

Abstract
Alcoholic liver disease (ALD) remains a leading cause of death from liver disease in the United States. In studies from the Veterans Administration, patients with cirrhosis and superimposed alcoholic hepatitis had greater than 60% mortality over a 4-year period, with most of those deaths occurring in the first month. Thus, the prognosis for this disease is more ominous than for many common types of cancer (eg, breast, prostate, and colon). Moreover, ALD imposes a significant economic burden from lost wages, health care costs, and lost productivity. Unfortunately, there is still no Food and Drug Administration–approved or widely accepted drug therapy for any stage of ALD. Thus, a pressing need exists for a more detailed understanding of mechanisms of liver injury. This article reviews recent advances in mechanisms and therapy related to five major areas of direct relevance to ALD: oxidative stress; gut-liver axis and cytokine signaling; malnutrition; fibrin/clotting; and stellate cell activation/fibrosis. We also review why therapies related to these mechanisms have performed well in experimental animals and in vitro systems, but have not necessarily translated into effective therapy for humans with ALD.
Keywords: Ethanol, Oxidative stress, Coagulation, Stress kinases, Lipopolysaccharide, Inflammation, Nutrition, Fibrosis/cirrhosis, Alcoholic liver disease
Introduction
Despite extensive research, alcohol abuse remains one of the most common causes of acute and chronic liver disease in the United States and worldwide. In Western countries, up to 50% of cases of end-stage liver disease have alcohol as a major etiologic factor. Excessive alcohol consumption is the third leading preventable cause of death in the United States. Alcohol-related deaths, excluding accidents/homicides, accounted for 22,073 deaths in the United States in 2006, with 13,000 of those specifically attributed to alcoholic liver disease (ALD). Moreover, the mortality of this liver disease is more than that of many major forms of cancer (eg, breast, colon, and prostate). Importantly, there is no Food and Drug Administration–approved therapy for any stage of ALD. This article reviews recent advances in mechanisms and therapy related to five major areas of direct relevance to ALD: oxidative stress; gut-liver axis and cytokine signaling; malnutrition; fibrin/clotting; and stellate cell activation/fibrosis (Fig. 1).
Fig. 1.

Mechanisms for alcoholic liver disease (ALD) are multiple and often interactive
Mechanisms of Injury and Potential Therapies
Oxidative Stress
Although free radical formation was originally considered an ex vivo phenomenon, later studies supported the concept that oxidants are produced by the cell under normal conditions [1]. An imbalance upregulating prooxidants and/or downregulating antioxidants, potentially leading to damage, was coined “oxidative stress” by Sies [2]. The evidence that alcohol causes oxidative stress in the human liver is extensive. In 1966, DiLuzio was the first to characterize lipid peroxidation following chronic exposure to alcohol [3]. Because alcoholics may receive up to 50% of their total daily calories from ethanol, it is not surprising that this high degree of dietary substitution leads to nutritional deficiencies (see section on gut-liver axis and cytokine signaling, below) via this route and via malabsorption [4, 5]. The net effect is that alcoholics often have lower levels of key antioxidant molecules [6]. Coupled with the fact that prooxidant production is increased during alcohol consumption, there is an imbalance between prooxidants and antioxidants in alcoholics, leading to oxidative stress.
Studies in experimental ALD have established a clear link between oxidative stress and liver damage [7]. Indeed, numerous antioxidants have been shown to protect against ethanol in models of ALD [8–10]. Oxidative stress is mediated by an increase in reactive oxygen species/reactive nitrogen species (ROS/RNS) production, and a decrease in antioxidant defenses [11]. In addition to nutritional deficiencies (see above), alcohol consumption renders the cell susceptible to oxidative stress via multiple mechanisms. For example, hypoxia caused by alcohol exposure can impair antioxidant defenses [12, 13]. Free iron is mobilized by alcohol [14], which can also lead to an increase in transition-metal catalysis to potent oxidants (eg, the Fenton reaction). Another example is that alcohol exposure inhibits the 26S proteosome in hepatocytes [15], which is responsible for degrading proteins damaged by ROS/RNS. Thus, when this complex is inhibited, proteins damaged by ROS/RNS accumulate in the cell [16]. Finally, a mass of proteins and systems are involved in the “antioxidant network.” This family does not directly block prooxidants, but serves to maintain the catalytic activity of antioxidant proteins or small molecules. These reactions are energy-dependent and the biochemical stress caused by alcohol exposure can impair this line of antioxidant defenses.
Chemical modification or damage to biologic molecules is a major mechanism by which ROS/RNS may cause cellular injury. It is also known that prooxidants alter and/or amplify their signal by modifying signaling cascades within the cell. Many reviews have focused on the role of signaling cascades in damage from oxidative stress [17–20]. Oxidant-sensitive signaling cascades include small molecules (eg, intracellular Ca++ [21]), stress-activated protein kinases [22], transcription factors [23], and modulators of apoptosis signaling [24]. Ethanol has been shown to alter the signal of many of these pathways in vitro and/or in experimental ALD [25, 26]; however, whether or not these effects are mediated by oxidative stress is unclear.
As mentioned above, although the experimental evidence supporting a role for oxidative stress in the development of ALD is extensive, this work has yet to translate to a successful clinical therapy based on targeting oxidative stress. For example, Mezey et al. [27•] tested the effect of vitamin E (1000 IU) administration on clinical and laboratory parameters of liver function and on markers of fibrogenesis in patients with mild to moderate alcoholic hepatitis (AH) in a double-blind, placebo-controlled, randomized trial. The authors found that although vitamin E treatment improved serum hyaluronic acid, it had no beneficial effects in patients with mild to moderate AH. Even studies with “antioxidant cocktails” have not shown much success in ALD. For example, patients with a severe AH received an antioxidant cocktail for 6 months in a double-blind, placebo-controlled study [28••]. The authors found that antioxidant therapy, alone or in combination with corticosteroids, did not improve survival. The causes of the apparent discrepancy between the experimental models and the clinical trials are unclear. It may reflect a limitation of the relative doses of antioxidants that can be used in humans, or may suggest that the models currently used do not sufficiently recapitulate the human disease.
Gut-Liver Axis and Cytokine Signaling
Endotoxin or lipopolysaccharide (LPS) is derived from the cell wall of intestinal gram-negative bacteria. Increased endotoxin levels are observed in patients with ALD and in rodent models of ALD. Elevated endotoxin levels in ALD may originate from 1) gram-negative bacterial overgrowth in the intestine; 2) increased intestinal permeability; and 3) impaired hepatic clearance of endotoxin [29]. Endotoxin then stimulates the production of tumor necrosis factor (TNF) and other proinflammatory cytokines through Toll-like receptor (TLR-4) signaling, which plays a critical role in the development and progression of ALD. Other bacterial-derived toxins that may impact TLR signaling and proinflammatory cytokine production include peptidoglycan and flagellin [29]. Indeed, injected peptidoglycan increases liver injury/inflammation in alcohol-fed compared to control-fed mice, and ethanol feeding increases peptidoglycan levels [29, 30].
It is well understood that the gut flora and gut-derived toxins play a critical role in the development of liver disease and its complications [29, 31–37]. Indeed, more than a half century ago, it was shown that germ-free rodents or rodents treated with antibiotics to “sterilize the gut” were resistant to nutritional and toxin-induced liver injury. Elegant studies by Broitman et al. [33] showed that rats fed a choline-deficient diet developed cirrhosis, which could be prevented by oral neomycin. However, when endotoxin was added to the water supply, neomycin could no longer prevent the development of liver injury and fibrosis [33]. Subsequently, antibiotics, prebiotics, and probiotics have all been used to prevent experimental alcohol-induced liver injury [38–41]. Increased plasma/hepatic concentrations of proinflammatory cytokines (eg, TNF-α) were noted in rodent models of ALD, and mice given anti-TNF antibodies or mice lacking TNF-R1 were protected against the development of experimental ALD [42, 43]. Moreover, chronic alcohol feeding sensitizes to the hepatotoxicity induced by gut-derived endotoxin and TNF, and specific components of the TLR-4 pathway responsible for alcohol-related liver injury are being defined [44, 45•]. TLR-4 activation by endotoxin results in recruitment of the adaptor molecules MyD88 and Toll/interleukin-1 receptor (TIR) domain–containing adapter-inducing interferon-β (TRIF), which each activate separate downstream signaling cascades. Recent data suggest that the MyD88-independent pathway (TRIF) is more important in the development of ALD, whereas nonalcoholic steatohepatitis appears to signal through the MyD88-dependent pathway [45•].
Concomitant studies in patients with AH and/or cirrhosis showed increased gut permeability and endotoxemia. More than 20 years ago, we first reported that in patients with AH, basal and endotoxin-stimulated monocyte TNF production levels are increased. Subsequent studies showed that plasma and monocyte proinflammatory cytokines correlated with the clinical course of AH and survival [46, 47]. Unfortunately, recent human studies have not demonstrated therapeutic efficacy for biologics such as anti-TNF antibody/TNF-soluble receptors in AH [48, 49]. Thus, it appears that complete TNF blockade is not a viable therapeutic option in ALD, possibly because of the necessary role of a basal level of TNF in liver regeneration. Another strategy is inhibiting TNF/proinflammatory cytokine production/activity with agents such as pentoxifylline, a broad phosphodiesterase inhibitor. Compelling animal data support a role for phosphodiesterase modulation in ALD, and two human trials support a role for pentoxifylline in AH [50, 51••]. The most recent compared prednisolone versus pentoxifylline therapy in AH, with greater benefit seen in the pentoxifylline group [51••]. This therapy seems to be especially beneficial in patients with marginal renal function.
Malnutrition
The most detailed reports on malnutrition in ALD come from two large studies from the Veterans Health Administration (VA) Cooperative Studies Program in patients having AH [52–55]. The first of these studies (study #119) demonstrated that virtually every patient with AH had some degree of malnutrition [53]. Almost 50% of patients’ energy intake came from alcohol. Although calorie intake was frequently not inadequate, intake of protein and critical micronutrients was often deficient. Importantly, the severity of liver disease correlated with malnutrition. Similar data were generated in a follow-up VA study on AH (study #275) [56]. In both studies, patients were given a balanced 2500-kcal hospital diet (monitored carefully by a dietitian) and encouraged to consume the diet. In the second study, patients in the therapy arm of the protocol also received an enteral nutritional support product high in branched-chain amino acids as well as the anabolic steroid oxandrolone (80 mg/day). Voluntary oral food intake correlated in a stepwise fashion with 6-month mortality data. Thus, patients who voluntarily consumed more than 3,000 kcal/day had virtually no mortality, whereas those consuming less than 1,000 kcal/day had greater than 80% 6-month mortality [52].
Compelling data supporting the use of nutrition therapy come from a multicenter study by Cabré et al. [57], which randomly assigned patients with severe AH to receive either prednisone, 40 mg/day, or a liver-specific formula containing 2,000 calories/day through a feeding tube. This polymeric enteral solution was enriched in branched-chain amino acids, energy dense (1.3 kcal/mL), and low in fat and sodium. The 1-year mortality was significantly lower in the enteral nutrition group compared to the glucocorticoid group, mainly because of fewer infectious complications. This study supports the importance of enteral nutrition in severe AH. We regularly place a nasogastric feeding tube as soon as AH patients are admitted to the hospital, if they are not consuming an adequate diet orally.
Patients with cirrhosis exhibit early onset of gluconeogenesis after short-term fasting. This accelerated metabolic reaction to starvation may result in increased protein requirements and muscle depletion. Thus, an approach to outpatient nutrition support is also necessary. A recent randomized controlled trial tested the hypothesis that provision of a late-evening nutritional supplement over a 12-month period would improve body protein stores in patients with cirrhosis. Total body protein was measured by neutron activation analysis at baseline, 3, 6, and 12 months. Consumption of a nighttime snack by patients with cirrhosis resulted in body protein accrual equivalent to about 2 kg of lean tissue sustained over 12 months; this benefit was not observed with daytime snacks. Thus, nighttime snacks are valuable nutritional interventions in outpatient cirrhotics [58••].
Zinc
Zinc is an essential trace element that participates in cellular function through hundreds of zinc proteins, including zinc metalloenzymes and critical zinc transcription factors [59]. Zinc deficiency is a frequent complication in ALD, a finding that has been well recognized for more than 50 years [59, 60]. Manifestations of zinc deficiency that are relevant to ALD include skin lesions, anorexia, depressed wound healing, hypogonadism, altered immune function, impaired night vision, and depressed mental function with possible encephalopathy [59, 60]. Mechanisms for altered zinc metabolism in ALD include inadequate intake, reduced absorption, and increased losses. Stress/inflammation caused by a variety of factors, including LPS/TNF, also cause an internal redistribution of zinc, with loss of zinc from some tissues (deficiency) and targeting to other tissues or organs such as the liver (redistribution). Importantly, zinc deficiency was recently shown to be induced by oxidative stress, in which thiol oxidation of zinc-finger transcription factors causes zinc loss, leading to loss of DNA-binding activity [61]. Thus, multiple defects ranging from nutritional deficiency to oxidative stress may cause critical impairment of zinc function in ALD.
Zinc deficiency has been associated with multiple forms of experimental liver injury, including alcohol-induced hepatotoxicity, and it sensitizes to experimental LPS-induced hepatotoxicity [59, 62, 63]. Recent studies from Zhou et al. [61] provide major new insights into the molecular mechanisms of altered zinc metabolism in the development and progression of experimental ALD. In both acute and chronic alcohol-induced hepatotoxicity, alcohol intake and oxidative stress disrupt tight junctions in the intestine, which leads to translocation of bacterial products, such as endotoxin [64]. Endotoxin activates TLR-4 and TNF production, with subsequent oxidative stress and liver injury. Endotoxin and TNF also play a critical role in liver fibrosis. Disruption of tight-junction proteins occurs not only in the intestine, but also in the lung and likely at the blood-brain barrier, thus potentially predisposing to lung injury and hepatic encephalopathy [65]. Zinc treatment in experimental animals with ALD attenuated the increased gut permeability, endotoxemia, TNF production, oxidative stress, and liver injury, while improving activity of key zinc transcription factors [59, 61]. Thus, zinc supplementation targets most postulated mechanisms for the development of ALD.
It is well documented that zinc supplementation corrects the manifestations of zinc deficiency in human ALD (eg, skin lesions and impaired night vision). A recent human pilot trial also suggests that zinc may stabilize or cause regression of hepatic fibrosis [60, 66]. Polaprezinc, a synthetic zinc-containing compound with 34 mg of elemental zinc, was administered daily for 24 weeks to patients with chronic hepatitis or cirrhosis (etiology, hepatitis C virus or alcohol). Zinc-supplemented patients had a significant increase in their serum zinc levels and a significant decrease in type IV collagen and serum tissue inhibitor of metalloproteinases 1 (TIMP1) levels. Liver biopsies were not performed. Further studies are required to evaluate multiple outcome markers of zinc therapy in ALD, ranging from changes in gut permeability to alterations in fibrosis.
Fibrin/Clotting
Homeostasis of fibrin metabolism is critical for normal organ function (Fig. 2); too little activity can lead to edema and clotting dysfunction, and too much activity can lead to hypercoagulation and hemostasis [67•]. Fibrin metabolism is regulated not only by fibrin deposition via coagulation, but also by degradation of the existing matrix via fibrinolysis [68]. Thrombin, the main protease of the coagulation system, catalyzes the conversion of fibrinogen to fibrin, which then mediates clot formation via the accumulation of extracellular matrix (ECM). Plasminogen activator inhibitor 1 (PAI-1) regulates fibrinolysis by inhibiting the conversion of plasminogen to plasmin via blocking plasminogen activators (uPA and tPA [69]). Inhibition of fibrinolysis by PAI-1 can cause fibrin ECM to accumulate, even in the absence of enhanced fibrin deposition by the thrombin cascade.
Fig. 2.
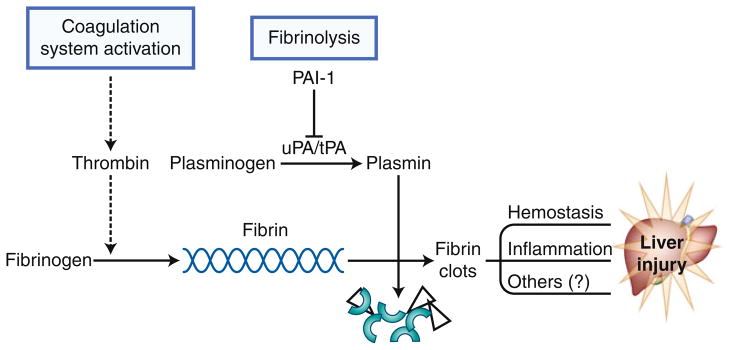
Fibrin metabolism. Activation of the coagulation cascade through thrombin initiates cross-linked fibrin deposition. Plasminogen activator inhibitor 1 (PAI-1) inhibits the activity of the plasminogen activators uPA and tPA by blocking the activation of plasmin. This process results in blunting fibrinolysis of fibrin matrices to fibrin degradation products (FDP)
Hepatic injury in models of liver disease often involves dysregulation of the coagulation cascade/fibrinolysis, leading to fibrin clots in the hepatic sinusoids [70, 71]. For example, ethanol enhances LPS-induced liver damage via mechanisms involving excess fibrin accumulation [71]. Exaggerated fibrin accumulation induced by ethanol was not associated with enhanced LPS-induced coagulation. Rather, the activity of PAI-1 was superinduced, indicating that impaired fibrinolysis was likely causal in the accumulation of fibrin ECM under these conditions. Blocking fibrin accumulation conferred protection against liver damage in this model [71].
It was shown in mice that acute ethanol exposure dramatically induced PAI-1 expression in the liver, and that steatosis under these conditions was prevented in PAI-1−/− mice or by pharmacologically inhibiting PAI-1 expression [72•]. Steatosis owing to chronic enteral alcohol exposure was also blunted by blocking PAI-1 induction, most likely via increasing hepatocyte growth factor (HGF) receptor (cMET) activation, leading to enhanced very low-density lipoprotein (VLDL) synthesis and export from the hepatocyte [72•]. Furthermore, PAI-1 has been shown to contribute to hepatic inflammation caused by alcohol [70, 71], as well as experimental hepatic fibrosis [73]. The latter effect is most likely via inhibiting ECM degradation during fibrogenesis [69]; the former effect is still being elucidated, but is likely mediated via fibrin matrix and/or fibrin signaling.
Fibrin ECM can mediate tissue damage via both direct and indirect means. Fibrin clots block the blood flow within the hepatic parenchyma (ie, hemostasis), causing microregional hypoxia and subsequent hepatocellular death [74, 75]. Hemostasis-induced hypoxia may also upregulate hypoxia-inducible factor-1α signaling in downstream areas. As mentioned above, fibrin(ogen) ECM not only serves as a physical structure, but it also binds/interacts with several biomolecules that can directly or indirectly alter responses. For example, integrins are a family of receptors for which fibrin(ogen) serves as a ligand. Integrins mediate attachment between a cell and the tissues surrounding it, which may be other cells or ECM. Integrin receptors transmit information from the ECM to the cell, allowing rapid and flexible responses to changes in the environment. Integrins play many roles within the body, including proliferation/angiogenesis, inflammation, and apoptosis [76, 77]. Fibrin(ogen) is a known ligand for several integrins, including integrin αIIbβ3, integrin αMβ2, and integrin αvβ3. These integrins are found on several non-parenchymal cells in the liver. Therefore, fibrin(ogen) ECM has the potential to alter intracellular signaling in liver via a variety of mechanisms.
Elevated PAI-1 levels and hypofibrinolysis are common during the development of ALD [78]. Indeed, PAI-1 levels during disease development are a predictor of later severity [79]. A recent human study further supports the hypothesis that PAI-1 plays a critical role in ALD [80]. Several PAI-1 inhibitors are being tested for the prevention of cardiovascular disease, and these drugs may have the potential to blunt ALD progression. However, few clinical studies have focused on the potential of inhibiting PAI-1 in the development of ALD. The multitude of cofactors influencing the coagulation/fibrinolysis system that are altered in cirrhotics make the routine assessment of bleeding or clotting risk in patients with ALD difficult. Routine diagnostic tests such as prothrombin time (PT) do not adequately reflect the hemostatic status in patients with liver disease, and bleeding cannot be predicted by PT [81]. Furthermore, because of the relatively uncommon occurrence of overt clinical thrombosis in patients with liver disease and the complexity of the coagulation cascade, the prevention and treatment of thrombosis or bleeding in patients with ALD is understudied. Clinical and basic science studies in the area of hypercoagulation are needed to help elucidate components of thrombophilia that might have a clinical impact on our understanding of coagulation in ALD. Therefore, although blocking coagulation may be protective during the development of ALD, it may actually be detrimental in patients with established endstage ALD (ie, cirrhotics). These points are discussed under Conclusions.
Stellate Cell Activation/Fibrosis
The final pathologic change in ALD is fibrosis/cirrhosis. Fibrosis is characterized by deposition of ECM. The main ECM to accumulate is collagen type I, but other ECM proteins also accumulate during fibrogenesis, such as laminin, fibronectin, and fibrin(ogen) [82]. The major cell type that contributes to fibrogenesis is the activated hepatic stellate cell; however, other cellular origins of myofibroblast-like cells are becoming increasingly recognized, such as peri-portal fibroblasts, fibrocytes, and transdifferentiated epithelia [83–86]. This stage has been referred to as end-stage liver disease, because it was assumed to be irreversible. However, more recent studies in animal models of fibrosis have demonstrated that even severe fibrotic changes may regress with time [87]. Furthermore, it was shown that in humans, HCV-induced liver fibrosis/cirrhosis can be reversed if the underlying infection is effectively treated [88••]. This exciting finding raises the prospect that advanced liver disease due to alcohol may also be reversible if the appropriate therapy can be identified. However, current clinical therapy for late-stage ALD focuses on the issues of decompensation and/or acute AH, with no universally accepted mechanism-based therapies to reverse the pathology. Nevertheless, candidate strategies have been identified in animal models that may show promise as emerging therapies, such as enhancing myofibroblast apoptosis [89], blocking transforming growth factor-β (TGF-β)/Sma- and Mad-related protein 3 (SMAD3) signaling [90], enhancing matrix resolution [91], and/or blocking inhibitors of matrix resolution [92].
A basic limitation in ALD research is that no rodent model completely recapitulates the human disease. Indeed, with rare exceptions [93], rodent alcohol models do not develop fibrotic changes. Thus, surrogate models of hepatic fibrosis (eg, bile duct ligation and carbon tetrachloride) are predominantly used [94]. Differences exist not only between the models and human fibrosis, but also between the models themselves [94]. To account for these limitations, multiple models are most appropriate to identify new mechanisms or therapeutic targets [95]. Furthermore, most models focus on the development of hepatic fibrosis, and few have studied the recovery from existing damage, although there are exceptions [87].
Conclusions
Major advances have been made in our understanding of the mechanisms for the development and progression of ALD. In experimental animals, one can block fatty liver and initial liver injury by blocking TNF/proinflammatory cytokine production, inhibiting oxidative stress, inhibiting PAI-1, or altering the clotting cascade, as outlined in this article. Moreover, many of these pathways are highly interactive. For example, major interactions occur between oxidative stress, proinflammatory cytokine production, and PAI-1 activity. Unfortunately, these seemingly straightforward mechanisms for ALD have not translated into effective therapy for human ALD.
Multiple potential reasons exist for this apparent lack of translation of therapeutic efficacy into humans. In most experimental models (both in vitro and in vivo), the experimental design focuses on preventing the development of liver disease rather than treating already-developed liver injury. In the clinical situation, patients present with AH or cirrhosis, and treatment instead of prevention is required.
Next, clinical trials have shown that small biologic molecules (eg, anti-TNF) appear to be ineffective in acute AH. Such therapy is highly effective for preventing experimental ALD, but may be less effective in treating established disease. A “baseline” low concentration of TNF appears to be required for liver regeneration, whereas excess TNF can be hepatotoxic. Thus, inhibiting all TNF activity with biologic therapy may not be an appropriate strategy for treatment of AH. Rather, drugs, such as pentoxifylline, that downregulate TNF production may be a more effective therapeutic approach.
Doses and formulations of agents may also play an important role, especially in antioxidant therapy. Agents such as vitamin E may impact terminal processes in oxidative stress and may not be potent enough to be effective. Potentially stimulating the body’s own endogenous antioxidant systems may be more beneficial than providing exogenous antioxidants. Moreover, we need to monitor therapeutic endpoints. Thus, if patients are receiving antioxidant therapy, is this therapy effective in decreasing biomarkers of oxidative stress? Such monitoring is not performed consistently.
Lastly, the correct patients need to be targeted in the study design. Pentoxifylline was recently shown not to improve survival in patients with Child’s C cirrhosis [96]. Pentoxifylline has both anti-inflammatory and anti-fibrotic properties and may be beneficial in less severe disease (Child’s A and B), whereas more severe disease may be too advanced for any medical therapy. Similarly, pentoxifylline was shown to be ineffective in rescuing patients with AH who did not respond to initial steroid therapy [97]. Again, these treatment-resistant patients may be too sick to respond to any drug therapy. On the other hand, combination therapy with pentoxifylline and prednisone may be more beneficial than treatment with either agent alone and warrants future investigation.
In summary, we have markedly expanded our understanding of all phases of ALD. This knowledge now needs to be translated into the development of effective therapy, and this will require close interactions among basic scientists, clinicians, and industry.
Acknowledgments
This work was supported by National Institutes of Health grants R01AA003624 (Arteel), R01AA015970 (McClain), R37AA010762 (McClain), R01DK071765 (McClain), P01AA017103 (McClain), R01AA018016 (McClain), RC2AA019385 (McClain), R01AA018869 (McClain), and the Veterans Administration (McClain).
Footnotes
Disclosure Conflicts of interest: J. Beier—none; G. Arteel—none; C. McClain—fees and honoraria from Vertex, Ocera, Gilead, Baxter, and Nestle, grants or contracts with Roche, Merck, Axcan, and Gilead.
Contributor Information
Juliane I. Beier, Department of Pharmacology and Toxicology, University of Louisville Health Sciences Center, Louisville, KY 40292, USA. University of Louisville Alcohol Research Center, University of Louisville Health Sciences Center, Louisville, KY 40292, USA
Gavin E. Arteel, Department of Pharmacology and Toxicology, University of Louisville Health Sciences Center, Louisville, KY 40292, USA. University of Louisville Alcohol Research Center, University of Louisville Health Sciences Center, Louisville, KY 40292, USA
Craig J. McClain, Email: craig.mcclain@louisville.edu, Department of Pharmacology and Toxicology, University of Louisville Health Sciences Center, Louisville, KY 40292, USA. University of Louisville Alcohol Research Center, University of Louisville Health Sciences Center, Louisville, KY 40292, USA. Department of Medicine, University of Louisville Health Sciences Center, Clinical and Translational Research Building, Room 503, 505 South Hancock Street, Louisville, KY 40292, USA. Robley Rex Veterans Administration Medical Center, Louisville, KY 40206, USA
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.McCord JM, Fridovich I. Superoxide dismutase: an enzymatic function of erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 2.Sies H. Oxidative stress: introductory remarks. In: Sies H, editor. Oxidative Stress. London: Academic Press; 1985. pp. 1–8. [Google Scholar]
- 3.Di Luzio NR. A mechanism of the acute ethanol-induced fatty liver and the modification of liver injury by antioxidants. Lab Invest. 1966;15:50–63. [PubMed] [Google Scholar]
- 4.Patek AJ., Jr Alcohol, malnutrition, and alcoholic cirrhosis. Am J Clin Nutr. 1979;32(6):1304–1312. doi: 10.1093/ajcn/32.6.1304. [DOI] [PubMed] [Google Scholar]
- 5.Bujanda L. The effects of alcohol consumption upon the gastrointestinal tract. Am J Gastroenterol. 2000;95(12):3374–3382. doi: 10.1111/j.1572-0241.2000.03347.x. [DOI] [PubMed] [Google Scholar]
- 6.Lieber CS. Alcohol: its metabolism and interaction with nutrients. Annu Rev Nutr. 2000;20:395–430. doi: 10.1146/annurev.nutr.20.1.395. [DOI] [PubMed] [Google Scholar]
- 7.Arteel GE. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology. 2003;124(3):778–790. doi: 10.1053/gast.2003.50087. [DOI] [PubMed] [Google Scholar]
- 8.Kono H, Arteel GE, Rusyn I, et al. Ebselen prevents early alcohol-induced liver injury in rats. Free Radic Biol Med. 2001;30 (4):403–411. doi: 10.1016/s0891-5849(00)00490-1. [DOI] [PubMed] [Google Scholar]
- 9.Kono H, Rusyn I, Uesugi T, et al. Diphenyleneiodonium sulfate, an NADPH oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G1005–G1012. doi: 10.1152/ajpgi.2001.280.5.G1005. [DOI] [PubMed] [Google Scholar]
- 10.Kono H, Rusyn I, Connor H, et al. Allopurinol prevents early alcohol-induced liver injury in rats. J Pharmacol Exp Therapeut. 2000;293(1):296–303. [PubMed] [Google Scholar]
- 11.Sies H. Biochemistry of oxidative stress. Angew Chem Int Ed Engl. 1986;25:1058–1071. [Google Scholar]
- 12.Jones DP. The role of oxygen concentration in oxidative stress: hypoxic and hyperoxic models. In: Sies H, editor. Oxidative Stress. London: Academic Press; 1985. pp. 151–195. [Google Scholar]
- 13.Tribble DL, Jones DP. Oxygen dependence of oxidative stress: Rate of NADPH supply for maintaining the GSH pool during hypoxia. Biochem Pharmacol. 1990;39(4):729–736. doi: 10.1016/0006-2952(90)90152-b. [DOI] [PubMed] [Google Scholar]
- 14.Shaw S, Jayatilleke E, Lieber CS. Lipid peroxidation as a mechanism of alcoholic liver injury: role of iron mobilization and microsomal induction. Alcohol. 1988;5:135–140. doi: 10.1016/0741-8329(88)90010-9. [DOI] [PubMed] [Google Scholar]
- 15.Bardag-Gorce F, Yuan QX, Li J, et al. The effect of ethanol-induced cytochrome p4502E1 on the inhibition of proteasome activity by alcohol. Biochem Biophys Res Commun. 2000;279 (1):23–29. doi: 10.1006/bbrc.2000.3889. [DOI] [PubMed] [Google Scholar]
- 16.Donohue TM., Jr The ubiquitin-proteasome system and its role in ethanol-induced disorders. Addict Biol. 2002;7(1):15–28. doi: 10.1080/135562101200100562. [DOI] [PubMed] [Google Scholar]
- 17.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 18.Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22(4–5):189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 19.Kamata H, Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11(1):1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 20.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28(3):463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 21.Ermak G, Davies KJ. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. 2002;38(10):713–721. doi: 10.1016/s0161-5890(01)00108-0. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22(1–2):269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 23.D’Angio CT, Finkelstein JN. Oxygen regulation of gene expression: a study in opposites. Mol Genet Metab. 2000;71(1–2):371–380. doi: 10.1006/mgme.2000.3074. [DOI] [PubMed] [Google Scholar]
- 24.Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol. 2002;27(1):63–68. doi: 10.1016/s0741-8329(02)00215-x. [DOI] [PubMed] [Google Scholar]
- 25.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16(11):1321–1329. doi: 10.3748/wjg.v16.i11.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50(6):1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Mezey E, Potter JJ, Rennie-Tankersley L, et al. A randomized placebo controlled trial of vitamin E for alcoholic hepatitis. J Hepatol. 2004;40(1):40–46. doi: 10.1016/s0168-8278(03)00476-8. In this study, the authors showed that a relatively high dose of vitamin E (1000 U) had no protective effect in patients with alcoholic hepatitis. The effectiveness of vitamin E alone was brought into question from the results of this study. [DOI] [PubMed] [Google Scholar]
- 28••.Stewart S, Prince M, Bassendine M, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol. 2007;47(2):277–283. doi: 10.1016/j.jhep.2007.03.027. The authors of this study used a “cocktail” of antioxidants in patients with alcoholic hepatitis and did not observe any protective effect. [DOI] [PubMed] [Google Scholar]
- 29.Purohit V, Bode JC, Bode C, et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol. 2008;42(5):349–361. doi: 10.1016/j.alcohol.2008.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gustot T, Lemmers A, Moreno C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43(5):989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- 31.Luckey TD, Reyniers JA, Gyorgy P, et al. Germfree animals and liver necrosis. Ann N Y Acad Sci. 1954;57(6):932–935. doi: 10.1111/j.1749-6632.1954.tb36472.x. [DOI] [PubMed] [Google Scholar]
- 32.Rutenburg AM, Sonnenblick E, Koven I, et al. The role of intestinal bacteria in the development of dietary cirrhosis in rats. J Exp Med. 1957;106(1):1–14. doi: 10.1084/jem.106.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broitman SA, Gottlieb LS, Zamcheck N. Influence of Neomycin and Ingested Endotoxin in the Pathogenesis of Choline Deficiency Cirrhosis in the Adult Rat. J Exp Med. 1964;119:633–642. doi: 10.1084/jem.119.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nolan JP. The contribution of gut-derived endotoxins to liver injury. Yale J Biol Med. 1979;52(1):127–133. [PMC free article] [PubMed] [Google Scholar]
- 35.Gabuzda GJ. Hepatic coma: clinical considerations, pathogenesis, and management. Adv Intern Med. 1962;11:11–73. [PubMed] [Google Scholar]
- 36.Zieve L. Pathogenesis of hepatic coma. Arch Intern Med. 1966;118(3):211–223. [PubMed] [Google Scholar]
- 37.McClain CJ, Zieve L. Portal Systemic Encephalopathy: Recognition and Variations. In: Davidson CS, editor. Problems in Liver Diseases. New York: Stratton Intercontinental Medical Book Corp; 1979. pp. 162–172. [Google Scholar]
- 38.Adachi Y, Moore LE, Bradford BU, et al. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology. 1995;108(1):218–224. doi: 10.1016/0016-5085(95)90027-6. [DOI] [PubMed] [Google Scholar]
- 39.Keshavarzian A, Choudhary S, Holmes EW, et al. Preventing gut leakiness by oats supplementation ameliorates alcohol-induced liver damage in rats. J Pharmacol Exp Ther. 2001;299(2):442–448. [PubMed] [Google Scholar]
- 40.Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc Soc Exp Biol Med. 1994;205(3):243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- 41.McClain CJ, Song Z, Barve SS, et al. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287(3):G497–502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 42.Iimuro Y, Gallucci RM, Luster MI, et al. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26(6):1530–1537. doi: 10.1002/hep.510260621. [DOI] [PubMed] [Google Scholar]
- 43.Yin M, Wheeler MD, Kono H, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117(4):942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 44.Honchel R, Ray MB, Marsano L, et al. Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res. 1992;16(4):665–669. doi: 10.1111/j.1530-0277.1992.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 45•.Szabo G, Bala S. Alcoholic liver disease and the gut-liver axis. World J Gastroenterol. 2010;16(11):1321–1329. doi: 10.3748/wjg.v16.i11.1321. The authors review the interactions of gut flora, gut-derived toxins, and liver injury, including the role of tolls in liver injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9(3):349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 47.Khoruts A, Stahnke L, McClain CJ, et al. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13(2):267–276. [PubMed] [Google Scholar]
- 48.Naveau S, Chollet-Martin S, Dharancy S, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology. 2004;39(5):1390–1397. doi: 10.1002/hep.20206. [DOI] [PubMed] [Google Scholar]
- 49.Boetticher NC, Peine CJ, Kwo P, et al. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology. 2008;135 (6):1953–1960. doi: 10.1053/j.gastro.2008.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:1637–1648. doi: 10.1053/gast.2000.20189. [DOI] [PubMed] [Google Scholar]
- 51••.De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World J Gastroenterol. 2009 Apr 7;15(13):1613–9. doi: 10.3748/wjg.15.1613. This article describes a randomized study that showed improved survival and renoprotection in alcoholic hepatitis with pentoxifylline compared to prednisolone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mendenhall C, Roselle GA, Gartside P, et al. Relationship of protein calorie malnutrition to alcoholic liver disease: a reexamination of data from two Veterans Administration Cooperative Studies. Alcohol Clin Exp Res. 1995;19(3):635–641. doi: 10.1111/j.1530-0277.1995.tb01560.x. [DOI] [PubMed] [Google Scholar]
- 53.Mendenhall CL, Anderson S, Weesner RE, et al. Protein-calorie malnutrition associated with alcoholic hepatitis. Veterans Administration Cooperative Study Group on Alcoholic Hepatitis. Am J Med. 1984;76(2):211–222. doi: 10.1016/0002-9343(84)90776-9. [DOI] [PubMed] [Google Scholar]
- 54.Mendenhall CL, Moritz TE, Roselle GA, et al. Protein energy malnutrition in severe alcoholic hepatitis: diagnosis and response to treatment. The VA Cooperative Study Group #275. JPEN J Parenter Enteral Nutr. 1995;19(4):258–265. doi: 10.1177/0148607195019004258. [DOI] [PubMed] [Google Scholar]
- 55.Mendenhall CL, Tosch T, Weesner RE, et al. VA cooperative study on alcoholic hepatitis. II: Prognostic significance of protein-calorie malnutrition. Am J Clin Nutr. 1986;43(2):213–218. doi: 10.1093/ajcn/43.2.213. [DOI] [PubMed] [Google Scholar]
- 56.Mendenhall CL, Moritz TE, Roselle GA, et al. A study of oral nutritional support with oxandrolone in malnourished patients with alcoholic hepatitis: results of a Department of Veterans Affairs cooperative study. Hepatology. 1993;17(4):564–576. doi: 10.1002/hep.1840170407. [DOI] [PubMed] [Google Scholar]
- 57.Cabre E, Rodriguez-Iglesias P, Caballeria J, et al. Short- and long-term outcome of severe alcohol-induced hepatitis treated with steroids or enteral nutrition: a multicenter randomized trial. Hepatology. 2000;32(1):36–42. doi: 10.1053/jhep.2000.8627. [DOI] [PubMed] [Google Scholar]
- 58••.Plank LD, Gane EJ, Peng S, et al. Nocturnal nutritional supplementation improves total body protein status of patients with liver cirrhosis: a randomized 12-month trial. Hepatology. 2008;48(2):557–566. doi: 10.1002/hep.22367. This article documents the importance of nighttime snacks in maintaining muscle mass in cirrhotics. Patients with cirrhosis can transition to a “starvation” state overnight. [DOI] [PubMed] [Google Scholar]
- 59.Kang YJ, Zhou Z. Zinc prevention and treatment of alcoholic liver disease. Mol Aspects Med. 2005;26(4–5):391–404. doi: 10.1016/j.mam.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 60.McClain CJ, Antonow DR, Cohen DA, et al. Zinc metabolism in alcoholic liver disease. Alcohol Clin Exp Res. 1986;10(6):582–589. doi: 10.1111/j.1530-0277.1986.tb05149.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhou Z, Kang X, Jiang Y, et al. Preservation of hepatocyte nuclear factor-4alpha is associated with zinc protection against TNF-alpha hepatotoxicity in mice. Exp Biol Med (Maywood) 2007;232(5):622–628. [PubMed] [Google Scholar]
- 62.Shea-Budgell M, Dojka M, Nimmo M, et al. Marginal zinc deficiency increased the susceptibility to acute lipopolysaccharide-induced liver injury in rats. Exp Biol Med (Maywood) 2006;231 (5):553–558. doi: 10.1177/153537020623100509. [DOI] [PubMed] [Google Scholar]
- 63.Lambert JC, Zhou Z, Kang YJ. Suppression of Fas-mediated signaling pathway is involved in zinc inhibition of ethanol-induced liver apoptosis. Exp Biol Med (Maywood) 2003;228 (4):406–412. doi: 10.1177/153537020322800411. [DOI] [PubMed] [Google Scholar]
- 64.Zhong W, McClain CJ, Cave M, et al. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am J Physiol Gastrointest Liver Physiol. 2010;298(5):G625–633. doi: 10.1152/ajpgi.00350.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Joshi PC, Mehta A, Jabber WS, et al. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am J Respir Cell Mol Biol. 2009;41(2):207–216. doi: 10.1165/rcmb.2008-0209OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi M, Saito H, Higashimoto M, et al. Possible inhibitory effect of oral zinc supplementation on hepatic fibrosis through downregulation of TIMP-1: A pilot study. Hepatol Res. 2007;37 (6):405–409. doi: 10.1111/j.1872-034X.2007.00065.x. [DOI] [PubMed] [Google Scholar]
- 67•.Northup PG, Sundaram V, Fallon MB, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost. 2008;6 (1):2–9. doi: 10.1111/j.1538-7836.2007.02772.x. This review is a state-of-the-art proceeding from the first Coagulopathy in Liver Disease conference held in Charlottesville, VA, USA. All authors presented at the conference on a separate topic concerning hypercoagulation in liver disease. [DOI] [PubMed] [Google Scholar]
- 68.Kolev K, Machovich R. Molecular and cellular modulation of fibrinolysis. Thromb Haemost. 2003;89(4):610–621. [PubMed] [Google Scholar]
- 69.Arteel GE. New role of plasminogen activator inhibitor-1 in alcohol-induced liver injury. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S54–59. doi: 10.1111/j.1440-1746.2007.05285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beier JI, Guo L, von Montfort C, et al. New role of resistin in lipopolysaccharide-induced liver damage in mice. J Pharmacol Exp Ther. 2008;325(3):801–808. doi: 10.1124/jpet.108.136721.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beier JI, Luyendyk JP, Guo L, et al. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology. 2009;49(5):1545–1553. doi: 10.1002/hep.22847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72•.Bergheim I, Guo L, Davis MA, et al. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006;130 (7):2099–2112. doi: 10.1053/j.gastro.2006.03.020. This article is the first to show that steatosis due to acute or chronic enteral alcohol exposure was blunted by blocking PAI-1 induction, independent of AMPK activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bergheim I, Guo L, Davis MA, et al. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther. 2006;316(2):592–600. doi: 10.1124/jpet.105.095042. [DOI] [PubMed] [Google Scholar]
- 74.Ganey PE, Luyendyk JP, Maddox JF, et al. Adverse hepatic drug reactions: inflammatory episodes as consequence and contributor. Chem Biol Interact. 2004;150(1):35–51. doi: 10.1016/j.cbi.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 75.Pearson JM, Schultze AE, Schwartz KA, et al. The thrombin inhibitor, hirudin, attenuates lipopolysaccharide-induced liver injury in the rat. J Pharmacol Exp Ther. 1996;278(1):378–383. [PubMed] [Google Scholar]
- 76.Hodivala-Dilke KM, Reynolds AR, Reynolds LE. Integrins in angiogenesis: multitalented molecules in a balancing act. Cell Tissue Res. 2003;314(1):131–144. doi: 10.1007/s00441-003-0774-5. [DOI] [PubMed] [Google Scholar]
- 77.Zhou HF, Chan HW, Wickline SA, et al. Alphavbeta3-targeted nanotherapy suppresses inflammatory arthritis in mice. FASEB J. 2009;23(9):2978–2985. doi: 10.1096/fj.09-129874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dimova EY, Kietzmann T. Metabolic, hormonal and environmental regulation of plasminogen activator inhibitor-1 (PAI-1) expression: lessons from the liver. Thromb Haemost. 2008;100 (6):992–1006. [PubMed] [Google Scholar]
- 79.Tran-Thang C, Fasel-Felley J, Pralong G, et al. Plasminogen activators and plasminogen activator inhibitors in liver deficiencies caused by chronic alcoholism or infectious hepatitis. Thromb Haemost. 1989;62(2):651–653. [PubMed] [Google Scholar]
- 80.Seth D, Hogg PJ, Gorrell MD, et al. Direct effects of alcohol on hepatic fibrinolytic balance: implications for alcoholic liver disease. J Hepatol. 2008;48(4):614–627. doi: 10.1016/j.jhep.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 81.Lisman T, Caldwell SH, Burroughs AK, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol. 2010;53(2):362–371. doi: 10.1016/j.jhep.2010.01.042. [DOI] [PubMed] [Google Scholar]
- 82.Schuppan D, Ruehl M, Somasundaram R, et al. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21 (3):351–372. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 83.Cassiman D, Libbrecht L, Desmet V, et al. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36(2):200–209. doi: 10.1016/s0168-8278(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 84.Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282(32):23337–23347. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 85.Robertson H, Kirby JA, Yip WW, et al. Biliary epithelial-mesenchymal transition in posttransplantation recurrence of primary biliary cirrhosis. Hepatology. 2007;45(4):977–981. doi: 10.1002/hep.21624. [DOI] [PubMed] [Google Scholar]
- 86.Omenetti A, Porrello A, Jung Y, et al. Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118(10):3331–3342. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gieling RG, Burt AD, Mann DA. Fibrosis and cirrhosis reversibility - molecular mechanisms. Clin Liver Dis. 2008;12 (4):915–937. xi. doi: 10.1016/j.cld.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 88••.Poynard T, McHutchison J, Manns M, et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122 (5):1303–1313. doi: 10.1053/gast.2002.33023. In this seminal work, the authors showed that “end-stage” liver disease may revert in some patients, if the underlying cause is effectively treated. This work sparked great interest in identifying new therapies to accelerate reversion of fibrosis/cirrhosis in experimental models and in patients. [DOI] [PubMed] [Google Scholar]
- 89.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102(3):538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu X, Hu H, Yin JQ. Therapeutic strategies against TGF-beta signaling pathway in hepatic fibrosis. Liver Int. 2006;26(1):8–22. doi: 10.1111/j.1478-3231.2005.01192.x. [DOI] [PubMed] [Google Scholar]
- 91.Iimuro Y, Brenner DA. Matrix metalloproteinase gene delivery for liver fibrosis. Pharm Res. 2008;25(2):249–258. doi: 10.1007/s11095-007-9311-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21(3):373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 93.Tsukamoto H, Horne W, Kamimura S, et al. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96 (1):620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117(3):539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lotersztajn S, Julien B, Teixeira-Clerc F, et al. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol Toxicol. 2005;45:605–628. doi: 10.1146/annurev.pharmtox.45.120403.095906. [DOI] [PubMed] [Google Scholar]
- 96.Lebrec D, Thabut D, Oberti F, et al. Pentoxifylline does not decrease short-term mortality but does reduce complications in patients with advanced cirrhosis. Gastroenterology. 2010;138 (5):1755–1762. doi: 10.1053/j.gastro.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 97.Louvet A, Diaz E, Dharancy S, et al. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol. 2008;48(3):465–470. doi: 10.1016/j.jhep.2007.10.010. [DOI] [PubMed] [Google Scholar]