

Background: Recent studies have linked synaptojanin 1 (synj1) with Alzheimer disease (AD).
Results: We report that synj1 reduction decreases amyloid plaque burden and attenuates cognitive deterioration in an AD mouse model. These effects are mediated through accelerating endosomal/lysosomal degradation of Aβ.
Conclusion: Our data suggest a novel mechanism by which synj1 reduction promotes Aβ clearance.
Significance: These studies implicate a therapeutic strategy for AD.
Keywords: Alzheimer Disease, Amyloid, Intracellular Trafficking, Protein Degradation, Transgenic Mice, PIP2, Clearance, Endosomal/Lysosomal Degradation, Synaptojanin 1
Abstract
Recent studies link synaptojanin 1 (synj1), the main phosphoinositol (4,5)-biphosphate phosphatase (PI(4,5)P2-degrading enzyme) in the brain and synapses, to Alzheimer disease. Here we report a novel mechanism by which synj1 reversely regulates cellular clearance of amyloid-β (Aβ). Genetic down-regulation of synj1 reduces both extracellular and intracellular Aβ levels in N2a cells stably expressing the Swedish mutant of amyloid precursor protein (APP). Moreover, synj1 haploinsufficiency in an Alzheimer disease transgenic mouse model expressing the Swedish mutant APP and the presenilin-1 mutant ΔE9 reduces amyloid plaque load, as well as Aβ40 and Aβ42 levels in hippocampus of 9-month-old animals. Reduced expression of synj1 attenuates cognitive deficits in these transgenic mice. However, reduction of synj1 does not affect levels of full-length APP and the C-terminal fragment, suggesting that Aβ generation by β- and γ-secretase cleavage is not affected. Instead, synj1 knockdown increases Aβ uptake and cellular degradation through accelerated delivery to lysosomes. These effects are partially dependent upon elevated PI(4,5)P2 with synj1 down-regulation. In summary, our data suggest a novel mechanism by which reduction of a PI(4,5)P2-degrading enzyme, synj1, improves amyloid-induced neuropathology and behavior deficits through accelerating cellular Aβ clearance.
Introduction
Alzheimer disease (AD)3 is neuropathologically characterized by senile plaques containing β-amyloid peptides (Aβ), as well as neurofibrillary tangles consisting of hyperphosphorylated Tau. The amyloidogenic Aβ peptide is proteolytically derived from the amyloid precursor protein (APP) by distinct enzymatic activities known as β-secretase (or BACE) and γ-secretase (1, 2). Overproduction and impaired Aβ clearance can both lead to Aβ accumulation. Recent evidence indicates that late onset AD cases are likely caused by an overall impairment in Aβ clearance (3).
Synaptojanin 1 (synj1), a major negative regulator of the levels of PI(4,5)P2 in the nervous tissue, has been implicated in the regulation of endocytic traffic at synapses (4). Trisomy of synj1 in Down syndrome mouse models causes a deficiency in PI(4,5)P2 as well as learning deficits (5), whereas synj1 haploinsufficiency is protective against Aβ-induced neurotoxicity in mouse models of AD (6, 7). In the current study, we link synj1 to regulation of cellular Aβ clearance.
We have found that down-regulation of synj1 leads to reduced Aβ accumulation and amyloid plaque load. synj1 knockdown promotes Aβ uptake and lysosomal trafficking, thereby facilitating cellular Aβ clearance without affecting BACE or γ-secretase expression or enzymatic activities to generate Aβ. These effects are dependent on PI(4,5)P2 that increases with synj1 down-regulation. As a consequence, reduction of synj1 attenuates amyloid-induced neuropathologic changes and behavior deficits in an AD transgenic mouse model. Our findings uncover a novel regulatory mechanism by a PI(4,5)P2-degrading enzyme synj1 that controls cellular Aβ degradation through the endosomal/lysosomal pathway.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following antibodies were used: 4G8 and 6E10 (Covance), anti-synj1 (mouse AC1; Novus), anti-amyloid (AB2454; Cell Signaling), anti-cleaved Notch1 Val1744 (D3B8; Cell Signaling), anti-BACE1 C terminus MAB5308 (clone 61; Millipore), anti-FLAG (Sigma), anti-Rab5 and anti-β-actin (Santa Cruz), anti-LAMP1 (Abcam), anti-mouse, anti-rabbit, and anti-goat HRP conjugates (Vector Laboratories), and Texas Red-conjugated anti-mouse IgG (Vector Laboratories Inc.). pAb369 (C-terminal APP antibody) was used to detect human and mouse holo-APP and C-terminal fragments (8). The amyloid specific luminescent conjugated oligothiophene (LCO) was used for histological staining of the presence of amyloid deposits in combination with immunofluorescence (9). AB14 (N-terminal PS1 antibody) (10) was used to detect PS1. Fluoro-conjugated Aβ42555 and Aβ42488 were purchased from AnaSpec (Fremont, CA). Aβ peptides were pretreated with trifluoroacetic acid that was distilled under nitrogen, washed with 1,1,1,3,3,3-hexafluoro-2-propanol, stored at −20 °C, and dissolved in Me2SO to 200 μm before use. Aβ42 solution was centrifuged at 17,000 × g for 30 s to remove aggregated Aβ. The lysosomal inhibitors leupeptin, pepstatin A, and E-64d were purchased from Sigma. The PIP2 modulator m-3m3FBS, which can activate phospholipase C and deplete PIP2 in cells, and its inactive analog o-3m3FBS (11) were purchased from Santa Cruz Biotechnology Inc.
Cell Lines
Mouse N2a neuroblastoma cells stably transfected with cDNAs encoding human Swedish mutant APP were maintained in medium containing 50% DMEM, 50% OPTI-MEM, supplemented with 5% fetal bovine serum, antibiotics, and 200 mg/ml G418 (Invitrogen). N2a cells were transfected with synj1 siRNA and maintained for 4–5 days to achieve ∼50–80% knockdown of synj1 protein levels. Alternatively, cells were treated with a γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) (12, 13) at 2 μm overnight before further analysis. In some experiments, cells after siRNA treatment were incubated with a PIP2 modulator (m-3-FBS or an inactive analog o-3-FBS) at 6.25 μm overnight before further analysis.
siRNA Oligonucleotides
To knock down synj1 protein expression, three siRNA duplexes specifically targeting synj1 (IDT Inc.) were synthesized. Sequences of the syn1siRNAs are as follows: siRNA1, forward, 5′-GUC UGG UUC UUG AAC GCU AUG CUG CGG-3′, and reverse, 5′-GCA GCA UAG CGU UCA AGA ACC AGAC-3′; siRNA2, forward, GGA CUU CAC GGA AAG AUA AUU AAGT-3′, and reverse, 5′-ACU UAA UUA UCU UUC CGU GAA GUC CAA-3′; and siRNA3, forward, 5′-AGC ACG GAA AGA UAA UAU AGG GCGT-3′, and reverse, 5′-ACG CCC UAU AUU AUC UUU CCG UGC UGU-3′. A scrambled universal negative control RNA duplex was used as a negative control (IDT Inc.).
Cell Transfection
For siRNA analysis, N2a cells were seeded at 50–60% confluence and transfected with 200 pmol of synj1 siRNA duplex versus control duplex (per well of a 6-well plate) using Lipofectamine RNAimax (Invitrogen) according to the manufacturer's instructions.
Cell Lysate Analysis
After transfection, the cells were harvested in lysis buffer (14). Equal amounts of total protein were loaded onto 10–20% Tricine SDS-PAGE gels for electrophoresis and transferred to PVDF membranes. The membranes were analyzed by Western blot using 6E10 to detect holoAPP and βCTF/C99. Aβ40 and Aβ42 levels in media were determined by human ELISA kits (WAKO), according to the manufacturer's instructions. In some experiments, the amount of Aβ in media and lysate with synj1 or control siRNA treatment were determined in the presence of lysosomal inhibitors (pepstatin A, 10 μm; leupeptin, 100 μm; E-64d, 50 μm) to block lysosomal degradation of Aβ. Alternatively, a PIP2 modulator m-3m3FBS or its inactive analog o-3m3FBS was added with or without lysosomal inhibitors to determine whether the degradation of Aβ with synj1 reduction is dependent upon elevated PIP2 levels.
Immunoprecipitation
Lysates were diluted with immunoprecipitation (IP) buffer (10) and immunoprecipitated using antibody 4G8 followed by immunoblotted with 6E10 for detection of intracellular Aβ and βCTF. Media were immunoprecipitated using 4G8 antibodies (Covance) and immunoblotted with 6E10 for detection of media Aβ as described before (15). In some experiments, after siRNA transfection, N2a cells were treated with Me2SO lysosomal inhibitors or PIP2 modulators overnight before analysis of media and lysate Aβ production.
Generation of synj1 Haploinsufficient Mice with AD Transgenic Mouse Background
Human Swedish APP and FAD-linked PS1 ΔE9 mutant transgenic mice (16, 17) were mated with heterozygous synj1 null mice (synj1+/−) (1). Double heterozygous F1s were then bred with heterozygous synj1 null mice only to generate offspring that express human Swedish mutant APP and FAD-linked PS1 ΔE9 in the synj1+/− background. Genotypes were determined by PCR amplification as described (1, 16, 17).
Brain Lysate Preparation and Analysis
Mouse brains were rapidly removed, hemisected, and snap frozen before further analysis. Each frozen hemi-brain was then processed via stepwise solubilization (14, 18). Lysates of hemi-brains derived from APP/PS1+/− synj1+/+ or APP/PS1+/− synj1+/− at 9 months of age were analyzed by SDS-PAGE and immunoblotted with 6E10 to determine levels of holoAPP and βCTF/C99. Levels of Aβ40 and Aβ42 were determined by human Aβ40 and high sensitivity human Aβ42 ELISA kits (Wako), according to the manufacturer's instructions. The results were normalized to wet brain weight.
Immunohistochemistry and LCO Staining of Amyloid Plaque
The hemi-brains of 9-month-old APP/PS1+/− synj1+/+ or APP/PS1+/− synj1+/− mice were processed, embedded, and sectioned at 10 μm. For amyloid plaque quantitation, the blocks were serial sectioned across the whole hippocampal regions, and every eight sections were used for staining (∼20 sections/animal). After deparaffination and antigen retrieval process, the brain sections were treated with anti-amyloid antibody AB2454 or 6E10 (1:200 dilution in TBS buffer) overnight at 4 °C. Following a thorough rinse in TBS buffer, the sections were incubated with secondary antibodies, i.e., biotinylated goat anti-mouse IgG or biotinylated goat anti-rabbit IgG (diluted 1:200 in TBS buffer), and then incubated with avidin biotinylated enzyme complex and 3,3′-diaminobenzidine. The amyloid plaque load density in the hippocampal region, as well as CA1/3 and dentate gyrus subregions, was measured using the Sinq Image Analysis System (Sinq Inc.) (19, 20). Alternatively, the brain sections were stained with anti-amyloid antibodies followed by incubated with secondary antibody Texas Red-conjugated anti-rabbit or anti-mouse IgG as well as LCO reagent (21) for double staining of amyloid before confocal microscopy analysis (LSM510; Zeiss).
Novel Object Recognition Task
Human Swedish APP and FAD-linked PS1 mutant transgenic mice show a deficit in the novel object recognition memory task (22). Littermates at 4–5 months of age were tested in novel object recognition, with onset of the exploration time defined as the moment the head of the animal approached the object within a 2-cm radius. Briefly, animals were habituated to testing arena for 10 min on day 1. After 24 h, the animals were tested with two trials (identical objects for 10 min followed by one novel per one familiar object for 4 min). The inter-trial interval was 1 h. Each animal was videotaped from overhead cameras and scored for total time spent investigating objects per trial. The objects were randomized left and right, as well as between animals. They were cleaned with alcohol between trials/animals to prevent odor cues.
Spatial Alternation/Y Maze Task
Briefly the Y maze test was performed in a Y-shaped maze with three white opaque plastic arms at a 120° angle from each other (23, 24). The test involves animals freely explored the three arms of the Y maze for 8 min (see Fig. 3B). Appropriate external cues were assigned for each arm. All animal activities were recorded by video camera, and the number of arm entries and the number of triads were determined to calculate the percentage of alternation. An entry occurs when all four limbs are within the arm.
FIGURE 3.

Several AD-related proteins remain unchanged with synj1 haploinsufficiency in AD transgenic mice. A, protein levels of holoAPP, C99/βCTF, PS1, BACE1, and synj1 in the hippocampal brain lysates of APP/PS1+/− synj1+/+ and APP/PS1+/− synj1+/− mice were determined by Western blot. B, results were normalized to actin and expressed as percentages of control. Reduction in synj1 protein levels by 60.2 ± 9.2% (*, p < 0.05) was observed in synj1 haploinsufficiency mice, as compared with control. C, the inositol lipid levels from hippocampal tissue of APP/PS1+/− synj1+/+ (n = 5) and APP/PS1+/− synj1+/− (n = 5) mice were determined by HPLC combined with suppressed conductivity.
In Vitro γ-Secretase Assays
In vitro γ-secretase assays using the recombinant C100-FLAG (kindly provided by Yue-Ming Li at Memorial Sloan Kettering Cancer Center) or N100-FLAG substrates (1696MYVAAAAFVLLFFVGCGVLLSRKRRRQGQLWFPEGFKVSEASKKKRREPLGEDSVGKPLKNASDGALMDDNQNEWGDEDLE1777DYKDDDDK-STOP). The membranes containing γ-secretase were prepared from N2a cells without overexpressing any transgene and solubilized in 2% CHAPSO. Incubation of γ-secretase membranes and substrates C100-FLAG or N100FLAG for 90 min was performed as previously reported (25, 26).
Aβ Uptake Assay
N2a Swedish mutant APP cells were transfected with synj1 siRNA or control siRNA duplex as indicated. At 4 day post-transfection, the cells were incubated with biotin-conjugated Aβ42 in PBS for 1 h at 37 °C as described previously (27, 28). The cells were subsequently washed by ice-cold PBS five times followed by incubation with 100 μm glutathione for 15 min at 4 °C. The cells were washed another two times with PBS and then incubated with 5 mg/ml iodoacetamide for 15 min followed by washing with ice-cold PBS five times before being harvested in cell lysis buffer (ThermoFisher Scientific) as described (14). Lysates were subsequently immunoprecipitated by streptavidin beads and analyzed by SDS-PAGE and Western blotting with 6E10 for internalized biotin-Aβ42. Similar experiments were performed in primary neuron culture derived from embryonic day 17 synj1+/+ versus synj1−/− animals. In some experiments, cells after siRNA treatment were incubated with a PIP2 modulator (m-3-FBS or an inactive analog o-3-FBS) at 6.25 μm overnight before being subjected to an Aβ uptake assay.
Cellular Aβ Turnover Rate Assay
For a time course of Aβ degradation, N2a Swedish mutant APP cells were transfected with synj1 siRNA or control siRNA duplex as indicated. At 4 days post-transfection, the cells were divided into 6-well plates coated with 100 μg/ml polyornithine and allowed to recover for 5 h. The cells were subsequently treated with 50 μg/ml cycloheximide (CHX) in serum-free and antibiotic-free DMEM (29). At the indicated time points, the cells were washed in ice-cold PBS and then harvested in radioimmune precipitation assay buffer as described (14). Lysates were subsequently analyzed by SDS-PAGE and Western blotting with 6E10 for Aβ and holoAPP.
Neuronal Culture and Confocal Microscopy
Primary hippocampal neurons were obtained from 17-day-old embryos of synj1+/+, synj1+/−, and synj1−/− mice and grown in neurobasal medium (Invitrogen) supplemented with 0.5 mm GlutaMAX (Invitrogen), 2% B27 (Invitrogen), and 1% penicillin-streptomycin (Invitrogen) in dishes containing poly-d-lysine coated cover glasses (Nalge Nunc International, Rochester, NY) for 7 days in vitro. They were then incubated with Alexa-Aβ555 or Alexa-Aβ488 for various time periods before fixation and double-stained for endosomal and lysosomal markers Rab5 and LAMP1 as well as a nuclear marker DAPI (blue) before confocal fluorescence microscopy analysis (Zeiss). Similar experiments were performed in N2a cells after control or synj1 siRNA transfection, followed by incubation with Alexa-Aβ555 or Alexa-Aβ488. Fluorescent colocalization analysis was quantified using the Zen program to calculate colocalized pixels (subtracted by background) and colocalization coefficiency as previously described (30, 31). The data are presented as percentages of control ± S.E.
Statistical Analysis
Densitometric analysis of Western blot bands (integrated density) was performed using Multigauge v3.1 software (Fujifilm). The levels of holoAPP, βCTF/C99, Aβ, synj1, BACE1, PS1, biotin-Aβ, and C100-/N100-FLAG were normalized to actin levels and expressed as percentages of control. Absolute Aβ40 and Aβ42 concentrations were quantitatively determined by sandwich ELISA (Wako) and expressed as percentages of control. For all analysis, independent sample t tests (parametric design) were used to determine significant mean differences (the threshold for significance is set at p < 0.05). Where two or more variables were compared, a one-way analysis of variance with post hoc tests were used to test group differences for multiple comparisons. All statistical analysis was performed using SPSS v21.0.
RESULTS
Knockdown of synj1 Reduces Aβ in N2a Swedish Mutant APP Cells
We first analyzed the effects of synj1 knockdown on Aβ levels in N2a cells stably expressing human Swedish mutant APP. Transfection with siRNA reduced synj1 protein levels by 50–80% after 4–5 days (Fig. 1, A and B). Under these conditions, both extracellular and intracellular Aβ levels were significantly decreased by 31.8 ± 11.0% and 53.5 ± 9.88%, respectively, compared with control siRNA transfection (Fig. 1B, top panel, p < 0.001). Similar effects on Aβ were achieved in three different synj1 siRNA transfection experiments (Fig. 1A, fourth, fifth, and sixth lanes). A γ-secretase inhibitor, DAPT (12, 13), was used as a control with 91.8 ± 2.12% reduction in extracellular and 90.4 ± 4.05% reduction in intracellular Aβ levels (Fig. 1A, seventh lane). There was 34.4 ± 13.6% reduction in Aβ40 and 47.7 ± 20.3% reduction in Aβ42 levels measured by sandwich ELISA with synj1 siRNA treatment (Fig. 1B, lower panel, p < 0.001). Levels of the APP C-terminal fragment C99/βCTF were determined by IP with antibody 4G8 followed by Western blotting with 6E10 (Fig. 1A, middle panel). No changes in levels of βCTF (95.8 ± 13.7%, p = 0.46; Fig. 1B, lower panel) or holoAPP (129 ± 14.5% of controls, p = 0.08; data not shown) were observed after synj1 knockdown.
FIGURE 1.

Knockdown of synj1 reduces Aβ in N2a Swedish mutant APP cells. A, N2a Swedish mutant APP cells were treated with synj1 or control siRNA duplex for 4–5 days before analysis. Levels of media and intracellular Aβ, as well as βCTF, were analyzed by immunoprecipitation with 4G8 followed by immunoblotting with 6E10. Levels of synj1 and β-actin were detected from cell lysates. Treatment with DAPT was included as a control. B, protein levels were normalized to actin and expressed as percentages of control. The data were collected in duplicate or triplicate from three independent experiments. Significant reductions (**, p < 0.001) of Aβ in media and lysate and reduction of synj1 protein levels were observed upon synj1 knockdown, as compared with control. Levels of Aβ40 and Aβ42 in the media were determined by sandwich ELISA analysis (**, p < 0.001). WB, Western blotting; ctrl, control.
synj1 Haploinsufficiency in AD Transgenic Mice Decreases Hippocampal Amyloid Plaque Load
We then investigated whether genetic reduction of synj1 affects Aβ generation in adult animals in vivo. The synj1 haploinsufficient (synj1+/−) mice (1) with AD transgenic background expressing human Swedish APP and PS1ΔE9 mutations (16, 17) were generated, and amyloid plaque burden, as well as Aβ40 and Aβ42 levels in the brains of 9-month-old mice, were analyzed. As shown in Fig. 2A, amyloid plaque load in hippocampus was determined by both immunoperoxidase (left panels) and immunofluorescence (right panels). There was an obvious reduction in the amount of dense core plaques (doubled stained by LCO and anti-amyloid antibody AB2454). The stereological quantitation of amyloid plaques in 9-month-old APP/PS1+/− synj1+/− male mouse brains showed that total plaque load in hippocampus, as well as in dentate gyrus subregions, was reduced by 34.3 ± 12.1% (p = 0.039) and 27.1 ± 11.1% (p = 0.014), respectively, compared with their APP/PS1+/− synj1+/+ littermates (Fig. 2B, left panel). However, total amyloid plaque load in the cortices of APP/PS1+/− synj1+/− mice was not clearly reduced when compared with wild type littermates, suggesting that partial reduction of synj1 may not be sufficient to alter heavy amyloid load in cortical regions at this stage of pathological progression.
FIGURE 2.

synj1 haploinsufficiency in AD transgenic mice decreases hippocampal amyloid plaque load. A, extracellular amyloid plaque deposits in hippocampal regions of transgenic mice expressing Swedish mutant APP/PS1ΔE9 (9 months old) with synj1+/+ (upper panels) or synj1+/− (lower panels) genetic background. Left panels, immunohistochemical staining of amyloid plaques with anti-amyloid antibody AB2454. Right panels, overlay fluorescent staining of amyloid plaques in dentate gyrus of hippocampus with 6E10 (red), LCO (green), and DAPI (blue). B, the total amyloid plaque load in hippocampus, as well as in the dentate gyrus (DG) subregion of animals, was determined by stereological quantification of brain sections through the whole hippocampus, and results were expressed as percentages of control (n = 3/group). Statistically significant reductions (*, p < 0.05) in amyloid plaque load were observed in synj1+/− mice, as compared with controls. Levels of Aβ40 and Aβ42 in the hippocampal brain lysates are determined by sandwich ELISA analysis (*, p < 0.05).
Furthermore, the levels of Aβ40 and Aβ42 determined by sandwich ELISA in hippocampal brain lysates from APP/PS1+/− synj1+/− male mice (n = 5) were decreased by 45.7 ± 12.2% (p = 0.047) and 19 ± 4.2% (p = 0.0018), respectively, compared with controls (n = 5; Fig. 2B, right panel). In brain lysates of APP/PS1+/− synj1+/− mice, the levels of holoAPP (106.3 ± 20.6%), C99/βCTF (determined by 6E10, 94.3 ± 21.9%), BACE1 (101.1 ± 6.3%), and PS1 (101.7 ± 11.8%) were comparable with levels of controls (Fig. 3, A and B). The synj1 protein levels in APP/PS1+/− synj1+/− mice were reduced by 60.2 ± 9.2% (p = 0.0013). Consistent with previously reported observations (1, 2), there were elevations in levels of PIP2 (known to contain mostly PI (4,5)P2) and phosphoinositol phosphate (known to contain mostly phosphoinositol 4-phosphate) of 66.2 ± 2.9% (p < 0.001) and 36.5 ± 10.6% (p = 0.003), respectively, as determined by HPLC with suppressed conductivity, without any changes in phosphoinositol levels (94.2 ± 9.5% of controls, p = 0.38) with synj1 haploinsufficiency in AD transgenic mice (Fig. 3C) as compared with synj1+/+ mice with AD transgenic background.
synj1 Haploinsufficiency in AD Transgenic Mice Attenuates Learning and Memory Deficits
Next, we investigated the effects of synj1 down-regulation on learning and memory impairments in AD transgenic mice. Previously studies have shown that one strain of human Swedish APP and FAD-linked PS1 double mutant transgenic mice (Swedish APP/PS1 M146V) exhibit deficits in the novel object recognition memory task (22). Similarly, our double transgenic mice (APP/PS1ΔE9+/−/synj1+/+) spent a comparable amount of time between novel and familiar objects (Fig. 4A; 45.6 ± 14.6% of total time with novel object, n = 5), indicating that they were unable to discriminate between these two objects. In contrast, APP/PS1ΔE9+/−/synj1+/− animals displayed improved exploratory behavior compared with APP/PS1ΔE9+/−/synj1+/+ littermates, spending more time with the novel object (66.0 ± 11.8%, n = 5, p = 0.019), similar to the degree of wild type and synj1+/− mice without AD transgenic background (74.5 ± 11.1%, n = 5, versus 68.6 ± 11.8%, n = 5). However, the total amount of time spent in exploring objects (seconds) was comparable among the four groups (data not shown).
FIGURE 4.
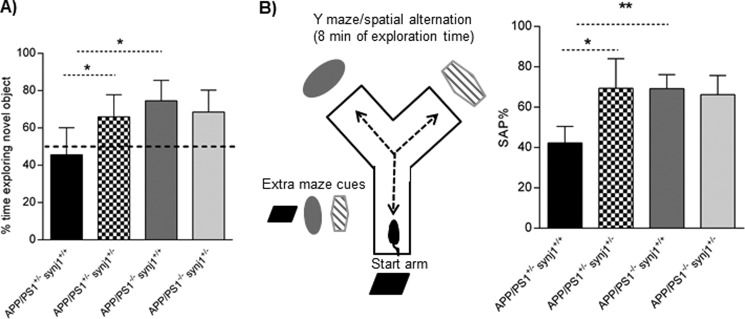
synj1 haploinsufficiency in AD transgenic mice attenuates learning and memory deficits. A, novel object recognition test; APP/PS1+/−/synj1+/+ APP/PS1+/−/synj1+/−, APP/PS1−/−/synj1+/+, and APP/PS1−/−/synj1+/− (n = 5/group). The novel object discrimination index is expressed as percentages of time exploring novel object = (time spent exploring novel object × 100)/total exploration time. B, Y maze test is used for analysis of spontaneous alteration: APP/PS1+/−/synj1+/+ (n = 5), APP/PS1+/−/synj1+/− (n = 6), APP/PS1−/−/synj1+/+ (n = 8), and APP/PS1−/−/synj1+/− (n = 9). The percentage of spontaneous alternation (% of SAP) = the number of triads × 100/(the number of total arm entries-2). *, p < 0.05; **, p < 0.001.
Next, the Y maze spontaneous alternation test was carried out to evaluate cognitive functions and learning in mice (23, 24). Again APP/PS1ΔE9+/−/synj1+/+ mice showed a statistically significant decrease in the percentage of spontaneous alterations (42.3 ± 8.2%, n = 4) when compared with wild type (69.2 ± 6.9%, n = 8, p = 0.0001) or synj1+/− mice without AD transgenic background (66.2 ± 9.5%, n = 9, p = 0.002). APP/PS1ΔE9+/−/synj1+/− animals, however, displayed improved levels of spontaneous alterations compared with APP/PS1ΔE9+/−/synj1+/+ mice (Fig. 4B, 69.4 ± 14.7%, n = 6, p = 0.004). There were no significant differences in the amount of total entry among the four groups (data not shown). In addition, no changes in general locomotor activities were noted among the four groups as assessed in an open field (data not shown). Together, our behavioral studies suggest that reduction of synj1 attenuates learning and memory deficits in a mouse model with AD double transgenes.
synj1 Haploinsufficiency Does Not Affect γ-Secretase Activity in Vitro
Previous reports indicate that Aβ generation and APP processing by PS1 can be modulated by PI(4,5)P2 (11). Because PI(4,5)P2 is a major substrate for synj1 and knockdown of synj1 elevates brain PI(4,5)P2 levels (Refs. 1 and 2 and Fig. 3C), we next investigated whether down-regulation of synj1 affected γ-secretase processing of APP.
Utilizing a well established in vitro γ-secretase assay (25, 26), we characterized the activity of solubilized γ-secretase derived from membrane fractions of N2a wild type cells transfected with synj1 siRNA or control siRNA toward the substrates C100-FLAG (Fig. 5A) and N100-FLAG (Fig. 5B). Synj1 knockdown did not affect γ-secretase cleavage of C100 to generate Aβ (101 ± 11.1% of controls, n = 6, p = 0.74) or of N100 to generate its cleavage product (recognized by Val1744 antibody only; 129.9 ± 27.4, n = 5, p = 0.11). A γ-secretase inhibitor, DAPT (12, 13), was used as a control, and reduced cleavage of both C100 and N100 substrates (69.3 ± 2.3% and 38.2 ± 16% of controls, respectively; Fig. 5, A and B, lane 4). It should also be noted that there was no change in levels of γ-secretase/PS1 total proteins in APP/PS1+/− synj1+/− mouse brains (Fig. 3A). Moreover, the levels of βCTF (precursor of Aβ) are not changed with synj1 knockdown in cells and in transgenic mouse brains (Figs. 1 and 3). These data altogether suggest that synj1 down-regulation does not affect γ-secretase cleavage of APP to generate Aβ in our system.
FIGURE 5.
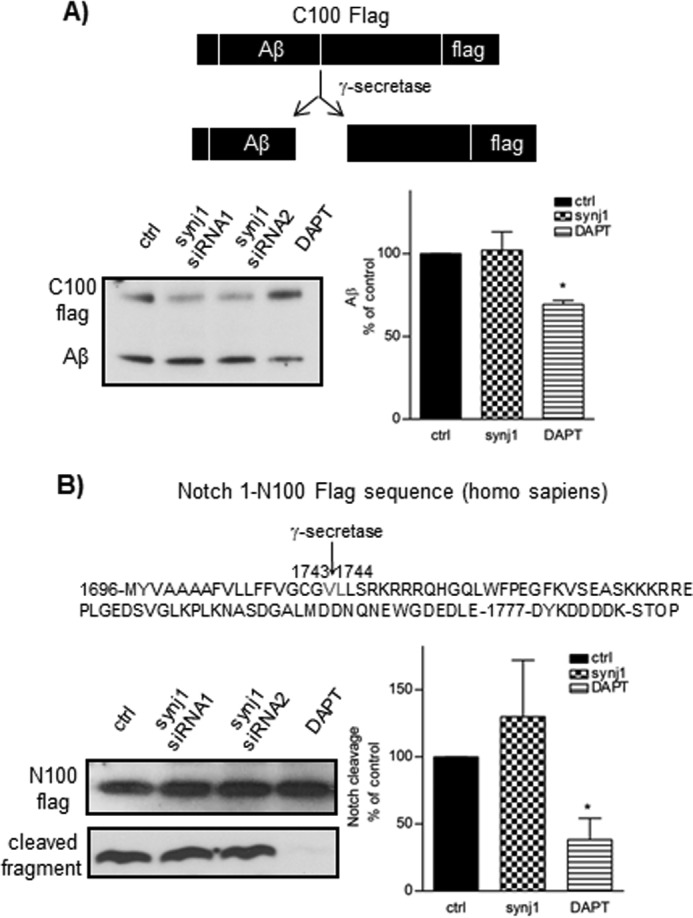
synj1 haploinsufficiency does not affect γ-secretase activity in vitro. A, C100-FLAG and Aβ generated by control or synj1 siRNA-treated membrane fractions were analyzed by immunoblot with 6E10. B, alternatively, N100-FLAG was analyzed by the in vitro γ-secretase assay. The Notch cleavage specific antibody Val1744 was used to detect the Notch cleavage fragment. The addition of DAPT to control membrane fractions was included as a control (69.3 ± 2.3% and 38.2 ± 16% of controls, respectively; lane 4). The results represent three independent experiments, and the data are presented as means ± S.D., *, p < 0.05. synj1 knockdown did not affect γ-secretase cleavage of C100 to generate Aβ (101 ± 11.1% of controls, n = 6, p = 0.74) or N100 to generate its cleavage product (recognized by Val1744 antibody only; 129.9 ± 27.4, n = 5, p = 0.11). ctrl, control.
Reduction of synj1 Accelerates Aβ Uptake into the Cells
We then investigated the rate of cellular Aβ uptake with reduced synj1 expression. As shown in Fig. 6A, the amount of biotin-Aβ42 after 1 h of incubation was increased by 25.2% in N2a cells with synj1 siRNA knockdown (125.2 ± 13.4% of controls, n = 3, p = 0.03). A similar increase in biotin-Aβ42 uptake was seen in primary neurons derived from embryonic day 17 synj1−/− animals in comparison to wild type cells (119.6 ± 9.0%, n = 4, p = 0.039).
FIGURE 6.

Reduction of synj1 accelerates Aβ uptake into cells. A, biotin-Aβ42 internalized into the cells after 1 h of incubation was analyzed by immunoprecipitation of lysates with streptavidin beads. The amount of Aβ42 taken up by the cells was increased in N2a cells with synj1 siRNA knockdown (middle panel) and primary neurons (right panel) derived from embryonic day 17 synj1−/− animals (125.2 ± 13.4% of controls, n = 3, p = 0.03; 119.6 ± 9.0% of synj1 WT neurons, n = 4, p = 0.039, respectively). B, N2a cells transfected with control siRNA or synj1 siRNA were incubated with Alexa488-Aβ42 (500 nm) for 1 h before analyzed by fluorescence confocal microscopy. Representative cells are shown. Upper panels, control siRNA-treated cells. Lower panels, synj1 siRNA-treated cells. Left panels, internalized Alexa488-Aβ42. Scale bar, 10 μm. Middle panels, overlay view of Alexa488-Aβ (green)/Rab5 (red) colocalization. DAPI for nuclear staining (blue). Scale bar, 10 μm. Right panels, enlarged view of colocalization of internalized Alexa488-Aβ42 and Rab5+ early endosomes. Scale bar, 5 μm. C, the increased amount of Aβ42 taken up by N2a cells with synj1 siRNA knockdown was unchanged with treatment of a PIP2-depleting reagent, m-3-FBS (149.6 ± 8.0% of controls in m-3-FBS-treated versus 144.3 ± 24.3% of controls in o-3-FBS-treated conditions, n = 3, p = 0.79). An inactive analog was used as a control (o-3-FBS). ctrl, control.
It has been reported that the majority of internalized Aβ traffics through Rab5- and Rab7-positive early and late endosomes, respectively. Most internalized Aβ is delivered to the lysosomal pathway for degradation (32). We then studied whether synj1 reduction would affect the amount of Alexa488-conjugated Aβ42 (green fluorescent signals) delivered to early endosomes (recognized by an early endosomal marker Rab5, red fluorescent signals) after 1 h of incubation. As shown in Fig. 6B, the amount of fluorescent-conjugated Aβ42 delivered to Rab5+ early endosomes was increased by 76.2 ± 43.4% (p = 0.03) with synj1 siRNA treatment when compared with controls. A magnified image of individual cells showed increased colocalization of Alexa488-Aβ and Rab5+ early endosomes (punctate structures shown in Fig. 6B, right panels). In addition, immunofluorescence staining of a second endosomal marker, EEA1, showed a similar pattern of increase in the amount of Aβ colocalized with EEA1+ endosomes with synj1 reduction. However, there were comparable amounts of Alexa488-conjugated Aβ42 colocalized with Rab11+ recycling endosomes after 3 h of incubation (data not shown), suggesting that the rate of Aβ recycling between endosomes and plasma membrane is not changed by syn1j reduction.
We next studied whether increased Aβ uptake with synj1 reduction is dependent upon elevated PIP2 levels, using a pharmacological reagent m-3m3FBS that activates phospholipase C to deplete PIP2 inside the cells (11). As shown in Fig. 6C, the amount of Aβ taken up by control cells was modestly decreased with reduction of PIP2 (86.4 ± 7.8%, p = 0.24). An inactive analog of m-3m3FBS was used as a control (o-3m3FBS). However, in synj1 knockdown cells, the amount of Aβ taken up by cells remained increased even with treatment to reduce PIP2 (149.6 ± 8.0% in m-3-FBS treated versus 144.3 ± 24.3% in o-3-FBS-treated conditions).
Together, our data suggest that reduction of synj1 accelerates Aβ uptake into cells and delivery to early endosomes. However, the increased Aβ uptake is independent from PIP2 increase induced by synj1 reduction.
Reduction of synj1 Increases Cellular Aβ Degradation
The rate of Aβ turnover was next determined in N2a cells with synj1 siRNA transfection by CHX time course experiments (Fig. 7A). synj1 knockdown resulted in decreased cellular Aβ levels at time point 0 (59.7 ± 13.9%; p = 0.015), consistent with prior observations (Fig. 1A). At 60 min of incubation, the amount of Aβ was diminished by 37.5% with synj1 knockdown (reduced to 22.2 ± 12.1% of control levels at time point 0), whereas Aβ levels in controls were only reduced by 11.5% (88.5 ± 15.2% of control levels at time point 0). However, synj1 reduction does not affect APP turnover rate. As shown in Fig. 7B, the amount of holoAPP in lysates was comparable between control and synj1 siRNA-treated conditions at all time points (0–180 min) after exposure of cells to CHX. These results in combination with previous data (Figs. 3 and 5) suggest that the reduced Aβ levels with synj1 down-regulation are likely due to accelerated degradation of Aβ instead of reduced generation.
FIGURE 7.

Reduction of synj1 increases cellular Aβ degradation. A, left panel, immunoblot analysis of Aβ turnover rate in the presence of CHX (50 μg/ml) in N2a cells transfected with either control or synj1 siRNA. Right panel, densitometric analysis of Aβ accumulation in the presence of CHX at 0, 30, 60, and 90 min. B, left panel, immunoblot analysis of holoAPP turnover rate in the presence of CHX (50 μg/ml) in N2a cells transfected with either control or synj1 siRNA. Right panel, densitometric analysis of holoAPP accumulation in the presence of CHX at 0, 60, 120, and 180 min. The results represent three independent experiments, and the data are presented as percentages of controls at time point 0; means ± S.E. *, p < 0.05. ctrl, control.
Delivery of Internalized Aβ42 to LAMP1+ Lysosomes Increases in synj1−/− Primary Neurons
We next investigated whether Aβ trafficking through the endosomal/lysosomal pathways was increased with synj1 reduction. Immunostaining with antibodies against LAMP1 was performed in mouse cultured cortical neurons after incubation with Alexa555-Aβ42 (500 nm) for 24 h. We found that Aβ42 was colocalized with a lysosomal marker LAMP1 (Fig. 8, A and B, top panels). However, the amount of Aβ42 colocalized with LAMP1+ lysosomes was significantly increased in both cell bodies and neurites of synj1+/− and synj1−/− neurons (Fig. 8, A and B, middle and bottom panels) compared with synj1+/+ cells. Upon quantification, we confirmed that the amounts of fluorescent-conjugated Aβ42 colocalized with LAMP1+ lysosomes was increased by 130.2 ± 42.4% in synj1−/− neurons if compared with synj1+/+ cells (p = 0.016). These results suggest that internalized Aβ42 is transported more rapidly to the endosome/lysosome degradation pathway in neurons with synj1 reduction.
FIGURE 8.

Delivery of internalized Aβ42 to LAMP1+ lysosomes increases in synj1−/− primary neurons. Embryonic cortical neurons (top panels, synj1+/+; middle panels, synj1+/−; bottom panels, synj1−/−) were incubated with Alexa555-Aβ42 (500 nm) for 24 h. Cells were then fixed and stained for a lysosomal marker LAMP1 (red) before analyzed by fluorescence confocal microscopy. Representative neurons are shown. A, left panels, internalized Alexa555-Aβ42 shown in red fluorescence. Right panels, a lysosomal marker LAMP1 shown in green fluorescence. Scale bar, 10 μm. B, enlarged view of colocalization of internalized Alexa555-Aβ42 with LAMP1+ lysosomes at cell body (left panels) and processes (right panels). Yellow, overlay view of Alexa555-Aβ/LAMP1 colocalization. Scale bar, 5 μm.
Reduction of synj1 Increases Aβ Degradation in Lysosomes Partially through Elevated PIP2 Levels
Furthermore, to assess lysosomal degradation of Aβ, we tested the effects of lysosomal inhibitors (leupeptin, pepstatin A, and E-64d), which block lysosomal enzyme activities (32, 33). N2a cells transfected with synj1 or control siRNA for 4 days were then incubated in the presence or absence of lysosomal inhibitors for 24 h. The amount of media and cell-associated Aβ in the presence of lysosomal inhibitors, determined by IP/Western blotting, were significantly increased (Fig. 9A, middle and right panels), indicating that lysosomal degradation of Aβ is a significant mechanism for elimination of Aβ following cellular Aβ uptake. More importantly, inhibition of Aβ degradation through lysosomal pathways abolishes the Aβ-lowering effects of synj1 reduction (with lysosomal inhibitors, media Aβ 192.5 ± 8.8% in controls versus 185.3 ± 2.6% in synj1 siRNA; lysate Aβ 156.1 ± 10.5% in controls versus 185.0 ± 14.0% in synj1 siRNA).
FIGURE 9.

Reduction of synj1 increases Aβ degradation in lysosomes partially through elevated PIP2 levels. A, N2a cells transfected with control or synj1 siRNA were incubated in the presence or absence of lysosomal inhibitors for 24 h. Media and lysate Aβ were quantified by IP/Western blot. The left panel shows one typical example of Western blot analysis. Right panel, data are plotted as percentages of controls, means ± S.D. (n = 3). *, p < 0.05. B, the amounts of degraded Aβ were calculated by subtracting the values of secreted or cell-associated Aβ in the absence of inhibitors from the values in the presence of inhibitors. The data are plotted as percentages of controls, means ± S.D. (n = 3). *, p < 0.05. C, N2a cells transfected with control or synj1 siRNA were incubated with a PIP2-depleting reagent m-3-FBS or its inactive analog o-3-FBS, in the presence or absence of lysosomal inhibitors for 24 h. Lysate Aβ were quantified by IP/Western blot. The data are plotted as percentages of controls, means ± S.D. (n = 3). *, p < 0.05. ctrl, control; DMSO, dimethyl sulfoxide; inh, inhibitor; N.S., not significant.
The amount of Aβ degraded by lysosomes was determined by subtracting the mean values of Aβ in the absence of lysosomal inhibitors from the values in the presence of inhibitors. Synj1 reduction increases lysosomal degradation of cellular Aβ by 112.6 ± 57.6% (p = 0.021) and media Aβ by 30.6 ± 17.5% (p = 0.043) if compared with controls (Fig. 9B). These data suggest that the Aβ-lowering effects of synj1 reduction are mainly mediated through promoting lysosomal degradation.
We next studied whether the increased Aβ degradation with synj1 reduction is dependent upon elevated PIP2 levels, using Synj1 knockdown N2a cells treated with a PIP2-depleting reagent (m-3-FBS). As shown in Fig. 9C, the addition of m-3-FBS completely abolished the Aβ-lowering effects induced by synj1 reduction (109.4 ± 13.6% in synj1 siRNA +m-3-FBS versus 94.6 ± 6.5% in control siRNA+m-3-FBS). Moreover, the amount of Aβ accumulated in the presence of lysosomal inhibitors was not increased further by the addition of a PIP2-depleting reagent (m-3-FBS/lyso inhibitor columns, 146.9 ± 11.8% in lyso inhibitors alone versus 138.5 ± 12.8% in m-3-FBS+lyso inhibitors), suggesting that the effects of PIP2 on Aβ levels act through the same endosomal/lysosomal degradation pathway. In summary, our data suggest that reduction of synj1 increases cellular Aβ uptake, endosomal/lysosomal trafficking and degradation, partially by increasing the levels of PIP2.
DISCUSSION
Recent studies indicate that late onset AD is correlated with an overall impairment in Aβ clearance (3). Furthermore, several studies report pathological changes of the lysosomal network, which develop in neurons as Alzheimer disease progresses and include dysregulation of endocytosis and progressive failure of lysosomal clearance mechanisms (34–36). A close connection between lysosomal protein clearance failure and mechanisms of neurodegeneration is also well documented (36–39).
In this study, we defined a novel mechanism by which a PI(4,5)P2-degrading enzyme, synj1, regulates the cellular itinerary of Aβ. We show that down-regulation of synj1 in the brain promotes Aβ uptake by neurons, accelerates Aβ delivery to the endosomal/lysosomal pathway, increases Aβ degradation, and thereby reduces amyloid plaque load and attenuates behavioral deficits in AD transgenic animals.
As proposed in our model (Fig. 10), in WT cells, extracellular Aβ is internalized into the early endosomes, and either recycled back to the plasma membrane through recycling endosomes or delivered to late endosomes/lysosomes for degradation. Under synj1 knockdown conditions, extracellular Aβ is rapidly internalized to the early endosomes and delivered to the late endosomal/lysosomal pathway for degradation. However, the amount of Aβ generated by APP processing/cleavage, as well as the rate of cellular Aβ recycling from the early endosomes to the plasma membrane, remains unchanged. As a consequence, Aβ is more rapidly degraded, whereas the rate of Aβ production is not affected. The amount of extracellular and intracellular Aβ at steady state is subsequently decreased by synj1 knockdown.
FIGURE 10.

Model for synj1-regulated intracellular Aβ trafficking and lysosomal degradation. In WT cells, secreted Aβ is internalized into early endosomes, followed by either recycling back to plasma membrane through recycling endosomes or delivered to late endosomes/lysosomes for degradation. With synj1 knockdown (KD), Aβ is more rapidly internalized to the early endosomes and delivered to the late endosomal/lysosomal pathway for degradation. As a consequence, the amount of secreted and cellular Aβ is decreased with synj1 reduction. The increased Aβ uptake and internalization to early endosomes with synj1 reduction are independent upon elevated PIP2, whereas the effects of accelerated Aβ degradation in lysosomes by synj1 reduction are PIP2-dependent.
Synj1 has been linked to AD and Down's syndrome (5–7). It is a phosphatase that catalyzes the dephosphorylation of the signaling phospholipid PI(4,5)P2, which is shown to control clathrin-mediated endocytosis (40, 41). Studies in the mouse and other model organisms have found that synj1 plays a critical role in synaptic vesicle recycling, actin regulation, and glutamate receptor trafficking (4, 42). Intriguingly, our results suggest that reduction of synj1 can promote Aβ uptake and internalization through the endosomal pathway (Fig. 6), and these effects are independent upon PI(4,5)P2 elevation (Fig. 6C). It is unclear to us whether this synj1-enhanced internalization is specific to Aβ. Further characterization in these aspects is currently ongoing.
Interestingly, others reported that haploinsufficiency of synj1 protects against Aβ-induced defects in long term potentiation (7) and ameliorates learning and memory deficits in a transgenic mouse model of AD (6). It was also suggested that increased PI(4,5)P2 caused by down-regulation of synj1 suppresses Aβ oligomer-induced neurotoxic effects (5–7). Our data suggest that the promotion of cellular Aβ degradation with synj1 reduction is partially dependent upon elevated PIP2 because pharmacological reduction of PIP2 abolishes the Aβ-lowering effects in synj1 knockdown conditions (Fig. 9C). Together, these findings support the notion that elevation of PI(4,5)P2 by synj1 reduction is neuroprotective against AD.
Our novel studies demonstrate that reduction of synj1 accelerates cellular Aβ uptake and delivery to lysosomes and thereby promotes Aβ degradation and reduces amyloid plaque load in an AD transgenic mouse model. It should be noted that the reduction in amyloid plaque load is most prominent in hippocampus, which could contribute to functional rescue of learning and memory deficits in our AD transgenic mouse model (Fig. 4) and is consistent with others' observations (42). The specific changes in hippocampal Aβ caused by synj1 reduction likely indicate potential sensitivity of hippocampal neurons to down-regulation of synj1. However, we did not see any significant difference in plaque load in neocortical regions, possibly because of heavy plaque burden at this stage. In contrast, we did observe a robust reduction of intracellular Aβ within both cortical and hippocampal neurons at the preplaque stage of young Swedish APP/PS1ΔE9+/− synj1+/− mice (data not shown), suggesting a potential therapeutic strategy targeting synj1 at early stages of AD.
It should be also noted that in studies by McIntire et al., no alterations in Aβ levels were evident in 6-month-old Tg2576 mice with synj1 haploinsufficiency (6), despite the fact that they previously reported that Aβ biogenesis is modulated by PI(4,5)P2 (11). They measured the whole brain lysates at 6 months of age, whereas we specifically quantified Aβ and amyloid plaque load from hippocampus in older mice (9–10 months of age). Moreover, our data suggest that the Aβ-lowering effects induced by synj1 down-regulation are mainly dependent on the promotion of cellular Aβ degradation without affecting Aβ generation through cleavage of APP by β- and γ-secretase. There was no change in levels of γ-secretase/PS1 or BACE1 total proteins in APP/PS1+/− synj1+/− mouse brains (Fig. 3, A and B). Furthermore, utilizing a well established in vitro γ-secretase assay (25, 26), we have found that synj1 knockdown did not affect γ-secretase cleavage of C100 to generate Aβ or N100 to generate its cleavage product (Fig. 5).
On the other hand, we observed that synj1 reduction promotes α-secretase cleavage of APP and up-regulates expression levels of α-secretase ADAM10.4 Promoting APP processing through the nonamyloidogenic pathway by synj1 reduction can further strengthen rescue of AD related changes (43, 44). For example, the α-secretase-derived soluble APP N-terminal fragment, sAPPα has been suggested to have neurotrophic and neuroprotective functions, further supporting the therapeutic value of reducing synj1 levels in the brain (45). Although α-secretase competes with BACE for APP cleavage and has the capacity to pre-empt Aβ generation, it is unlikely that this is the main mechanism underlying synj1 knockdown-induced Aβ-lowering effects. Our data (Figs. 1 and 3) show that the levels of βCTF, the immediate precursor of Aβ, are not affected by synj1 reduction, suggesting that the BACE cleavage of APP is not affected under our conditions.
More importantly, increased synj1 expression has been functionally linked to the enlargement of early endosomes (46). It has been reported that endosome anomalies are the earliest specific pathology reported in AD brain tissue (47, 48). We here show that synj1 regulates endosomal trafficking and lysosomal degradation of Aβ. Reduction of synj1 can promote intracellular Aβ degradation through the endosomal/lysosomal pathway. Therefore, it would be interesting to investigate whether synj1 reduction can reverse early AD pathological changes such as enlargement of early endosomes.
In summary, our studies demonstrate a novel mechanism by which a PI(4,5)P2-degrading enzyme, synj1, regulates the cellular itinerary of Aβ. We show for the first time that endosomal/lysosomal degradation of Aβ can be modulated by phosphoinositol homeostasis. Importantly, endosomal anomalies are considered as one of the earliest AD pathologies, and increased function of synj1 is linked to enlargement of early endosomes (46–48). Thus, our findings uncover a new therapeutic direction for AD aiming at modulation of cellular Aβ clearance by a phosphoinositol regulator, in the hopes of reversing amyloid-induced pathological changes and cognitive deficits.
Acknowledgments
We thank Dr. Pietro De Camilli (Yale School of Medicine) for scientific discussion and providing critical research tools for our studies. We thank Dr. Yueming Li (Memorial Sloan-Kettering Cancer Center) for the C100-FLAG construct. Antibodies p369 and AB14 were generously provided by Dr. Paul Greengard. We also thank Drs. Paul Greengard, Christopher Cardozo, Gregory Elder, Rachel Lane, and John Steele for critical reading of the manuscript. We thank the Icahn School of Medicine at Mount Sinai Microscopy Shared Resource Facility for performing confocal laser scanning microscopy for confocal image studies. We also thank Saijai Simavijai for helping with graphic design of the Fig. 10 model.
This work was supported, in whole or in part, by National Institutes of Health Grant NS047229 (to N. R.). This work was also supported by a seed fund provided to Dongming Cai by the Department of Neurology and Friedman Brain Institute of Icahn Mount Sinai School of Medicine, funds from the Ellison Foundation (to P. D.), National Institutes of Health-National Cancer Institute Shared Resources Grant 5R24CA095823-04, National Science Foundation Major Research Instrumentation Grant DBI-9724504, and National Institutes of Health Shared Instrumentation Grant 1S10RR09145-01.
L. Zhu, J. Zhao, N. K. Robakis, and D. Cai, manuscript in preparation.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- APP
- amyloid precursor protein
- holoAPP
- full-length APP
- CTF
- C-terminal fragment
- synj1
- synaptojanin 1
- PI(4,5)P2
- phosphoinositol (4,5)-biphosphate phosphatase
- PS1
- presenilin 1
- IP
- immunoprecipitation
- CHX
- cycloheximide
- LCO
- luminescent conjugated oligothiophene
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- CHAPSO
- 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonic acid
- PIP2
- phosphoinositol biphosphate.
REFERENCES
- 1. Barthet G., Georgakopoulos A., Robakis N. K. (2012) Cellular mechanisms of r-secretase substrate selection, processing and toxicity. Prog. Neurobiol. 98, 166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang H., Ma Q., Zhang Y. W., Xu H. (2012) Proteolytic processing of Alzheimer's β-amyloid precursor protein. J. Neurochem. 120, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mawuenyega K. G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J. C., Yarasheski K. E., Bateman R. J. (2010) Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cremona O., Di Paolo G., Wenk M. R., Lüthi A., Kim W. T., Takei K., Daniell L., Nemoto Y., Shears S. B., Flavell R. A., McCormick D. A., De Camilli P. (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99, 179–188 [DOI] [PubMed] [Google Scholar]
- 5. Voronov S. V., Frere S. G., Giovedi S., Pollina E. A., Borel C., Zhang H., Schmidt C., Akeson E. C., Wenk M. R., Cimasoni L., Arancio O., Davisson M. T., Antonarakis S. E., Gardiner K., De Camilli P., Di Paolo G. (2008) Synaptojanin 1-linked phosphoinositide dyshomeostasis and cognitive deficits in mouse models of Down's syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 9415–9420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McIntire L. B., Berman D. E., Myaeng J., Staniszewski A., Arancio O., Di Paolo G., Kim T. W. (2012) Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer's disease. J. Neurosci. 32, 15271–15276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berman D. E., Dall'Armi C., Voronov S. V., McIntire L. B., Zhang H., Moore A. Z., Staniszewski A., Arancio O., Kim T. W., Di Paolo G. (2008) Oligomeric amyloid-β peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat. Neurosci. 11, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Buxbaum J. D., Gandy S. E., Cicchetti P., Ehrlich M. E., Czernik A. J., Fracasso R. P., Ramabhadran T. V., Unterbeck A. J., Greengard P. (1990) Processing of Alzheimer β/A4 amyloid precursor protein. Modulation by agents that regulate protein phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 87, 6003–6006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aslund A., Sigurdson C. J., Klingstedt T., Grathwohl S., Bolmont T., Dickstein D. L., Glimsdal E., Prokop S., Lindgren M., Konradsson P., Holtzman D. M., Hof P. R., Heppner F. L., Gandy S., Jucker M., Aguzzi A., Hammarström P., Nilsson K. P. (2009) Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem. Biol. 4, 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cai D., Netzer W. J., Zhong M., Lin Y., Du G., Frohman M., Foster D. A., Sisodia S. S., Xu H., Gorelick F. S., Greengard P. (2006) Presenilin-1 uses phospholipase D1 as a negative regulator of β-amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 103, 1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Landman N., Jeong S. Y., Shin S. Y., Voronov S. V., Serban G., Kang M. S., Park M. K., Di Paolo G., Chung S., Kim T. W. (2006) Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. U.S.A. 103, 19524–19529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., Hu K. L., Johnson-Wood K. L., Kennedy S. L., Kholodenko D., Knops J. E., Latimer L. H., Lee M., Liao Z., Lieberburg I. M., Motter R. N., Mutter L. C., Nietz J., Quinn K. P., Sacchi K. L., Seubert P. A., Shopp G. M., Thorsett E. D., Tung J. S., Wu J., Yang S., Yin C. T., Schenk D. B., May P. C., Altstiel L. D., Bender M. H., Boggs L. N., Britton T. C., Clemens J. C., Czilli D. L., Dieckman-McGinty D. K., Droste J. J., Fuson K. S., Gitter B. D., Hyslop P. A., Johnstone E. M., Li W. Y., Little S. P., Mabry T. E., Miller F. D., Audia J. E. (2001) Functional gamma-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- 13. Lanz T. A., Himes C. S., Pallante G., Adams L., Yamazaki S., Amore B., Merchant K. M. (2003) The gamma-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester reduces Aβ levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice. J. Pharmacol. Exp. Ther. 305, 864–871 [DOI] [PubMed] [Google Scholar]
- 14. Lane R. F., Raines S. M., Steele J. W., Ehrlich M. E., Lah J. A., Small S. A., Tanzi R. E., Attie A. D., Gandy S. (2010) Diabetes-associated SorCS1 regulates Alzheimer's amyloid-β metabolism. Evidence for involvement of SorL1 and the retromer complex. J. Neurosci. 30, 13110–13115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu L., Su M., Lucast L., Liu L., Netzer W. J., Gandy S. E., Cai D. (2012) Dynamin 1 regulates amyloid generation through modulation of BACE1. PLoS One 7, e45033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., Younkin S. G., Borchelt D. R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo. Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170 [DOI] [PubMed] [Google Scholar]
- 17. Jankowsky J. L., Slunt H. H., Ratovitski T., Jenkins N. A., Copeland N. G., Borchelt D. R. (2001) Co-expression of multiple transgenes in mouse CNS. A comparison of strategies. Biomol. Eng. 17, 157–165 [DOI] [PubMed] [Google Scholar]
- 18. Kawarabayashi T., Younkin L. H., Saido T. C., Shoji M., Ashe K. H., Younkin S. G. (2001) Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J. Neurosci. 21, 372–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Calhoun M. E., Kurth D., Phinney A. L., Long J. M., Hengemihle J., Mouton P. R., Ingram D. K., Jucker M. (1998) Hippocampal neuron and synaptophysin-positive bouton number in aging C57BL/6 mice. Neurobiol. Aging 19, 599–606 [DOI] [PubMed] [Google Scholar]
- 20. Shamir L., Delaney J. D., Orlov N., Eckley D. M., Goldberg I. G. (2010) Pattern recognition software and techniques for biological image analysis. PLoS Comput. Biol. 6, e1000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Berg I., Nilsson K. P., Thor S., Hammarström P. (2010) Efficient imaging of amyloid deposits in Drosophila models of human amyloidoses. Nat. Protoc. 5, 935–944 [DOI] [PubMed] [Google Scholar]
- 22. Howlett D. R., Richardson J. C., Austin A., Parsons A. A., Bate S. T., Davies D. C., Gonzalez M. I. (2004) Cognitive correlates of Aβ deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. 1017, 130–136 [DOI] [PubMed] [Google Scholar]
- 23. Holcomb L., Gordon M. N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D., Sanders S., Zehr C., O'Campo K., Hardy J., Prada C. M., Eckman C., Younkin S., Hsiao K., Duff K. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100 [DOI] [PubMed] [Google Scholar]
- 24. Lalonde R. (2002) The neurobiological basis of spontaneous alternation. Neurosci. Biobehav. Rev. 26, 91–104 [DOI] [PubMed] [Google Scholar]
- 25. Cacquevel M., Aeschbach L., Osenkowski P., Li D., Ye W., Wolfe M. S., Li H., Selkoe D. J., Fraering P. C. (2008) Rapid purification of active γ-secretase, an intramembrane protease implicated in Alzheimer's disease. J. Neurochem. 104, 210–220 [DOI] [PubMed] [Google Scholar]
- 26. Li Y. M., Lai M. T., Xu M., Huang Q., DiMuzio-Mower J., Sardana M. K., Shi X. P., Yin K. C., Shafer J. A., Gardell S. J. (2000) Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. Proc. Natl. Acad. Sci. U.S.A. 97, 6138–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaufty J., Sullivan S. E., Ho A. (2012) Intracellular amyloid precursor protein sorting and amyloid-β secretion are regulated by Src-mediated phosphorylation of Mint2. J. Neurosci. 32, 9613–9625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prabhu Y., Burgos P. V., Schindler C., Farías G. G., Magadán J. G., Bonifacino J. S. (2012) Adaptor protein 2-mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol. Biol. Cell 23, 2339–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vieira S. I., Rebelo S., Esselmann H., Wiltfang J., Lah J., Lane R., Small S. A., Gandy S., da Cruz E. S., da Cruz E. S. (2010) Retrieval of the Alzheimer's amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener. 5, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zinchuk V., Grossenbacher-Zinchuk O. (2011) Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr. Protoc. Cell Biol. Chapter 4, Unit 4.19 [DOI] [PubMed] [Google Scholar]
- 31. Jaskolski F., Mulle C., Manzoni O. J. (2005) An automated method to quantify and visualize colocalized fluorescent signals. J. Neurosci. Methods 146, 42–49 [DOI] [PubMed] [Google Scholar]
- 32. Li J., Kanekiyo T., Shinohara M., Zhang Y., LaDu M. J., Xu H., Bu G. (2012) Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 287, 44593–44601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iwabu Y., Fujita H., Kinomoto M., Kaneko K., Ishizaka Y., Tanaka Y., Sata T., Tokunaga K. (2009) HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 284, 35060–35072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ihara Y., Morishima-Kawashima M., Nixon R. (2012) The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nixon R. A., Yang D. S., Lee J. H. (2008) Neurodegenerative lysosomal disorders. A continuum from development to late age. Autophagy 4, 590–599 [DOI] [PubMed] [Google Scholar]
- 36. Nixon R. A., Cataldo A. M. (2006) Lysosomal system pathways. Genes to neurodegeneration in Alzheimer's disease. J. Alzheimers Dis. 9, 277–289 [DOI] [PubMed] [Google Scholar]
- 37. Abrahams J. P., Lutter R., Todd R. J., van Raaij M. J., Leslie A. G., Walker J. E. (1993) Inherent asymmetry of the structure of F1-ATPase from bovine heart mitochondria at 6.5 A resolution. EMBO J. 12, 1775–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee S., Sato Y., Nixon R. A. (2011) Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy 7, 1562–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cherra S. J., 3rd, Dagda R. K., Chu C. T. (2010) Review. Autophagy and neurodegeneration. Survival at a cost? Neuropathol. Appl. Neurobiol. 36, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McPherson P. S., Garcia E. P., Slepnev V. I., David C., Zhang X., Grabs D., Sossin W. S., Bauerfeind R., Nemoto Y., De Camilli P. (1996) A presynaptic inositol-5-phosphatase. Nature 379, 353–357 [DOI] [PubMed] [Google Scholar]
- 41. Slepnev V. I., De Camilli P. (2000) Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat. Rev. Neurosci. 1, 161–172 [DOI] [PubMed] [Google Scholar]
- 42. Frere S. G., Chang-Ileto B., Di Paolo G. (2012) Role of phosphoinositides at the neuronal synapse. Subcell. Biochem. 59, 131–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Postina R., Schroeder A., Dewachter I., Bohl J., Schmitt U., Kojro E., Prinzen C., Endres K., Hiemke C., Blessing M., Flamez P., Dequenne A., Godaux E., van Leuven F., Fahrenholz F. (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim M. L., Zhang B., Mills I. P., Milla M. E., Brunden K. R., Lee V. M. (2008) Effects of TNFα-converting enzyme inhibition on amyloid β production and APP processing in vitro and in vivo. J. Neurosci. 28, 12052–12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vingtdeux V., Marambaud P. (2012) Identification and biology of α-secretase. J. Neurochem. 120, 34–45 [DOI] [PubMed] [Google Scholar]
- 46. Cossec J. C., Lavaur J., Berman D. E., Rivals I., Hoischen A., Stora S., Ripoll C., Mircher C., Grattau Y., Olivomarin J. C., de Chaumont F., Lecourtois M., Antonarakis S. E., Veltman J. A., Delabar J. M., Duyckaerts C., Di Paolo G., Potier M. C. (2012) Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 21, 3156–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cataldo A. M., Barnett J. L., Pieroni C., Nixon R. A. (1997) Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease. Neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J. Neurosci. 17, 6142–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cataldo A. M., Peterhoff C. M., Troncoso J. C., Gomez-Isla T., Hyman B. T., Nixon R. A. (2000) Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and Down syndrome. Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]