

Abstract
HIV infection is characterized by rapid and error-prone viral replication resulting in genetically diverse virus populations. The rate of accumulation of diversity and the mechanisms involved are under intense study to provide useful information to understand immune evasion and the development of drug resistance. To characterize the development of viral diversity after infection, we carried out an in-depth analysis of single genome sequences of HIV pro-pol to assess diversity and divergence and to estimate replicating population sizes in a group of treatment-naive HIV-infected individuals sampled at single (n = 22) or multiple, longitudinal (n = 11) time points. Analysis of single genome sequences revealed nonlinear accumulation of sequence diversity during the course of infection. Diversity accumulated in recently infected individuals at rates 30-fold higher than in patients with chronic infection. Accumulation of synonymous changes accounted for most of the diversity during chronic infection. Accumulation of diversity resulted in population shifts, but the rates of change were low relative to estimated replication cycle times, consistent with relatively large population sizes. Analysis of changes in allele frequencies revealed effective population sizes that are substantially higher than previous estimates of approximately 1,000 infectious particles/infected individual. Taken together, these observations indicate that HIV populations are large, diverse, and slow to change in chronic infection and that the emergence of new mutations, including drug resistance mutations, is governed by both selection forces and drift.
INTRODUCTION
Infection with human immunodeficiency virus type 1 (HIV) results in lifelong persistent infection. In most cases, HIV infection results from expansion of a single or limited number of viral variants (1–4), producing an initially uniform virus population. From the time of infection, HIV genetic diversity emerges as a function of mutation, drift, recombination, selection, and population size. Early in infection, genetic diversity increases in a linear fashion (5), at rates somewhat lower than that predicted by the rapid (generation time, 1 to 2 days) and error-prone replication program (with the unselected reverse transcription mutation rate of 3 × 10−5 to 5 × 10−5 mutations/base/replication cycle [6]). HIV variants emerge with mutations at a number of positions; distribution of genetic distances in these early populations largely approximates a Poisson distribution, suggesting that in general, sites undergo mutation at random. Emergence of variants with mutations at specific cytotoxic T-lymphocyte (CTL) sites is relatively frequent and suggests that although mutations may occur at random, individual variants emerge as escape mutations (1, 5). These data (1, 5) demonstrate a strong role for both mutation and selection in the formation of initial populations in infected individuals. After years of infection, substantial genetic diversity accumulates, and this highly diverse population can rapidly respond to selective pressures, facilitating immune escape and resistance to antiviral drugs. Understanding how new mutations emerge and become fixed in HIV populations is critical to designing effective strategies for the prevention and suppression of these sequelae (7–14).
Although much has been learned regarding establishing HIV infection in vivo, critical gaps in our understanding persist that limit our understanding of the dynamics of HIV populations and the emergence of drug resistance. In particular, the size of the replicating HIV population in vivo remains uncertain. Relatively small population sizes (<1,000 infected cells/replication cycle/infected individual) have been reported, implying that stochastic effects and genetic drift predominate, with the potential for rapid emergence of mutations and shifts in population structure. In contrast, we and others have suggested a relatively large replicating population in vivo, with considerable contribution of deterministic effects, including slow shifts in population structure (15, 16). Most prior studies of HIV diversity in infected patients focused on env. env data sets are characterized by high diversity and are rich in strongly selected immune response sites, but they do not offer potential to understand detailed emergence of antiretroviral drug resistance. In addition, high genetic diversity presents substantial challenges in obtaining data sets that are not biased by selective amplification; genetic diversity and an excess of insertions and deletions also render env data sets difficult to align with confidence, complicating detailed phylogenetic and population genetic analyses.
To investigate HIV population genetics parameters, including population size in regions relevant to antiviral drug resistance to the majority of antiretrovirals, we investigated a 1.3-kb amplicon in pro-pol, which includes positions where mutations conferring escape from the CTL response as well as resistance to commonly used treatment regimens are found (12, 17). This region has a degree of genetic diversity (0.8% to 2% average pairwise difference [18–20]) in chronically infected individuals that is suitable for detailed fine-structure analyses of HIV populations using phylogenetic (18–20) and population genetics (15, 19, 21–23) approaches. We investigated pro-pol diversity in 33 treatment-naive individuals by analyzing large collections of individual HIV sequences, in many cases at multiple time intervals after infection. pro-pol diversity varied almost 100-fold, from 0.02% in recently infected individuals to more than 2% in individuals infected for more than 15 years. This new data set permitted a detailed analysis of HIV genetic variation, from which robust measures of diversity, divergence, and population size were obtained. Total sequence diversity (including synonymous and nonsynonymous changes) was strongly correlated with duration of infection even during chronic infection. Increases in genetic diversity over time correlated with increases in synonymous but not nonsynonymous mutations and did not correlate with plasma HIV RNA level or CD4+ T cell count. Studies of 11 patients sampled over 1 to 14 years revealed that the genetic composition of HIV populations changed slowly; significant shifts in HIV populations occurred only after 100 to 1,000 viral generations. We estimated that the effective replicating virus population is at least 10-fold larger than previous measurements derived using analysis of env sequences. The size and diversity of the replicating populations suggest that both selection and drift are important mechanisms leading to the emergence of HIV variants in vivo.
MATERIALS AND METHODS
Patients.
All HIV-infected patients were enrolled in studies of HIV infection at the NIH Clinical Center; patients donated blood samples after giving written informed consent. Duration of infection was estimated for recently infected patients using time of onset of symptoms. All such patients were enrolled in natural history studies of recent HIV infection; all were at least 18 years old and had a recent (<8 weeks) history of an acute febrile illness, consistent with symptomatic HIV infection syndrome following exposure. They also had a history of a nonreactive HIV-1/2 enzyme-linked immunosorbent assay (ELISA) within a year prior to enrollment or were documented to have plasma HIV levels of >100,000 copies/ml of plasma with an evolving or negative HIV Western blot following exposure. Of the remaining patients, none had a recent history of seroconversion syndrome, and the date of the first positive Western blot was used to estimate the minimum duration of infection. HIV RNA levels were determined using bDNA Versant, version 3.0 (Bayer, Inc.) as previously described (24). CD4 cell subsets were determined by standard clinical immunophenotyping.
Ethics statement.
All participants in this study were enrolled in clinical protocols (00-I-0110, 97-I-0082, and 95-I-0072) approved by the NIAID Institutional Review Board (FWA00005897) administered at the NIH Clinical Center in Bethesda, MD. Individuals underwent an informed-consent process and provided written consent for participation.
SGS.
Plasma from patients was frozen within 5 h of phlebotomy. Specimens were subjected to single-genome sequencing (SGS) as described previously (25). An amplicon encompassing 297 nucleotides (nt) of protease and ca. 700 to 1,200 nt of reverse transcriptase (RT) was sequenced. As previously demonstrated (20, 25), pro-pol sequences obtained by SGS from each individual patient were highly correlated and were clearly distinguishable from other patient SGS data sets (data not shown).
Alignments and analyses.
Sequences were aligned with Clustal W using DNASTAR/Megalign (DNASTAR, Inc.; gap penalty, 2.00; gap length penalty, 2.00). Neighbor-joining (NJ) trees were constructed through Megalign and confirmed in gap-stripped NJ trees in PAUP using pNL4-3 as an outgroup. Nodes were tested for significance in PAUP using 1,000 bootstrap replicates; nodes with >75% bootstrap significance were identified. Measures of diversity (average pairwise distances [APD] expressed as a percent, using p distances to determine pairwise differences; p distance is defined as the number of nucleotide differences between two single-genome sequences/total nucleotide sequences) (26). In all of these studies, intrapatient p distance determinations were relatively small (<0.03); as described by Nei and Kumar (27) and Nei (28), in the setting of such low p distances, phylogenetic trees using uncorrected p distance provide greater accuracy than trees constructed using more complicated models because of substantial increases in the variance of more complicated models. As expected, therefore, calculating genetic diversity by p distance and Jukes-Cantor-corrected p distance yielded nearly identical results that were highly correlated throughout the range of APD (r2 = 0.9999).
We obtained an average of 22 (range, 9 to 51) sequences for each time point. To investigate the precision of genetic diversity by this method, we generated model populations with comparable genetic diversities and obtained random samples for genetic diversity determinations.
All polymorphisms (excluding indels) in individual patients were identified, and the positions of polymorphisms in each patient alignment were tabulated. Allele frequencies were analyzed with Microsoft Excel-based programs.
Replicating population sizes were compared for 11 study patients with longitudinal sampling available. Coalescent estimation of HIV replicating effective population size (Ne) was performed as previously described (29) using the formula ϴ = 2Neμ, where ϴ is the neutral mutation parameter that defines a neutral coalescence process; for these calculations, ϴ is estimated by the nucleotide diversity π, defined in reference 26 as the average number of nucleotide substitutions per site between two sequences, and μ is the neutral mutation rate per sequence per generation (using 3.4 × 10−5 as the per-site mutation rate).
Changes in allele frequency were also used to estimate Ne (30–32) using the following formula:
where n = number of alleles per locus, xi and yi represent the allele frequencies at the two time points, t is the number of generations between sampling (1 day/generation), and S1 and S2 are the sample sizes at time 1 and time 2, respectively. Ne was calculated for each site from each patient data set, and quartile summary statistics were generated.
The geographic subdivision test was carried out as described by Achaz et al. (21). The test statistic, P, is determined by comparing the genetic distance between populations sampled at different times with the distances obtained after repeated shuffling of the same two sets of sequences and determining how often the distance between the randomized sets exceeded the observed distance. The lower the value of P, the less the chance that two populations arose from the same population (panmixia), with P values of <1 × 10−9 indicating that population shift has occurred. Statistical tests for significance of correlation coefficient were performed. In analysis of temporally spaced samples, the Fisher exact test was used to determine whether differences in allele frequencies between time points were significant. To determine whether an allele was fixed or newly emerged, we studied all positions that were polymorphic at one time point and monomorphic at the other. To identify only those changes that were due to true fixation or emergence, we eliminated those positions in which sampling error could have been responsible for the absence of the minor allele. To eliminate sampling error, we determined the allele frequency at the time the allele was polymorphic and then calculated the Poisson probability that an allele frequency of zero (not finding polymorphism at that position) at the second time point. For example, if the allele frequency at time 1 is a, then the Poisson probability of finding an allele frequency at time 2 of 0 is calculated as p(0) = e−a. If p(0) < 0.05, then it was statistically unlikely that sampling error was responsible for the absence of polymorphism and we concluded that the polymorphism had arisen or had undergone fixation.
Nucleotide sequence accession numbers.
Sequences have been deposited in GenBank under accession numbers KF469371 to KF470757.
RESULTS
We used single-genome sequencing (SGS) (20, 25, 33) to study pro-pol evolution in 33 HIV-infected treatment-naive patients, all with unprotected sex as their risk category (Table 1). Study patients were predominantly male, with an average age of 35.1 years. Patients were infected from an estimated 9 days to more than 15 years prior to the first sample based on patient history and laboratory studies; 22 patients were infected for <1 year. All but one (patient 1) had a positive Western blot at the time of phlebotomy. All patients had CD4 lymphopenia with a median of 401 CD4 cells/μl blood, and viral RNA levels ranged from 3.1 to 6.1 log10 copies/ml of plasma. As described previously (20, 25), SGS produces a data set of individual sequences derived from single HIV genomes that is ideally suited to investigate genetic diversity because of its low error rate, undetectable assay-based recombination, and absence of founder effects due to resampling. We obtained an average of 22 (range, 9 to 51) sequences for each time point. To investigate the precision of these determinations, we constructed theoretical populations, which we sampled with multiple replicates of increasing sample sizes. As shown in Fig. 1, increasing sample sizes above 10 sequences yielded adequately precise measurements of genetic diversity (to within 1% of theoretical value, with a standard error of mean of 0.11). This level of sampling yields reproducible measurements of genetic diversity.
Table 1.
Patients in the study
| Patient no. | Sexa | Age (yr) | Duration of infection (days) | CD4 cells/μl | RNA (log10 copies/ml) | APD (%) |
|---|---|---|---|---|---|---|
| 1 | F | 43.1 | 9 | 495 | 5.67 | 0.08 |
| 2 | M | 35.8 | 20 | 628 | 4.66 | 0.03 |
| 3 | M | 33.2 | 21 | 205 | 4.60 | 0.52 |
| 4 | M | 29.8 | 22 | 790 | 5.16 | 0.02 |
| 5 | M | 39.5 | 39 | 298 | 5.37 | 0.07 |
| 6 | M | 30.8 | 42 | 494 | 3.89 | 0.29 |
| 7 | M | 33.3 | 57 | 311 | 5.31 | 0.21 |
| 8 | M | 51.1 | 62 | 579 | 4.86 | 0.24 |
| 9 | F | 38.2 | 64 | 6 | 5.00 | 0.68 |
| 10 | M | 29.7 | 71 | 546 | 4.69 | 0.35 |
| 11 | M | 46.5 | 101 | 18 | 5.46 | 1.17 |
| 12 | M | 37.9 | 104 | 226 | 5.31 | 0.09 |
| 13 | M | 29.3 | 123 | 617 | 3.99 | 0.23 |
| 14 | M | 43.5 | 131 | 424 | 4.22 | 0.72 |
| 15 | M | 43.5 | 147 | 11 | 5.52 | 1.10 |
| 16 | M | 23.9 | 154 | 315 | 4.47 | 1.67 |
| 17 | M | 28.5 | 154 | 440 | 4.34 | 0.39 |
| 18 | M | 30.4 | 183 | 222 | 5.82 | 1.05 |
| 19 | M | 35.8 | 249 | 890 | 4.78 | 0.56 |
| 20 | M | 26.5 | 263 | 548 | 4.18 | 0.80 |
| 21 | M | 51.1 | 336 | 672 | 4.30 | 1.20 |
| 22 | M | 19.5 | 337 | 327 | 4.94 | 0.37 |
| 23 | M | 29.9 | 592 | 334 | 3.63 | 0.48 |
| 24 | M | 26.2 | 691 | 401 | NAb | 1.32 |
| 25 | M | 48.3 | 798 | 207 | 4.02 | 1.39 |
| 26 | M | 32.4 | 804 | 678 | 4.04 | 1.01 |
| 27 | M | 40.5 | 1,195 | 177 | 4.71 | 1.24 |
| 28 | M | 33.1 | 1,540 | 297 | 3.12 | 0.92 |
| 29 | M | 28.5 | 2,536 | 407 | 4.44 | 0.82 |
| 30 | M | 35.5 | 3,457 | 653 | 3.45 | 1.84 |
| 31 | M | 41.9 | 3,907 | 506 | 4.32 | 1.21 |
| 32 | M | 51.5 | 5,396 | 30 | 5.34 | 1.79 |
| 33 | M | 40.1 | 5,521 | 211 | 6.20 | 2.03 |
F, female; M, male.
NA, not available.
Fig 1.
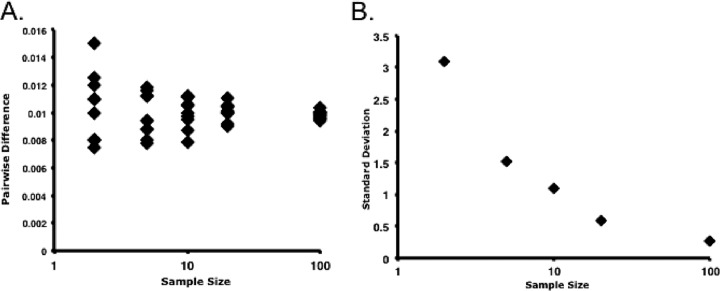
Determining precision of SGS. (A) Theoretical Poisson-distributed populations of 1,000 sequences with an average pairwise difference of 1% were generated. Seven replicate samples of increasing numbers of sequences from 2 to 100 sequences per sample were obtained and APDs determined. (B) Standard deviation of the APD determinations.
Among these study patients, pro-pol nucleotide diversity, as measured by percent average pairwise distance (APD), ranged nearly 100-fold from 0.02% in early (<1 year's duration) infection to slightly more than 2% after 15 years of infection (Table 1). Notably, all but one (patient 1) of the early infection patients had positive Western blots, demonstrating that a strong serologic response was already present. We first compared the minimum duration of infection with genetic diversity of each patient sample tested. Overall, there was a significant correlation between the minimum duration of infection and diversity, measured as APD (r2 = 0.47, P < 0.001), indicating a progressive increase in diversity with time. Detailed analyses revealed that the rate of accumulation of APD was not uniform, however. As shown in Fig. 2A, analysis of all samples from the 33 patients revealed that early in infection (patients 1 to 17), APD increased relatively rapidly, at an overall rate (0.006%/day; r2 = 0.45, P = 0.002) which approximated that expected from the mutation rate of reverse transcriptase (corresponding to an increase of 0.004%/day, assuming 1 replication cycle/day [6, 34, 35]) (Fig. 2B) and which was similar to previously published data (5). As Keele et al. (1) and others have reported, analysis of HIV genetic diversity in early infections revealed that pairwise differences were Poisson distributed, indicating that overall, mutations occurred randomly throughout the sequence. Consistent with earlier findings, we identified several individuals with recent HIV infection with HIV populations with higher genetic diversity than expected assuming a single infecting virus, indicating infection with more than one founder (Fig. 1A).
Fig 2.

Nonlinear accumulation of HIV diversity over time. (A) Overall diversity, expressed as percent average pairwise difference, was determined from alignments of pro-pol sequences obtained from all samples from patients 1 to 33 and are presented as a function of minimum duration of infection as defined in Materials and Methods. Patients for whom only a single sample was available for analysis are shown in black. (B) Accumulation of mutations in recently infected individuals (patients 1 to 17). (C) Accumulation of mutations in chronically infected individuals (patients 18 to 33). (D) Accumulation of mutations during 0.5 to 3 years' time. (E) Diversity measurements were obtained separately for synonymous and nonsynonymous sites from SGS data sets using DNASP and are presented as a function of time after infection. To avoid overweighting of patients with multiple samples, only the earliest time point for each patient was included for analyses in panels B to E.
In contrast to recent infections, when analyses were restricted to the patients infected for more than 1 year, APD increased 0.0002%/day (r2 = 0.49, P = 0.005) (Fig. 2C), indicating ongoing accumulation of new mutations, albeit at a rate about 30-fold lower than in early infection. During the period where accumulation of diversity slowed (1 to 2 years), we noted considerable range in diversity among patients (Fig. 1A and Fig. 2D), suggesting variable effects of selection and drift.
The period approximating 1 year of infection included samples with a relatively wide spectrum of genetic diversity. To investigate whether mutations were distributed randomly throughout pro-pol, we analyzed the distribution of pairwise differences. As previously described, random accumulation of mutations yields distributions according to Poisson statistics, while nonrandom mutation results in skewed pairwise differences. Analysis of the distribution of pairwise differences in HIV populations from chronically infected individuals revealed distributions with strong Poisson characteristics, but with deviations from ideal Poisson populations (F. Maldarelli, unpublished data). These data suggest that mutations continue to accumulate in random fashion during chronic infection, but specific changes may occur as well.
To further characterize the accumulation of new mutations, we compared changes in synonymous and nonsynonymous diversity over time. As shown in Fig. 2E, both nonsynonymous and synonymous diversity increased sharply during early months of infection; however, approximately 8 months later, nonsynonymous diversity stabilized and synonymous diversity continued to increase. These data indicate that PR and RT are undergoing change largely under purifying selection most likely as a result of constraints on protein structure.
Although we detected a significant correlation between genetic diversity and duration of infection, the correlation coefficients for recent and chronic infection (r2 = 0.45 and 0.49, respectively) indicated that duration of infection explained only a portion of the variability in genetic diversity. To look for other correlates, we compared virologic and immunologic measures. No correlation was found between diversity and plasma HIV RNA or CD4 count in individuals with established HIV infection (duration of infection of >3 months, r2 = 0.04 and 0.07, respectively) (Fig. 3A), indicating that overall HIV pro-pol genetic variation was not associated with the level of viremia or extent of immunodeficiency.
Fig 3.
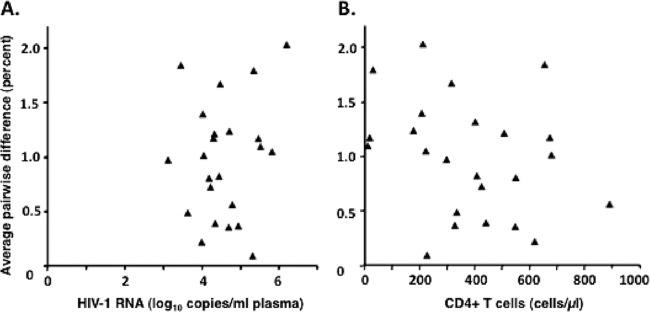
No correlation was found between HIV genetic diversity and level of viremia (A) or CD4 cell concentration (B). Overall diversity, expressed as percent average pairwise difference, was determined from alignments of pro-pol sequences obtained from all samples from patients 1 to 27 and is presented as a function of minimum duration of infection as defined in Materials and Methods. Only the earliest time point from each patient was included for analysis.
We further investigated the relationship between genetic diversity and time with longitudinal sampling of 12 patients with HIV infection and various baseline diversities. To determine the relative tempo of HIV variation, we compared sequences from samples obtained on daily, monthly, and yearly bases by phylogenetic analysis. As we and others have shown (1–5), HIV population structure was relatively monomorphic during early infection (Fig. 4, patient 2, panel D), which arose from the few mutations that appeared over the relatively short period of observation. Early in infection (<1 year), diversity increased approximately as predicted by the mutation rate (Fig. 4, patient 10), as previously noted (5). In contrast, during chronic HIV infection, diversity remained relatively stable (Fig. 4, patients 14, 24, 29, 30, and 31, panels B), even during progressive decline of CD4 cell counts (patients 24 and 30) and more than 10-fold increases in HIV RNA levels (patient 30). As expected from cross-sectional data (Fig. 2A), increases in diversity were nonetheless detectable in temporally spaced samples obtained from individual patients, although consistent rates of divergence among all patients were, in general, not discernible (data not shown). Analysis of daily samples from two patients revealed no variation in HIV diversity over a 10-day observation period (Fig. 4, patient 29, daily samples, panel B; data for the second patient are not shown), excluding rapid fluctuation due, for example, to differential seeding of the population from genetically distinct tissue sites of replication.
Fig 4.

HIV pro-pol diversity and population shifts in HIV-infected patients. Each patient enrolled in the study underwent phlebotomy at the study days indicated. (A) The levels of viremia (boxes) and CD4 lymphopenia (diamonds) were determined. Samples indicated by colored circles were subjected to SGS. (B) Sequences obtained by SGS at the indicated times were aligned, and APD was determined. (C) Sequences from the indicated time points were compared to the sequence set from the earliest time point in the patient data set, and the probability of panmixia was calculated (21). (D) Neighbor-joining trees of the entire data set were constructed from the alignments, with each sequence colored to correspond to the sample time in panel A. Trees were subjected to bootstrap analysis (1,000 replicates). The branches having bootstrap support values of >75% are highlighted in bold using the color of the sampling date. The outgroup in each case is pNL4-3; for ease of display, the distance to the outgroup for some phylogenetic trees is reduced as indicated.
Neighbor-joining analysis revealed that temporally spaced pro-pol sequences remained highly related to one another. Samples obtained on a daily basis (Fig. 4, patient 29) or over 5 years revealed that only a few (1 to 6) sequences or clusters of sequences from individual times had bootstrap values (>75%) sufficient to support the observed branching (Fig. 4, patients 10, 14, 29, 30, and 31, panels D, thick colored bars). Of the 12 patients with longitudinal sampling, one (Fig. 4, patient 30, panel D, blue branches) had evidence for divergence in a subset of 6 sequences after a sampling interval exceeding 5 years, and a second (patient 24) had evidence of emergence of a distinct lineage after nearly 14 years. In the remaining patients, phylogenetic topologies of temporally spaced samples suggested a shared common ancestry for HIV sequences; the most recent sequences did not demonstrate progressive accumulation of diversity compared to the earliest sequences.
Temporally spaced data were also useful in providing a detailed view of HIV polymorphisms and identify changes in individual allele frequencies over time. As shown in Table 2, for 10/11 patients, a relatively small number of alleles underwent change during the observation period (median, 9%; range, 0 to 18%). None of the alleles that emerged or underwent fixation were linked to alleles that underwent significant change in allele frequency, indicating that fixation did not result in a selective sweep that carried other alleles. Rather, the occurrence of unlinked polymorphisms emerging or undergoing fixation in this fashion indicates that populations are highly diverse, and certain lineages were simply lost or emerged as result of new mutation. Most sites did not undergo changes in allele frequency, suggesting that selection at these sites was not sufficiently strong to change the frequency. HIV from one patient (patient 24) underwent significant change during a prolonged observation period (5,099 days), with 43% of 90 polymorphisms undergoing significant change, 21 of which were new or lost alleles, a number of which were linked (Table 2). As shown in Fig. 4, the HIV population structure in this patient was distinct, with all of the sequences from the later time point on a distinct lineage with strong bootstrap support, accounting for the number of new changes. Patient 30 also had a new bootstrap-supported lineage emerge after a long period (5.7 years) but also had a number of variants present.
Table 2.
Polymorphism analysis
| Patient no. | HIV-1 RNA (copies/ml) | Duration of interval between sampling (days) | Polymorphisms with change in allele frequency (%)a | No. of polymorphisms that arose or underwent fixationb |
|---|---|---|---|---|
| 10 | 4.7 | 1,017 | 3.6 | 3 |
| 13 | 4.0 | 339 | 14 | 4 |
| 14 | 4.2 | 383 | 5.9 | 0 |
| 18 | 5.8 | 72 | 0 | 0 |
| 20 | 4.2 | 520 | 10 | 3 |
| 21 | 4.3 | 168 | 8 | 1 |
| 24 | NAc | 5,099 | 43.3 | 21 |
| 28 | 3.1 | 422 | 13 | 0 |
| 29 | 4.4 | 2,112 | 9 | 0 |
| 30 | 3.5 | 2,085 | 5.1 | 3 |
| 31 | 4.3 | 1,373 | 18 | 0 |
| Median | 4.3 | 520 | 9.0 | 1.0 |
Polymorphisms were identified and allele frequencies determined. Polymorphisms with a significant change in allele frequency (Fisher exact test, P < 0.05) were determined.
Determined as described in Materials and Methods.
NA, not available.
Recombination is a common phenomenon in HIV replication; as we previously reported, approximately 6% of infected cells are likely infected with more than one provirus (36), providing the opportunity for recombination to occur. In HIV-infected patients, recombinants accrue during the entire course of infection. As a result, demonstration of recombination using standard phylogenetic techniques (37) detected frequent evidence of recombination with recombination intervals of 36 to 120 nt (Maldarelli, unpublished).
Despite the absence of clear phylogenetic evidence of divergence and the relatively stable intrapatient viral diversity, substantial population shifts were detectable when we applied an adaptation of the geographic subdivision test (21) to identify patterns of population structure. Population shift is indicated by a loss of panmixia (a population characteristic in which all sequences in the sample comparison belong to a single replicating group); in comparing sequences from two different time points, a low (1 × 10−9) probability of panmixia indicates population divergence. In contrast to the relatively homogeneous populations indicated by the NJ analyses, the geographic subdivision test showed clear evidence of population shift in pro-pol sequences from patients with HIV infection sampled over prolonged periods (Fig. 4, patients 10, 14, 24, 29, 30, and 31, panels C), whereas at short intervals (Fig. 4, patient 2 or patient 29, daily samples), no evidence of population shift was detectable. Cumulative analysis of all intrapatient pairwise comparisons revealed that the median time to population shift (defined as a probability of panmixia of <10−9) was 1,017 days, and the minimum duration before shift was detected was 193 days (Fig. 5). These data are consistent with our initial report of the population subdivision adaptation (21) and indicate that significant change in HIV pro-pol population structure takes place with a time scale that is 100- to 1,000-fold longer than the replication cycle time of HIV in vivo (1 to 2 days).
Fig 5.
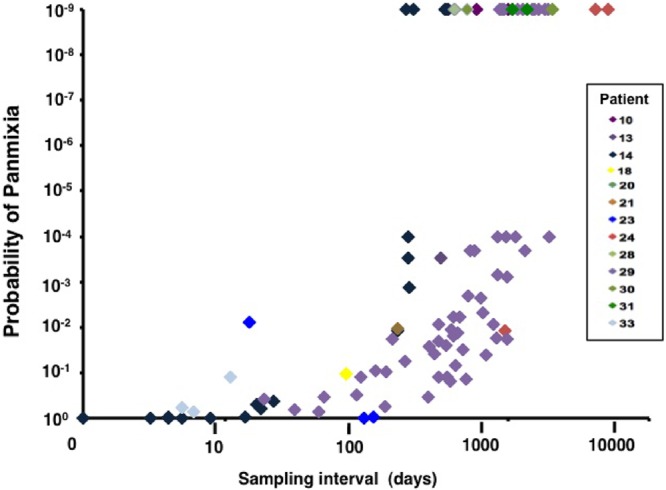
Shifts in HIV populations with time. Plasma HIV RNA sequences were obtained from individual time points. The population subdivision test was performed for all pairwise combinations of samples for each patient data set, and the probability of panmixia is reported here as a function of the time between the sample pairs. Data for a series of 8 patients and 101 pairwise comparisons are presented. The median time to achieve a low probability of panmixia (10−9) was 1,017 days.
The relatively low rate of population shift in HIV population structure implies relatively large replicating populations in vivo. Therefore, we used two tests to investigate further the effective size (Ne) of the HIV populations. As shown in Fig. 6, coalescent analyses (diamonds) yielded uniformly low measures of effective population size, on the order of 100 to 1,000, similar to estimates previously reported (33, 38–41), a surprising result in light of the slow change in population structure detected by the population subdivision analyses. This difference may be due to the fact that this method ignores the contributions of selection and recombination, both of which can lead to underestimation of population size (15, 42). Therefore, we next determined population size using a phylogeny-independent method described by Tajima and Nei (31) and Waples (32). This method estimates population size based on the rate of change of individual allele frequencies over time and thus yields a range of population sizes; assuming no selection, large changes in allele frequencies yield the smallest estimates of population size, and relatively small changes in allele frequency yield the largest population sizes. As shown in Fig. 6A (whisker plot), Ne estimates varied more than 10- to 100-fold among individual patients, reflecting a wide range of changes in allele frequency among HIV pro-pol polymorphisms. HIV populations from two individuals (patients 10 and 14) had relatively narrow quartile distributions of population sites, reflecting a restricted range of allele frequency changes.
Fig 6.
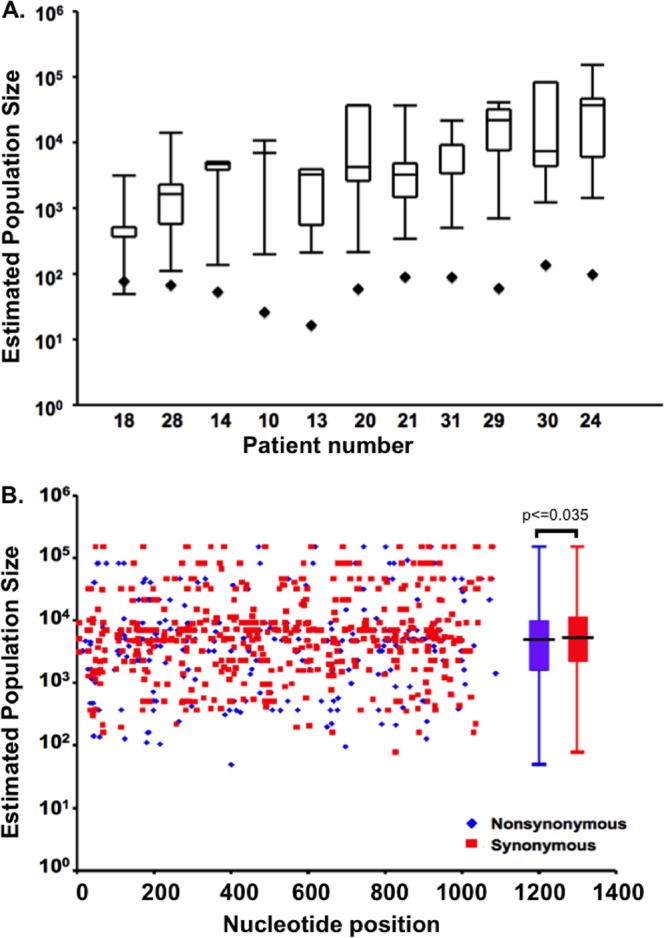
Estimates of HIV replicating effective population size (Ne) in vivo using two methods. (A) Ne was calculated for the virus population in each of the 10 patients shown as described in Materials and Methods using a coalescence-based method (diamonds). In addition, Ne was determined by measuring the change in allele frequencies for each polymorphic allele in pro-pol and is presented as box-and-whisker plots, with the box extending one quartile from the mean value and the ends of the whiskers indicating the extreme values. (B) Population size estimated from changes in allele frequency at each individual site for all patients as a function of position in the pro-pol amplicon. Population sizes determined from allele frequency changes at synonymous and nonsynonymous sites are indicated; box-and-whisker plots summarizing the average population size estimates for all patients are presented adjacent to the distribution.
The median Ne estimates obtained using the latter method were in the range of 103 to 104 (Fig. 6B), or >30-fold higher than that measured by coalescence-based methods, and are more consistent with, although still less than, population sizes estimated from linkage equilibrium analyses (15). Even the minimum estimates of allele frequencies obtained by this method were, in general, greater than those estimated by coalescent methods. To investigate the relative contributions of selection and drift on Ne, we further analyzed the type of variability on a site-by-site basis (Fig. 6B). We expected that nonsynonymous polymorphisms resulting in changes in amino acids would be subject to greater selective forces and would yield smaller values for Ne, whereas estimates of Ne using synonymous polymorphisms would be less subject to selection and more influenced by genetic drift and would yield large values for Ne. Consistent with this expectation, the overall population size measured using synonymous sites was greater than that measured using nonsynonymous sites; the difference, however, was modest and of marginal statistical significance (5,600 versus 4,500 transmitting cells per generation for nonsynonymous and synonymous sites, respectively; two-sided t test, P = 0.035). We investigated the estimates of population sizes by nucleotide position of polymorphisms within pro-pol to investigate the role of synonymous and nonsynonymous sites and to determine whether there were region-specific effects of drift or selection. As shown in Fig. 6B, the nonsynonymous and synonymous alleles that contributed to large and small population size estimates were distributed throughout pro-pol and were not localized by gene or domain.
The observation that some nonsynonymous sites yielded high population sizes suggests that some sites are not undergoing selection; alternatively, it is possible that such polymorphisms are maintained by frequent mutation at specific sites. If individual sites were undergoing frequent mutation, we would expect to identify such sites as repeatedly polymorphic in several individuals. However, of 56 nonsynonymous sites yielding population estimates of >20,000, only 2 (3.6%) were present more than once. As a result, it is unlikely that frequent mutation at individual sites is responsible for persistence of stable polymorphisms; these polymorphisms are more likely to be stably maintained over time because of relatively large population sizes. Taken together, these data indicate that measurements of HIV effective population sizes are heavily influenced by variations in allele frequency and change over time and from one site to the next due to variation in selection and drift. Our estimates should, therefore, be taken as a lower bound, and the true values are likely to be much higher.
DISCUSSION
HIV genetic diversity within individuals is the substrate upon which immune and antiretroviral drug selection act. Previous studies (1–5, 13) have reported that diversity in most recently infected individuals is very low, consistent with initiation of infection with a single variant. In patients with established infection, pro-pol diversity accumulated at a much lower rate than in recently infected individuals, and over the course of infection, diversity increased in a nonlinear fashion (Fig. 2A). The strength of the correlation between diversity and time for both early and established HIV infection (r2 = 0.47 to 0.55) suggests that duration of infection explains only a portion of the variability in diversity. All of the participants in this study were infected with subtype B virus; a recent study sequencing single genomes from early-postinfection subtype C-infected individuals has identified a similar increase in genetic diversity in nonstructural genes, including vif, vpu, tat, and rev (43).
The absence of association between pro-pol diversity and viral RNA level that we observed is similar to a previous analysis of env diversity and viral RNA levels (44) and implies that despite 100- to 1,000-fold differences in the level of viremia, the number of productively infected cells must be sufficiently large to sustain a highly diverse population of virus. Furthermore, we found no instances of a sudden shift in the HIV population that would suggest a bottleneck due to a selective sweep or other strong limitation on the infected-cell population size. Additionally, the absence of short-term fluctuations in diversity implies that the virus in blood is a well-mixed population derived from a constant, steady source, rather than localized bursts of virus from sites infected with genetically distinct populations. Finally, in a related study, we have found that diversity of the virus population is maintained throughout the course of infection, even following reductions in the number of productively infected cells 10,000-fold following antiretroviral therapy, indicating a large population of infected cells (M. Kearney, J. Spindler, S. Yu, W. Shao, A. O'Shea, C. Rehm, C. Poethke, J. W. Mellors, J. M. Coffin, and F. Maldarelli, presented at the 17th Conference on Retroviruses and Opportunistic Infections, San Francisco CA, 16 to 19 February 2010). As previously observed (5), genetic diversity early in HIV infection accumulated at a rate approximating that expected from its mutation rate. In contrast, accumulation of diversity slowed more than 30-fold in chronically infected individuals, suggesting a restriction on accumulation of new mutations. Differential accumulation of synonymous and nonsynonymous mutations is consistent with limitation of diversity due to purifying selection. In general, only a small proportion of polymorphisms underwent change over time, fewer still were fixed, and in only one patient (patient 19), with strong phylogenetic evidence of emergence of a distinct variant more than 13 years after infection, were these fixed polymorphisms linked (Table 2). Previous reports of accumulation of variation in env according to a strict (17) or relaxed (45) molecular clock were not reflected in our overall analysis of pro-pol. Instead, diversity increased asymptotically, with maximum APD values on the order of 2% about 15 years after infection, suggesting a limit to the amount of diversity that can accumulate within an individual. Similar conclusions (15) on the lack of temporal structure in HIV sequences have been drawn from analyses of env sequences in several patients (46). Maximum intrapatient pro-pol diversity during chronic infection was still substantially lower than the corresponding interpatient pairwise comparisons, which typically exceeded 5% (reference 5 and data not shown). In addition, it is not clear why accumulation of diversity slowed markedly after 9 to 18 months of infection. It is unlikely that slowing in diversity accumulation was the result of onset of immune responses, as accumulation of diversity occurred after development of serologic and cellular immune responses. These data indicate that within an individual, HIV genetic variation remains restricted by strong purifying selective forces.
All of the participants in this study were infected with subtype B virus. It will be of great interest to determine whether other subtypes have similar intrapatient diversity and accumulate diversity at rates comparable to subtype B. Recently, Rossenkhan and coworkers (43) conducted a detailed analysis of subtype C-infected individuals, sequencing single genomes from early-postinfection individuals to obtain diversity estimates for HIV accessory genes, including vif, vpu, tat, and rev. Similar to the case with subtype B, genetic diversity was restricted in these early infection samples and accumulated over time. A comprehensive analysis of subtype-specific genetic variation will yield new insights in understanding HIV pathogenesis.
The relative size of the replicating HIV population (Ne) remains uncertain but is a critical parameter in understanding the spread of new mutations conferring resistance and immune escape (8, 9, 13). In relatively small populations (≪1/mutation rate or ≪3 × 104), new mutations spread in stochastic fashion, while in large populations (≫1/mutation rate or ≫3 × 104), emergence of new variants approaches a deterministic limit (16). Estimating replicating population sizes typically uses coalescent approaches. Coalescent theory is an inherently retrospective approach rooted in neutral population genetics theory that reconstructs a genetic history based on present population structure. The model assumes that mutations arise according to a constant mutation rate in a strict molecular clock-like fashion; all alleles are neutral and reassort in random mating in populations that remain constant in size. Using a contemporaneous set of polymorphisms with measured allele frequencies in populations, coalescence uses probability analyses to reconstruct an entire population history, identifies times when genealogies “coalesce” to a most recent common ancestor (MRCA) of the population, and describes the most probable pathway to the ancestor, depicted in dendrograms that are measured in time (rather that genetic distances present in phylogenetic analyses). Based on genetic diversity determinations, a replicating population size can be estimated. Coalescence theory generally underestimates population size but represents a powerful approach to reconstructing genetic histories of diverse variants, including HIV (47), over long periods, where genetic diversity is substantial. In analysis of intrapatient data, however, the genetic diversity is more restricted, and coalescent approaches may be more sensitive to the effects of selection, yielding lower estimates for population size. In our estimates, standard coalescent approaches yielded uniformly low replicating population sizes, in the range of 10 to 100 (Fig. 3). Additional analyses using allele frequency variation to estimate Ne yielded replicating population sizes that were 30-fold greater than coalescence-based estimates, and these estimates varied greatly from one site to the next.
Site-by-site analysis also revealed that both synonymous and nonsynonymous polymorphisms underwent relatively slow change, indicating that some nonsynonymous sites are subject to relatively little selection. In addition, we also observed nonsynonymous and synonymous sites that underwent change at relatively high rates, suggesting that such sites were undergoing selection compared to others. Constraints on nonsynonymous sites have been well described: additional selective forces, including RNA structure and codon preference, may affect the allele frequency of synonymous sites. One consequence of large population sizes is a relatively long time to detectable genetic shift. The median time of approximately 1,000 days (corresponding to about 1,000 virus generations) for population genetic shift to appear suggests that prior to therapy, HIV replication proceeds as a large, well-mixed population without selective sweeps or rapid changes in composition. Since many, if not most, of the nonsynonymous changes in HIV that become fixed during all phases of infection are in sites recognized by the cellular or humoral immune response (5, 48–50), the absence of detectable bottlenecks in the population associated with their appearance implies that the selective force imposed by the immune response to any given epitope, although readily detectable by the selection of escape mutations, is not sufficiently strong to influence the overall population size or structure.
Our finding of relatively large population sizes contrasts sharply with previous studies that concluded the existence of relatively small population sizes using env sequences for analyses. Earlier env data sets available for study, such as those of Shankarappa et al. (17), are extensive but have relatively few individual plasma-derived sequences compared to the larger numbers of sequences used here to determine population size. For comparison purposes, we did carry out a site-by-site analysis on two patients in the data set of Shankarappa et al. with 10 or 11 sequences/time point. Our analysis revealed median population sizes of 2,736 (range, 2,362 to 53,702) and 5,688 (range, 3,197 to 62,571), similar to what we have identified in pro-pol; the high upper boundaries of these determinations represent the contribution of alleles with relatively stable allele frequency over time and reflect the presence of a relatively large population size. New studies with more single-genome sequences will be useful in directly estimating population sizes using env and pro-pol sequences.
Population sizes in the range of 1 × 104 to 1 × 105 approximate the inverse of the estimated unselected mutation rate of 3 × 10−5 to 4 × 10−5 (6); the HIV mutation rate in vivo has not been well studied, and actual mutation frequencies are likely to be strongly influenced by both selection and genetic drift (15, 39, 51–53). This conclusion is consistent with the detection of alleles with rapid (selection) and slow (drift) change and with the overall slowing in accumulation of diversity in chronic HIV infection. The issue becomes particularly important in considering the frequency of drug resistance mutations in untreated individuals. The rapid and reproducible appearance of such mutations following monotherapy with antiviral drugs such as lamivudine (3TC) (11) and single-dose nevirapine (54) implies their presence in the replicating virus population in most or all infected individuals prior to therapy. Their frequency will be determined by the balance between mutation, counterselection, and drift (16) but must be at least the inverse of the replicating population size, on average. Studies to date using sensitive allele-specific PCR methods, however, have failed to reproducibly detect such mutations, suggesting that the population size may be substantially larger than estimated here. Further development of very sensitive mutation detection technology as well as advances in mathematical modeling will be needed to resolve this important issue and provide critical tests of the selection-drift hypothesis and a better understanding of the virus population size and structure, which can be directly applied to understanding the emergence of drug resistance.
The population studies reported here have broad implications for understanding the pathogenesis and therapeutic responses in other chronic viral infections, especially so for those viruses with constantly replicating populations in chronic infection and new and expanding therapeutic agents, such as hepatitis B and C viruses. Hepatitis B virus has a number of effective therapeutic agents, although determinants of viral control and resistance are poorly understood. Genetic diversity is substantial, but the relationships between genetic diversity, population size, and emergence of resistance have not been extensively investigated (55, 56). Therapy for hepatitis C has expanded with additional targets and therapeutic agents; cure rates have improved, but the virologic correlates of eradication are incompletely understood. Population genetics studies have demonstrated that hepatitis C virus populations are highly genetically diverse, even relative to HIV, so it is likely that, similar to the case with HIV, drug-resistant mutations will preexist prior to therapy. Intrapatient genetic variation has been investigated (57–61), although population sizes have not been extensively investigated and it is not known how fast new drug-resistant mutations may be expected to emerge. Additional studies such as those reported here will have direct applications in the design of clinical trials and the composition of combination therapy necessary to eradicate viral infection.
ACKNOWLEDGMENTS
We thank Marguerite Van Houtte, Brendan Larder, Werner Verbiest, Wei-Shau Hu, Vinay Pathak, Brendan Larsen, and Justin Palmer for discussions and technical assistance. We are indebted to H. Masur and H. C. Lane for support and insightful discussions.
J.M.C. was a research professor of the American Cancer Society with support from the F.M. Kirby Foundation.
Footnotes
Published ahead of print 15 May 2013
REFERENCES
- 1. Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH, Shaw GM. 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 105:7552–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poss M, Martin HL, Kreiss JK, Granville L, Chohan B, Nyange P, Mandaliya K, Overbaugh J. 1995. Diversity in virus populations from genital secretions and peripheral blood from women recently infected with human immunodeficiency virus type 1. J. Virol. 69:8118–8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J. Virol. 82:3952–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wolinsky SM, Wike CM, Korber BT, Hutto C, Parks WP, Rosenblum LL, Kunstman KJ, Furtado MR, Munoz JL. 1992. Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science 255:1134–1137 [DOI] [PubMed] [Google Scholar]
- 5. Kearney M, Maldarelli F, Shao W, Margolick JB, Daar ES, Mellors JW, Rao V, Coffin JM, Palmer S. 2009. Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J. Virol. 83:2715–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mansky LM, Temin HM. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Althaus CL, Bonhoeffer S. 2005. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. J. Virol. 79:13572–13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coffin JM. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483–489 [DOI] [PubMed] [Google Scholar]
- 9. Daar ES, Richman DD. 2005. Confronting the emergence of drug-resistant HIV type 1: impact of antiretroviral therapy on individual and population resistance. AIDS Res. Hum. Retroviruses 21:343–357 [DOI] [PubMed] [Google Scholar]
- 10. Frost SD, McLean AR. 1994. Quasispecies dynamics and the emergence of drug resistance during zidovudine therapy of HIV infection. AIDS 8:323–332 [DOI] [PubMed] [Google Scholar]
- 11. Frost SD, Nijhuis M, Schuurman R, Boucher CA, Brown AJ. 2000. Evolution of lamivudine resistance in human immunodeficiency virus type 1-infected individuals: the relative roles of drift and selection. J. Virol. 74:6262–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mullins JI, Jensen MA. 2006. Evolutionary dynamics of HIV-1 and the control of AIDS. Curr. Top. Microbiol. Immunol. 299:171–192 [DOI] [PubMed] [Google Scholar]
- 13. Overbaugh J, Bangham CR. 2001. Selection forces and constraints on retroviral sequence variation. Science 292:1106–1109 [DOI] [PubMed] [Google Scholar]
- 14. Rong L, Gilchrist MA, Feng Z, Perelson AS. 2007. Modeling within-host HIV-1 dynamics and the evolution of drug resistance: trade-offs between viral enzyme function and drug susceptibility. J. Theor. Biol. 247:804–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rouzine IM, Coffin JM. 1999. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proc. Natl. Acad. Sci. U. S. A. 96:10758–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rouzine IM, Rodrigo A, Coffin JM. 2001. Transition between stochastic evolution and deterministic evolution in the presence of selection: general theory and application to virology. Microbiol. Mol. Biol. Rev. 65:151–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL, Mullins JI. 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73:10489–10502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jordan MR, Kearney M, Palmer S, Shao W, Maldarelli F, Coakley EP, Chappey C, Wanke C, Coffin JM. 2010. Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J. Virol. Methods 168:114–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mens H, Kearney M, Wiegand A, Shao W, Schonning K, Gerstoft J, Obel N, Maldarelli F, Mellors JW, Benfield T, Coffin JM. 2010. HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J. Virol. 84:12971–12981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Jr, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. 2005. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 43:406–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Achaz G, Palmer S, Kearney M, Maldarelli F, Mellors JW, Coffin JM, Wakeley J. 2004. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol. 21:1902–1912 [DOI] [PubMed] [Google Scholar]
- 22. Batorsky R, Kearney MF, Palmer SE, Maldarelli F, Rouzine IM, Coffin JM. 2011. Estimate of effective recombination rate and average selection coefficient for HIV in chronic infection. Proc. Natl. Acad. Sci. U. S. A. 108:5661–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dykes C, Najjar J, Bosch RJ, Wantman M, Furtado M, Hart S, Hammer SM, Demeter LM. 2004. Detection of drug-resistant minority variants of HIV-1 during virologic failure of indinavir, lamivudine, and zidovudine. J. Infect. Dis. 189:1091–1096 [DOI] [PubMed] [Google Scholar]
- 24. Elbeik T, Alvord WG, Trichavaroj R, de Souza M, Dewar R, Brown A, Chernoff D, Michael NL, Nassos P, Hadley K, Ng VL. 2002. Comparative analysis of HIV-1 viral load assays on subtype quantification: Bayer Versant HIV-1 RNA 3.0 versus Roche Amplicor HIV-1 Monitor version 1.5. J. Acquir. Immune Defic. Syndr. 29:330–339 [DOI] [PubMed] [Google Scholar]
- 25. Kearney M, Palmer S, Maldarelli F, Shao W, Polis MA, Mican J, Rock-Kress D, Margolick JB, Coffin JM, Mellors JW. 2008. Frequent polymorphism at drug resistance sites in HIV-1 protease and reverse transcriptase. AIDS 22:497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497 [DOI] [PubMed] [Google Scholar]
- 27. Nei M, Kumar S. 2000. Molecular evolution and phylogenetics. Oxford University Press, Inc, New York, NY [Google Scholar]
- 28. Nei M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY [Google Scholar]
- 29. Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7:256–276 [DOI] [PubMed] [Google Scholar]
- 30. Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tajima F, Nei M. 1984. Note on genetic drift and estimation of effective population size. Genetics 106:569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waples RS. 1989. A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121:379–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown AJ. 1997. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. U. S. A. 94:1862–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191 [DOI] [PubMed] [Google Scholar]
- 35. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586 [DOI] [PubMed] [Google Scholar]
- 36. Josefsson L, King MS, Makitalo B, Brannstrom J, Shao W, Maldarelli F, Kearney MF, Hu WS, Chen J, Gaines H, Mellors JW, Albert J, Coffin JM, Palmer SE. 2011. Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc. Natl. Acad. Sci. U. S. A. 108:11199–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hudson RR. 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23:183–201 [DOI] [PubMed] [Google Scholar]
- 38. Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W. 2002. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161:1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frost SD, Dumaurier MJ, Wain-Hobson S, Brown AJ. 2001. Genetic drift and within-host metapopulation dynamics of HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 98:6975–6980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seo TK, Thorne JL, Hasegawa M, Kishino H. 2002. Estimation of effective population size of HIV-1 within a host: a pseudomaximum-likelihood approach. Genetics 160:1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shriner D, Shankarappa R, Jensen MA, Nickle DC, Mittler JE, Margolick JB, Mullins JI. 2004. Influence of random genetic drift on human immunodeficiency virus type 1 env evolution during chronic infection. Genetics 166:1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Y, Mittler JE. 2008. Selection dramatically reduces effective population size in HIV-1 infection. BMC Evol. Biol. 8:133. 10.1186/1471-2148-8-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rossenkhan R, Novitsky V, Sebunya TK, Musonda R, Gashe BA, Essex M. 2012. Viral diversity and diversification of major non-structural genes vif, vpr, vpu, tat exon 1 and rev exon 1 during primary HIV-1 subtype C infection. PLoS One 7:e35491. 10.1371/journal.pone.0035491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bello G, Casado C, Garcia S, Rodriguez C, del Romero J, Borderia AV, Lopez-Galindez C. 2004. Plasma RNA viral load is not associated with intrapatient quasispecies heterogeneity in HIV-1 infection. Arch. Virol. 149:1761–1771 [DOI] [PubMed] [Google Scholar]
- 45. Lemey P, Kosakovsky Pond SL, Drummond AJ, Pybus OG, Shapiro B, Barroso H, Taveira N, Rambaut A. 2007. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput. Biol. 3:e29. 10.1371/journal.pcbi.0030029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bello G, Casado C, Garcia S, Rodriguez C, del Romero J, Carvajal-Rodriguez A, Posada D, Lopez-Galindez C. 2007. Lack of temporal structure in the short term HIV-1 evolution within asymptomatic naive patients. Virology 362:294–303 [DOI] [PubMed] [Google Scholar]
- 47. Yusim K, Peeters M, Pybus OG, Bhattacharya T, Delaporte E, Mulanga C, Muldoon M, Theiler J, Korber B. 2001. Using human immunodeficiency virus type 1 sequences to infer historical features of the acquired immune deficiency syndrome epidemic and human immunodeficiency virus evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289 [DOI] [PubMed] [Google Scholar]
- 49. Liu Y, McNevin JP, Holte S, McElrath MJ, Mullins JI. 2011. Dynamics of viral evolution and CTL responses in HIV-1 infection. PLoS One 6:e15639. 10.1371/journal.pone.0015639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Troyer RM, McNevin J, Liu Y, Zhang SC, Krizan RW, Abraha A, Tebit DM, Zhao H, Avila S, Lobritz MA, McElrath MJ, Le Gall S, Mullins JI, Arts EJ. 2009. Variable fitness impact of HIV-1 escape mutations to cytotoxic T lymphocyte (CTL) response. PLoS Pathog. 5:e1000365. 10.1371/journal.ppat.1000365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Edwards CT, Holmes EC, Pybus OG, Wilson DJ, Viscidi RP, Abrams EJ, Phillips RE, Drummond AJ. 2006. Evolution of the human immunodeficiency virus envelope gene is dominated by purifying selection. Genetics 174:1441–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rouzine IM, Coffin JM. 2005. Evolution of human immunodeficiency virus under selection and weak recombination. Genetics 170:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rouzine IM, Coffin JM. 1999. Search for the mechanism of genetic variation in the pro gene of human immunodeficiency virus. J. Virol. 73:8167–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johnson JA, Li JF, Morris L, Martinson N, Gray G, McIntyre J, Heneine W. 2005. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 192:16–23 [DOI] [PubMed] [Google Scholar]
- 55. Sheldon J, Ramos B, Garcia-Samaniego J, Rios P, Bartholomeusz A, Romero M, Locarnini S, Zoulim F, Soriano V. 2007. Selection of hepatitis B virus (HBV) vaccine escape mutants in HBV-infected and HBV/HIV-coinfected patients failing antiretroviral drugs with anti-HBV activity. J. Acquir. Immune Defic. Syndr. 46:279–282 [DOI] [PubMed] [Google Scholar]
- 56. Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608.e2. 10.1053/j.gastro.2009.08.063 [DOI] [PubMed] [Google Scholar]
- 57. Bernini F, Ebranati E, De Maddalena C, Shkjezi R, Milazzo L, Lo Presti A, Ciccozzi M, Galli M, Zehender G. 2011. Within-host dynamics of the hepatitis C virus quasispecies population in HIV-1/HCV coinfected patients. PLoS One 6:e16551. 10.1371/journal.pone.0016551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Honda M, Kaneko S, Sakai A, Unoura M, Murakami S, Kobayashi K. 1994. Degree of diversity of hepatitis C virus quasispecies and progression of liver disease. Hepatology 20:1144–1151 [PubMed] [Google Scholar]
- 59. Liu L, Fisher BE, Dowd KA, Astemborski J, Cox AL, Ray SC. 2010. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J. Virol. 84:5067–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu Z, Netski DM, Mao Q, Laeyendecker O, Ticehurst JR, Wang XH, Thomas DL, Ray SC. 2004. Accurate representation of the hepatitis C virus quasispecies in 5.2-kilobase amplicons. J. Clin. Microbiol. 42:4223–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Netski DM, Mao Q, Ray SC, Klein RS. 2008. Genetic divergence of hepatitis C virus: the role of HIV-related immunosuppression. J. Acquir. Immune Defic. Syndr. 49:136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]