

Abstract
Proper circuit function in the mammalian nervous system depends on the precise assembly and development of excitatory and inhibitory synaptic connections between neurons. Through a loss-of-function genetic screen in cultured hippocampal neurons, we previously identified the class 4 Semaphorin Sema4D as being required for proper GABAergic synapse development. Here we demonstrate that Sema4D is sufficient to promote GABAergic synapse formation in rodent hippocampus and investigate the kinetics of this activity. We find that Sema4D treatment of rat hippocampal neurons increases the density of GABAergic synapses as detected by immunocytochemistry within 30 min, much more rapidly than has been previously described for a prosynaptogenic molecule, and show that this effect is dependent on the Sema4D receptor PlexinB1 using PlxnB1−/− mice. Live imaging studies reveal that Sema4D elicits a rapid enhancement (within 10 min) in the rate of addition of synaptic proteins. Therefore, we demonstrate that Sema4D, via PlexinB1, acts to initiate synapse formation by recruiting molecules to both the presynaptic and the postsynaptic terminals; these nascent synapses subsequently become fully functional by 2 h after Sema4D treatment. In addition, acute treatment of an organotypic hippocampal slice epilepsy model with Sema4D reveals that Sema4D rapidly and dramatically alters epileptiform activity, which is consistent with a Sema4D-mediated shift in the balance of excitation and inhibition within the circuit. These data demonstrate an ability to quickly assemble GABAergic synapses in response to an appropriate signal and suggest a potential area of exploration for the development of novel antiepileptic drugs.
Introduction
Biochemical and candidate gene approaches over the past four decades have led to the identification of molecules that function to regulate excitatory, glutamatergic synapse formation and synaptic transmission (Li and Sheng, 2003; Kang et al., 2008). In contrast, far less is known about inhibitory, GABAergic synapse formation and function. Previously, we discovered that knock-down of the transmembrane class 4 Semaphorin Sema4D in the postsynaptic neuron led to a decrease in the density of GABAergic synapses formed onto that neuron, without an effect on glutamatergic synapse density (Paradis et al., 2007). These experiments identified Sema4D as one of only a few molecules described thus far that preferentially regulate GABAergic synapse formation.
The hallmark of a Semaphorin family member is the extracellular Sema domain, a conserved, cysteine-rich region of ∼500 amino acids at the N terminus of the protein (Yazdani and Terman, 2006). Sema4D is a transmembrane protein with a short intracellular domain in addition to its extracellular Sema domain. Our studies are the first to implicate Semaphorin signaling in GABAergic synapse formation, but other studies have implicated other Semaphorin family members in glutamatergic synapse formation or elimination (Sahay et al., 2005; Morita et al., 2006; Paradis et al., 2007; O'Connor et al., 2009; Tran et al., 2009; Ding et al., 2012). Although it is becoming clear that Semaphorins play a necessary role in synapse development and function, it remains an open question as to which step in the assembly of synapses Semaphorins act.
Recently, time-lapse imaging studies have provided some insight into the cell biology of GABAergic synapse development (Wierenga et al., 2008; Dobie and Craig, 2011). Live-imaging of GABAergic synapse formation in hippocampal slices revealed that GABAergic synapses form at preexisting axodendritic crossings without the involvement of axonal or dendritic protrusions (Wierenga et al., 2008). Time-lapse imaging in maturing neuronal cultures of labeled components of GABAergic synapses revealed that synaptic components are transported in mobile packets to synaptic sites along dendrites (Maas et al., 2006; Twelvetrees et al., 2010; Dobie and Craig, 2011). However, remarkably little is known about the molecular signals that initiate GABAergic synapse formation.
To gain insight into these unanswered questions, we treated hippocampal neurons with the soluble, extracellular domain of the Sema4D protein and assayed morphological and functional GABAergic synapse formation. We observed a surprisingly rapid and robust increase in functional GABAergic synapse density that was entirely dependent on PlexinB1 receptor expression. In addition, we monitored GABAergic synapse assembly by time-lapse imaging of the fluorescently tagged, GABAergic synapse-specific scaffolding protein Gephyrin in cultured neurons. We report that Sema4D treatment increased the rate of addition of GFP-Gephyrin along dendrites through a previously underappreciated mechanism: splitting of preexisting Gephyrin puncta. These experiments suggest that Sema4D/PlexinB1 signaling acts in the earliest stages of synapse development. Last, we report that Sema4D treatment of an organotypic hippocampal slice model of epilepsy dramatically suppressed neuronal hyperexcitability through a shift in the excitation–inhibition balance. The ability of Sema4D to suppress network hyperexcitability through increased inhibition suggests its possible use as a novel treatment for epilepsy.
Materials and Methods
Mice
PlxnB1−/− mice were generated as described by Friedel et al. (2005). Mice were cared for in accordance with Brandeis University Institutional Animal Care and Use Committee. Timed pregnancies were set up between PlxnB1+/− males and females in which the day of vaginal plug observation was designated as E0 and hippocampi were dissected at E16. Day of birth was designated as P0. Genotyping of all mice, including all embryos, was performed by PCR.
Culture and organotypic slices
Neurons were dissociated and cultured at low density on an astrocyte feeder layer. Astrocytes were isolated from P0 rat cortex by plating dissociated cells at low density in DMEM + 10% FBS on 10 cm tissue culture dishes. Once confluent, glia were trypsinized and plated on 12-mm glass coverslips in 24-well plates that had been coated overnight at 37°C with poly-d-lysine (20 μg/ml) and laminin (3.4 μg/ml). Dissociated hippocampal neurons from E18 rats or E16 mice were plated at a density of 80,000/well onto the confluent glia and grown in neurobasal medium (Invitrogen) with NS21 supplementation (Chen et al., 2008). E18 rat and E16 mouse hippocampal dissections yield cultures that are neuron rich and contain primarily glutamatergic, pyramidal neurons and GABAergic interneurons. AraC (Sigma-Aldrich) was added to a final concentration of 5 μm 4–24 h after plating, when glia were confluent. For experiments using cultured hippocampal neurons from embryos isolated from PlxnB1+/− female mice, littermates were dissociated side by side and plated on 24-well plates on top of a glia feeder layer. After genotyping, neurons that had been isolated from PlxnB1−/− and wildtype pups were used in experiments.
For time-lapse imaging, rat neurons were grown at 400,000 neurons/35-mm glass bottom dish (MatTek, catalog #P35G-1.5–14-C) coated with poly-d-lysine (20 μg/ml) and laminin (3.4 μg/ml). These neurons were grown without a glia feeder layer and therefore were not treated with AraC.
Hippocampal organotypic slices were harvested from P6 rat pups (Hayashi et al., 2009), cut on a tissue chopper to 380 μm thickness, and then placed on cell culture inserts (0.4 μm pore size; Millipore) and incubated in slice culture media for 7 DIV at 35°C, 5% CO2. Beginning at 2 DIV, slices were treated with TTX (1 μm) in slice culture medium or with slice culture medium alone (control). Cutting solution consisted of the following (in mm): 4 KCl, 1 CaCl2, 8 MgCl2, 26 NHCO3, 200 sucrose, 30 d-glucose, and 25 HEPES free acid, 320 mOsm, oxygenated. Slice culture medium in MEM salts powder + glutamine (Invitrogen) contained the following (in mm): 25 HEPES free acid, 26 NaHCO3, 30 d-glucose, 0.5 l-ascorbate, 2 MgSO4, 2 Glutamax, 0.2 CaCl2, 1 μg/ml insulin (Sigma-Aldrich), and 20% horse serum (Invitrogen), 320 mOsm, pH 7.2.
Sema4D treatments
Neurons were treated for varying amounts of time (see Results) with either Sema4D-Fc or Fc (both R&D Systems) and later assayed for synaptic phenotypes (1 nm treatments) or for growth cone collapse (10 nm). EphrinA1-Fc (43 nm; Richter et al., 2007; R&D Systems) was used as a positive control in the growth cone collapse assays. For the Sema4D-AP experiments, an N-terminal translational fusion of Sema4D with alkaline phosphatase (gift from Alain Chedotal; Flanagan and Leder, 1990; Flanagan et al., 2000) or the empty AP5 vector (1 nm) was used. The alkaline phosphatase constructs were transfected by calcium phosphate transfection (Xia et al., 1996) into HEK 293T cells and allowed to be expressed for 3–5 d. The Sema4D-AP and AP only were secreted from the cells and the media was harvested and concentrated with Centricon filters (0.4 μm pore size; Millipore) and the concentration was determined via a colorimetric reaction (Flanagan et al., 2000). For this, concentrated HEK293T media (1%) was added to lysis buffer and 2× SEAP buffer (OD 405 nm) was measured at 0 and 60 s. Concentration was calculated as follows: (OD60 − OD0)/0.04 × 100(9.3 × 10−12). AP constructs were then bath applied to neurons for experiments (1 nm Sema4D-AP). Lysis buffer contained 1% Triton X-100, 10 mm Tris-HCl, pH 8, 2×. SEAP buffer contained 2 m diethanolamine, pH 9.8, 1 mm MgCl2, 20 mm l-homoarginine, 40.6 mm p-nitrophenylphosphate, and 10% BSA.
Growth cone collapse assay
Neurons were cultured without an astrocyte feeder layer on 35 mm dishes and transfected with GFP at 1 DIV by calcium phosphate transfection. Neurons were then treated with Sema4D-Fc (10 nm), Fc alone (10 nm), or EphrinA1-Fc (43 nm; R&D Systems; Richter et al., 2007) for 1.5 h at 37°C. EphrinA1-Fc was preclustered with an anti-Fc antibody (α-IgG-AP, 1:4 IgG:EphrinA1-Fc; Jackson ImmunoResearch Laboratories) for 1 h before treatment of neurons, as is commonly done for this molecule (Richter et al., 2007). Neurons were imaged on an inverted microscope (IX70; Olympus) with a 60 × oil-immersion objective using Volocity image acquisition software (PerkinElmer). Images were acquired as a z-stack with a 0.5 μm step size. Growth cones were considered collapsed if they did not contain filopodial extensions and visible lamellipodia.
Immunostaining
Neurons were fixed and stained for synaptic markers at 11 DIV. Neuronal media was replaced with 1 × PBS and neurons were then fixed with 4% paraformaldehyde/4% sucrose for 8 min at room temperature. Coverslips were then washed 3 times with 1× PBS for 5 min each and incubated overnight at 4°C in a humidified chamber with primary antibody. All antibody dilutions were prepared in 1× GDB buffer (0.1% gelatin, 0.3% Triton X-100, 4.2% 0.4 m phosphate buffer, and 9% 5 m NaCl). After overnight incubation, coverslips were washed 3 times with 1× PBS for 5 min each and then incubated with appropriate Cy3- and Cy5-conjugated secondary antibodies (1:500 each; Jackson ImmunoResearch Laboratories) in 1× GDB for 2 h at room temperature. Coverslips were then washed 3 times with 1× PBS for 10 min each, dipped in dH2O, and mounted on glass slides with Aquamount (Lerner Laboratories). The following antibodies were used: ms α-Glutamic Acid Decarboxylase 65 (α-GAD65, 1:1000; Millipore), rb α-GABAA receptor subunit γ2 (GABAAR γ2, 1:100; Millipore), ms α-Gephyrin (1:500; Synaptic Systems), rb α-Synapsin I (1:1000; Millipore), ms α-GluA2 (1:500; NeuroMab), ms α-Microtubule Associated Protein 2 (α-MAP2, 1:1000; Sigma-Aldrich), α-ms 488 (1:1000; Invitrogen), α-ms Cy3 (1:500; Jackson Immuno), and α-rb Cy5 (1:500; Jackson ImmunoResearch Laboratories).
Live imaging
Cells were plated on 35 mm dishes and transfected with GFP-Gephyrin (gift from M. Kneussel; Maas et al., 2006) at 2 DIV. At 8 DIV, neurons were imaged with a spinning disc confocal (3i Marianas spinning disk confocal system with Yokogawa CSU-X1 confocal head mounted on a ZeissAxio-Observer inverted microscope with a 63 × oil objective). Images were acquired every 15 s for 10 min before treatment with either Sema4D-Fc (1 nm) or Fc control (1 nm, Fc only), and then imaged again after treatment for 30 min (15 s image interval). Neurons were imaged on a stage heater (∼33°C, QE-1; Warner Instruments) in HEPES buffered imaging solution containing the following (in mm): 117 NaCl, 5.3 KCl, 1.8 CaCl2, 0.814 MgSO4, 1 NaH2PO4, 20 HEPES, 50 d-glucose, 0.1% BSA, 320 mOsm, pH 7.2 (Wierenga et al., 2005). SlideBook acquisition software was used to acquire z-stack images (0.5 μm step size). For analysis, time-lapse imaging series were separated into 10 min increments. Images were exported and registered to eliminate any drifting that occurred over time (“StackReg” plugin from ImageJ). Dendrites were then traced and kymographs were made (“MultipleKymograph” plugin from ImageJ) for all dendrites in each condition. Although kymographs were used to analyze the movement of every GFP-Gephyrin puncta in each neuron, any movement observed in the kymograph was subsequently characterized in the raw time-lapse images (Movie 1). Only those movements that were consistent between the kymograph and the image series were analyzed. Using this program, we followed each GFP-Gephyrin puncta across the time-lapse series. GFP-Gephyrin puncta dynamics were broken down into two different categories. In the first, we quantified the instantaneous velocity of each puncta. This is defined as the velocity from one frame to the next. Any change in direction of movement or velocity was recorded. For example, in the right panel of Figure 3B, the moving puncta at 10′, the puncta splits from the original puncta by moving to the left (retrograde track 1), pauses, and then continues moving to the left (retrograde track 2). The instantaneous velocity was measured once per continuous movement. In addition, the duration (seconds) and the distance (micrometers) of each continuous movement, defined as the distance and time that the puncta moves without changing direction or velocity, was determined. The average of all puncta in each condition at each time point was calculated. The average total distance each puncta moved during a 10 min time-lapse series was also quantified. In addition to quantifying the movement characteristics of the GFP-Gephyrin puncta, the percentage of stable versus mobile puncta were determined for each neuron. Quantifications are presented in Table 1.
Figure 3.

Sema4D-Fc drives formation of functional GABAergic synapses. A, Representative whole-cell voltage-clamp recordings of mIPSCs from primary hippocampal neurons treated with Fc control (left) or Sema4D-Fc (right) for 0.5, 1, 2, and 4 h. B, Quantification of mIPSC (top) frequency and (bottom) amplitude, *p < 0.05, Student's t test compared with corresponding Fc control. Data plotted as mean ± SEM. C, D, Cumulative distribution plots of mIPSC interevent intervals (C) and mIPSC amplitude (D) at 0.5, 1, 2, and 4 h of Sema4D-Fc treatment. n = 14 neurons for all conditions from three experiments. *p < 0.02, Kolmogorov–Smirnov test.
Table 1.
GFP-Gephyrin dynamics
| Fc control | Sema4D | |
|---|---|---|
| % Stable | 0.945 ± 0.19 | 0.934 ± 0.019 |
| Instantaneous velocity (μm/min) | 1.005 ± 0.06 | 0.925 ± 0.18 |
| Average track distance (μm) | 0.752 ± 0.07 | 0.897 ± 0.16 |
| Average track duration (s) | 59.464 ± 4.76 | 74.498 ± 14.09 |
| Average puncta total distance (μm) | 2.090 ± 1.07 | 2.078 ± 0.47 |
| Average puncta total duration (s) | 141.016 ± 54.38 | 186.045 ± 24.55 |
Sema4D treatment does not alter the baseline dynamics of GFP-Gephyrin puncta. Dynamics of GFP-Gephyrin puncta after 10 min of Sema4D-Fc or Fc control treatment are shown. See Materials and Methods for details of calculations. Sema4D-Fc: n = 5 neurons, 1025 puncta, average 205 puncta/neuron (±22.75); Fc control: n = 3 neurons, 446 puncta, average 148.67 puncta/neuron (±33.9). All data are shown as ± SEM; p > 0.05 in all conditions, t test.
Movie 1.
Mobile GFP-Gephyrin puncta in living neurons. Time-lapse movie of the same stretch of dendrite shown in Figure 7B immediately after the neuron was treated with Sema4D-Fc (far right, 10 min kymograph). Neuron is expressing GFP-Gephyrin. There are 7 frames/s, images acquired every 15 s, 10 min total, 9.66 μm H × 17.7 μm W.
Figure 7.

Application of soluble Sema4D-Fc leads to a rapid increase in the rate of GFP-Gephyrin addition in cultured hippocampal neurons. A, Stretches of dendrite from cultured rat hippocampal neurons expressing GFP-Gephyrin and treated with Fc control (left) or Sema4D-Fc (right). Scale bars, 2 μm. Below each dendrite is a kymograph of the region highlighted by the red box above that visualizes the movement of puncta over time. B indicates before treatment; 10, 0–10 min after treatment; 20, 10–20 min after treatment; 30, 20–30 min after treatment; blue arrows, puncta splitting event. B, Additional representative kymographs from sample stretches of different dendrites from the same neurons as in A. Scale bars: y-axis = 3 min, x-axis = 3 μm. C, Top: The number of puncta added (average per neuron) during each imaging session in either Fc control (light gray) or Sema4D-treated neurons (dark gray). *p < 0.05, Student's t test. Bottom: The average number of GFP-Gephyrin puncta added normalized to the total number of GFP-Gephyrin puncta per neuron. *p < 0.05, Student's t test. D, Top: The number of GFP-Gephyrin puncta removed (average per neuron) during the imaging session in either Fc control (light gray) or Sema4D-treated neurons (dark gray). Bottom: The average number of GFP-Gephyrin puncta removed normalized to the total number of GFP-Gephyrin puncta per neuron. The average number of GFP-Gephyrin puncta was not different between conditions. Sema4D-Fc: n = 5 neurons, 1025 puncta, average 205 puncta/neuron (±22.75); Fc control: n = 3 neurons, 446 puncta, average 148.67 puncta/neuron (±33.9). All data are plotted as mean ± SEM.
Electrophysiology
Cultured neurons.
Whole-cell voltage-clamp recordings were obtained from primary hippocampal cultured neurons at 11 DIV. Neurons were first treated with Fc control (Fc construct alone) or Sema4D-Fc at 1 nm for 0.5, 1, 2, or 4 h by removing the existing media and replacing with 0.5 ml of fresh media plus Fc alone or Sema4D-Fc. After treatment, glass coverslips were transferred to a recording chamber attached to an Olympus upright microscope with continual perfusion of ACSF (95% O2:5% CO2) containing the following (in mm): 125 NaCl, 26 NaHCO3, 2.3 KCl, 1.26 KH2PO4, 2 CaCl2, 2 MgSO4, 10 glucose, and 1 nm Fc or Sema4D-Fc, pH 7.4, maintained at 32°C. Separate holding reservoirs and tubing were used to avoid any cross-contamination between Fc and Sema4D-Fc treatments perfused with a peristaltic pump. Measures of mIPSCs were done in the presence of 1 μm TTX (Abcam), 5 μm DL-2-amino-5-phosphonopentanoic acid (APV; Sigma-Aldrich), and 10 μm DNQX (Sigma-Aldrich). Patch pipettes (3–5 MΩ) were filled with intracellular solution containing the following (in mm): 120 CsCl, 10 HEPES, 1 EGTA, 0.1 CaCl2, 1.5 MgCl2, 4 Na2ATP, and 0.3 Na2GTP, pH 7.3, adjusted with CsOH. A Multiclamp700B amplifier and a Digidata 1440A digitizer controlled by pClamp10 software were used for recordings. Rs and RIN were monitored throughout experiments using the Lab Bench function of Clampex 10.2, and changes of >30% throughout the course of the recording were discarded. To record mIPSCs, the membrane potential was held at −70 mV and events were filtered at 1 kHz. Data were recorded in 3 epochs at 100 s each for a total duration of 300 s per cell. mIPSCs were then evaluated offline in Clampfit 10.2 (Molecular Devices).
Acute hippocampal slice.
Wildtype or PlxnB1−/− mice (P11–P12) were anesthetized with isoflurane, decapitated, and the brains removed with the head immersed in ice-cold, choline-based cutting solution. The choline cutting solution contained the following (in mm): 25 NaHCO3, 1.25 NaH2PO4·H2O, 2.5 KCl, MgCl2·6H2O, 25 glucose, 0.5 CaCl2, 110 C5H14ClNO, 11.6 ascorbic acid, and 3.1 pyruvic acid. Coronal slices (300 μm) were cut from the posterior to anterior hippocampus. Slices were moved to a recovery chamber for 40 min at 36°C. After recovery, slices were moved to room temperature and placed in a separate holding chamber containing either 1 nm Fc or Sema4D-Fc for 1.5 h before recordings. After incubation, acute hippocampal slices were then transferred to the recording chamber at 34°C and perfused with ACSF (see above). Whole-cell recordings were made from visually identified CA1 pyramidal neurons and measures of mIPSCs were performed as described above (cultured neurons).
Organotypic slice cultures.
Hippocampal organotypic slice cultures (control or TTX treated) were transferred to the recording chamber and continuously perfused with aerated ACSF containing 1 nm Fc or Sema4D-Fc. Culture slices were incubated at 27°C for 30 min during TTX washout to prevent excitotoxicity and then recovered at 32°C for 1.5 h. Whole-cell patch-clamp recordings were performed in current clamp to measure action potential firing with a set of step pulses from −50 to +100 pA in 25 pA increments from rest. Voltage monitoring of spontaneous activity was performed on cells with resting membrane potentials ≤ −50 mV for a duration of 100 s. Voltage monitoring allowed confirmation of the presence of spontaneous epileptic activity in TTX-treated slices. sEPSCs and sIPSCs were recorded in the same cells by voltage clamping the membrane potential at the reversal potential of one of the two postsynaptic currents (−65 to −55 mV for GABAergic and 5–15 mV for glutamatergic currents) measured independently in each cell by clamping the membrane in 5 mV increments. The intracellular solution for spontaneous events contained the following (in mm): 120 CsMe, 10 KCl, 2 MgSO4, 10 HEPES, 0.5 EGTA, 3 ATP, 0.3 GTP, 10 phosphocreatine, and 1 QX-314. Only experiments in which TTX treatment resulted in increased spontaneous activity compared with control were used.
Data analysis and statistics
Imaging/analysis for fixed cell experiments.
Image acquisition and quantification were performed in a blinded manner. Twelve-bit images of neurons were acquired on an Olympus Fluoview300 confocal microscope using a 60 × objective. Within each experiment, images were acquired with identical settings for laser power, detector gain, and amplifier offset. Images were acquired as a z-stack (8–13 optical sections and 0.5 μm step size), and maximum intensity projections were created from the stacks (ImageJ).
For synapse density experiments, synapse density was quantified as the overlap of MAP2, the presynaptic antibody, and the postsynaptic antibody using MetaMorph image analysis software. For each experiment, the threshold for the Cy3 and Cy5 channels was determined visually using three images of control neurons. The threshold was chosen such that all punctate structures would be included in the analysis. This threshold was then applied across all images within the experiment. The threshold for MAP2 was determined independently for each image. A binary mask including all pixels above the threshold was created for all channels for each image. Except where indicated (see below), the cell body was then manually deleted from the MAP2 mask. The “logical and” function was used to determine regions of triple colocalization at least one pixel in size. To calculate synapse density, this number was divided by the area of the neuron as measured using the MAP2 mask minus the cell body. Presynaptic puncta density was quantified by determining the density of either GAD65 or Synapsin puncta that overlapped with MAP2. Similarly, postsynaptic puncta density was quantified by calculating the density of GABAARγ2, Gephyrin, GluA2, or PSD95 puncta overlapping with MAP2. Approximately 10–30 images from at least two separate coverslips were acquired and analyzed for each condition within an experiment for a total of three experiments.
For experiments analyzing the synapse density on somas of cultured neurons, images were processed similarly to those used in dendrite analysis except, instead of the morphology of the cell being determined by automated detection of MAP2 fluorescence, the soma was defined by an ROI manually drawn around the cell soma. Synapses were counted within this ROI. Here, the “logical and” function was used to determine regions of colocalization of at least one pixel in size between the Cy3 and Cy5 channel (presynaptic and postsynaptic makers). Synapse density was calculated as this number divided by the area of the ROI. GABAergic interneurons were excluded from this analysis.
For experiments measuring GAD65 or GABAARγ2, the average puncta area and average puncta immunofluorescence intensity were quantified as the overlap between either GAD65 or GABAARγ2 and MAP2 immunostaining. For each experiment, the thresholds for the Cy3 and Cy5 channels were the same as that used in the synapse density quantification and applied across all images within the experiment. The “logical and” function was used to find areas of colocalization between either all three channels (“synaptic”) or between either GAD65 or GABAARγ2 and MAP2 (all puncta, including extrasynaptic and synaptic). The area (in micrometers) and fluorescence intensity (in arbitrary units) of each GAD65 or GABAARγ2 puncta was measured using the “reconstruct from above” function in MetaMorph. Similar to the synapse density analyses, 10–30 images from at least 2 separate coverslips were acquired and analyzed for each condition with an experiment for a total of 2–4 experiments.
Synapse density, puncta area, and puncta intensity values within each experiment were normalized to account for the variation in antibody staining and neuronal density from experiment to experiment before combining data from separate experiments. Within an experiment, the average synapse density value was obtained for the control and experimental conditions. The normalized value of each experiment is the experimental average value divided by the control average value (for details of this conversion, see Paradis et al., 2007). Statistical analysis was performed comparing each experimental condition to control on the combined raw data from all experiments using SPSS software to run a two-way between-effect ANOVA (factors were transfection group and date of experiment), followed by Tukey's post hoc test for significance. Error bars in the figures denote SEM.
Electrophysiology data analysis.
The average spike frequency was measured by counting the number of action potentials fired for a duration of 125 s (if no action potentials were present during the 125 s recording for a given neuron, a zero value was included in the average). The frequency and amplitude of mIPSCs was measured using a template generated in Clampfit 10.2 by selecting 50 random mIPSC events. Only single event mIPSCs with a stable baseline, sharp rise phase, and exponential decay were chosen. Double and multiple peak mIPSCs were excluded. Spontaneous EPSCs and IPSCs were measured using the threshold search function in Clampfit 10.2 measuring the area of all negative or positive going events from baseline with a noise rejection of 2 ms. All data are plotted as mean ± SEM. Significance was determined with SPSS Statistics 19 software with a one-way ANOVA to determine significance between conditions plotted on the x-axis, followed by a Student's t test to determine significance between Fc control and Sema4D treatment for each condition. Cumulative distribution plots were created in Excel software using 100–500 randomly selected points for each cell, and significance was determined based on a two-sample Kolmogorov–Smirnov test.
Results
Extracellular domain of Sema4D promotes the rapid formation of GABAergic synapses
To determine whether Sema4D is sufficient to promote GABAergic synapse formation, we treated 11 DIV cultured rat hippocampal neurons with the extracellular domain of mouse Sema4D (amino acids 24–711) conjugated to the Fc region of mouse IgG2A or Fc control for 0.5, 1, 2, and 4 h. Neurons were subsequently fixed and immunostained for MAP2 to visualize dendrites and proteins that localize specifically to GABAergic synapses, the presynaptic protein GAD65 and GABAARγ2. Next, we quantified synapse density in these neurons using confocal microscopy; synapse density was defined as the number of opposing GAD65/GABAARγ2 puncta divided by the dendritic area. Because our hippocampal cultures were composed of ∼75% glutamatergic principal cells and ∼25% GABAergic interneurons (data not shown), the majority of synapses quantified were formed onto the dendrites of principal cells. Using this assay, we found that 0.5 h of Sema4D-Fc treatment led to a 50% increase in GABAergic synapse density that was also observed with 1, 2, and 4 h treatments (Fig. 1A). Interestingly, Sema4D-Fc treatment increased the density of both presynaptic GAD65 puncta and postsynaptic GABAARγ2 puncta at all time points (data not shown). We verified the biological activity of the Sema4D-Fc protein by demonstrating that it is capable of collapsing hippocampal growth cones (Fig. 1B), as reported previously (Swiercz et al., 2002).
Figure 1.
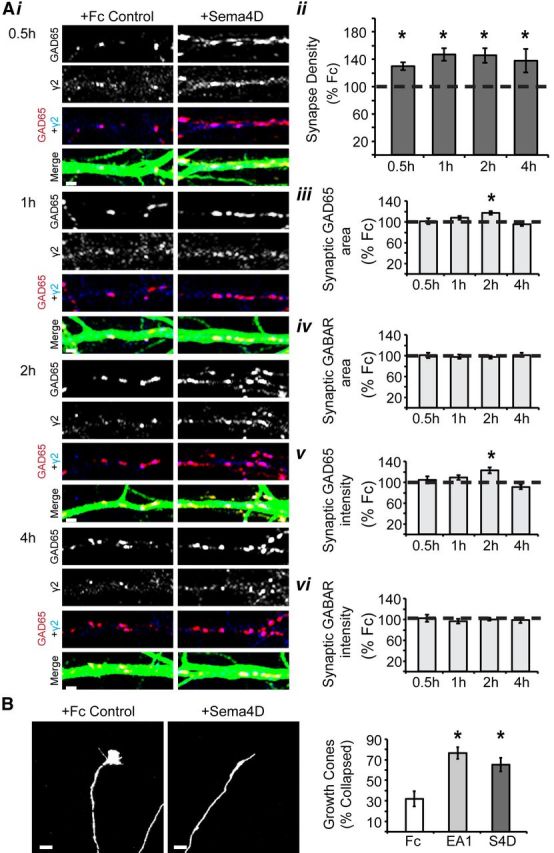
Soluble Sema4D-Fc treatment rapidly increases the density of inhibitory synapses. Ai, Representative stretches of dendrite from neurons treated with either Fc control or Sema4D-Fc and immunostained for GAD65 (red), GABAARγ2 (blue), and MAP2 (green) to visualize dendrites at 0.5, 1, 2, and 4 h of treatment. Scale bars, 2 μm. Aii, Quantification of inhibitory synapse density (GAD65/γ2) as percentage synapse density of Fc control treated neurons (100%, represented by dashed line). Aiii, Aiv, Quantification of the average area of GAD65 (Aiii) and GABAARγ2 (Aiv) puncta as the percentage of Fc control treated neurons (100%, represented by dashed line). Av, Avi, Quantification of the average fluorescence intensity of GAD65 (Av) and GABAARγ2 (Avi) puncta as the percentage of Fc control treated neurons (100%, represented by dashed line; n > 50 neurons in each condition, 3+ experiments; *p < 0.05, two-way ANOVA). B, Left: Representative images of growth cones from cultured hippocampal neurons treated with Fc control (10 nm, left) or Sema4D-Fc (10 nm, right). Right: Quantification of the percentage collapsed growth cones (n > 84 growth cones per treatment; scale bar, 5 μm; *p < 0.05 Student's t test). All data are plotted as mean ± SEM.
We sought further insight into possible changes to GABAergic synaptic components by quantifying the average size and average intensity (see Materials and Methods) of synaptic GAD65- and GABAARγ2-immunopositive puncta on the cultured neurons. We found no effect of Sema4D-Fc treatment on these parameters at 0.5, 1, or 4 h of treatment (Fig. 1Aiii–Avi). Interestingly, there was a significant, yet transient, increase in the synaptic GAD65 puncta area and intensity after 2 h of Sema4D-Fc treatment (Fig. 1Aiii,Av), with no corresponding change in synaptic GABAARγ2 puncta area or intensity (Fig. 1Aiv,Avi). In addition, we quantified the average size and average intensity of total GAD65- and GABAARγ2-immunopositive puncta (i.e., without demanding colocalization with a synaptic marker; see Materials and Methods) in the cultures. We found no effect of Sema4D-Fc treatment on these parameters at any time point (data not shown), leading us to conclude that the transient increase in GAD65 puncta size and intensity was confined to synaptically localized GAD65. Overall, our results indicate that the primary effect of Sema4D-Fc treatment on our cultures was to promote the formation of new inhibitory synapses, as evidenced by increased synaptic puncta density.
In addition, we detected a similarly rapid increase in GABAergic synapse density in response to Sema4D-Fc treatment by immunostaining with antibodies that recognize an entirely different set of synaptic proteins: Gephyrin, a postsynaptic scaffolding protein localized exclusively to inhibitory synapses (Sassoè-Pognetto et al., 2000; Fritschy et al., 2008) and Synapsin I, a presynaptic-vesicle-associated protein (Cesca et al., 2010). We found a significant increase in the density of Gephryin/Synapsin-colocalized puncta within 0.5 h of Sema4D-Fc treatment that was also observed at 1, 2, and 4 h of treatment (Fig. 2A).
Figure 2.

Sema4D-Fc treatment increases the density of inhibitory but not excitatory synapses. A, Top: Representative stretches of dendrite from neurons treated with either Fc control or Sema4D-Fc for 0.5 or 1 h and immunostained for Synapsin I (red), Gephyrin (blue), and MAP2 (green). Scale bars, 5 μm. Bottom: Quantification of inhibitory (Gephyrin/Synapsin) synapse density of neurons treated with Sema4D-Fc for 0.5, 1, 2, and 4 h plotted as percentage of Fc control treated neurons (100% represented by dashed line; n > 40 neurons in each condition, 2+ experiments). B, Top: Representative stretches of dendrite from neurons treated with either Fc control or Sema4D-Fc for 0.5 or 1 h and immunostained for Synapsin I (red), GluA2 (blue), and MAP2 (green). Bottom: Quantification of excitatory synapse density (measured by GluA2/Synapsin I staining) of neurons treated 0.5, 1, 2 and 4 h plotted as a percentage of Fc control treated neurons (100% represented by dashed line; n > 45 neurons in each condition from at least 3 experiments for 0.5, 1 and 2 h; n>20 neurons from 1 experiment for 4 h). Scale bars, 5 μm.*p < 0.05, two-way ANOVA.
We also investigated whether the rapid action of Sema4D-Fc treatment affects excitatory synapse formation. Hippocampal cultures that had been treated with Sema4D-Fc or Fc control were immunostained with antibodies that specifically recognize a postsynaptic component of glutamatergic synapses, the glutamate receptor GluA2, and the presynaptic protein Synapsin I to quantify glutamatergic synapse density (Fig. 2B). Interestingly, we observed that Sema4D-Fc treatment caused a small but significant decrease in excitatory synapse density at the 0.5 h time point, while having no effect at subsequent time points (Fig. 2B). We verified these results by immunostaining with an antibody that specifically recognizes a different postsynaptic component of glutamatergic synapses, the postsynaptic protein PSD95, in combination with anti-Synapsin I. We again found a small but significant decrease in excitatory synapse density at the 0.5 h time point (data not shown). We hypothesize that this transient decrease in excitatory synapse density was the result of the rapid assembly of GABAergic synapses in response to Sema4D-Fc treatment, which temporarily interfered with ongoing assembly of glutamatergic synapses. Overall, and consistent with our loss-of-function studies (Paradis et al., 2007), we conclude that addition of exogenous Sema4D-Fc to cultured hippocampal neurons does not promote glutamatergic synapse formation.
Next, we determined the time course of functional GABAergic synapse formation in response to Sema4D-Fc treatment. We performed whole-cell voltage-clamp recordings to measure the frequency and amplitude of GABAAR-mediated mIPSCs (Fig. 3). We found a significant increase in the frequency of mIPSCs after both 2 and 4 h of Sema4D-Fc treatment (Fig. 3B, top, C). This time frame suggests a model in which the arrival of synaptic components (as detected by immunocytochemistry; Fig. 1A, Fig. 2A) precedes functionality at nascent GABAergic synapses (Fig. 3). These data indicate that Sema4D signaling rapidly and simultaneously recruits scaffolding molecules, GABAARs, and presynaptic active zone components to form fully functional GABAergic synapses within 2 h.
Our analysis of GABAergic synaptic transmission also revealed a transient decrease in average mIPSC amplitude (∼35%) at 0.5 h, followed by a transient increase in mIPSC amplitude (∼50%) that was only detectable at 2 h of Sema4D-Fc treatment (Fig. 3B, bottom, D). We hypothesize that the transient decrease in mIPSC amplitude observed at 0.5 h was due to recruitment of GABAARs from preexisting synapses to nascent synapses. Subsequently, as the neuron rapidly assembles more GABAergic synapses, a transient overshoot in receptor abundance occurs, as evidenced by the increased mIPSC amplitude observed at 2 h that is absent after 4 h of Sema4D-Fc treatment. Somewhat surprisingly, we did not observe a concomitant change in GABAARγ2 puncta size or intensity by immunostaining (Fig. 1Aiv,Avi) at these time points. Because mammalian GABAARs are heteropentamers that can be assembled from 18 distinct, homologous subunits (Jacob et al., 2008), it is possible that changes in GABAAR abundance are not revealed by immunoreactivity against the γ2 subunit. Interestingly, we did observe an increase in GAD65 puncta immunofluorescence intensity and size after 2 h of treatment (Fig. 1Aiii,Av). Although changes in miniature event amplitude are generally thought to be due to changes in postsynaptic receptor abundance or function, in theory, changes in presynaptic neurotransmitter release could also modulate miniature event amplitude (Vautrin and Barker, 2003). In support of this hypothesis, alterations in vesicular GABA content have been implicated in changes in mIPSC amplitude (Lau and Murthy, 2012), although whether the intensity of GAD65 immunostaining is a relevant proxy for vesicular GABA content remains to be determined (Tian et al., 1999; Hartman et al., 2006).
PlexinB1 is necessary for the effect of Sema4D on GABAergic synaptogenesis in cultured neurons and acute hippocampal slices
We subsequently sought to identify the relevant receptor through which Sema4D signals to regulate GABAergic synapse formation. Sema4D has been shown to bind to multiple receptors through its extracellular domain, including PlexinB family members and CD72 (Tamagnone et al., 1999; Kumanogoh et al., 2000; Kikutani and Kumanogoh, 2003), with PlexinB1 suggested to be the highest affinity receptor (Tamagnone et al., 1999). To begin, we investigated whether PlexinB1 was the functional receptor in Sema4D-mediated synaptogenesis by using Sema4D-Fc to promote GABAergic synapse formation either in the presence or absence of PlexinB1 expression. We cultured hippocampal neurons isolated from E16 mouse PlxnB1−/− or wildtype littermates (Friedel et al., 2005). Application of Sema4D-Fc at 11 DIV for 2 h significantly increased synapse density onto wildtype neurons, as measured by the overlap of anti-GAD65 and anti-GABAARγ2 puncta (Fig. 4A,B) and as previously demonstrated in rat neuronal cultures (Fig. 1, Fig. 2). The ability of Sema4D-Fc treatment to drive GABAergic synapse formation was completely abolished in neuronal cultures isolated from the PlxnB1−/− mice (Fig. 4). Similar to the time course experiments in rat neurons (Fig. 1), the density of both presynaptic (GAD65) and postsynaptic (GABAARγ2) components increased in response to Sema4D-Fc treatment in wildtype neurons, but there was no change in either presynaptic or postsynaptic density in the PlxnB1−/− neurons (data not shown).
Figure 4.
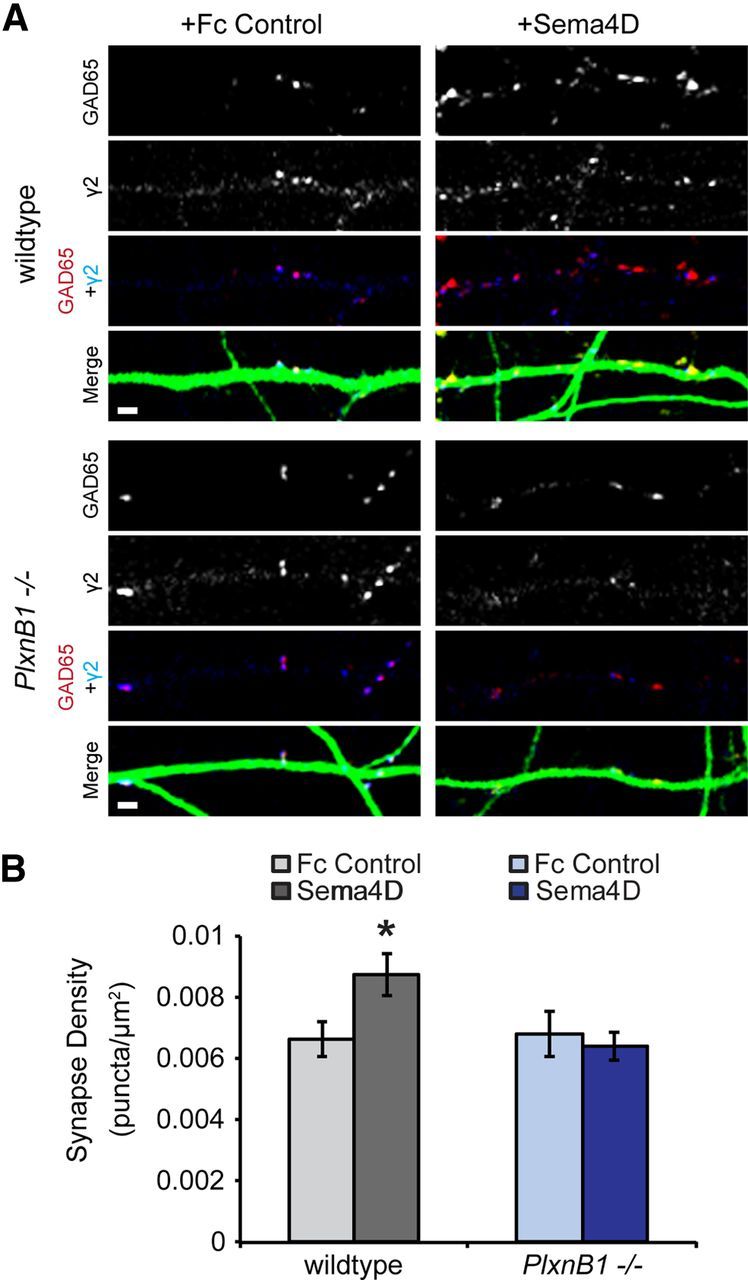
Sema4D-Fc triggers an increase in GABAergic synapse density in a PlexinB1-dependent manner. A, Representative stretches of dendrites from neurons (wildtype, top; PlxnB1−/−, bottom) treated with Fc control (left) or Sema4D-Fc (right) immunostained for the presynaptic protein GAD65 (red), the postsynaptic protein GABAARγ2 (blue), and MAP2 (green) to visualize dendrites. Scale bars, 2 μm. B, Quantification of inhibitory synapse density. n > 58 neurons for each condition from three experiments; *p < 0.05, two-way ANOVA. Data are plotted as mean ± SEM.
Next, we investigated whether Sema4D-Fc treatment could rapidly promote functional GABAergic synapse formation in an acute hippocampal slice preparation and, if so, if this effect was also dependent on PlexinB1. We performed whole-cell voltage-clamp recordings to assay mIPSC frequency and amplitude from CA1 pyramidal neurons in acute hippocampal brain slices derived from either P11–P12 wildtype or PlxnB1−/− mice (Fig. 5). Upon 2 h of Sema4D-Fc treatment, we observed a significant increase in mIPSC frequency (Fig. 5B,D) with no change in mIPSC amplitude (Fig. 5C,E) in wildtype hippocampal slices. Consistent with our immunocytochemical analysis in cultured neurons (Fig. 4), we found that the ability of Sema4D-Fc treatment to promote functional GABAergic synapse formation in acute hippocampal slices was completely dependent on the expression of PlexinB1 (Fig. 5B,D). Unexpectedly, we observed that mIPSC amplitude was slightly but significantly decreased in recordings from CA1 neurons in acute slices isolated from PlxnB1−/− mice treated with Fc control compared with acute slices isolated from wildtype mice treated with Fc control (Fig. 5C,E). Interestingly, the decreased mIPSC amplitude was rescued by Sema4D-Fc treatment (Fig. 5C,E), suggesting a Sema4D-dependent effect on mIPSC amplitude that is PlexinB1 independent.
Figure 5.

The formation of functional GABAergic synapses in response to Sema4D-Fc treatment is PlexinB1 dependent. A, Representative mIPSCs recorded from wildtype (top) or PlxnB1−/− (bottom) CA1 neurons in acute hippocampal slice treated with either Fc control or Sema4D-Fc for 2 h. B, C, Quantification of mIPSC frequency (B) and mIPSC amplitude (C); n = 29 neurons per condition; *p < 0.05 compared with wildtype Fc control treatment, Student's t test. For A–C, All data are plotted as mean ± SEM. D, E, Cumulative distribution plots of mIPSC interevent intervals (D) and amplitude (E) in wildtype (gray lines) and PlxnB1−/− mice (blue lines) in the absence (Fc Control) or presence of Sema4D-Fc. n = 29 neurons each condition from four experiments; *p < 0.02, Kolmogorov–Smirnov test.
We also examined whether treatment of cultured neurons with an entirely different source of Sema4D protein, the extracellular domain of Sema4D (amino acids 27–490) fused to the alkaline phosphatase enzyme (Sema4D-AP) and secreted from HEK 293T cells, could drive a robust increase in GABAergic synapse density. We found that Sema4D-AP was capable of promoting GABAergic synaptogenesis (Fig. 6, “wildtype” condition), similar to our results with Sema4D-Fc (Fig. 1). Using this same assay, we sought to determine whether Sema4D treatment increases GABAergic synapse density at discrete locations on the postsynaptic neuron and, if so, if this effect was PlexinB1 dependent. Different classes of interneurons synapse onto pyramidal cells at stereotyped locations: for example, parvalbumin-positive fast-spiking interneurons synapse exclusively onto the perisomatic region of pyramidal cells (Kullmann et al., 2005). Therefore, such a finding would suggest that Sema4D promotes GABAergic synapse formation between particular cell types. We found that Sema4D-AP treatment caused an increase in GABAergic synapse density onto both the soma and dendrites of glutamatergic neurons (Fig. 6, “wildtype” dendritic and somatic). Similarly, we found no change in the rise time of our mIPSC events recorded from either cultured neurons or acute slice after Sema4D-Fc treatment (data not shown), a change that would have suggested a bias in location of synapse formation. Therefore, we conclude that Sema4D is a synaptogenic factor that generally promotes GABAergic synapse formation between a variety of interneuron classes and principal cells. Consistent with this result, when we analyzed GABAergic synapse density onto either the dendritic arbor or the soma of PlxnB1−/− pyramidal neurons in response to Sema4D-AP treatment, we found that the absence of PlexinB1 abrogated the Sema4D-dependent increase in GABAergic synapse density onto both the dendrites and soma (Fig. 6). These data strongly suggest that that Sema4D-PlexinB1 signaling generally promotes GABAergic synapse formation regardless of subcellular location.
Figure 6.

Sema4D-AP treatment promotes GABAergic synapse formation onto both dendrites and somas of hippocampal neurons. A, Stretches of dendrites from cultured hippocampal neurons isolated from PlxnB1−/− or wildtype littermates (11 DIV) treated with AP control (AP alone) or Sema4D-AP for 4 h. Neurons were immunostained for GAD65 (red), GABAARγ2 (blue), and MAP2 (green). Scale bar, 5 μm B, Inhibitory synapses (GAD65/GABAAR γ2) on the somas of neurons treated with AP control or Sema4D-AP were analyzed by tracing somas (white dashed lines) and synapse density was quantified within these regions of interest. Scale bar, 5 μm. C, Quantification of synapse density from A and B plotted as a percentage of AP control (100% represented by dashed line; data are plotted as mean ± SEM; n > 20, 2 experiments; *p < 0.05, two-way ANOVA).
Time-lapse imaging studies of synapse formation during Sema4D-Fc treatment reveal increased rate of addition of Gephyrin puncta
Given the rapid nature of the Sema4D effect on GABAergic synapse formation, we next considered which step(s) in GABAergic synapse formation might be influenced by Sema4D signaling. We began our studies by imaging the dynamics of GFP-tagged Gephyrin protein. We chose Gephyrin based on our observed increase in Gephyrin puncta density in response to Sema4D-Fc treatment (Fig. 2A), the critical role of Gephyrin in forming the postsynaptic specialization of GABAergic synapses (Maas et al., 2006; Fritschy et al., 2008; Dobie and Craig, 2011; Kuzirian and Paradis, 2011), and its previous use in time-lapse imaging experiments (Maas et al., 2006; Dobie and Craig, 2011). We performed time-lapse imaging of cultured hippocampal neurons at 8 DIV that had been transfected with a GFP-Gephryin construct at 2 DIV (Maas et al., 2006). We acquired images every 15 s for 10 min before the addition of either Fc control or Sema4D-Fc and every 15 s for 30 min (divided into three sets of 10 min videos for analysis) immediately after Sema4D-Fc or Fc addition.
To follow the behavior of each individual GFP-Gephyrin puncta over time (Fig. 7), we constructed kymographs of all imaged dendrites. We observed a significant increase in the rate of addition of GFP-Gephyrin puncta after Sema4D-Fc treatment compared with Fc control (Fig. 7A–C). This increase occurred without a change in the number of GFP-Gephyrin puncta removed over the imaging session in Sema4D-Fc-treated neurons compared with Fc control (Fig. 7D), indicating that there was a net increase in the number of GFP-Gephyrin puncta as opposed to simply an increase in the turnover rate of GFP-Gephyrin puncta. The vast majority of GFP-Gephyrin puncta additions or removals were the result of either splitting (95 ± 2.9%) or merging (94.6 ± 3.9%) of existing GFP-Gephyrin puncta (Fig. 7A,B).
We then sought to determine whether Sema4D-Fc treatment affects other aspects of GFP-Gephyrin dynamics over our 40 min imaging session. To this end, we quantified the percentage of GFP-Gephyrin puncta that did not move over the imaging period (“% Stable”) and found no difference in the percentage of stable puncta between Fc control and Sema4D-Fc-treated neurons. We also determined the instantaneous velocity of GFP-Gephyrin puncta in Fc control versus Sema4D-Fc treatment and did not observe any differences between conditions (Table 1). Similarly, we quantified total distance and duration of puncta travel from the start to finish of the imaging period, as well as the average distance and duration of each individual puncta movement (Table 1) and, again, found no differences. Therefore, the major effect of Sema4D signaling is to drive the addition of new postsynaptic assemblies of scaffolding proteins by splitting preexisting assemblies of these proteins.
Sema4D-Fc treatment suppresses network hyperexcitability in an in vitro model of epilepsy
We hypothesized that because Sema4D rapidly and selectively drives GABAergic synapse formation, perhaps it could functionally restore inhibition in the context of enhanced excitability in a neural network. To explore the functional consequence of Sema4D-mediated synapse formation on network excitation, we used an in vitro model of epileptiform activity (Bausch et al., 2006; Wong, 2011). Chronic treatment with TTX to block voltage-gated sodium channels can lead to synaptic homeostasis that subsequently promotes epileptic activity via an increase in network excitation after TTX removal (Turrigiano et al., 1998; Bausch et al., 2006; Kim and Tsien, 2008). Therefore, hippocampal slices were cultured from P7 rat pups and chronically treated with TTX (1 μm) for 6–7 d, followed by a 30 min TTX withdrawal period at 27°C. We refer to these slices as TTX-EA, for TTX-induced epileptic activity.
Immediately after TTX withdrawal, we observed a significant and sustained (up to 6 h after TTX removal) increase in activity in the form of spontaneous action potential firing that was absent from untreated control slices (Fig. 8A,B, compare untreated Fc control, TTX-EA-Fc control; note different scale for y-axis untreated vs TTX-EA in Fig. 8B). This activity was not observed in slices that had been treated with Sema4D-Fc for just 2 h after TTX withdrawal (Fig. 8A,B, compare TTX-EA-Fc control, TTX-EA-Sema4D). A comparison between Fc control and Sema4D-Fc-treated slices revealed a 90% reduction in average spike frequency in both untreated and TTX-EA slices (Fig. 8A,B), suggesting that Sema4D drives GABAergic synapse formation independent of the initial level of network activity.
Figure 8.

Sema4D-Fc treatment rapidly suppresses TTX-induced epileptic activity. A, Representative traces of spontaneous activity observed in untreated (left) or TTX-treated slices (TTX-EA, right) with either Fc control (top) or Sema4D-Fc treatment (bottom). Note that for these representative traces, spikes are only present in the TTX-EA Fc control condition. B, Quantification of average spike frequency (top) and total area (bottom) measured from baseline (dashed red line) for each condition; n > 12 neurons for each condition; *p < 0.05, Student's t test. C, D, Representative traces of sIPSCs (C) or sEPSCs (D) observed in untreated (left) or TTX-EA slices (1 μm, right) with either Fc control (1 nm, top) or Sema4D-Fc treatment (1 nm, bottom). Shown is the quantification of total inhibitory synaptic charge (C) or total excitatory synaptic charge (D) measured from the same cell for each experimental condition. For C and D, n = 19 neurons for each condition; *p < 0.05, Student's t test. All data are plotted as mean ± SEM.
To begin to examine the mechanism underlying the reduced hyperexcitability observed in the hippocampal slices after Sema4D-Fc treatment, we measured sIPSCs and sEPSCs in the same cell (Fig. 8C,D). Due to a high degree of event overlap, individual events were not measured. Rather, the total synaptic charge was calculated for both inhibitory and excitatory events by integrating the total area of the events from baseline over 100 s. We found that Sema4D-Fc treatment led to an increase in total inhibitory synaptic charge in both control and TTX-treated slices (Fig. 8C). In addition, there was a significant decrease in excitatory synaptic charge observed in the TTX-EA Sema4D-Fc-treated slices compared with TTX-EA-Fc control slices. These results suggest that Sema4D treatment alters the balance between excitation and inhibition (E–I balance) in these slices, resulting in reduced hyperexcitability. However, our experiments do not distinguish between a role for Sema4D in shifting E–I balance by driving increased inhibition, perhaps through the formation of new GABAergic synapses, and some other mechanism, such as elimination of excitatory synapses. Interestingly, the fact that a Sema4D-Fc dependent decrease in excitatory synaptic charge was not observed in the untreated slices (Fig. 8D) suggests that, in the context of ongoing network hyperexcitability, Sema4D treatment triggers additional compensatory network alterations, leading to an overall reduction in network excitability.
Discussion
In this study, we demonstrate that treatment of cultured hippocampal neurons or acute hippocampal slice with the extracellular domain of the protein Sema4D causes a rapid increase (i.e., within 0.5–2 h) in the density of functional GABAergic synapses in a PlexinB1-dependent manner. Time-lapse, live imaging studies of Sema4D-Fc-treated neurons revealed an increased rate of GFP-Gephryin addition via splitting of preexisting GFP-Gephyrin puncta within 10 min of Sema4D-Fc addition. Further, using organotypic hippocampal slice culture as an in vitro model of epileptiform activity, we demonstrated that acute Sema4D-Fc treatment rapidly and dramatically altered the hyperexcitability found in these slices in a manner consistent with a Sema4D-mediated shift in E–I balance. Therefore, we show that Sema4D, acting through the PlexinB1 receptor, initiates GABAergic synaptogenesis by recruiting components of the synapse to both the presynaptic and postsynaptic termini. These newly assembled synapses become competent to release and receive neurotransmitter shortly after recruitment of the synaptic components.
To our knowledge, our study is the first to observe functional, GABAergic synapse formation in real time in response to a synaptogenic signal. Previous studies have observed either glutamatergic or GABAergic synapse formation in situ during development (Li and Sheng, 2003; McAllister, 2007; Kuzirian and Paradis, 2011) or on a much longer time scale in fixed neurons after application of a synaptogenic factor (Sahay et al., 2005; Zhang et al., 2006; Tran et al., 2009; Terauchi et al., 2010). Therefore, we have made a number of key observations. First, the action of Sema4D is surprisingly rapid. A change in the behavior of synaptic components was apparent within minutes of Sema4D-Fc application (Fig. 7). This observation suggests that GABAergic synaptic components are poised and ready to respond to reception of a prosynaptogenic signal. Second, the mechanism by which Sema4D-Fc treatment promotes synapse formation appears to be through splitting preexisting synaptic components, a process that was observed previously during live imaging studies of GABAergic synaptic development (Maas et al., 2006; Dobie and Craig, 2011). This finding indicates that application of exogenous Sema4D accelerates or co-opts ongoing mechanisms of GABAergic synapse development, and further suggests a mechanism by which endogenous Sema4D-PlexinB1 signaling promotes GABAergic synapse formation. Last, our experiments using Sema4D treatment demonstrate that there is a lag between recruitment of GABAergic synaptic components and the synapses becoming fully competent to release and respond to neurotransmitter.
Our studies are consistent with a model in which Sema4D on the postsynaptic principal neuron engages PlexinB1 on the presynaptic inhibitory neuron, generating a transsynaptic signal that regulates GABAergic synapse formation. In support of this model, Sema4D is expressed in the principal cells of the hippocampus during embryonic and postnatal development (Magdaleno et al., 2006; Lein et al., 2007; Henry and Hohmann, 2012; Aram J. Raissi and S.P., unpublished observations; Allen Developing Mouse Brain Atlas, http://developingmouse.brain-map.org/) and is required in these postsynaptic neurons to mediate GABAergic synapse formation (Paradis et al., 2007). Immunohistochemical analysis of hippocampi isolated from mice in which the Sema4D gene was constitutively deleted (Shi et al., 2000) revealed a modest but significant decrease in the intensity of GABA-synthesizing enzyme GAD67 immunoreactivity in the neuropil (Paradis et al., 2007). This result is consistent with a deficit in GABAergic synapse development in the absence of Sema4D in vivo (Paradis et al., 2007). Further experiments will be required to uncover additional deficits in inhibitory synapse formation and/or function in the hippocampi of Sema4D−/− and PlxnB1−/− mice.
In addition to Sema4D, a few other secreted or transmembrane proteins affect GABAergic synapse formation and function, including BDNF/TrkB (Rico et al., 2002; Hong et al., 2008; Chen et al., 2011), Neuroligin-2/β-Neurexin (Varoqueaux et al., 2004; Chubykin et al., 2007; Gibson et al., 2009), FGF7/FGFR2 (Terauchi et al., 2010), and Neuregulin 1/ErbB4 (Fazzari et al., 2010). The ligands Neuroligin-2 and Neuregulin 1 both function in the postsynaptic neuron to mediate GABAergic synapse formation via an interaction with their cognate receptors, β-Neurexin and ErbB4, respectively, in presynaptic interneurons (Varoqueaux et al., 2004; Gibson et al., 2009; Fazzari et al., 2010; Fu and Huang, 2010), similar to our working model of Sema4D-PlexinB1 signaling. It will be interesting to determine whether other GABAergic synaptogenic factors, such as FGF7, have a previously unappreciated capacity for driving GABAergic synapse formation on a similarly rapid time scale as Sema4D and, further, if the mechanisms of synapse assembly in response to other signals is the same or different from that of Sema4D.
Another possible model is that Sema4D and PlexinB1 interact in cis in the postsynaptic neuron to promote synapse formation. Interestingly, an increasing number of studies demonstrate cis interactions between cell adhesion ligands and their receptors, including Semaphorin family members (Taniguchi et al., 2007; Haklai-Topper et al., 2010). However, the only described function of these cis interactions to date is inhibition of their cognate receptor-mediated signaling pathways (Yaron and Sprinzak, 2012). Our data demonstrate that Sema4D-Fc treatment of cultured neurons caused splitting of GFP-Gephyrin puncta within 10 min of Sema4D-Fc addition, a time course that may be more consistent with a role for PlexinB1 signaling in the postsynaptic neuron. Determination of the location of PlexinB1 expression and the subcellular localization of the PlexinB1 protein in the early postnatal hippocampus will aid in discriminating between these two models.
There is ample evidence to suggest that decreased inhibition in neuronal circuits contributes to many forms of epilepsy (Bernard et al., 2000; Cossart et al., 2001). For example, mutations in GABAARs have been shown to be the cause of a subset of familial epileptic syndromes (McNamara et al., 2006; Fernandez and Garner, 2007). Moreover, the mechanism of action of many antiseizure medications is to enhance GABA-mediated inhibition (Czapinski et al., 2005). One conceivable anti-epilepsy treatment would be to restore normal network activity by increasing the number of inhibitory inputs onto principal neurons by stimulating the formation of GABAergic synapses. In support of this idea, a recent study demonstrated that intracerebral infusion of Neuregulin 1 acted as an anti-epileptic agent in a mouse kindling model of limbic epilepsy (Tan et al., 2012). This type of treatment could be particularly useful in the developing nervous system, such as in children with infantile spasms, who have a significant risk of going on to develop epilepsy as adults (Paciorkowski et al., 2011). Building new GABAergic synapses in the developing brain could halt the progression to runaway excitation and turn the network back to a normal physiological range. We hypothesize that the ability of Sema4D to quickly increase inhibition via addition of new GABAergic synapses could represent just such an approach to the treatment of epilepsy, one that may be able to halt the progression of the disorder as opposed to merely treating the symptoms.
Footnotes
This work was supported in part by the March of Dimes (Research Grant #5-FY09-125), the National Institutes of Health (Grant #R01NS065856 to S.P. and Grant #F31NS078901 to M.S.K.; A.R.M. supported by Grant #T32NS007292 to Eve Marder), and Core Facilities for Neurobiology at Brandeis University (Grant #P30NS45713). We thank Dr. Alain Chedotal for the gift of the Sema4D-AP construct, Dr. Marc Tessier-Lavigne for the gift of the PlexinB1−/− mice, and Drs. Paul Garrity and Sacha Nelson for experimental advice and critical reading of this manuscript.
M.S.K., A.R.M. and S.P. are listed as coinventors on a provisional patent application entitled “Methods of Modulating GABAergic Inhibitory Synapse Formation and Function Using Sema4D.” The remaining authors declare no competing financial interests.
References
- Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J Neurophysiol. 2006;96:2151–2167. doi: 10.1152/jn.00355.2006. [DOI] [PubMed] [Google Scholar]
- Bernard C, Cossart R, Hirsch JC, Esclapez M, Ben-Ari Y. What is GABAergic inhibition? How is it modified in epilepsy? Epilepsia. 2000;41:S90–S95. doi: 10.1111/j.1528-1157.2000.tb01564.x. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chen AI, Nguyen CN, Copenhagen DR, Badurek S, Minichiello L, Ranscht B, Reichardt LF. TrkB (tropomyosin-related kinase B) controls the assembly and maintenance of GABAergic synapses in the cerebellar cortex. J Neurosci. 2011;31:2769–2780. doi: 10.1523/JNEUROSCI.4991-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC. Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron. 2007;54:919–931. doi: 10.1016/j.neuron.2007.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci. 2001;4:52–62. doi: 10.1038/82900. [DOI] [PubMed] [Google Scholar]
- Czapinski P, Blaszczyk B, Czuczwar SJ. Mechanisms of action of antiepileptic drugs. Curr Top Med Chem. 2005;5:3–14. doi: 10.2174/1568026053386962. [DOI] [PubMed] [Google Scholar]
- Ding JB, Oh WJ, Sabatini BL, Gu C. Semaphorin 3E-Plexin-D1 signaling controls pathway-specific synapse formation in the striatum. Nat Neurosci. 2012;15:215–223. doi: 10.1038/nn.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie FA, Craig AM. Inhibitory synapse dynamics: coordinated presynaptic and postsynaptic mobility and the major contribution of recycled vesicles to new synapse formation. J Neurosci. 2011;31:10481–10493. doi: 10.1523/JNEUROSCI.6023-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, Lerma J, Marin O, Rico B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature. 2010;464:1376–1380. doi: 10.1038/nature08928. [DOI] [PubMed] [Google Scholar]
- Fernandez F, Garner CC. Over-inhibition: a model for developmental intellectual disability. Trends Neurosci. 2007;30:497–503. doi: 10.1016/j.tins.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-T. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, Cheng HJ, Feldheim DA, Hattori M, Lu Q, Vanderhaeghen P. Alkaline phosphatase fusions of ligands or receptors as in situ probes for staining of cells, tissues, and embryos. Methods Enzymol. 2000;327:19–35. doi: 10.1016/S0076-6879(00)27264-9. [DOI] [PubMed] [Google Scholar]
- Friedel RH, Plump A, Lu X, Spilker K, Jolicoeur C, Wong K, Venkatesh TR, Yaron A, Hynes M, Chen B, Okada A, McConnell SK, Rayburn H, Tessier-Lavigne M. Gene targeting using a promoterless gene trap vector (“targeted trapping”) is an efficient method to mutate a large fraction of genes. Proc Natl Acad Sci U S A. 2005;102:13188–13193. doi: 10.1073/pnas.0505474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritschy JM, Harvey RJ, Schwarz G. Gephyrin: where do we stand, where do we go? Trends Neurosci. 2008;31:257–264. doi: 10.1016/j.tins.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Fu Y, Huang ZJ. Differential dynamics and activity-dependent regulation of alpha- and beta-neurexins at developing GABAergic synapses. Proc Natl Acad Sci U S A. 2010;107:22699–22704. doi: 10.1073/pnas.1011233108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson JR, Huber KM, Südhof TC. Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci. 2009;29:13883–13897. doi: 10.1523/JNEUROSCI.2457-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haklai-Topper L, Mlechkovich G, Savariego D, Gokhman I, Yaron A. Cis interaction between Semaphorin6A and Plexin-A4 modulates the repulsive response to Sema6A. EMBO J. 2010;29:2635–2645. doi: 10.1038/emboj.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
- Hayashi MK, Tang C, Verpelli C, Narayanan R, Stearns MH, Xu RM, Li H, Sala C, Hayashi Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell. 2009;137:159–171. doi: 10.1016/j.cell.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry AM, Hohmann JG. High-resolution gene expression atlases for adult and developing mouse brain and spinal cord. Mamm Genome. 2012;23:539–549. doi: 10.1007/s00335-012-9406-2. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Zhang X, Dobie F, Wu H, Craig AM. Induction of GABAergic postsynaptic differentiation by alpha-neurexins. J Biol Chem. 2008;283:2323–2334. doi: 10.1074/jbc.M703957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikutani H, Kumanogoh A. Semaphorins in interactions between T cells and antigen-presenting cells. Nat Rev Immunol. 2003;3:159–167. doi: 10.1038/nri1003. [DOI] [PubMed] [Google Scholar]
- Kim J, Tsien RW. Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron. 2008;58:925–937. doi: 10.1016/j.neuron.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Ruiz A, Rusakov DM, Scott R, Semyanov A, Walker MC. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog Biophys Mol Biol. 2005;87:33–46. doi: 10.1016/j.pbiomolbio.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, Hirata H, Iwahori K, Uchida J, Yasui T, Matsumoto M, Yoshida K, Yakura H, Pan C, Parnes JR, Kikutani H. Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity. 2000;13:621–631. doi: 10.1016/S1074-7613(00)00062-5. [DOI] [PubMed] [Google Scholar]
- Kuzirian MS, Paradis S. Emerging themes in GABAergic synapse development. Prog Neurobiol. 2011;95:68–87. doi: 10.1016/j.pneurobio.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Murthy VN. Activity-dependent regulation of inhibition via GAD67. J Neurosci. 2012;32:8521–8531. doi: 10.1523/JNEUROSCI.1245-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li Z, Sheng M. Some assembly required: the development of neuronal synapses. Nat Rev Mol Cell Biol. 2003;4:833–841. doi: 10.1038/nrm1242. [DOI] [PubMed] [Google Scholar]
- Maas C, Tagnaouti N, Loebrich S, Behrend B, Lappe-Siefke C, Kneussel M. Neuronal cotransport of glycine receptor and the scaffold protein gephyrin. J Cell Biol. 2006;172:441–451. doi: 10.1083/jcb.200506066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdaleno S, Jensen P, Brumwell CL, Seal A, Lehman K, Asbury A, Cheung T, Cornelius T, Batten DM, Eden C, Norland SM, Rice DS, Dosooye N, Shakya S, Mehta P, Curran T. BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006;4:e86. doi: 10.1371/journal.pbio.0040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Morita A, Yamashita N, Sasaki Y, Uchida Y, Nakajima O, Nakamura F, Yagi T, Taniguchi M, Usui H, Katoh-Semba R, Takei K, Goshima Y. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J Neurosci. 2006;26:2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor TP, Cockburn K, Wang W, Tapia L, Currie E, Bamji SX. Semaphorin 5B mediates synapse elimination in hippocampal neurons. Neural Dev. 2009;4:18. doi: 10.1186/1749-8104-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011;45:355–367. doi: 10.1016/j.pediatrneurol.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Harrar DB, Lin Y, Koon AC, Hauser JL, Griffith EC, Zhu L, Brass LF, Chen C, Greenberg ME. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron. 2007;53:217–232. doi: 10.1016/j.neuron.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Murai KK, Bourgin C, Pak DT, Pasquale EB. The EphA4 receptor regulates neuronal morphology through SPAR-mediated inactivation of Rap GTPases. J Neurosci. 2007;27:14205–14215. doi: 10.1523/JNEUROSCI.2746-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF. TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat Neurosci. 2002;5:225–233. doi: 10.1038/nn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Kim CH, Sepkuty JP, Cho E, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins modulate synaptic transmission in the adult hippocampus. J Neurosci. 2005;25:3613–3620. doi: 10.1523/JNEUROSCI.5255-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoè-Pognetto M, Panzanelli P, Sieghart W, Fritschy JM. Colocalization of multiple GABA(A) receptor subtypes with gephyrin at postsynaptic sites. J Comp Neurol. 2000;420:481–498. doi: 10.1002/(SICI)1096-9861(20000515)420:4<481::AID-CNE6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Shi W, Kumanogoh A, Watanabe C, Uchida J, Wang X, Yasui T, Yukawa K, Ikawa M, Okabe M, Parnes JR, Yoshida K, Kikutani H. The class IV semaphorin CD100 plays nonredundant roles in the immune system: defective B and T cell activation in CD100-deficient mice. Immunity. 2000;13:633–642. doi: 10.1016/S1074-7613(00)00063-7. [DOI] [PubMed] [Google Scholar]
- Swiercz JM, Kuner R, Behrens J, Offermanns S. Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron. 2002;35:51–63. doi: 10.1016/S0896-6273(02)00750-X. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/S0092-8674(00)80063-X. [DOI] [PubMed] [Google Scholar]
- Tan GH, Liu YY, Hu XL, Yin DM, Mei L, Xiong ZQ. Neuregulin 1 represses limbic epileptogenesis through ErbB4 in parvalbumin-expressing interneurons. Nat Neurosci. 2012;15:258–266. doi: 10.1038/nn.3005. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, Gollan L, Scholl FG, Mahadomrongkul V, Dobler E, Limthong N, Peck M, Aoki C, Scheiffele P. Silencing of neuroligin function by postsynaptic neurexins. J Neurosci. 2007;27:2815–2824. doi: 10.1523/JNEUROSCI.0032-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H. Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature. 2010;465:783–787. doi: 10.1038/nature09041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, Kolodkin AL. Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature. 2009;462:1065–1069. doi: 10.1038/nature08628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, Humbert S, Triller A, Saudou F, Yan Z, Kittler JT. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. 2010;65:53–65. doi: 10.1016/j.neuron.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- Vautrin J, Barker JL. Presynaptic quantal plasticity: Katz's original hypothesis revisited. Synapse. 2003;47:184–199. doi: 10.1002/syn.10161. [DOI] [PubMed] [Google Scholar]
- Wierenga CJ, Ibata K, Turrigiano GG. Postsynaptic expression of homeostatic plasticity at neocortical synapses. J Neurosci. 2005;25:2895–2905. doi: 10.1523/JNEUROSCI.5217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga CJ, Becker N, Bonhoeffer T. GABAergic synapses are formed without the involvement of dendritic protrusions. Nat Neurosci. 2008;11:1044–1052. doi: 10.1038/nn.2180. [DOI] [PubMed] [Google Scholar]
- Wong M. Epilepsy in a dish: an in vitro model of epileptogenesis. Epilepsy Curr. 2011;11:153–154. doi: 10.5698/1535-7511-11.5.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A, Sprinzak D. The cis side of juxtacrine signaling: a new role in the development of the nervous system. Trends Neurosci. 2012;35:230–239. doi: 10.1016/j.tins.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]