

Abstract
Over the last two decades, an increasing number of SCN5A mutations have been described in patients with long QT syndrome type 3 (LQT3), Brugada syndrome, (progressive) conduction disease, sick sinus syndrome, atrial standstill, atrial fibrillation, dilated cardiomyopathy, and sudden infant death syndrome (SIDS). Combined genetic, electrophysiological and molecular studies have provided insight into the dysfunction and dysregulation of the cardiac sodium channel in the setting of SCN5A mutations identified in patients with these inherited arrhythmia syndromes. However, risk stratification and patient management is hindered by the reduced penetrance and variable disease expressivity in sodium channelopathies. Furthermore, various SCN5A-related arrhythmia syndromes are known to display mixed phenotypes known as cardiac sodium channel overlap syndromes. Determinants of variable disease expressivity, including genetic background and environmental factors, are suspected but still largely unknown. Moreover, it has become increasingly clear that sodium channel function and regulation is more complicated than previously assumed, and the sodium channel may play additional, as of yet unrecognized, roles in cardiac structure and function. Development of cardiac structural abnormalities secondary to SCN5A mutations has been reported, but the clinical relevance and underlying mechanisms are unclear. Increased insight into these issues would enable a major next step in research related to cardiac sodium channel disease, ultimately enabling improved diagnosis, risk stratification and treatment strategies.
 |
Carol Ann Remme studied Medicine at Utrecht University, The Netherlands, and obtained her PhD at the University of Amsterdam, The Netherlands. Her current research at the Department of Experimental Cardiology of the Academic Medical Center, Amsterdam, focuses on transgenic models of cardiac arrhythmias and sudden cardiac death. Other research topics include sodium channelopathies, cardiac sodium channel (dys)function, and genetic determinants and modifiers of cardiac electrophysiology.
Introduction
The influx of sodium ions through cardiac voltage-gated sodium channels is responsible for the initial fast upstroke of the cardiac action potential, thereby triggering the initiation and propagation of action potentials throughout the myocardium. Cardiac sodium channels thus play a central role in excitability of myocardial cells and proper conduction of the electrical impulse within the heart. Sodium channel dysfunction caused by mutations in the SCN5A gene, encoding the major cardiac sodium channel NaV1.5, is associated with a number of inherited arrhythmia syndromes including long QT syndrome type 3 (LQT3), cardiac conduction disease and Brugada syndrome. Over the last 20 years, (combined) genetic, electrophysiological and molecular studies have provided insight into the (dys)function and (dys)regulation of the cardiac sodium channel under physiological circumstances and in the setting of SCN5A mutations identified in patients with inherited arrhythmia syndromes.
Although our understanding of these sodium channelopathies has increased substantially, important issues remain incompletely understood. It has become increasingly clear that sodium channel function and regulation is more complicated than traditionally assumed, caused at least in part by the many newly identified proteins interacting with the channel. The fact that SCN5A mutations have more recently also been associated with dilated cardiomyopathy suggests that the sodium channel may play additional, as of yet unrecognized roles in cardiomyocyte function. Development of cardiac structural abnormalities secondary to SCN5A mutations has been reported, but the clinical relevance and underlying mechanisms are unclear. Similarly, the environmental and genetic factors determining variable disease severity in SCN5A mutation carriers are yet to be established. Increased knowledge of these issues will be essential for further improvement of diagnosis, risk stratification and treatment in patients with sodium channelopathies.
Here, an overview is provided of both established and more recent insights into the genetic, electrophysiological and molecular aspects of sodium channel dysregulation and dysfunction in the setting of SCN5A mutations.
The cardiac sodium channel: structure, function and localization
Sodium channel structure and function
The voltage-dependent cardiac sodium channel consists of a transmembrane pore-forming α-subunit protein of about 220 kDa associated with a small (30–40 kDa) ancillary modulatory β-subunit and several regulatory proteins (the stoichiometry of α- and β-subunits in heart is as yet incompletely known). The α-subunit protein NaV1.5 (encoded by the SCN5A gene located on chromosome 3p21) is made up of a cytoplasmic N terminus, four internally homologous domains (DI–DIV, each consisting of six transmembrane α-helical segments S1–S6) interconnected by cytoplasmic linkers, and a cytoplasmic C terminal domain (for details, see legend to Fig. 1). Channel activation (opening of the pore) is triggered by the outward movement of the positively charged S4 segments allowing for sodium ion influx to commence. Simultaneously, the processes of fast and slow inactivation are initiated leading to closure of the channel pore. Fast inactivation of the sodium current involves an inactivation gate formed by a cluster of three hydrophobic amino acids (isoleucine, phenylalanine and methionine, or IFM-motif; see Fig. 1) in the intracellular DIII–DIV linker, together with two docking sites located on the intracellular linkers between S4 and S5 of DIII and DIV (West et al. 1992; Kass, 2006). In addition, the carboxy (COOH-) terminal domain is also involved in the inactivation process and acts through stabilization of the closed gate, thus minimizing channel re-opening (Motoike et al. 2004). During prolonged periods of depolarization, the sodium channel enters the state of slow inactivation, which requires much longer recovery times than fast inactivation. Slow inactivation is an important determinant of sodium channel availability, and most likely involves conformational changes in the S5–S6 P-loops, although the DIII S4–S5 linker may also be of importance (Kass, 2006; Casini et al. 2007). Activation and inactivation properties of sodium channels are tightly regulated during physiological conditions but may be altered in the setting of genetic and acquired sodium channel dysfunction. As a consequence, sodium channel availability and peak sodium current may be decreased, or the channel is not properly inactivated, resulting in a persistent (sustained), non-inactivating sodium current during the action potential plateau (George, 2005). These alterations in sodium channel function may have profound consequences for cardiac electrophysiology and arrhythmogenesis.
Figure 1. Schematic representation of the structure of the cardiac sodium channel.
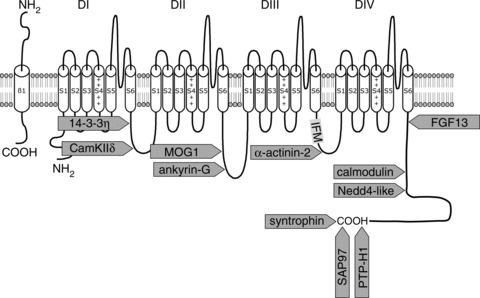
The locations are indicated where interacting proteins bind to the various regions of the channel. For a number of proteins known to modulate sodium channel function, their putative binding site is unknown. The DI–DIV domains each consist of six transmembrane α-helical segments (S1–S6), which in turn are interconnected by extracellular and cytoplasmic loops. The four domains fold around an ion-conducting pore, which is lined by the extracellular loops (P-loops) between S5 and S6 segments; the P-loops are considered to contain the channels’ selectivity filter for sodium ions (see Kass, 2006). The fourth transmembrane segment, S4, is highly charged and acts as the voltage sensor responsible for increased channel permeability (channel activation) during membrane depolarization (Balser, 2001). The locations are indicated where interacting proteins bind to the various regions of the channel. Redrawn from Shy et al. 2013, with permission.
Expression and distribution of sodium channels in the heart
Cardiac sodium channels show inhomogeneous expression within the cardiac conduction system and across the ventricular wall. NaV1.5 protein expression is low to absent in the sinoatrial and atrioventricular nodes, but abundant in the His bundle, bundle branches and Purkinje fibres (Yoo et al. 2006; Remme et al. 2009b). Furthermore, a transmural gradient is observed in left and right ventricle, with lower NaV1.5 labelling intensity and decreased functional sodium channel availability in the subepicardium as compared to the subendocardium (Ashamalla et al. 2001; Cordeiro et al. 2008; Remme et al. 2009b). In addition, differences in sodium channel (in)activation properties and pharmacological responsiveness have been described between atrial and ventricular myocytes (Li et al. 2002; Burashnikov et al. 2007).
Distinct pools of sodium channels within the cardiomyocyte: intercalated disc versus lateral membrane
More recently, the existence of two distinct functional pools of sodium channels within the cardiomyocyte has been demonstrated. NaV1.5 localization is observed in both the intercalated discs and the lateral membrane of the myocyte, and different interacting proteins (discussed in more detail below) appear to associate with NaV1.5 at these distinct areas (Fig. 2A). Sodium channels located at the lateral membrane are associated with the syntrophin–dystrophin complex, and dystrophin-deficient mdx mice display reduced lateral membrane NaV1.5 expression levels and concomitant changes in conduction velocity (Petitprez et al. 2011). In contrast, NaV1.5 channels in the intercalated disc region (which is devoid of syntrophin) interact with synapse-associated protein 97 (SAP97), a member of the membrane-associated guanylate kinase (MAGUK) family (Petitprez et al. 2011). Recently, Milstein and colleagues demonstrated that SAP97 allows NaV1.5 to interact with the Kir2.1 potassium channel in the heart, further underlining the relevance of the macromolecular complex (Milstein et al. 2012). The functional relevance of the two separate pools of sodium channels within myocytes has been investigated by macropatch measurements. Differences in peak sodium current amplitude and kinetics between channels located at the intercalated discs and at the cardiomyoyte lateral membrane have been observed (Fig. 2B–E), although species-specific differences probably exist (Verkerk et al. 2007; Lin et al. 2011). Nevertheless, these findings suggest that sodium channels are differentially regulated at various subcellular regions within the myocyte (Mohler & Hund, 2011). As yet, little is known about the specific roles for these separate pools of channels or their functional relevance during (patho)physiological conditions.
Figure 2.
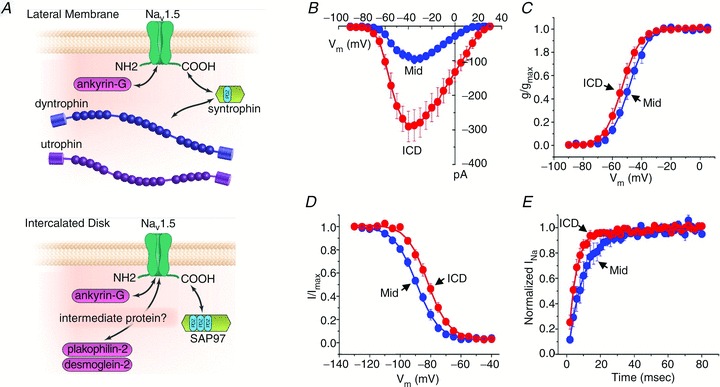
A, NaV1.5 resides in distinct macromolecular complexes at two different subcellular domains of the cardiac cell: (top) at the lateral membrane where it interacts with the dystrophin/syntrophin complex; and (bottom) at the intercalated discs with the MAGUK protein SAP97 (reproduced from Shy et al. 2013, with permission). B–E, macropatch measurements in rat ventricular myocytes reveal significant differences in peak sodium current magnitude (B), steady-state activation (C), steady-state inactivation (D) and reactivation (E) between the intercalated disc region (ICD) and the lateral side (Mid) of the cell (reproduced from Lin et al. 2011, with permission).
Regulation of sodium channel expression and function
(Post-)transcriptional regulation of SCN5A
On the transcriptional level, SCN5A mRNA expression is likely to be regulated by various enhancer and repressor sequences close to or within the promoter region of the gene (Arnolds et al. 2012; van den Boogaard et al. 2012). Furthermore, a circadian expression pattern of cardiac Scn5a mRNA under control of the molecular clock transcription factor Bmal1 was recently demonstrated in mice (Schroder et al. 2013). Post-transcriptionally, alternative splicing of the SCN5A gene produces transcript variants with different functional characteristics (see Schroeter et al. 2010). In human heart, two main alternatively spliced SCN5A variants are expressed, which vary in the presence or absence of a glutamine at position 1077 (SCN5A-Q1077 versus SCN5A-Q1077del; Makielski et al. 2003). These splice variants may be of significant functional relevance for disease expressivity in sodium channelopathy, as evidenced by the differential effect of these variants on reduced sodium channel membrane expression due to defective trafficking of the Brugada syndrome mutation SCN5A-G1406R (Tan et al. 2006). Moreover, SCN5A splicing is also developmentally regulated, and a specific neonatal isoform has been identified which is downregulated after birth (Chioni et al. 2005; Schroeter et al. 2010). Sodium channel dysfunction may be greatly enhanced in the setting of this neonatal SCN5A splice isoform, as demonstrated recently for a mutation identified in a fetus with severe long QT syndrome (LQTS; Murphy et al. 2012). Finally, during pathophysiological conditions such as human heart failure, increased expression of abnormal C-terminal SCN5A splicing variants may result in the formation of truncated, non-functional sodium channels, thereby predisposing to cardiac conduction slowing and arrhythmogenesis (Shang et al. 2007).
Post-translational regulation of sodium channels
Post-translational modifications of NaV1.5 expression and function include phosphorylation, glycosylation, S-nitrosylation, ubiquitination and methylation (reviewed by Rook et al. 2012). NaV1.5 harbours conserved amino acid motifs for N-glycosylation in its extracellular domain, and glycosylation may affect sodium channel gating (Zhang et al. 1999; Rook et al. 2012). Furthermore, aberrant expression of glycogens during, for instance, heart failure may modulate voltage-dependent sodium channel-mediated electrical signalling during such pathophysiological conditions (Montpetit et al. 2009). Protein kinase A (PKA)-dependent phosphorylation of NaV1.5 increases peak sodium current through facilitation of channel transport from intracellular storage compartments to the cell surface (Yarbrough et al. 2002). In contrast, the effect most frequently reported in relation to protein kinase C (PKC) activation concerned a reduction in sodium channel membrane trafficking and current amplitude (Murray et al. 1997; Hallaq et al. 2012). Sodium channel density and kinetics are furthermore also regulated by intracellular calcium levels (Casini et al. 2009), extracellular protons and pH (Jones et al. 2011), reactive oxygen species (Liu et al. 2010), temperature (Amin et al. 2005) and stretch (Beyder et al. 2010).
Sodium channel regulation by interacting proteins within the sodium channel macromolecular complex
NaV1.5 is known to interact with a large number of proteins which regulate sodium channel expression, trafficking and/or function (Table 1). For many of these regulatory proteins, their interaction site within various domains of NaV1.5 has been identified (Fig. 1). The intracellular C-terminal part of NaV1.5 contains several protein–protein interaction sites, including the calmodulin-binding IQ motif, the PY motif, and the PDZ domain-binding motif. Proteins such as PTPH1, SAP97 and syntrophins have been described to interact with the PDZ domain-binding motif (Jespersen et al. 2006; Gavillet et al. 2006; Petitprez et al. 2011). The ubiquitin-protein ligase Nedd4-2 directly binds to the PY motif and regulates NaV1.5 function by ubiquitylation of the channel (van Bemmelen et al. 2004). The IQ motif constitutes a binding site for calmodulin (CaM), a ubiquitous calcium-binding protein. Although discrepant effects have been described, CaM may confer sensitivity to intracellular calcium levels on NaV1.5 (see Shy et al. 2013). Fibroblast growth factor homologous factors (FHF) family members can also interact with the C-terminus of NaV1.5, and fibroblast growth factor homologous factor 13 (FGF13) has been shown to regulate sodium channels and conduction velocity in murine hearts (Wang et al. 2011). The cytosolic adaptor protein 14-3-3η interacts with the DI–DII linker region, thereby modulating steady-state inactivation of the channel (Allouis et al. 2006). This region furthermore contains multiple PKA and PKC phosphorylation sites in addition to an interaction site for the calcium/calmodulin-dependent protein kinase II (CAMKII), which has multiple regulatory effects on both sodium channel availability and persistent current magnitude (Wagner et al. 2006; Ashpole et al. 2012). Ankyrin-G and MOG1 have been identified to interact with the II–III linker segment of NaV1.5, and both proteins regulate cell surface expression of sodium channels (Lemaillet et al. 2003; Mohler et al. 2004; Kattygnarath et al. 2011). A member of the superfamily of F-actin cross-linking proteins, α-actinin-2, interacts with the cytoplasmic loop connecting domains III and IV, thereby increasing sodium current density without affecting gating properties (Ziane et al. 2010). Furthermore, β-subunits (β1–β4 encoded by the SCN1B–SCN4B genes) probably bind to the extracellular connecting loops between S5 and S6, allowing them to modulate sodium channel density and kinetics (Malhotra et al. 2001; Ko et al. 2005; Meadows & Isom, 2005; Medeiros-Domingo et al. 2007). Other proteins that may directly or indirectly interact with NaV1.5 and functionally regulate this channel include caveolin-3 (Vatta et al. 2006), glycerol-3-phosphate dehydrogenase 1-like protein (GPD1L; London et al. 2007), plakophilin-2 (Sato et al. 2009), desmoglein-2 (Rizzo et al. 2012), telethonin (Mazzone et al. 2008), and Z-band-alternatively spliced-PDZ motif protein (ZASP; Li et al. 2010; Table 1).
Table 1.
Clinical cardiac phenotypes of the cardiac sodium channel and its interacting proteins
| Gene | Protein | Clinical cardiac phenotypes |
|---|---|---|
| Cardiac sodium channel | ||
| SCN5A | NaV1.5 | LQT3, Brugada syndrome, cardiac conduction disease, sick sinus disease, overlap syndrome, atrial standstill, dilated cardiomyopathy, atrial fibrillation, SIDS |
| NaV1.5 interacting proteins | ||
| SCN1B | β1 | Brugada syndrome, conduction disease, atrial fibrillation |
| SCN2B | β2 | Atrial fibrillation |
| SCN3B | β3 | Brugada syndrome, conduction disease, atrial fibrillation, idiopathic ventricular fibrillation |
| SCN4B | β4 | Long QT syndrome type 10 (LQT10) |
| SNTA | α1-syntrophin | Long QT syndrome type 12 (LQT12) |
| MOG1 | MOG1 | Brugada syndrome |
| PTPH1 | Protein tyrosine phosphatase H1 | — |
| NEDD4L | Nedd4-2/Nedd4-like | — |
| CALM | Calmodulin | — |
| SAP97 | SAP97 | — |
| YWHAH | 14-3-3η | — |
| FGF13 | Fibroblast growth factor 13 (FGF13) | — |
| CAMK2D | CAMKIId | — |
| ANK3 | Ankyrin-G | — |
| ACTN2 | α-Actinin-2 | — |
| CAV3 | Caveolin-3 | Long QT syndrome type 9 (LQT9) |
| GPD1L | Glycerol-3-phosphate dehydrogenase 1-like | Brugada syndrome |
| PKP2 | Plakophilin-2 | Arrhythmogenic cardiomyopathy |
| DSG2 | Desmoglein-2 | Arrhythmogenic cardiomyopathy |
| TCAP | Telethonin | — |
| ZASP | Z-band alternatively spliced PDZ motif protein | — |
Functional and clinical relevance of NaV1.5-interacting proteins
Thus, sodium channels are not isolated units within the myocyte membrane, but are functional components of a macromolecular complex through which they associate with proteins that participate in cell adhesion, signal transduction, and cytoskeleton anchoring. The functional relevance of such interactions is evidenced by the fact that mutations in these modulatory proteins are associated with sodium channel dysfunction and arrhythmia (Table 1). Of the sodium channel interacting proteins, caveolin-3 and α1-syntrophin have been implicated in LQTS; Vatta et al. 2006; Wu et al. 2008) whereas mutations in GPD1L and MOG1 have been associated with Brugada syndrome (London et al. 2007; Abriel, 2010; Kattygnarath et al. 2011). Furthermore, mutations in sodium channel β-subunits have been identified in patients presenting with Brugada syndrome (BrS) and/or conduction disease (β1 and β3), atrial fibrillation (β1, β2 and β3), LQTS (β4), and idiopathic ventricular fibrillation (IVF) in the absence of a BrS phenotype (β3) (Medeiros-Domingo et al. 2007; Watanabe et al. 2008, 2009; Hu et al. 2009; Valdivia et al. 2010; Olesen et al. 2011, 2012).
More recently, mutations in the sarcolemmal membrane-associated protein (SLMAP), a protein of unknown function localizing at T-tubules and sarcoplasmic reticulum, were identified in BrS patients. SLMAP was found to modulate intracellular trafficking of hNaV1.5 channels, but no direct interaction between SLMAP and hNaV1.5 was observed (Ishikawa et al. 2012). In addition, the anticancer drug taxol, which changes the properties of the cytoskeletal component tubulin, affects sodium current density and gating in vitro and is associated with increased occurrence of cardiac conduction disorders and arrhythmias in vivo (Rowinsky et al. 1991; Casini et al. 2010).
Diseases and syndromes associated with SCN5A mutations
From a biophysical point of view, different SCN5A mutations can lead to multiple rhythm disturbances by modulating various gating and other functional properties of the sodium channel (Viswanathan & Balser, 2004; Remme et al. 2008). Indeed, mutations in SCN5A have been implicated in multiple inherited arrhythmia syndromes (Table 1). Although caused by mutations in the same ion channel, these clinical syndromes each display distinct phenotypical characteristics, as is discussed in more detail below.
Long QT syndrome type 3 (LQT3)
Long QT syndrome (LQTS) is characterized by prolonged QT intervals on the ECG and increased risk for sudden death due to ventricular tachyarrhythmias, in particular torsades de pointes. Various subtypes of LQTS exist, each associated with distinct clinical features and underlying genetic defect. Patients with LQTS type 3 (LQT3, associated with SCN5A mutations) display arrhythmias predominantly during rest or sleep (at slow heart rates), and they are often relatively bradycardic (Schwartz et al. 2001; Schwartz, 2006). Compared to other LQT subtypes, LQT3 patients are particularly at risk for sudden death, and cardiac arrest (rather than syncope) is often the first clinical event (Zareba et al. 2001; Schwartz et al. 2001). Most SCN5A mutations associated with LQT3 typically disrupt fast inactivation of the sodium current, allowing for sodium channels to re-open, resulting in a persistent (late) inward current during the action potential plateau phase (Bennett et al. 1995). Consequently, delayed repolarization and action potential prolongation occurs, and early after-depolarizations may subsequently trigger torsades de pointes and sudden death. Alternatively, SCN5A mutations less commonly cause LQT3 through slowed inactivation (resulting in channel openings of longer duration), faster recovery from inactivation (causing increased sodium channel availability), and increased peak sodium current density (Wedekind et al. 2001; Rivolta et al. 2001; Clancy et al. 2003). Therapeutic options for LQT3 include β-blockers, sodium channel blockers mexiletine or flecainide (depending on gene mutation), and implantable cardioverter defibrillator in high risk patients (see Remme & Bezzina, 2010). However, β-blockers may not be ideal in LQT3 (since cardiac events occur mostly during rest or sleep), and the effects of sodium channel blockers are probably mutation specific. More recently, the late sodium current inhibitor ranolazine has been proposed as a potential pharmacological therapy for cardiac sodium channel disease related to gain of sodium current function, including LQT3 (Fredj et al. 2006, Moss et al. 2008; Remme & Wilde, 2013). Indeed, in a small set of five patients carrying the SCN5A-ΔKPQ mutation, short-term ranolazine infusion significantly decreased QTc-intervals in the absence of adverse effects (Moss et al. 2008), indicating a potential beneficial effect of late sodium current inhibition in LQT3 (discussed further below). In general, however, efficacy of pharmacological treatment strategies in LQT3 has not been extensively studied due to small patient population sizes, and variation in results is probably further increased by genotype-specific differences in response to treatment.
Brugada syndrome
Brugada syndrome (BrS) is a familial disorder characterized by ventricular arrhythmias and sudden cardiac death occurring in otherwise healthy individuals at a relatively young age (<40 years). On ECG analysis, a typical pattern is observed comprising ST-segment elevation in the right-precordial leads V1–V3, which is unmasked or increased after administration of Class 1A or 1C anti-arrhythmic sodium channel-blocking drugs (ajmaline, flecainide), or during exercise (see Meregalli et al. 2005). In most cases signs of conduction disease are also present on the ECG, e.g. prolonged PQ- and QRS-duration in the presence of a normal QT-interval (Antzelevitch, 2001). Arrhythmias and sudden death occur mostly during rest or sleep, and arrhythmia incidence is higher in males than females (Antzelevitch, 2006). Furthermore, episodes of fever and other factors may provoke or exacerbate the typical ECG pattern and promote onset of arrhythmias (Dumaine et al. 1999; Chockalingam et al. 2011).
In general, SCN5A mutations associated with BrS are loss-of-function mutations, leading to reduced sodium channel availability, either through decreased trafficking and membrane surface channel expression, or through altered channel gating properties (Tan et al. 2003; Viswanathan & Balser, 2004; Kapplinger et al. 2010). Reduced sodium current decreases action potential upstroke velocity, leading to atrial and ventricular conduction slowing accompanied by prolongation of PR- and QRS-intervals on the ECG. The right-precordial ST segment elevation and its relation to arrhythmogenesis is less well understood, but two major hypotheses have been proposed. The repolarization disorder hypothesis involves increased transmural heterogeneity in action potential duration, which occurs preferentially in the right ventricle due to the more pronounced presence of the transient outward potassium current (Ito) in the right ventricular epicardium (Antzelevitch, 2006). Alternatively, conduction slowing in the right ventricular outflow tract may be exacerbated in the setting of decreased sodium current (depolarization hypothesis; Meregalli et al. 2005). These characteristics may render the right ventricle more susceptible to the deleterious effects secondary to sodium channel dysfunction, thereby promoting right ventricular arrhythmogenesis observed in BrS patients (Tukkie et al. 2004; Antzelevitch et al. 2005). Structural discontinuities in the epicardium of the right ventricle may further contribute to the typical BrS ECG pattern as well as the onset of ventricular arrhythmias (Hoogendijk et al. 2010).
Progressive cardiac conduction defect (PCCD) and sick sinus syndrome
PCCD, also called Lenègre or Lev disease, is characterized by progressive conduction slowing through the His-Purkinje system, with right and/or left bundle branch block and QRS-widening, leading to complete atrioventricular block, syncope and sudden death. In some cases, inherited PCCD is associated with mutations in SCN5A (Schott et al. 1999; Wolf & Berul, 2006). From a mechanistic point of view, SCN5A mutations underlying PCCD lead to reduced sodium channel availability (loss-of-function) similar to BrS, and considerable overlap exists between these two clinical entities (see also below) (Kyndt et al. 2001). Similarly, inherited sick sinus syndrome has also been associated with loss-of-function mutations in SCN5A (Benson et al. 2003; Smits et al. 2005), in addition to atrial standstill (Groenewegen et al. 2003; Takehara et al. 2004; Lopez et al. 2011). Although sodium channels are not considered essential for sinoatrial nodal pacemaking, they do, however, contribute to cardiac automaticity through the depolarizing effects of inward sodium current (albeit relatively small). In addition to reduced automaticity of sinoatrial pacemaking tissue, decreased sodium channel availability may also cause bradycardia by slowing or block of conduction from the central sinoatrial region to the surrounding atrial tissue (see Lei et al. 2008). Furthermore, sinus bradycardia and/or arrest may also occur in LQT3 patients with SCN5A gain-of-function mutations, as a consequence of action potential prolongation caused by increased persistent inward sodium current (Veldkamp et al. 2003).
Atrial fibrillation
Atrial fibrillation is the most prevalent clinical arrhythmia affecting mostly elderly patients with underlying structural cardiac abnormalities, but it may also occur as a hereditary disease in young patients with structurally normal hearts. Both loss-of-function and gain-of-function mutations in SCN5A have been described in this familial form, which are thought to induce atrial fibrillation through decreased atrial conduction velocity and increased atrial action potential duration and excitability, respectively (Darbar et al. 2008; Ellinor et al. 2008; Makiyama et al. 2008; Li et al. 2009). In addition, a role for structural remodelling of the atria secondary to sodium channel dysfunction is suspected. Mutations in the β-subunits causing sodium current reduction have also been associated with atrial fibrillation (Watanabe et al. 2009; Olesen et al. 2011).
Dilated cardiomyopathy
Familial forms of dilated cardiomyopathy (DCM) are mostly associated with mutations in cytoskeletal proteins, but have also been reported in patients with SCN5A mutations (Bezzina et al. 2003; McNair et al. 2004), often in combination with atrial arrhythmias and/or fibrillation (Olson et al. 2005). The question remains whether arrhythmias and structural defects are a direct effect of sodium current alterations, or merely secondary to long-standing cardiac abnormalities. Biophysical properties consistent with both loss and gain of sodium channel function have been observed in SCN5A mutations associated with DCM (McNair et al. 2004; Ge et al. 2008; Nguyen et al. 2008; Bezzina & Remme, 2008). Very few commonalities between various SCN5A mutations related to DCM exist (Bezzina & Remme, 2008), although they appear to be preferentially localized to the region of the channel containing the voltage sensor (McNair et al. 2011). The mechanisms underlying SCN5A mutation-related DCM remain unclear, and may involve a complex interplay of altered sodium channel current, (pre-existent) myocardial structural abnormalities, and occurrence of long-standing arrhythmias (Fig. 3). Recently, it was demonstrated that the SCN5A-R219H mutation causes a proton leak current, suggesting that intracellular acidification may contribute to the DCM (and arrhythmias) observed in carriers of this mutation (Gosselin-Badaroudine et al. 2012).
Figure 3.
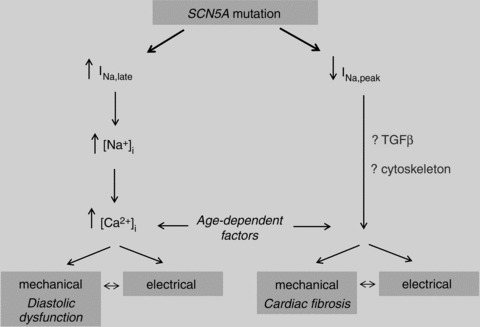
Potential mechanisms and processes involved in development of cardiac structural abnormalities secondary to SCN5A mutations.
Overlap syndromes of cardiac sodium channelopathy
In some cases, one single SCN5A mutation can result in multiple rhythm disturbances within one family. The first such mutation, the SCN5A-1795insD mutation, was characterized by our group in a large Dutch family presenting with extensive variability in type and severity of symptoms, including ECG features of sinus node dysfunction, bradycardia, conduction disease, Brugada syndrome (ST-segment elevation), and long QT syndrome (QT-interval prolongation), either in isolation or in combinations thereof (Bezzina et al. 1999; van den Berg et al. 2001). Therefore, an intriguing clinical and genetic overlap exists between these arrhythmia syndromes, previously considered separate entities. Similar findings for other mutations were subsequently reported for other SCN5A mutations by others (Kyndt et al. 2001; Grant et al. 2002; Rossenbacker et al. 2004). Today, extensive clinical and biophysical overlap among the various types of SCN5A mutations is known to exist, now referred to as ‘overlap syndromes’ of cardiac sodium channel disease (see Remme et al. 2008).
The simultaneous presence of long QT syndrome and Brugada syndrome or conduction disease in one patient was considered improbable prior to our description of the 1795insD family. Expression studies of the 1795insD mutation in Xenopus oocytes and the human embryonic kidney cell line HEK293 revealed opposing and inconclusive effects on sodium current density and kinetics (Bezzina et al. 1999; Veldkamp et al. 2000), necessitating the analysis of the biophysical properties of this mutation in native myocytes. Therefore, we generated transgenic mice carrying the heterozygous Scn5a1798insD/+ mutation, equivalent to human SCN5A-1795insD (Remme et al. 2006). In Scn5a1798insD/+ myocytes, a drastic reduction in peak sodium current density was found, underlying the observed conduction slowing in mice with the mutation. A small but relevant persistent inward sodium current was measured in myocytes from Scn5a1798insD/+ mice, explaining the presence of increased action potential duration and QTc prolongation. Disruption of fast inactivation of the sodium current, as evidenced by a delayed time course of sodium current decay in Scn5a1798insD/+ myocytes, potentially underlies the observed persistent current. These findings confirmed that the presence of a single SCN5A mutation is indeed sufficient to cause an overlap syndrome of cardiac sodium channel disease.
Variable disease severity in sodium channelopathy: clinical and genetic factors
Disease variability in SCN5A mutation carriers
Similar to most primary electrical disorders and Mendelian disorders in general, patients harbouring mutations in SCN5A may display profound variability in the clinical expression of the disease (Probst et al. 2009). This variability may stem from the different severities of the biophysical defect associated with the various mutations. For instance, in a study conducted among patients with loss-of-function mutations in SCN5A, Meregalli and co-workers demonstrated that truncating mutations were associated with more severe conduction disease (Meregalli et al. 2009). However, variability in disease severity is also observed among family members with the same primary genetic defect. Here, while some family members display electrocardiographic signs of the disease, others may be unaffected in spite of having the same mutation, as was observed in the SCN5A-1795insD family (Bezzina et al. 1999; Fig. 4A). Moreover, among the electrocardiographically affected family members, some individuals present with arrhythmia while others remain symptom-free throughout life. Thus, identification of mutation carriers at risk for serious cardiac events is essential.
Figure 4.
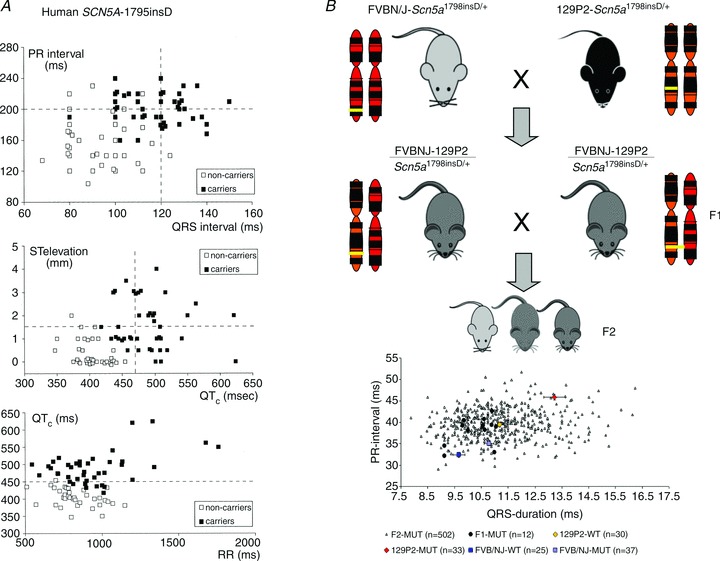
A, variable disease severity in SCN5A-1795insD mutation carriers as demonstrated by a large variation in ECG parameters (upper limit of normal values is indicated by dashed lines). B, spectrum of conduction disease severity among F2-MUT, F1-MUT, 129P2-WT, 129P2-MUT, FVB/NJ-WT and FVB/NJ-MUT mice. WT, wild-type; MUT, Scn5a1798insD/+. Reproduced in part from Bezzina et al. 1999 and Scicluna et al. 2011, with permission.
Modifiers of disease expression: age and sex
Kyndt and colleagues suggested that the patient's sex is a possible modifier of disease expression in a family carrying the SCN5A-G1406R mutation. In this family females displayed predominantly with conduction defects whereas males presented mostly with clinical features pertaining to the Brugada syndrome (Kyndt et al. 2001). The preponderance of Brugada syndrome in males also suggested sex as a modifier in sodium channelopathy. The contribution of age in modulation of the phenotype in the setting of sodium channelopathy is most elegantly observed in patients with progressive cardiac conduction disease (Probst et al. 2003), where age-related fibrosis is thought to underlie increased cardiac conduction delay with advancing age. This hypothesis is supported by experimental findings in Scn5a+/− mice that are haploinsufficient for the cardiac sodium channel (Royer et al. 2005). Interestingly, fibrotic changes were most pronounced in aged male Scn5a+/− mice, in line with the male predominance observed in Brugada syndrome patients (Jeevaratnam et al. 2012).
Genetic modifiers
While evidence points to genetic background effects on the severity of ECG manifestations and arrhythmia risk, genetic modifiers of clinical variability in sodium channelopathy remain largely unknown (Kolder et al. 2012). We have previously investigated the effects of genetic factors on disease severity by comparing the phenotype in two distinct strains of mice carrying the Scn5a1798insD/+ mutation equivalent to SCN5A-1795insD in humans. We found that phenotype severity was more pronounced in 129P2 mice as compared to FVBN/J mice, providing the first conclusive evidence that genetic modifiers determine disease expressivity in cardiac sodium channelopathy (Remme et al. 2009a). By comparing cardiac gene expression between these two strains of mice we subsequently identified Scn4b, encoding a sodium channel β-subunit, as a modifier of conduction disease severity in these mice (Remme et al. 2009a). More recently, in a systems genetics approach on F2 progeny arising from the 129P2 and FVBN/J mouse strains carrying the Scn5a1798insD/+ mutation, we have identified Tnni3k encoding troponin 1-interacting kinase as a novel modulator of cardiac conduction (Scicluna et al. 2011, Lodder et al. 2012; Fig. 4B). We are currently correlating findings from these mouse studies to clinical and genetic data from carriers and non-carriers in the large family segregating the SCN5A-1795insD mutation.
Although a few studies have investigated the effects of certain candidate genetic variants in the form of single nucleotide polymorphisms (SNPs) on disease severity in small families with cardiac sodium channelopathy (Groenewegen et al. 2003), systematic genome-wide searches for such genetic modifiers are as yet unavailable. SNPs recently identified in large genome-wide association studies conducted in the general population as modulators of heart rate and ECG indices of conduction and repolarization (reviewed in Kolder et al. 2012), constitute ideal candidates for modulatory effects.
Development of cardiac structural abnormalities secondary to SCN5A mutations
Cardiac sodium channelopathies were originally considered pure electrical entities occurring in the absence of structural heart disease, and the presence of myocardial abnormalities in these disorders was originally actually excluded by definition. However, evidence is now accumulating that sodium channelopathy can also be associated with the development of cardiac fibrosis, dilatation, and hypertrophy. A number of studies have now shown that sodium channel dysfunction may result in (progressive) structural abnormalities in the myocardium, both in patients with SCN5A mutations (Bezzina et al. 2003; Coronel et al. 2005; Frustaci et al. 2005) and in Scn5a mouse models (Royer et al. 2005; Zhang et al. 2011; Jeevaratnam et al. 2012). Interestingly, fibrotic changes were recently found to be most pronounced in the right ventricle of aged male haplo-insufficient Scn5a+/− mice, in line with the male predominance and right ventricular involvement observed in BrS patients (Jeevaratnam et al. 2012). The available clinical, molecular and biophysical information on this issue is still limited, and the mechanisms involved are as yet unknown. Although it is unclear how a mutation in a cardiac ion channel may ultimately lead to structural changes in the myocardial tissue, a number of mechanisms may be proposed.
Sodium channels and cardiac development/structure
Through its macromolecular complex, the sodium channel interacts with many cytoskeletal proteins and components of the extracellular matrix, and defects in some of these interacting proteins have been shown to negatively affect sodium channel function (see Abriel, 2010). It is currently unknown whether sodium channel dysfunction in turn can also destabilize cytoskeletal integrity. However, studies in embryos of mice carrying homozygous Scn5a mutations (which are embryonically lethal) indicate that sodium channels are essential for maintaining cardiac structure during development (Nuyens et al. 2001; Papadatos et al. 2002). Similarly, knockdown of Scn5a in zebrafish resulted in abnormal heart development, which appeared to be independent of electrophysiological effects (Chopra et al. 2010).
Potential role for late sodium current and/or CAMKII
Murine models of LQT3 have demonstrated that the increased late or persistent sodium current associated with gain-of-function Scn5a mutations can disrupt intracellular calcium homeostasis, thereby providing a pro-arrhythmic substrate and a potential mechanism for development of fibrosis and/or hypertrophy (Lindegger et al. 2009; Remme et al. 2010). Interestingly, the late sodium current blocker ranolazine was effective in reducing or preventing intracellular calcium overload in vitro (Lindegger et al. 2009). Thus, pharmacological late sodium current inhibition by compounds such as ranolazine may prove a useful and beneficial therapeutic approach in LQT3 patients, not only through stabilizing repolarization abnormalities, but also by preventing more long-term detrimental effects of intracellular calcium overload (see Remme & Wilde, 2013). Moreover, increased sodium influx with subsequent activation of CAMKII was observed in cardiomyocytes from mice overexpressing the human SCN5A-N1325S gain-of-function mutation (Yao et al. 2011), providing another potential therapeutic target.
Involvement of the TGFβ pathway
Electrical activity-dependent stimulation of pro-fibrotic factors of the transforming growth factor β (TGFβ) pathway may play a role in sodium channel dysfunction (Leask, 2007). In skeletal muscle, inhibition of sodium current was shown to increase levels of TGFβ1 receptor levels, indicating a potential regulatory effect of electrical activity on TGFβ signalling (Ugarte & Brandan, 2006). Indeed, TGFβ1 transcripts were upregulated in aged heterozygous Scn5a knockout mice, suggesting development of TGFβ1-mediated cardiac fibrosis secondary to sodium channel dysfunction (Hao et al. 2011).
Novel role for sodium channels in regulating myocardial structure and function?
Taken together, these observations are intriguing since they imply that sodium channels not only determine electrophysiological characteristics of the myocardium, but also exert as yet unknown regulatory effects on myocardial structure and function, implying a functional contribution of cardiac sodium channels to signal transduction. Different types of SCN5A mutations (gain- versus loss-of-function) may lead to structural abnormalities through various mechanisms (Fig. 3). Regardless of the underlying mechanism, cardiac structural abnormalities secondary to sodium channel dysfunction may further predispose to arrhythmogenesis, and will determine overall cardiac function, thereby contributing significantly to disease severity, progression and prognosis. Thus, novel treatment strategies aimed at reduction and/or prevention of structural abnormalities secondary to sodium channel dysfunction may prove more effective than existing therapy.
The way forward: incorporating established and new research strategies
Traditionally, heterologous expression systems have been used to study the biophysical properties of SCN5A mutations and correlating them to the clinical disease phenotype. While these studies were certainly informative, results obtained in such cell lines may not always be relevant to the cardiomyocyte environment. Findings from a number of transgenic mouse studies have indeed demonstrated the danger of over-interpretation of biophysical properties of a certain mutation based solely on studies in heterologous expression systems. As mentioned before, functional analysis of the SCN5A-1795insD mutation in Xenopus oocytes and HEK293 revealed opposing and inconclusive effects on sodium current density and kinetics (Bezzina et al. 1999; Veldkamp et al. 2000). Furthermore, the SCN5A-D1275N mutation displayed no significant functional differences in vitro, but was associated with DCM and arrhythmias in vivo in both humans and mice (Watanabe et al. 2011). While the generation of transgenic mouse models of each identified mutation would clearly be impossible, investigation of cardiomyocytes derived from human induced pluripotent stem cells (hiPSC) have so far proven useful for studying mutations in a more physiological environment (see Hoekstra et al. 2012). We and others have shown that hiPSC-derived cardiomyocytes from human SCN5A mutation carriers and Scn5a transgenic mice recapitulate the disease phenotype (Malan et al. 2011; Davis et al. 2012; Terrenoire et al. 2013), making them suitable candidates for investigation of patient- and disease-specific consequences for electrophysiology and pharmacology, in addition to providing a tool for studying the role of genetic background and modifiers. Despite these advances, in vivo, ex vivo and in vitro studies in transgenic mouse models will remain important for assessment of the differential effects of sodium channel defects in different regions and cell types of the heart, as well as investigation of disease progression and development of structural abnormalities with age.
Future directions
While significant progress has been made in understanding the genetic and biophysical aspects of cardiac sodium channelopathy, there are still many issues requiring further investigation. Identification of genetic modifiers of disease severity is essential to facilitate risk stratification in patients. iPSC technology may enable identification of patient- and mutation-specific treatment strategies. The search for mutations in novel genes in patients with inherited arrhythmia syndromes remains crucial, in addition to identification of genetic variants modulating cardiac conduction and/or repolarization. Combining such population studies with genetic, molecular and functional studies in animals will help identify and prioritize novel genes and pathways potentially relevant for cardiac electrophysiology (Scicluna et al. 2011; Kolder et al. 2012; Lodder et al. 2012). Furthermore, ongoing molecular studies are addressed at identifying novel components and functions of the sodium channel macromolecular complex, which may in turn provide new targets for genetic screening and/or future development of therapeutic strategies (Abriel, 2010; Shy et al. 2013). Ultimately, these combined clinical, genetic and translational studies should lead to improved diagnosis, risk stratification, treatment and outcome in patients with cardiac sodium channelopathy.
Glossary
- BrS
Brugada syndrome
- CAMKII
calcium/calmodulin-dependent protein kinase II
- DCM
dilated cardiomyopathy
- ICD
intercalated disc
- LQT3
long QT syndrome type 3
- LQTS
long QT syndrome
- PCCD
progressive cardiac conduction defect
- SIDS
sudden infant death syndrome
- SNP
single nucleotide polymorphism
Additional information
Competing interests
None.
Funding
This research was supported by the Netherlands Heart Institute (grant 061.02), and the Division for Earth and Life Sciences (ALW; project 836.09.003) with financial aid from the Netherlands Organization for Scientific Research (NWO).
References
- Abriel H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010;48:2–11. doi: 10.1016/j.yjmcc.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Allouis M, Le Bouffant F, Wilders R, Péroz D, Schott JJ, Noireaud J, Le Marec H, Mérot J, Escande D, Baró I. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ Res. 2006;98:1538–1546. doi: 10.1161/01.RES.0000229244.97497.2c. [DOI] [PubMed] [Google Scholar]
- Amin AS, Verkerk AO, Bhuiyan ZA, Wilde AA, Tan HL. Novel Brugada syndrome-causing mutation in ion-conducting pore of cardiac Na+ channel does not affect ion selectivity properties. Acta Physiol Scand. 2005;185:291–301. doi: 10.1111/j.1365-201X.2005.01496.x. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C. The Brugada syndrome: diagnostic criteria and cellular mechanisms. Eur Heart J. 2001;22:356–363. doi: 10.1053/euhj.2000.2461. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29:1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, Le Marec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2:429–440. doi: 10.1016/j.hrthm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J Clin Invest. 2012;122:2509–2518. doi: 10.1172/JCI62617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashamalla SM, Navarro D, Ward CA. Gradient of sodium current across the left ventricular wall of adult rat hearts. J Physiol. 2001;536:439–443. doi: 10.1111/j.1469-7793.2001.0439c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, Bers DM, Hudmon A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–19869. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR. The cardiac sodium channel: gating function and molecular pharmacology. J Mol Cell Cardiol. 2001;33:599–613. doi: 10.1006/jmcc.2000.1346. [DOI] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL., Jr Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A. J Clin Invest. 2003;112:1019–1028. doi: 10.1172/JCI18062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol. 2010;588:4969–4985. doi: 10.1113/jphysiol.2010.199034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzina CR, Remme CA. Dilated cardiomyopathy due to sodium channel dysfunction: what is the connection. Circ Arrhythm Electrophysiol. 2008;1:80–82. doi: 10.1161/CIRCEP.108.791434. [DOI] [PubMed] [Google Scholar]
- Bezzina CR, Rook MB, Groenewegen WA, Herfst LJ, van der Wal AC, Lam J, Jongsma HJ, Wilde AAM, Mannens MMAM. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ Res. 2003;92:159–168. doi: 10.1161/01.res.0000052672.97759.36. [DOI] [PubMed] [Google Scholar]
- Bezzina CR, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini S, Tan HL, Bhuiyan ZA, Bezzina CR, Barnett P, Cerbai E, Mugelli A, Wilde AA, Veldkamp MW. Characterization of a novel SCN5A mutation associated with Brugada syndrome reveals involvement of DIIIS4-S5 linker in slow inactivation. Cardiovasc Res. 2007;76:418–429. doi: 10.1016/j.cardiores.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Casini S, Tan HL, Demirayak I, Remme CA, Amin AS, Scicluna BP, Chatyan H, Ruijter JM, Bezzina CR, van Ginneken AC, Veldkamp MW. Tubulin polymerization modifies cardiac sodium channel expression and gating. Cardiovasc Res. 2010;85:691–700. doi: 10.1093/cvr/cvp352. [DOI] [PubMed] [Google Scholar]
- Casini S, Verkerk AO, van Borren MM, van Ginneken AC, Veldkamp MW, de Bakker JM, Tan HL. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc Res. 2009;81:72–81. doi: 10.1093/cvr/cvn274. [DOI] [PubMed] [Google Scholar]
- Chioni AM, Fraser SP, Pani F, Foran P, Wilkin GP, Diss JK, Djamgoz MB. A novel polyclonal antibody specific for the Nav1.5 voltage-gated Na+ channel ‘neonatal’ splice form. J Neurosci Methods. 2005;147:88–98. doi: 10.1016/j.jneumeth.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Chockalingam P, Rammeloo LA, Postema PG, Hruda J, Clur SA, Blom NA, Wilde AA. Fever-induced life-threatening arrhythmias in children harboring an SCN5A mutation. Pediatrics. 2011;127:e239–e244. doi: 10.1542/peds.2010-1688. [DOI] [PubMed] [Google Scholar]
- Chopra SS, Stroud DM, Watanabe H, Bennett JS, Burns CG, Wells KS, Yang T, Zhong TP, Roden DM. Voltage-gated sodium channels are required for heart development in zebrafish. Circ Res. 2010;106:1342–1350. doi: 10.1161/CIRCRESAHA.109.213132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy CE, Tateyama M, Liu H, Wehrens XH, Kass RS. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 2003;107:2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD. [DOI] [PubMed] [Google Scholar]
- Cordeiro JM, Mazza M, Goodrow R, Ulahannan N, Antzelevitch C, Di Diego JM. Functionally distinct sodium channels in ventricular epicardial and endocardial cells contribute to a greater sensitivity of the epicardium to electrical depression. Am J Physiol Heart Circ Physiol. 2008;295:H154–H162. doi: 10.1152/ajpheart.01327.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJG, Verkerk AO, de Groot JR, Bhuiyan Z, Bezzina CR, Veldkamp MW, Linnenbank AC, van der Wal AC, Tan HL, Brugada P, Wilde AA, de Bakker JM. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiologic, genetic, histopathologic and computational study. Circulation. 2005;112:2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614. [DOI] [PubMed] [Google Scholar]
- Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, George AL, Jr, Roden DM. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008;117:1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C, Salvatori D, Oostwaard DW, Wilde AA, Bezzina CR, Verkerk AO, Freund C, Mummery CL. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]
- Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Nam EG, Shea MA, Milan DJ, Ruskin JN, Macrae CA. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008;5:99–105. doi: 10.1016/j.hrthm.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frustaci A, Priori SG, Pieroni M, Chimenti C, Napolitano C, Rivolta I, Sanna T, Bellocci F, Russo MA. Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation. 2005;112:3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999. [DOI] [PubMed] [Google Scholar]
- Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- Ge J, Sun A, Paajanen V, Wang S, Su C, Yang Z, Li Y, Wang S, Jia J, Wang K, Zou Y, Gao L, Wang K, Fan Z. Molecular and clinical characterization of a novel SCN5A mutation associated with atrioventricular block and dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2008;1:83–92. doi: 10.1161/CIRCEP.107.750752. [DOI] [PubMed] [Google Scholar]
- George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, Brink M, Chahine M. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One. 2012;7:e38331. doi: 10.1371/journal.pone.0038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AO, Carboni MP, Neplioueva V, Starmer CF, Memmi M, Napolitano C, Priori S. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–1209. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewegen WA, Firouzi M, Bezzina CR, Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB, Jongsma HJ, Wilde AA. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003;92:14–22. doi: 10.1161/01.res.0000050585.07097.d7. [DOI] [PubMed] [Google Scholar]
- Hallaq H, Wang DW, Kunic JD, George AL, Jr, Wells KS, Murray KT. Activation of protein kinase C alters the intracellular distribution and mobility of cardiac Na+ channels. Am J Physiol Heart Circ Physiol. 2012;302:H782–H789. doi: 10.1152/ajpheart.00817.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X, Zhang Y, Zhang X, Nirmalan M, Davies L, Konstantinou D, Yin F, Dobrzynski H, Wang X, Grace A, Zhang H, Boyett M, Huang CL-H, Lei M. TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ Arrhythm Electrophysiol. 2011;4:397–406. doi: 10.1161/CIRCEP.110.960807. [DOI] [PubMed] [Google Scholar]
- Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol. 2012;3:346. doi: 10.3389/fphys.2012.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogendijk MG, Potse M, Linnenbank AC, Verkerk AO, den Ruijter HM, van Amersfoorth SC, Klaver EC, Beekman L, Bezzina CR, Postema PG, Tan HL, Reimer AG, van der Wal AC, Ten Harkel AD, Dalinghaus M, Vinet A, Wilde AA, de Bakker JM, Coronel R. Mechanism of right precordial ST-segment elevation in structural heart disease: excitation failure by current-to-load mismatch. Heart Rhythm. 2010;7:238–248. doi: 10.1016/j.hrthm.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in theβ3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270–278. doi: 10.1161/CIRCGENETICS.108.829192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Sato A, Marcou CA, Tester DJ, Ackerman MJ, Crotti L, Schwartz PJ, On YK, Park JE, Nakamura K, Hiraoka M, Nakazawa K, Sakurada H, Arimura T, Makita N, Kimura A. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ Arrhythm Electrophysiol. 2012;5:1098–1107. doi: 10.1161/CIRCEP.111.969972. [DOI] [PubMed] [Google Scholar]
- Jeevaratnam K, Rewbury R, Zhang Y, Guzadhur L, Grace AA, Lei M, Huang CL. Frequency distribution analysis of activation times and regional fibrosis in murine Scn5a+/− hearts: the effects of ageing and sex. Mech Ageing Dev. 2012;133:591–599. doi: 10.1016/j.mad.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespersen T, Gavillet B, van Bemmelen MX, Cordonier S, Thomas MA, Staub O, Abriel H. Cardiac sodium channel Nav1.5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem Biophys Res Commun. 2006;348:1455–1462. doi: 10.1016/j.bbrc.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Jones DK, Peters CH, Tolhurst SA, Claydon TW, Ruben PC. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys J. 2011;101:2147–2156. doi: 10.1016/j.bpj.2011.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AA, Brugada R, Schott JJ, Ackerman MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS. Sodium channel inactivation in heart: a novel role of the carboxyterminal domain. J Cardiovasc Electrophysiol. 2006;17(Suppl. 1):S21–S25. doi: 10.1111/j.1540-8167.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- Kattygnarath D, Maugenre S, Neyroud N, Balse E, Ichai C, Denjoy I, Dilanian G, Martins RP, Fressart V, Berthet M, Schott JJ, Leenhardt A, Probst V, Le Marec H, Hainque B, Coulombe A, Hatem SN, Guicheney P. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet. 2011;4:261–268. doi: 10.1161/CIRCGENETICS.110.959130. [DOI] [PubMed] [Google Scholar]
- Ko SH, Lenkowski PW, Lee HC, Mounsey JP, Patel MK. Modulation of Nav1.5 byβ1- and β3-subunit co-expression in mammalian cells. Pflugers Arch. 2005;449:403–412. doi: 10.1007/s00424-004-1348-4. [DOI] [PubMed] [Google Scholar]
- Kolder IC, Tanck MW, Bezzina CR. Common genetic variation modulating cardiac ECG parameters and susceptibility to sudden cardiac death. J Mol Cell Cardiol. 2012;52:620–629. doi: 10.1016/j.yjmcc.2011.12.014. [DOI] [PubMed] [Google Scholar]
- Kyndt F, Probst V, Potet F, Demolombe S, Chevallier JC, Baró I, Moisan JP, Boisseau P, Schott JJ, Escande D, Le Marec H. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation. 2001;104:3081–3086. doi: 10.1161/hc5001.100834. [DOI] [PubMed] [Google Scholar]
- Leask A. TGFβ, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74:207–212. doi: 10.1016/j.cardiores.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Lei M, Huang CL, Zhang Y. Genetic Na+ channelopathies and sinus node dysfunction. Prog Biophys Mol Biol. 2008;98:171–178. doi: 10.1016/j.pbiomolbio.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channelα subunits. J Biol Chem. 2003;278:27333–27339. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- Li GR, Lau CP, Shrier A. Heterogeneity of sodium current in atrial vs epicardial ventricular myocytes of adult guinea pig hearts. J Mol Cell Cardiol. 2002;34:1185–1194. doi: 10.1006/jmcc.2002.2053. [DOI] [PubMed] [Google Scholar]
- Li Q, Huang H, Liu G, Lam K, Rutberg J, Green MS, Birnie DH, Lemery R, Chahine M, Gollob MH. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem Biophys Res Commun. 2009;380:132–137. doi: 10.1016/j.bbrc.2009.01.052. [DOI] [PubMed] [Google Scholar]
- Li Z, Ai T, Samani K, Xi Y, Tzeng HP, Xie M, Wu S, Ge S, Taylor MD, Dong JW, Cheng J, Ackerman MJ, Kimura A, Sinagra G, Brunelli L, Faulkner G, Vatta M. A ZASP missense mutation, S196L, leads to cytoskeletal and electrical abnormalities in a mouse model of cardiomyopathy. Circ Arrhythm Electrophysiol. 2010;3:646–656. doi: 10.1161/CIRCEP.109.929240. [DOI] [PubMed] [Google Scholar]
- Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–1930. doi: 10.1016/j.hrthm.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindegger N, Hagen BM, Marks AR, Lederer WJ, Kass RS. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J Mol Cell Cardiol. 2009;47:326–334. doi: 10.1016/j.yjmcc.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu H, Dudley SC., Jr Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107:967–974. doi: 10.1161/CIRCRESAHA.110.220673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder EM, Scicluna BP, Milano A, Sun AY, Tang H, Remme CA, Moerland PD, Tanck MW, Pitt GS, Marchuk DA, Bezzina CR. Dissection of a quantitative trait locus for PR interval duration identifies Tnni3k as a novel modulator of cardiac conduction. PLoS Genet. 2012;8:e1003113. doi: 10.1371/journal.pgen.1003113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez KN, Decker JA, Friedman RA, Kim JJ. Homozygous mutation in SCN5A associated with atrial quiescence, recalcitrant arrhythmias, and poor capture thresholds. Heart Rhythm. 2011;8:471–473. doi: 10.1016/j.hrthm.2010.10.014. [DOI] [PubMed] [Google Scholar]
- McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, Mestroni L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–2167. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L. Familial Cardiomyopathy Registry Research Group. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J Am Coll Cardiol. 2011;57:2160–2168. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–828. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, Ohno S, Nishio Y, Tsuji K, Itoh H, Kimura T, Kita T, Horie M. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008;52:1326–1334. doi: 10.1016/j.jacc.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011;109:841–847. doi: 10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- Malhotra DJ, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS, Isom LL. Characterization of sodium channelα and β-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–1310. doi: 10.1161/01.cir.103.9.1303. [DOI] [PubMed] [Google Scholar]
- Mazzone A, Strege PR, Tester DJ, Bernard CE, Faulkner G, De Giorgio R, Makielski JC, Stanghellini V, Gibbons SJ, Ackerman MJ, Farrugia G. A mutation in telethonin alters Nav1.5 function. J Biol Chem. 2008;283:16537–16544. doi: 10.1074/jbc.M801744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows LS, Isom LL. Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res. 2005;67:448–458. doi: 10.1016/j.cardiores.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusié-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channelβ4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli PG, Tan HL, Probst V, Koopmann TT, Tanck MW, Bhuiyan ZA, Sacher F, Kyndt F, Schott JJ, Albuisson J, Mabo P, Bezzina CR, Le Marec H, Wilde AA. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm. 2009;6:341–348. doi: 10.1016/j.hrthm.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more. Cardiovasc Res. 2005;67:367–378. doi: 10.1016/j.cardiores.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Milstein ML, Musa H, Balbuena DP, Anumonwo JM, Auerbach DS, Furspan PB, Hou L, Hu B, Schumacher SM, Vaidyanathan R, Martens JR, Jalife J. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc Natl Acad Sci U S A. 2012;109:E2134–E2143. doi: 10.1073/pnas.1109370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohler PJ, Hund TJ. Membrane-select regulation of cardiac Nav channel isoforms. Heart Rhythm. 2011;8:1931–1932. doi: 10.1016/j.hrthm.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Mohler PJ, Rivolta I, Napolitano C, Lemaillet G, Lambert S, Priori SG, Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit ML, Stocker PJ, Schwetz TA, Harper JM, Norring SA, Schaffer L, North SJ, Jang-Lee J, Gilmartin T, Head SR, Haslam SM, Dell A, Marth JD, Bennett ES. Regulated and aberrant glycosylation modulate cardiac electrical signalling. Proc Natl Acad Sci U S A. 2009;106:16517–16522. doi: 10.1073/pnas.0905414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol. 2008;19:1289–1293. doi: 10.1111/j.1540-8167.2008.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike HK, Liu H, Glaaser IW, Yang AS, Tateyama M, Kass RS. The Na+ channel inactivation gate is a molecular complex: a novel role of the COOH-terminal domain. J Gen Physiol. 2004;123:155–165. doi: 10.1085/jgp.200308929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LL, Moon-Grady AJ, Cuneo BF, Wakai RT, Yu S, Kunic JD, Benson DW, George AL., Jr Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm. 2012;9:590–597. doi: 10.1016/j.hrthm.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KT, Hu N, Daw JR, Shin HG, Watson MT, Mashburn AB, George AL., Jr Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ Res. 1997;80:370–376. doi: 10.1161/01.res.80.3.370. [DOI] [PubMed] [Google Scholar]
- Nguyen TP, Wang DW, Rhodes TH, George AL., Jr Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res. 2008;102:364–371. doi: 10.1161/CIRCRESAHA.107.164673. [DOI] [PubMed] [Google Scholar]
- Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- Olesen MS, Holst AG, Svendsen JH, Haunso S, Tfelt-Hansen J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm. 2012;9:770–773. doi: 10.1016/j.hrthm.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Olesen MS, Jespersen T, Nielsen JB, Liang B, Møller DV, Hedley P, Christiansen M, Varró A, Olesen SP, Haunsø S, Schmitt N, Svendsen JH. Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res. 2011;89:786–793. doi: 10.1093/cvr/cvq348. [DOI] [PubMed] [Google Scholar]
- Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99:6210–6215. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- Probst V, Kyndt F, Potet F, Trochu JN, Mialet G, Demolombe S, Schott JJ, Baró I, Escande D, Le Marec H. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenègre disease. J Am Coll Cardiol. 2003;41:643–652. doi: 10.1016/s0735-1097(02)02864-4. [DOI] [PubMed] [Google Scholar]
- Probst V, Wilde AAM, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scouarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson J, Meregalli PG, Le Marec H, Tan HL, Schott JJ. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009;2:552–557. doi: 10.1161/CIRCGENETICS.109.853374. [DOI] [PubMed] [Google Scholar]
- Remme CA, Baartscheer A, Verkerk AO, van Rijen HV, Zeng D, Belardinelli L, Wilde AA, de Bakker JMT, Bezzina CR. Late sodium current block by ranolazine attenuates intracellular Na+ and Ca2+ dysregulation in myocytes from Scn5a-1798insD/+ mice. Heart Rhythm. 2010;7:S160. [Google Scholar]
- Remme CA, Bezzina CR. Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther. 2010;28:287–294. doi: 10.1111/j.1755-5922.2010.00210.x. [DOI] [PubMed] [Google Scholar]
- Remme CA, Scicluna BP, Verkerk AO, Amin AS, van Brunschot S, Beekman L, Deneer VH, Chevalier C, Oyama F, Miyazaki H, Nukina N, Wilders R, Escande D, Houlgatte R, Wilde AA, Tan HL, Veldkamp MW, de Bakker JM, Bezzina CR. Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ Res. 2009a;104:1283–1292. doi: 10.1161/CIRCRESAHA.109.194423. [DOI] [PubMed] [Google Scholar]
- Remme CA, Verkerk AO, Hoogaars WM, Aanhaanen WT, Scicluna BP, Annink C, van den Hoff MJ, Wilde AA, van Veen TA, Veldkamp MW, de Bakker JM, Christoffels VM, Bezzina CR. The cardiac sodium channel displays differential distribution in the conduction system and transmural heterogeneity in the murine ventricular myocardium. Basic Res Cardiol. 2009b;104:511–522. doi: 10.1007/s00395-009-0012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme CA, Verkerk AO, Nuyens D, van Ginneken AC, van Brunschot S, Belterman CN, Wilders R, van Roon MA, Tan HL, Wilde AA, Carmeliet P, de Bakker JM, Veldkamp MW, Bezzina CR. Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation. 2006;114:2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949. [DOI] [PubMed] [Google Scholar]
- Remme CA, Wilde AA. Late sodium current inhibition in acquired and inherited ventricular (dys)function and arrhythmias. Cardiovasc Drugs Ther. 2013;27:91–101. doi: 10.1007/s10557-012-6433-x. [DOI] [PubMed] [Google Scholar]
- Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Rivolta I, Abriel H, Tateyama M, Liu H, Memmi M, Vardas P, Napolitano C, Priori SG, Kass RS. Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J Biol Chem. 2001;276:30623–30630. doi: 10.1074/jbc.M104471200. [DOI] [PubMed] [Google Scholar]
- Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, Basso C, Remme CA, Thiene G, Bezzina CR. Intercalated disc abnormalities, reduced Na+ current density and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–418. doi: 10.1093/cvr/cvs219. [DOI] [PubMed] [Google Scholar]
- Rook MB, Evers MM, Vos MA, Bierhuizen MF. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc Res. 2012;93:12–23. doi: 10.1093/cvr/cvr252. [DOI] [PubMed] [Google Scholar]
- Rossenbacker T, Carroll SJ, Liu H, Kuipéri C, de Ravel TJ, Devriendt K, Carmeliet P, Kass RS, Heidbüchel H. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm. 2004;1:610–615. doi: 10.1016/j.hrthm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Rowinsky EK, McGuire WP, Guarnieri T, Fisherman JS, Christian MC, Donehower RC. Cardiac disturbances during the administration of taxol. J Clin Oncol. 1991;9:1704–1712. doi: 10.1200/JCO.1991.9.9.1704. [DOI] [PubMed] [Google Scholar]
- Royer A, van Veen TA, Le Bouter S, Marionneau C, Griol-Charhbili V, Léoni AL, Steenman M, van Rijen HV, Demolombe S, Goddard CA, Richer C, Escoubet B, Jarry-Guichard T, Colledge WH, Gros D, de Bakker JM, Grace AA, Escande D, Charpentier F. Mouse model of SCN5A-linked hereditary Lenègre's disease: age-related conduction slowing and myocardial fibrosis. Circulation. 2005;111:1738–1746. doi: 10.1161/01.CIR.0000160853.19867.61. [DOI] [PubMed] [Google Scholar]
- Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM, Le Marec H. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet. 1999;23:20–21. doi: 10.1038/12618. [DOI] [PubMed] [Google Scholar]
- Schroder EA, Lefta M, Zhang X, Bartos DC, Feng HZ, Zhao Y, Patwardhan A, Jin JP, Esser KA, Delisle BP. The cardiomyocyte molecular clock, regulation of Scn5a and arrhythmia susceptibility. Am J Physiol Cell Physiol. 2013:304. doi: 10.1152/ajpcell.00383.2012. C954–C965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter A, Walzik S, Blechschmidt S, Haufe V, Benndorf K, Zimmer T. Structure and function of splice variants of the cardiac voltage-gated sodium channel Nav1.5. J Mol Cell Cardiol. 2010;49:16–24. doi: 10.1016/j.yjmcc.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47. doi: 10.1111/j.1365-2796.2005.01583.x. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- Scicluna BP, Tanck MW, Remme CA, Beekman L, Coronel R, Wilde AA, Bezzina CR. Quantitative trait loci for electrocardiographic parameters and arrhythmia in the mouse. J Mol Cell Cardiol. 2011;50:380–389. doi: 10.1016/j.yjmcc.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM, Dudley SC., Jr Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–1154. doi: 10.1161/CIRCRESAHA.107.152918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: The multiple pool model. Biochim Biophys Acta. 2013;1833:886–894. doi: 10.1016/j.bbamcr.2012.10.026. [DOI] [PubMed] [Google Scholar]
- Smits JPP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan ZA, Mannens MM, Balser JR, Tan HL, Bezzina CR, Wilde AA. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J Mol Cell Cardiol. 2005;38:969–981. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Takehara N, Makita N, Kawabe J, Sato N, Kawamura Y, Kitabatake A, Kikuchi K. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J Intern Med. 2004;255:137–142. doi: 10.1046/j.0954-6820.2003.01247.x. [DOI] [PubMed] [Google Scholar]
- Tan BH, Valdivia CR, Song C, Makielski JC. Partial expression defect for the SCN5A missense mutation G1406R depends on splice variant background Q1077 and rescue by mexiletine. Am J Physiol Heart Circ Physiol. 2006;291:H1822–H1828. doi: 10.1152/ajpheart.00101.2006. [DOI] [PubMed] [Google Scholar]
- Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003;57:961–973. doi: 10.1016/s0008-6363(02)00714-9. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, Jean JC, Omari A, Sampson KJ, Kotton DN, Keller G, Kass RS. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukkie R, Sogaard P, Vleugels J, de Groot I, Wilde AA, Tan HL. Delay in right ventricular activation contributes to Brugada syndrome. Circulation. 2004;109:1272–1277. doi: 10.1161/01.CIR.0000118467.53182.D1. [DOI] [PubMed] [Google Scholar]
- Ugarte G, Brandan E. Transforming growth factor β (TGF-β) signalling is regulated by electrical activity in skeletal muscle cells. J Biol Chem. 2006;281:18473–18481. doi: 10.1074/jbc.M600918200. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Medeiros-Domingo A, Ye B, Shen WK, Algiers TJ, Ackerman MJ, Makielski JC. Loss-of-function mutation of the SCN3B-encoded sodium channelβ3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc Res. 2010;86:392–400. doi: 10.1093/cvr/cvp417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bemmelen MX, Rougier JS, Gavillet B, Apothéloz F, Daidié D, Tateyama M, Rivolta I, Thomas MA, Kass RS, Staub O, Abriel H. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res. 2004;95:284–291. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- Van den Berg MP, Wilde AAM, Viersma JW, Brouwer J, Haaksma J, van der Hout AH, Stolte-Dijkstra I, Bezzina TCR, Van Langen IM, Beaufort-Krol GC, Cornel JH, 2nd, Crijns HJ. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:630–636. doi: 10.1046/j.1540-8167.2001.00630.x. [DOI] [PubMed] [Google Scholar]
- Van den Boogaard M, Wong LY, Tessadori F, Bakker ML, Dreizehnter LK, Wakker V, Bezzina CR, ‘t Hoen PA, Bakkers J, Barnett P, Christoffels VM. Genetic variation in T-box binding element functionally affects SCN5ASCN10A enhancer. J Clin Invest. 2012;122:2519–2530. doi: 10.1172/JCI62613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- Veldkamp MW, Viswanathan PC, Bezzina C, Baartscheer A, Wilde AAM, Balser JR. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ Res. 2000;86:e91–e97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- Veldkamp MW, Wilders R, Baartscheer A, Zegers JG, Bezzina CR, Wilde AA. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ Res. 2003;92:976–983. doi: 10.1161/01.RES.0000069689.09869.A8. [DOI] [PubMed] [Google Scholar]
- Verkerk AO, van Ginneken AC, van Veen TA, Tan HL. Effects of heart failure on brain-type Na+ channels in rabbit ventricular myocytes. Europace. 2007;9:571–577. doi: 10.1093/europace/eum121. [DOI] [PubMed] [Google Scholar]
- Viswanathan PC, Balser JR. Inherited sodium channelopathies: a continuum of channel dysfunction. Trends Cardiovasc Med. 2004;14:28–35. doi: 10.1016/j.tcm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006;116:3127–3128. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011;109:775–782. doi: 10.1161/CIRCRESAHA.111.247957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ, Roden DM. Mutations in sodium channelβ1 and β2 subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009;2:268–275. doi: 10.1161/CIRCEP.108.779181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Koopmann TT, Le Scouarnec S, Yang T, Ingram CR, Schott JJ, Demolombe S, Probst V, Anselme F, Escande D, Wiesfeld AC, Pfeufer A, Kääb S, Wichmann HE, Hasdemir C, Aizawa Y, Wilde AA, Roden DM, Bezzina CR. Sodium channelβ1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260–2268. doi: 10.1172/JCI33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, Kupershmidt S, Chopra N, Magnuson MA, Tanabe N, Knollmann BC, George AL, Jr, Roden DM. Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation. 2011;124:1001–1011. doi: 10.1161/CIRCULATIONAHA.110.987248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedekind H, Smits JP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, Borggrefe M, Brinkmann B, Warnecke I, Funke H, Bhuiyan ZA, Wilde AA, Breithardt G, Haverkamp W. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. 2001;104:1158–1164. doi: 10.1161/hc3501.095361. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf CM, Berul CI. Inherited conduction system abnormalities–one group of diseases, many genes. J Cardiovasc Electrophysiol. 2006;17:446–455. doi: 10.1111/j.1540-8167.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, Abbasi S, Purevjav E, Samani K, Ackerman MJ, Qi M, Moss AJ, Shimizu W, Towbin JA, Cheng J, Vatta M. α-1-syntrophin mutation and the long QT syndrome: a disease of sodium channel disruption. Circ Arrhythm Electrophysiol. 2008;1:193–201. doi: 10.1161/CIRCEP.108.769224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Fan P, Jiang Z, Viatchenko-Karpinski S, Wu Y, Kornyeyev D, Hirakawa R, Budas GR, Rajamani S, Shryock JC, Belardinelli L. Nav1.5-dependent persistent Na+ influx activates CaMKII in rat ventricular myocytes and N1325S mice. Am J Physiol Cell Physiol. 2011;301:C577–C586. doi: 10.1152/ajpcell.00125.2011. [DOI] [PubMed] [Google Scholar]
- Yarbrough TL, Lu T, Lee H-C, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- Yoo S, Dobrzynski H, Fedorov VV, Xu SZ, Yamanushi TT, Jones SA, Yamamoto M, Nikolski VP, Efimov IR, Boyett MR. Localization of Na+ channel isoforms at the atrioventricular junction and atrioventricular node in the rat. Circulation. 2006;114:1360–1371. doi: 10.1161/CIRCULATIONAHA.106.613182. [DOI] [PubMed] [Google Scholar]
- Zareba W, Sattari MN, Rosero S, Couderc JP, Moss AJ. Altered atrial, atrioventricular, and ventricular conduction in patients with the long QT syndrome caused by the ΔKPQ SCN5A sodium channel gene mutation. Am J Cardiol. 2001;88:1311–1314. doi: 10.1016/s0002-9149(01)02097-5. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hartmann HA, Satin J. Glycosylation influences voltage-dependent gating of cardiac and skeletal muscle sodium channels. J Membr Biol. 1999;171:195–207. doi: 10.1007/s002329900571. [DOI] [PubMed] [Google Scholar]
- Zhang T, Yong SL, Drinko JK, Popović ZB, Shryock JC, Belardinelli L, Wang QK. LQTS mutation N1325S in cardiac sodium channel gene SCN5A causes cardiomyocyte apoptosis, cardiac fibrosis and contractile dysfunction in mice. Int J Cardiol. 2011;147:239–245. doi: 10.1016/j.ijcard.2009.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M. Cell membrane expression of cardiac sodium channel Nav1.5 is modulated byα-actinin-2 interaction. Biochemistry. 2010;49:166–178. doi: 10.1021/bi901086v. [DOI] [PMC free article] [PubMed] [Google Scholar]