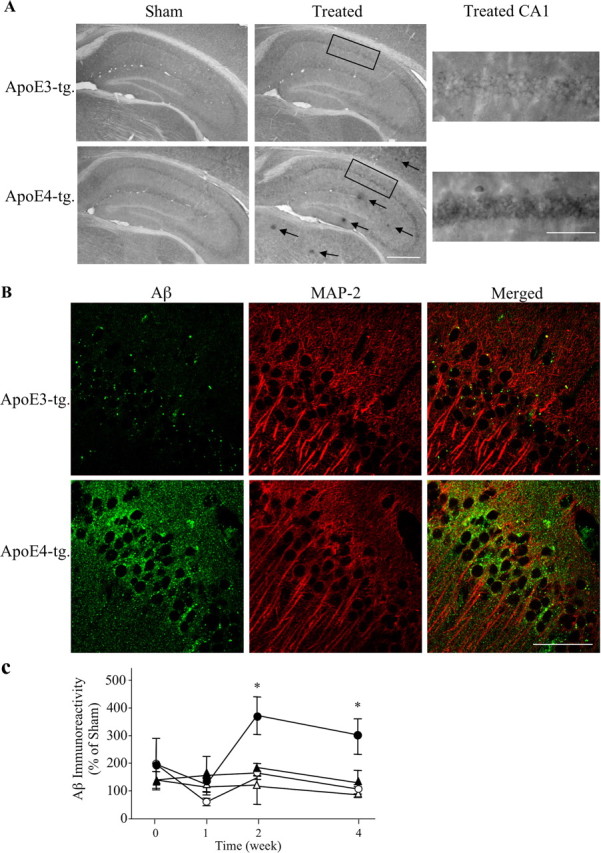
Accumulation of extracellular and intracellular Aβ deposits in apoE3 and apoE4 mice after inhibition of neprilysin. ApoE3 and apoE4 mice were injected intracerebroventricularly using Alzet mini pumps with the neprilysin inhibitor thiorphan for 2 weeks or sham injected, after which the brains were processed for immunohistochemistry and confocal microscopy, as described in Materials and Methods. A, Coronal sections of sham- and thiorphan-treated apoE3 and apoE4 mice that were stained immunohistochemically with the anti-Aβ mAb 4G8. The left panels depict extracellular Aβ deposits (arrows), which are present specifically in the thiorphan-treated apoE4 mice. Scale bar, 500 μm. The rectangles indicate the CA1 subfield shown at a higher magnification in the right panels. Scale bar, 100 μm. They depict the specific accumulation of Aβ in the CA1 hippocampal field of the thiorphan-treated apoE4 mice. B, Double-labeling confocal microscopy of Aβ (left), the cytoplasmic marker MAP-2 (middle), and their merged images (right) in the thiorphan-treated apoE3 and apoE4 mice. Scale bar, 50 μm. C, Kinetics analysis and quantitation of the levels of Aβ in CA1 hippocampal neurons of apoE3 and apoE4 mice after inhibition of neprilysin. Results were obtained from coronal sections at bregma −2.5 stained immunohistochemically with mAb 4G8 and were analyzed at the indicated time points in terms of the fraction of the CA1 field stained for Aβ as described in Materials and Methods. They are presented (mean ± SE of n = 5 per group × treatment) as the percentage of the sham-treated apoE3 mice. The symbols ▴ and • represent the thiorphan-treated apoE3 and apoE4 mice, whereas ▵ and ○ represent the corresponding sham-treated mice (*p < 0.04 for comparing the results of the thiorphan-treated apoE4 with those of the other mouse groups).
Kinetic analysis of the accumulation of neuronal Aβ in the perikerya of the CA1 neurons was performed on coronal sections at bregma −2.5, which is where the staining was most pronounced. This revealed that the levels of CA1 Aβ peaked and reached a plateau by 2 weeks, which was maintained for at least the 4 week duration of the thiorphan treatment (Fig. 1C). Statistical analysis of these results revealed significant effects of group × treatment at 2 and 4 weeks (p < 0.001) and that the levels of Aβ in CA1 neurons of the thiorphan-treated apoE4 mice were significantly higher than those of the other mouse groups (p < 0.04). Similar results were observed in the dendritic fields of the CA1 neurons, except that Aβ staining was maximal at 2 weeks and declined back to basal levels by 4 weeks (data not shown). Quantitation of the levels of Aβ in the perikerya of hippocampal CA1 neurons was also performed by stereological volumetric estimation using brain sections of mice that were treated with thiorphan for 2 weeks. This revealed that the presence of apoE4 resulted in an approximately fourfold elevation in the levels of Aβ in CA1 neurons of the thiorphan-treated apoE4 mice (380 ± 50% of the corresponding sham-treated mice) and a much smaller (160 ± 30%) elevation in the treated apoE3 mice. Statistical analysis of these results revealed a significant effect of group × treatment (p < 0.05), and that the Aβ levels of the thiorphan-treated apoE4 mice were significantly higher than those of the other mouse groups (p < 0.04).
Additional immunohistochemical experiments using polyclonal Abs directed specifically at Aβ42 and Aβ40 revealed that the CA1 neurons of the thiorphan-treated apoE4 mice contained elevated levels of Aβ42, and that the Aβ42 levels in the corresponding apoE3 mice and in the sham-treated apoE3 and apoE4 mice were low and similar (Fig. 2). In contrast, the levels of Aβ40 were slightly elevated in the apoE3 but not in the apoE4 mice after the thiorphan treatment (Fig. 2). Statistical analysis revealed a significant effect of group × treatment for Aβ42 (p < 0.005), and that the Aβ42 levels of the thiorphan-treated apoE4 mice were higher than those of all the other mouse groups (p < 0.005). In contrast, the increase in Aβ40 in the treated apoE3 mice was insignificant. This indicates that the increase in CA1 Aβ, which is observed with the pan Aβ mAb 4G8 in the apoE4 mice after the thiorphan treatment (Fig. 1), is mainly caused by the accumulation of Aβ42 in these neurons. Immunoblot experiments revealed, in accordance with our previous findings (Dolev and Michaelson, 2004), that the thiorphan-treated apoE3 and apoE4 mice contained similarly elevated levels of Aβ (data not shown), suggesting that the elevation of Aβ levels in CA1 neurons is attributable to specific effects of ApoE4 on the accumulation of Aβ in these neurons and not on the overall levels of Aβ. We next examined the extent to which the accumulation of Aβ in CA1 neurons of the apoE4 mice is associated with parallel and isoform-specific accumulation of apoE4 in these neurons. Confocal microscopy revealed that apoE accumulated in CA1 neurons after the thiorphan treatment, and that this effect was markedly more pronounced in the apoE4 than in the apoE3 mice (Fig. 3A). Double-labeling confocal microscopy revealed that many of the apoE4-containing CA1 neurons also had Aβ. There were, however, CA1 neurons that stained positively for Aβ but had no detectable apoE4 and vice versa. Additional examination of the confocal images revealed that in the cells that stained positively for both Aβ and apoE4, only a fraction of these molecules was colocalized. Immunoblot experiments revealed that the total brain levels of apoE in the apoE3 and apoE4 mice were similarly elevated by the thiorphan treatment (Fig. 3B), suggesting that the observed preferential accumulation of apoE4 in the CA1 neurons of the apoE4 mice is not caused by differences in the total apoE levels of the apoE3 and apoE4 mice but instead is driven by an isoform-specific property of apoE4.
Figure 2.

Accumulation of Aβ42 and Aβ40 in CA1 neurons of apoE3 and apoE4 mice after inhibition of neprilysin. The mice were injected intracerebroventricularly, using Alzet mini pumps, with the neprilysin inhibitor thiorphan for 2 weeks or sham injected, after which the brains were processed for immunohistochemistry, as described in Materials and Methods. Results shown are from coronal sections at bregma −2.5, which were stained with anti-Aβ42 (Chemicon) and anti-Aβ40 antisera (Chemicon). Scale bar, 100 μm. Quantitation of the results (mean ± SE; n = 5 per group × treatment) was performed by computerized densitometry, as described in Materials and Methods, and is presented for each of the Aβ stainings as a percentage of the sham-treated apoE3 mice. The striped and dotted bars correspond, respectively, to Aβ42 and Aβ40 (*p < 0.005 for comparison of the thiorphan-treated apoE4 with the other mouse groups).
Figure 3.
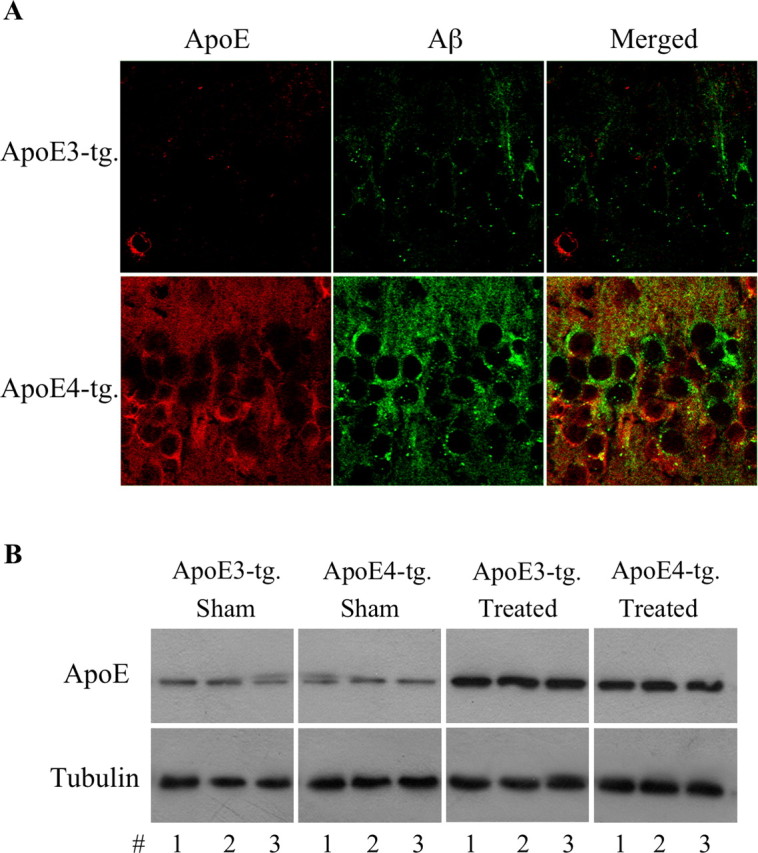
The levels of brain apoE in apoE3 and apoE4 mice after inhibition of neprilysin. ApoE3 and apoE4 mice were injected intracerebroventricularly, using Alzet mini pumps, with the neprilysin inhibitor thiorphan for 2 weeks or sham injected, after which the brains were processed for confocal microscopy and immunoblot assays, as described in Materials and Methods. A, Double-labeling confocal microscopy of apoE (left) and Aβ (middle) of CA1 neurons and their merged images (right) in the thiorphan-treated apoE3 and apoE4 mice. Scale bar, 50 μm. As shown, the thiorphan-treated apoE4 mice contain higher levels of apoE in CA1 neurons than do the corresponding apoE3 mice. Importantly, the Aβ and apoE of the apoE4 mice do not fully overlap in the CA1 neurons. B, Immunoblots of brain apoE of thiorphan- and sham-treated apoE3 and apoE4 mice, including tubulin gel-loading controls.
Neuropathology
According to the amyloid hypothesis, the apoE4-induced increase in the accumulation of Aβ is expected to cause neurodegeneration. Indeed, treatment of the apoE4 mice with thiorphan for 2 weeks resulted in decreased density of NeuN-positive CA1 neurons, whereas the corresponding CA1 neurons of the apoE3 mice were not affected (Fig. 4A). A similar decrease in the CA1 neurons in the thiorphan-treated apoE4 mice was also observed in hematoxylin-stained hippocampal sections (Fig. 4A), suggesting that the observed decrease in NeuN staining is indeed a result of neuronal loss and not to shrinkage of the nucleus or loss of the NeuN antigen. Kinetic experiments revealed that the decreased NeuN staining in the neprilysin-inhibited apoE4 mice, like that of the accumulation of Aβ in these neurons (Fig. 1C), was apparent 1 week after initiation of the inhibition of neprilysin and reached a plateau by 2 weeks (Fig. 4B). In contrast, the levels of NeuN staining in the treated apoE3 mice were not affected during the first 2 weeks of the thiorphan treatment and decreased partially at 4 weeks (Fig. 4B). Therefore, the neprilysin inhibition paradigm also affects the CA1 neurons in the apoE3 mice, but this effect is less pronounced and delayed relative to that obtained in the presence of apoE4. Statistical analysis of these results revealed significant effects of group × treatment at 1 and 2 weeks (p < 0.005) and that the levels of NeuN staining in CA1 neurons of the thiorphan-treated apoE4 mice were significantly lower than those of the other mouse groups (p < 0.03). Quantitation of the observed neuropathology by stereological volumetric estimation was performed at the 2 week time point. This revealed a decrease of 26 ± 5% in the levels of NeuN-positive CA1 neurons in the thiorphan-treated apoE4 mice, and no change at this time point in the corresponding NeuN levels in the apoE3 mice, which were similar to those of the sham-treated apoE3 and apoE4 mice (p < 0.03 for group × treatment; p < 0.03 for treated apoE4 vs the other mouse groups). Confocal microscopy measurements of the effects of the thiorphan treatment on hippocampal synapses were performed using the presynaptic marker synaptophysin (Levi et al., 2003). In accordance with the NeuN results, there was a significant decrease in synaptophysin staining of the thiorphan-treated apoE4 mice and no such change in the corresponding apoE3 mice (p < 0.02 for group × treatment; p < 0.05 for treated apoE4 vs the other mouse groups) (Fig. 5).
Figure 4.

Neuronal loss in the hippocampus of apoE3 and apoE4 mice after inhibition of neprilysin. ApoE3 and apoE4 mice were injected intracerebroventricularly, using Alzet mini pumps, with thiorphan- or sham-injected for the indicated times. Their brains were then excised and sectioned, after which they were stained immunohistochemically, with antisera directed against the neuronal marker NeuN, and histochemically with hematoxylin, as described in Materials and Methods. A, Representative NeuN-stained sections of the CA1 hippocampal subfield of thiorphan- and sham-treated apoE3 and apoE4 mice. The panels on the right depict sections of CA1 neurons of the thiorphan-treated mice that were stained histochemically with hematoxylin. Scale bar, 100 μm. B, Computerized densitometric analysis of the kinetics of the effects of apoE and thiorphan on the loss of hippocampal CA1 neurons. Results were obtained from coronal sections at bregma −2.5, which were stained immunohistochemically for NeuN and were analyzed, as described in Materials and Methods, in terms of the fraction of the CA1 field stained for NeuN. They are presented (mean ± SE of n = 5 per group × treatment) as the percentage of sham-treated apoE3 mice. The symbols ▴ and • represent the thiorphan-treated apoE3 and apoE4 mice, whereas ▵ and ○ represent the corresponding sham-treated mice (*p < 0.03 for comparing the kinetics and volumetric results of the thiorphan-treated apoE4 mice with those of the other mouse groups).
Figure 5.
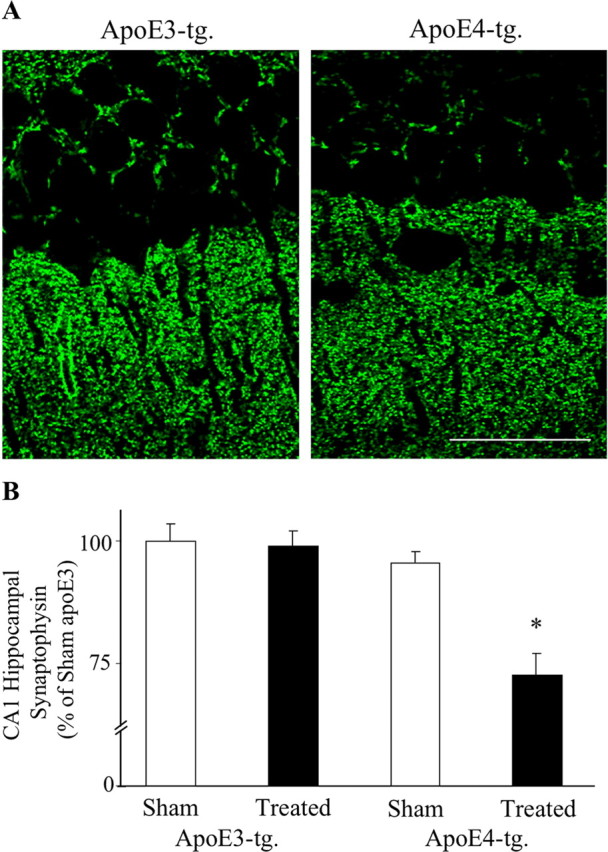
Synaptic loss in the hippocampus of apoE3 and apoE4 mice after inhibition of neprilysin. ApoE3 and apoE4 mice were injected intracerebroventricularly, using Alzet mini pumps, with thiorphan for 2 weeks or were sham injected. The brains were then excised and sectioned, after which they were stained immunohistochemically and visualized by confocal microscopy, as described in Materials and Methods. A, Representative sections of the CA1 hippocampal subfield (bregma −2.5) of thiorphan-treated apoE3 and apoE4 mice. Scale bar, 50 μm. B, Computerized densitometric analysis of the synaptic levels of thiorphan- and sham-treated apoE3 and apoE4 mice. Results shown were obtained from coronal sections at bregma −2.5, as described in Materials and Methods, and are presented (mean ± SE; n = 5 per group × treatment) as → percentage of the sham-treated apoE3 mice (*p < 0.05 for comparing the thiorphan-treated apoE4 with the other mouse groups).
Next, ultrastructural alterations in the CA1 region of the hippocampus in the thiorphan-treated apoE mice were examined. The results indicated the presence of abundant flat vacuoles and pathological inclusions in the distal dendrites of the apoE4 mice at 2 weeks after the thiorphan treatment, and that such structural alterations were markedly less frequent in the corresponding apoE3 mice (Fig. 6). These effects increased by 4 weeks, after the thiorphan treatment, and continued to be more pronounced in the apoE4 than in the apoE3 mice at this time point (Fig. 6). Vacuoles and pathological inclusions were also observed in CA1 cell bodies. This effect was, however, apparent only at the 4 week time point and was also more pronounced in the apoE4 mice.
Figure 6.

Ultrastructural alterations in CA1 neurons of thiorphan-treated apoE3 and apoE4 mice. The apoE transgenic mice were injected intracerebroventricularly with thiorphan or were sham treated for the indicated time, after which their brains were processed and visualized by electron microscopy (magnification, 10,000×), as described in Materials and Methods. At 2 weeks after thiorphan treatment of the apoE3 mice, scattered flat vacuoles and inclusions in the distal dendrites (top middle) were observed, whereas in the apoE4 mice, at this time point, abundant flat vacuoles and inclusions in the distal dendrites (top right) were observed. At 4 weeks after thiorphan treatment of apoE3 mice, flat vacuoles and inclusions in the distal dendrites (bottom middle) were observed, whereas in the apoE4 mice, at this time point, there was further elevation in the levels of flat vacuoles and inclusions in the distal dendrites (bottom right). The neuronal inclusions in the different sections are indicted by arrows, whereas dendrites and vacuole are indicated by d and v, respectively. Sham controls are depicted in the left panels. Semiquantitative analysis of the vacuolar neuronal pathology at 2 and 4 weeks was performed as described in Materials and Methods, and the results obtained are depicted in the graphs on the right. The dark and light bars correspond, respectively, to the thiorphan-treated apoE3 and apoE4 mice.
Brain area specificity
The extent to which the observed effects of ApoE4 are specific to CA1 neurons was examined by measuring the effects of the thiorphan treatment on other hippocampal neurons (e.g., DG granular neurons), on brain areas that are directly connected to the hippocampus (e.g., entorhinal cortex and septum), and on other brain areas. The results thus obtained are depicted in Figure 7A. As can be seen, the levels of NeuN-positive neurons of the CA1, entorhinal cortex, and the septum decreased in the ApoE4 transgenic mice after the thiorphan treatment, whereas the corresponding brain areas of the ApoE3 mice were not affected. In contrast, DG and cortical neurons that are not connected directly to the hippocampus (e.g., the visual cortex adjacent to and on the same section as the assessed entorhinal cortex) were not affected by the thiorphan treatment in either the ApoE4 or ApoE3 mice. Two-way ANOVA revealed a significant effect of group × treatment in the CA1 and entorhinal cortex (p < 0.03) and in the septum (p < 0.002), which was associated with a significant reduction in the neuronal levels of the CA1 (26 ± 5%; p < 0.03), entorhinal cortex (25 ± 6%; p < 0.03), and septum (44 ± 5%; p < 0.002) of the treated ApoE4 mice.
Figure 7.
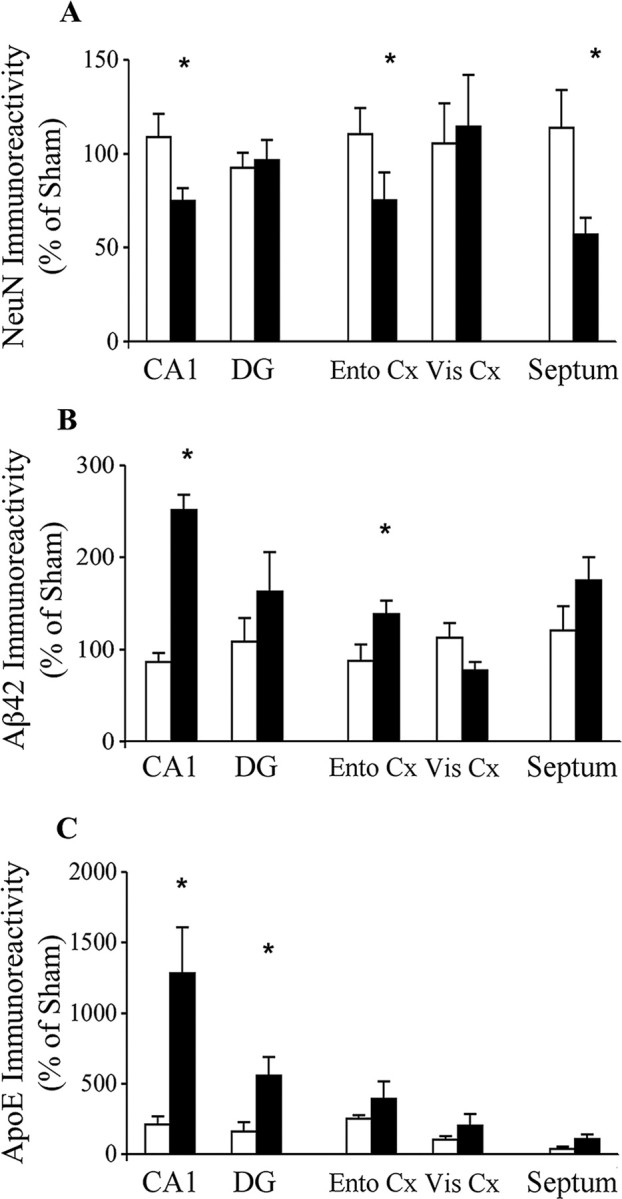
Brain area specificity of the effects of thiorphan on NeuN, intracellular Aβ42, and ApoE in distinct brain areas of apoE4 and ApoE3 transgenic mice. The mice were injected using Alzet mini pumps with the neprilysin inhibitor thiorphan for 2 weeks or were sham injected, after which their brains were sectioned and stained immunohistochemically (NeuN and Aβ42) and for immunofluorescence (ApoE), and the results were quantified, as described in Materials and Methods. Results of the CA1 and DG neurons and of the entorhinal (Ento Cx) and visual cortex (Vis Cx) were obtained from the same section (bregma −2.5). However, the septum was stained using sections of bregma 0.6. Results shown (mean ± SE of 5 per group × treatment for each brain area) are presented as a percentage of sham-treated ApoE3 mice. The black and white bars correspond, respectively, to thiorphan-treated apoE4 and apoE3 mice (*p < 0.03 for comparing the results of the thiorphan-treated apoE4 with those of the other mouse groups).
Measurements of the levels of cellular Aβ42 revealed an elevation in all the examined brain areas except the visual cortex of the thiorphan-treated ApoE4 mice. This effect was significant in the CA1 and entorhinal cortex (p < 0.03; two-way ANOVA) and close to being significant in the septum (p = 0.06; two-way ANOVA) but, because of larger variations, was not significant in the DG. Post hoc analysis revealed that the elevation in the levels of Aβ42-containing neurons of the ApoE4 mice was most pronounced and significant in CA1 neurons (250 ± 15%; p < 0.001), and that it was also significant in the entorhinal cortex (138 ± 10%; p < 0.01).
Neuronal ApoE levels were also elevated isoform specifically in the thiorphan-treated ApoE4 mice (Fig. 7C). This effect was statistically significant only in CA1 and DG neurons (p < 0.001 and p < 0.05 by two-way ANOVA, respectively) and, similar to that observed with Aβ42, was most pronounced in CA1 neurons (1300 ± 330%; p < 0.001).
Lysosomal activation
Previous studies of AD brains revealed generalized upregulation of the lysosomal system in neurons of affected brain areas (Nixon and Cataldo, 2006), and that apoE4 exacerbates endosomal and lysosomal dysfunction (Cataldo et al., 1997). In addition, cell culture in vitro studies revealed that apoE4 potentiates Aβ-induced neuronal death by enhancing lysosomal activation and leakage (Ji et al., 2002, 2006). We therefore examined the possibility that the presently observed synergistic pathological effects of apoE4 and Aβ in the neprilysin inhibition model are associated with lysosomal activation. The experiments focused on CA1 neurons in which the accumulation of intraneuronal ApoE and Aβ were most pronounced. Double-labeling confocal microscopy, using the lysosomal marker cathepsin D, revealed that the CA1 neurons of the thiorphan-treated apoE4 mice contained intracellular punctate cathepsin D-positive lysosomes, the size and levels of which were markedly higher than those of the corresponding apoE3 mice, as well as increased levels of diffused granular cathepsin D staining (Fig. 8). Furthermore, a large fraction of the Aβ and punctate cathepsin D staining of the CA1 neurons in the apoE4 mice colocalize (Fig. 8), suggesting that at least some of the accumulated Aβ is associated with lysosomes.
Figure 8.

Lysosomal localization of Aβ in CA1 neurons of apoE3 and apoE4 mice after inhibition of neprilysin. The apoE transgenic mice were injected intracerebroventricularly with thiorphan for 2 weeks, after which their brains were excised, sectioned, and subjected to cathepsin D and Aβ double-labeling confocal microscopy, as described in Materials and Methods. Laser confocal images in red are of the lysosomal marker cathepsin-D (left), whereas images in green are of Aβ (mAb 4G8) (middle), and their merged images are depicted in the right panels. Images are from the hippocampal CA1 region. Scale bar, 50 μm. As shown, apoE4-treated mice have higher punctated and diffuse lysosomal staining than the corresponding apoE3 mice. Moreover, thiorphan-treated apoE4 mice but not apoE3 mice exhibit extensive accumulation of intracellular Aβ that colocalizes with cathepsin D.
Behavioral experiments
We next investigated the extent to which the brain neuropathology of the ApoE4 mice is associated with impaired cognitive performance. This was first assessed by monitoring spatial navigation, which is known to be affected by hippocampal dysfunction (Morris and Frey, 1997), using a dry maze paradigm that monitors the ability of the mice to locate a small water-filled well in an open field (Van der Zee et al., 1992). As described in Materials and Methods, the mice were first tested for 6 d (five trials per day) with the water-filled well at a fixed position. Both the sham- and the thiorphan-treated apoE3 and apoE4 mice improved their performance during the training period and similarly shortened the length of their trajectories to the water-filled well (p < 0.001 for the effect of days on all mouse groups) (Fig. 9A). Similar improvements were noted when the performances of the mice were assessed in terms of the time it took them to find the water-filled well (data not shown). Analysis of the speed of locomotion revealed that it was similar in the sham-treated apoE3 and apoE4 mice (7.6 ± 0.8 and 7.7 ± 0.8 cm/s, respectively) and the corresponding thiorphan-treated mice (8.8 ± 0.4 and 8 ± 1.4 cm/s, respectively).
Figure 9.

The effects of apoE and thiorphan on learning and memory. ApoE3 and apoE4 mice were injected intracerebroventricularly with thiorphan or sham treated for up to 3 weeks and subjected to a spatial navigation learning and memory test and to an object recognition test, as described in Materials and Methods. A, The mice were tested, starting at day 5, after initiation of the thiorphan treatment, for their ability to locate a water-filled well in an open field (5 trials/d for 6 consecutive days). Results shown represent the path length traversed per trial by the mice at the indicated days (mean ± SE; n = 5 per group × treatment). Representative trajectories obtained on days 1 and 5 are depicted on the left. The positions of the starting point of the mice and of the water-filled wells are denoted by a filled square and an arrowhead, respectively. B, The position of the water-filled well was changed, and the performance of the mice was then monitored for 3 consecutive days (5 trials/d), during which the position of the water-filled well was unchanged. Results shown (mean ± SE of n = 5 mice per group × treatment) were obtained on the first and third days after the position of the well was changed. The symbols ▴ and • represent the thiorphan-treated apoE3 and apoE4 mice, whereas ▵ and ○ represent the corresponding sham-treated mice. C, Object recognition. The black bars correspond to thiorphan-treated mice, whereas the white bars represent the corresponding sham-treated mice. As shown, the thiorphan-treated apoE4 mice did not exhibit a preference to either the novel or the previously seen object, whereas all the other groups had a preference toward the new object. Two-way ANOVA revealed p = 0.02 for group × treatment, which was associated with a significant difference between the performance of the thiorphan-treated apoE4 mice and those of the other mouse groups (p < 0.007; t test, post hoc analysis). D, A three-dimensional chart summary of the effects of apoE and thiorphan on the neurodegeneration and Aβ contents of CA1 hippocampal neurons and on learning and memory, of the individual sham- and thiorphan-treated apoE3 and apoE4 mice. Results shown were adapted from Figures 2B, 4C, and 9B. The symbols ▴ and • correspond to thiorphan-treated apoE3 and apoE4 mice, whereas ▵ and ○ represent the corresponding sham-treated mice. The results of the apoE4 mice are attached to black lines, whereas those of the apoE3 mice are attached to gray lines.
In the second part of the experiment, the water was placed in a different well positioned in the opposite quadrant of the field, and the mice were tested for three consecutive days for their ability to reach the new location. As expected, the performance of all the mouse groups was poor in the first trial after the position of the water-filled well was changed (Fig. 9B). However, in subsequent trials during the first day, the performances of the sham- and thiorphan-treated apoE3 mice improved markedly, whereas those of the sham- and thiorphan-treated apoE4 mice improved only slightly and did not differ from each other (Fig. 9B). The performance of the sham-treated apoE4 mice improved markedly during the next 2 d of testing, as did that of the sham- and thiorphan-treated apoE3 mice. In contrast, the thiorphan-treated apoE4 mice did not improve their performance, and the length of their trajectories to the water-filled well on day 3 after the position of the well was changed was similar to their performance during the very first trial (Fig. 9B). Repeated-measurements analysis of the results obtained on day 3 revealed that group and treatment had a significant effect (p < 0.001), and that the trajectory of the thiorphan-treated apoE4 mice was significantly longer than that of the other groups (p < 0.001; t test post hoc analysis). Regression analysis revealed an inverse linear correlation between the performance of the thiorphan- and sham-treated apoE3 and apoE4 mice at day 3 after the relocation of the water-filled well, and the Aβ content of their CA1 neurons (r2 = 0.45 and p = 0.02). Similar results were obtained when the performance of the mice was analyzed in terms of the time to reach the water-filled well, which at day 3 was 17.4 ± 4 and 12.6 ± 4 s, respectively, for the sham-treated apoE3 and apoE4 mice and 9.7 ± 0.7 and 98 ± 5 s, respectively, and for the corresponding thiorphan-treated apoE3 and apoE4 mice. Next, the mice were subjected to an object recognition test, which monitors the ability of the mice to distinguish between a new and a previously seen object (Chen et al., 2000). According to the results, the sham- and thiorphan-treated apoE3 mice and the sham-treated apoE4 mice performed similarly and had a preference toward the new object, whereas the thiorphan-treated apoE4 mice had no such preference and reacted similarly to the old and new objects (Fig. 9C). A summary of the correlation between Aβ, the levels of CA1 neurons, and the cognitive performances of the individual neprilysin- or sham-treated apoE3 and apoE4 mice is depicted in Figure 9D. As shown, the thiorphan-treated apoE4 mice form a cluster characterized by low densities of CA1 neurons, high levels of Aβ in CA1 neurons, and impaired learning and memory that is clearly distinguishable from the other mouse groups.
Discussion
This study revealed that activating the amyloid cascade by inhibiting the Aβ-degrading enzyme neprilysin triggers the specific degeneration of hippocampal CA1 neurons and of entorhinal cortex and septal neurons of ApoE4 mice but does not affect the corresponding neurons of ApoE3 mice. This was associated with accumulation of Aβ in these neurons in ApoE4 but not in ApoE3 mice, which was most pronounced in CA1 neurons. Furthermore, accumulation of Aβ in the affected neurons was associated with elevated intracellular ApoE levels that were also most pronounced in CA1 neurons of the treated ApoE4 mice, and with enhanced lysosomal activation. These neuropathological effects correlate with impairments in learning and memory of the ApoE4 mice.
The thiorphan treatment also induced deposition of extracellular Aβ, the levels of which were higher in the apoE4 than in the corresponding apoE3 mice, which, however, do not correlate either spatially or temporarily with the observed neuropathology (compare Figs. 1A and 4–7.). This suggests that the extracellular Aβ deposits do not play a primary role in the observed neuronal and cognitive deficits of the apoE4 mice.
The presently observed specific vulnerability of the hippocampal CA1 neurons and of entorhinal cortex and septal neurons to the thiorphan treatment and the ability of the hippocampal DG neurons to withstand this treatment, parallel the situation in AD (West et al., 1994; Geula and Mesulam, 1999; Hof and Morrison, 1999). Furthermore, several lines of APP transgenic mice are associated with the specific degeneration of CA1 neurons (Oddo et al., 2003; Casas et al., 2004; Tseng et al., 2004). In addition, the observed correlation between the cognitive impairments of the apoE4 mice and loss of their CA1 entorhinal and septal neurons (Figs. 7, 9D) is in accordance with the role of these neurons in learning and memory, suggesting that these pathological and behavioral effects may be causally related.
The rapid rate at which the apoE4-dependent Aβ-mediated neurodegeneration and behavioral deficits evolve (t1/2 ≈ 1 week) render this in vivo model particularly suitable for studying the mechanisms underlying the pathological cross talk between apoE4 and the amyloid cascade. The rapidity of the model is a particular advantage, because it enables a detailed dissection of the interactions of apoE4 with the different stages of the amyloid cascade. In addition, this model, unlike the APP transgenic mice, is not associated with elevated levels of either APP or of its proteolytic breakdown peptides. This model can also be used for studying aspects of the amyloid cascade that are not related to apoE4. However, because neurodegeneration in the absence of apoE4 is slower (Fig. 4), this may require longer durations than with the apoE4 mice.
The observed kinetic and spatial correlation between the accumulation of Aβ in CA1 and entorhinal neurons and their degeneration (Figs. 1, 4, 7) suggests that the pathological effects of apoE4 are initiated by intraneuronal accumulation of Aβ, which then triggers neuronal loss and cognitive impairments. These results are in accordance with previous findings from AD and transgenic animal model studies, which revealed that Aβ accumulates intraneuronally and can thereby induce neuronal dysfunction (for review, see LaFerla et al., 2007). The finding that DG and septal neurons have comparable levels of Aβ after thiorphan treatment, but that the DG neurons, unlike those of the septum, are not affected by this treatment (Fig. 7), suggests that DG neurons are less susceptible to Aβ. The finding that only a fraction of the CA1, entorhinal, and septal neurons are affected by thiorphan (Fig. 7), suggests that subpopulations of these neurons are differentially susceptible to the thiorphan treatment and to Aβ.
Interestingly, the deposition of extracellular Aβ in APP × apoE4 double transgenic mice is not associated with accumulation of intraneuronal Aβ and neurodegeneration of CA1 neurons (Holtzman et al., 2000; Fagan et al., 2002; Holtzman, 2004). This difference between the double transgenic mice and the presently described thiorphan-treated apoE4 mice may be related to the different kinetics of activation of the amyloid cascade in these systems. Furthermore, the effects of apoE4 in the double transgenic mice are apparent only in aged mice and may thus be masked by age-related processes.
Accumulation of intracellular Aβ in the ApoE4 mice was associated with a parallel elevation in the levels of intracellular ApoE4, both of which were most pronounced in CA1 neurons (Fig. 7). We next discuss the factors that determine the specific ability of the ApoE4 neurons to accumulate Aβ and ApoE4, and possible mechanisms that may mediate the resulting pathological effects. Theoretically, the levels of intraneuronal Aβ after the thiorphan treatment can be determined by the synthesis and degradation of Aβ, by the ability of the cells to accumulate Aβ, and by experimental factors related to the distribution of thiorphan in the brain. Immunohistochemistry using anti-APP N- and C-terminal antibodies suggests that the rank order of the levels of neuronal APP is CA1 > entorhinal cortex > DG (data not shown), suggesting that the specifically high levels of interneuronal Aβ in CA1 neurons may be related to increased production of Aβ in these neurons. Furthermore, because neprilysin levels are particularly elevated in the molecular layer of the DG (Fukami et al., 2002), it is possible that residual neprilysin activity, which remains after thiorphan treatment, can play a role in lowering the levels of Aβ in the DG. Because Aβ levels in CA1, DG, and entorhinal neurons were analyzed in the same sections (bregma −2.5), it is unlikely that caudodorsal differences in the distribution of thiorphan contributed much to the observed differences in the Aβ levels of these neurons. This could, however, affect the relative levels of Aβ in septal neurons, which reside caudally to the other brain areas. Last, because the septum and entorhinal cortex are directly connected to CA1 neurons, possibly some of their Aβ is transported from the CA1 neurons.
ApoE is synthesized by glia and neurons (Xu et al., 1999). The accumulated neuronal ApoE in the thiorphan-treated ApoE4 mice may therefore be attributable to uptake of ApoE4 produced and secreted by glia and/or to neuronal synthesis. Recent studies suggest that accumulation of Aβ in hippocampal neurons is apoE dependent and is mediated via the low-density lipoprotein receptor-related protein (LRP) (Zerbinatti et al., 2006). Furthermore, preliminary findings from our laboratory revealed that LRP levels are elevated specifically in the hippocampus of the apoE4 mice (data not shown), suggesting that accumulation of Aβ and apoE in the CA1 neurons of the thiorphan-treated apoE4 mice may be mediated via LRP receptors. Additional studies are required for unraveling the contribution of neuronal ApoE uptake and synthesis to the observed difference between the levels of ApoE in distinct brain neurons of the thiorphan-treated apoE4 mice (Fig. 7).
The study of the intracellular processes that mediate the neuropathological effects of apoE4 in the thiorphan-treated mice focused on CA1 neurons and the interactions among Aβ, ApoE4, and the lysosomes. This revealed that degeneration of CA1 neurons in thiorphan-treated apoE4 mice was associated with elevated levels of enlarged lysosomes and with increased diffused staining of the lysosomal marker cathepsin D (Fig. 8). Furthermore, the intracellular staining of Aβ was also punctate, and it colocalized with the lysosomes (Fig. 8). In contrast, ApoE staining in the CA1 neurons is more homogeneous and does not specifically colocalize with the punctate staining of Aβ and the lysosomes (Fig. 3). This observed difference between the interneuronal staining patterns of Aβ and ApoE may be attributable to differential uptake and/or intracellular trafficking of ApoE4 and Aβ. Lysosomal activation is associated with endocytosis (Nixon and Cataldo, 2006), and it is therefore possible that the large vacuolar inclusions, which appear in the distal dedrites of the CA1 neurons after thiorphan treatment of the ApoE4 mice result from lysosomal activation via the endocytotic pathway. Previous immuno-electron microscopic studies of AD and transgenic mice, which used the same anti Aβ42 antisera as presently used, revealed that distal dendrites are a major site of Aβ accumulation (Takahashi et al., 2002). It is thus possible that presently observed initiation of neuropathology of the distal neurites of the CA1 neurons (Fig. 6) is driven by the accumulation of Aβ at this site. Previous cell culture studies revealed that apoE4 potentiates lysosomal activation and leakage in an Aβ-dependent process, resulting in neuronal death (Ji et al., 2002, 2006). It is possible that the presently observed synergistic pathological effects of apoE4 and Aβ are caused by a similar process. However, additional studies are required for examining this possibility and the role of additional mechanisms not directly linked to the lysosomes (Huang et al., 2004) in mediating the pathological effects of ApoE4 and Aβ.
In conclusion, this study describes a novel model of neurodegeneration and AD, based on activating the amyloid cascade in vivo by inhibiting the Aβ-degrading enzyme neprilysin. Using this model, we showed that apoE4 stimulates the accumulation of intraneuronal Aβ and the degeneration of distinct hippocampal, entorhinal, and septal neurons, which are associated with cognitive impairments.
Footnotes
This work was supported in part by grants from the United States–Israel Binational Foundation and the Joseph and Inez Eichenbaum Foundation (D.M.M.) and by National Institutes of Health Grants AG10435, AG022074, and AG48440 (E.M.). D.M.M. is the incumbent of the Myriam Lebach Chair in Molecular Neurodegeneration. We thank Steve Manch for his editorial assistance and Prof. T. Hartmann for kindly providing AbR7334.
References
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde.”. Clin Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- Arendt T, Schindler C, Bruckner MK, Eschrich K, Bigl V, Zedlick D, Marcova L. Plastic neuronal remodeling is impaired in patients with Alzheimer's disease carrying apolipoprotein ε4 allele. J Neurosci. 1997;17:516–529. doi: 10.1523/JNEUROSCI.17-02-00516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendza RP, Bales KR, Paul SM, Holtzman DM. Role of apoE/Abeta interactions in Alzheimer's disease: insights from transgenic mouse models. Mol Psychiatry. 2002;7:132–135. doi: 10.1038/sj.mp.4001006. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- DeMattos RB. Apolipoprotein E dose-dependent modulation of beta-amyloid deposition in a transgenic mouse model of Alzheimer's disease. J Mol Neurosci. 2004;23:255–262. doi: 10.1385/JMN:23:3:255. [DOI] [PubMed] [Google Scholar]
- Dolev I, Michaelson DM. A nontransgenic mouse model shows inducible amyloid-beta (Abeta) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc Natl Acad Sci USA. 2004;101:13909–13914. doi: 10.1073/pnas.0404458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolev I, Michaelson DM. The nucleation growth and reversibility of Amyloid-beta deposition in vivo. J Alzheimers Dis. 2006;10:291–301. doi: 10.3233/jad-2006-102-313. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. Mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-β 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami S, Watanabe K, Iwata N, Haraoka J, Lu B, Gerard NP, Gerard C, Fraser P, Westaway D, St George-Hyslop P, Saido TC. Abeta-degrading endopeptidase, neprilysin, in mouse brain: synaptic and axonal localization inversely correlating with Abeta pathology. Neurosci Res. 2002;43:39–56. doi: 10.1016/s0168-0102(02)00015-9. [DOI] [PubMed] [Google Scholar]
- Geula C, Mesulam MM. Cholinergic system in Alzheimer disease. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer disease. Ed 2. Philadelphia: Lippincott, Williams, and Wilkins; 1999. p. 457. [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Grimm HS, Beher D, Lichtenthaler SF, Shearman MS, Beyreuther K, Hartmann T. gamma-Secretase cleavage site specificity differs for intracellular and secretory amyloid beta. J Biol Chem. 2003;278:13077–13085. doi: 10.1074/jbc.M210380200. [DOI] [PubMed] [Google Scholar]
- Hardy J. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Laurer H, Longhi L, Bales KR, Paul SM, McIntosh TK, Holtzman DM. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer's disease. J Neurosci. 2002;22:10083–10087. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Morrison JH. The cellular basis of cortical disconnection in Alzheimer disease and related dementing conditions. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer Disease. Ed 2. Philadelphia: Lippincott, Williams, and Wilkins; 1999. p. 457. [Google Scholar]
- Holtzman DM. In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology. J Mol Neurosci. 2004;23:247–254. doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Weisgraber KH, Mucke L, Mahley RW. Apolipoprotein E: diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer's disease. J Mol Neurosci. 2004;23:189–204. doi: 10.1385/JMN:23:3:189. [DOI] [PubMed] [Google Scholar]
- Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002;277:21821–21828. doi: 10.1074/jbc.M112109200. [DOI] [PubMed] [Google Scholar]
- Ji ZS, Mullendorff K, Cheng IH, Miranda RD, Huang Y, Mahley RW. Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem. 2006;281:2683–2692. doi: 10.1074/jbc.M506646200. [DOI] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ. Isoform-specific effect of apolipoprotein E on cell survival and β-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Levi O, Jongen-Relo AL, Feldon J, Roses AD, Michaelson DM. ApoE4 impairs hippocampal plasticity isoform-specifically and blocks the environmental stimulation of synaptogenesis and memory. Neurobiol Dis. 2003;13:273–282. doi: 10.1016/s0969-9961(03)00045-7. [DOI] [PubMed] [Google Scholar]
- Masters CL, Beyreuther K. Alzheimer's centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain. 2006;129:2823–2839. doi: 10.1093/brain/awl251. [DOI] [PubMed] [Google Scholar]
- Matsumori Y, Hong SM, Fan Y, Kayama T, Hsu CY, Weinstein PR, Liu J. Enriched environment and spatial learning enhance hippocampal neurogenesis and salvages ischemic penumbra after focal cerebral ischemia. Neurobiol Dis. 2006;22:187–198. doi: 10.1016/j.nbd.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Mori E, Lee K, Yasuda M, Hashimoto M, Kazui H, Hirono N, Matsui M. Accelerated hippocampal atrophy in Alzheimer's disease with apolipoprotein E epsilon4 allele. Ann Neurol. 2002;51:209–214. doi: 10.1002/ana.10093. [DOI] [PubMed] [Google Scholar]
- Morris RG, Frey U. Hippocampal synaptic plasticity: role in spatial learning or the automatic recording of attended experience? Philos Trans R Soc Lond B Biol Sci. 1997;352:1489–1503. doi: 10.1098/rstb.1997.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9:277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Puttfarcken PS, Manelli AM, Falduto MT, Getz GS, LaDu MJ. Effect of apolipoprotein E on neurite outgrowth and beta-amyloid-induced toxicity in developing rat primary hippocampal cultures. J Neurochem. 1997;68:760–769. doi: 10.1046/j.1471-4159.1997.68020760.x. [DOI] [PubMed] [Google Scholar]
- Roses AD. On the discovery of the genetic association of apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–366. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng BP, Kitazawa M, LaFerla FM. Amyloid beta-peptide: the inside story. Curr Alzheimer Res. 2004;1:231–239. doi: 10.2174/1567205043332045. [DOI] [PubMed] [Google Scholar]
- Van der Zee EA, Compaan JC, de Boer M, Luiten PG. Changes in PKCγ immunoreactivity in mouse hippocampus induced by spatial discrimination learning. J Neurosci. 1992;12:4808–4815. doi: 10.1523/JNEUROSCI.12-12-04808.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- Xu PT, Gilbert JR, Qiu HL, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE. Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol. 1999;154:601–611. doi: 10.1016/S0002-9440(10)65305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wahrle SE, Kim H, Cam JA, Bales K, Paul SM, Holtzman DM, Bu G. Apolipoprotein E and low density lipoprotein receptor-related protein facilitate intraneuronal Abeta42 accumulation in amyloid model mice. J Biol Chem. 2006;281:36180–36186. doi: 10.1074/jbc.M604436200. [DOI] [PubMed] [Google Scholar]