

Background: Osteocytes, the most abundant cells in adult bone, express PPR.
Results: Mice with constitutive PPR deletion in osteocytes demonstrate blunted anabolic and catabolic bone responses and the inability to recruit osteoblasts and osteoclasts upon PTH administration.
Conclusion: PPR in osteocytes is needed for a full skeletal response to PTH administration.
Significance: PPR signaling in osteocytes is necessary for PTH-driven anabolic effects during osteoporosis therapy.
Keywords: Bone, Mouse Genetics, Osteocyte, Osteoporosis, Parathyroid Hormone, Anabolic, Catabolic
Abstract
Parathyroid hormone (PTH) is the only Food and Drug Administration-approved anabolic agent to treat osteoporosis; however, the cellular targets of PTH action in bone remain controversial. PTH modulates bone turnover by binding to the PTH/PTH-related peptide (PTHrP) type 1 receptor (PPR), a G-protein-coupled receptor highly expressed in bone and kidneys. Osteocytes, the most abundant cells in adult bone, also express PPR. However, the physiological relevance of PPR signaling in osteocytes remains to be elucidated. Toward this goal, we generated mice with PPR deletion in osteocytes (Ocy-PPRKO). Skeletal analysis of these mice revealed a significant increase in bone mineral density and trabecular and cortical bone parameters. Osteoblast activities were reduced in these animals, as demonstrated by decreased collagen type I α1 mRNA and receptor activator of NF-κB ligand (RANKL) expression. Importantly, when subjected to an anabolic or catabolic PTH regimen, Ocy-PPRKO animals demonstrated blunted skeletal responses. PTH failed to suppress SOST/Sclerostin or induce RANKL expression in Ocy-PPRKO animals compared with controls. In vitro, osteoclastogenesis was significantly impaired in Ocy-PPRKO upon PTH administration, indicating that osteocytes control osteoclast formation through a PPR-mediated mechanism. Taken together, these data indicate that PPR signaling in osteocytes is required for bone remodeling, and receptor signaling in osteocytes is needed for anabolic and catabolic skeletal responses.
Introduction
Parathyroid hormone (PTH)2 is an 84-amino acid single-chain polypeptide synthesized and secreted by the parathyroid glands in a calcium-regulated manner. PTH maintains serum calcium homeostasis, controls renal phosphate resorption, and vitamin D 1α-hydroxylation (1) by activating the PTH/PTH-related peptide (PTHrP) type 1 receptor (PPR), a G-protein-coupled receptor capable of activating multiple G protein-coupled pathways, including those signaling through cAMP/protein kinase A (PKA), phospholipase C (PLC)/protein kinase C (PKC), and non-PLC-dependent PKC and Cai2+ (1). The first 34 amino acids of PTH are necessary and sufficient to fully activate the receptor (1), and the amino-terminal region received Food and Drug Administration approval in 2002 as the first anabolic agent to treat osteoporosis, a health disorder estimated to affect 200 million people worldwide (2, 3). In a 2002 prevalence report by the National Osteoporosis Foundation, it was estimated that over 10 million people suffered from osteoporosis in the United States, and ∼80% of these were women. According to the World Health Organization, osteoporotic fractures are a major cause of morbidity and disability in older people, and hip fractures could lead to premature death (3). Therefore, understanding the cellular targets of PTH action in bone is central for the development of future osteoporosis therapeutics.
The PPR is abundantly expressed in bone and kidney, and its expression has also been reported in a variety of other tissues where it likely reflects the local paracrine role of PTHrP (4). In bone, PPR is expressed in cells of the osteoblast lineage, including osteocytes. Osteocytes are terminally differentiated osteoblasts and comprise 90–95% of all cells in adult bone (5). They survive for decades in their mineralized microenvironment and have the longest life span among all skeletal cells. Recent evidence suggests direct or indirect interactions of osteocytes with organs, such as kidneys, muscles, heart, and bone, through various molecules, such as FGF-23 and sclerostin (5, 6). Moreover, osteocytes might play important roles in diseases, such as hypophosphatemic rickets, osteopenia, and osteopetrosis (5, 6). Emerging literature also supports crucial roles of osteocytes in normal physiological processes, such as lactation and bone modeling and remodeling (4, 7, 8).
It has been previously proposed that osteocytes may mediate, in part, the anabolic effects of PTH by suppressing Sclerostin expression through a Mef2C-mediated pathway (9–12). Sclerostin, encoded by the SOST gene, is secreted by osteocytes and inhibits osteoblast function and bone formation by binding to the low density lipoprotein receptor-5 and -6 and suppressing the Wnt signaling pathway (13–16). Moreover, PPR on osteoblasts is known to regulate RANKL expression and thereby control osteoclast formation and bone resorption (17, 18). Importantly, activities of both osteoblasts and osteoclasts are needed for bone remodeling and anabolic and catabolic bone responses.
Recently osteocytes have been shown to modulate osteoclast function(s) through a RANKL-mediated mechanism (19, 20). Mice lacking RANKL specifically in osteocytes showed an increase in bone mineral density and osteopetrosis (19, 20), suggesting a role of osteocyte-derived RANKL in bone remodeling.
We previously generated transgenic mice in which the PPR was conditionally ablated from osteocytes post-natally upon tamoxifen injection (10-kb DMP1-Cre-Ert2). These mice demonstrated mild osteopenia by 4–6 weeks of age, characterized by a reduction in trabecular bone, and accompanied by a tonic elevation of SOST and Sclerostin and a lack of PTH-induced SOST and Sclerostin suppression (4). To further investigate the physiological role of PPR in osteocytes, we have generated mice with constitutive PPR ablation in osteocytes by using the constitutive 10-kb DMP1 promoter to drive Cre recombinase expression in PPR-floxed mice. Osteocyte-PPR KO mice (Ocy-PPRKO) showed a significant increase in bone mineral density (BMD) and trabecular and cortical bone parameters at 12 weeks of age, indicating that PPR on osteocytes is required for normal bone remodeling. Interestingly, Ocy-PPRKO displayed normal serum calcium, phosphate, and PTH, suggesting that under physiological conditions PPR signaling in osteocytes is not needed to maintain normal mineral homeostasis. When subjected to intermittent or continuous PTH administration, Ocy-PPRKO generated blunted anabolic and catabolic skeletal responses, indicating that PPR signaling in osteocytes is necessary for full skeletal responses. Upon PTH administration, Ocy-PPRKO failed to increase collagen type I α1 (Col1α1) mRNA expression in osteoblasts or suppress SOST/Sclerostin expression in osteocytes. Moreover, reduced RANKL expression in osteocytes was observed in Ocy-PPRKO, and PTH failed to induce a catabolic response in these animals. Altogether, these data highlight the necessity of PPR signaling in osteocytes for proper bone remodeling and full skeletal responses to PTH.
EXPERIMENTAL PROCEDURES
Mice
To generate Ocy-PPRKO animals, mice in which the 10-kb DMP1 promoter drives the expression of Cre-recombinase (10-kb DMP1-Cre, kindly provided by Dr. J. Feng) were mated with mice in which the E1 exon of the PPR gene is flanked by Lox-P sites (control, kindly provided by Dr. T. Kobayashi). The genotypes of the mice were determined by PCR analysis of the genomic DNA extracted from tail biopsies. For the 10-kb DMP1-Cre transgene, the forward Cre primer (5′-CGCGGTCTGGCAGTAAAAACTATC-3′) and the reverse Cre primer (5′-CCCACCGTCAGTACGTGAGATATC-3′) were used to generate a PCR product of ∼400 bp. For the floxed PPR allele, the P1 primer (5′-ATG AGG TCT GAG GTA CAT GGC TCT GA-3′) and the P2 primer (5′-CCT GCT GAC CTC TCT GAA AGA ATG T-3′) were used, which recognized the sequence spanning the 3′lox-P site, as reported previously (4). Wild-type and mutant alleles give ∼210- and 290-bp products, respectively. The Institutional Animal Care and Use Committee, Subcommittee on Research Animal Care, at Massachusetts General Hospital approved all animal protocols.
Allele-specific DNA recombination was performed on DNA isolated from osteocyte-enriched long bones (generated by sequential collagenase and EDTA digestions as described previously (4)), calvaria, kidney, liver, lung, heart, skeletal muscle, and spleen. Multiplex PCR analysis was performed using allele-specific primers, P1, P2, and P3 (5′ACA TGG CCA TGC CTG GGT CTG AGA 3′) following the manufacturer's protocol (QiagenTM Multiplex PCR kit, Valencia, CA).
Histology
Tibiae and vertebrae were fixed in 10% formalin/PBS solution at 4 °C, overnight, decalcified in 20% EDTA, pH 8.0, for 7–15 days, processed, embedded in paraffin, and sectioned. Sections were stained with hematoxylin and eosin, or used for immunohistochemistry. Undecalcified femurs, fixed in 70% ethanol, were embedded in methyl methacrylate (Aldrich), sectioned with a diamond-embedded wire saw, and stained by the Von Kossa method.
Immunohistochemistry (IHC)
Immunohistochemical RANKL expression detection was performed as described previously (19) with minor modifications. On deparaffinized tibiae, endogenous peroxidase activity was inhibited by 3% H2O2 treatment for 10 min. Subsequently, antigen retrieval was performed with Tris/EDTA buffer (10 mm Tris base, 1 mm EDTA, 0.05% Tween 20, pH 9.0) in a boiling water bath at 95 °C for 12 min. Slides were cooled to RT, blocked with TNB (TSATM biotin tyramide kit, PerkinElmer Life Sciences) for 30 min at RT, and incubated with anti-RANKL antibody (N-19, Santa Cruz Biotechnology), 4 °C, overnight in Can Get Signal immunostain solution A (Toyobo, Japan). Slides were washed and incubated with Biotin-SP-conjugated AffiniPure rabbit anti-goat IgG (H+L) (Jackson ImmunoResearch) for 30 min at RT. Afterward, the slides were washed, incubated for 30 min with streptavidin-conjugated horseradish peroxidase (HRP) and tyramide following the manufacturer's protocol (TSATM biotin tyramide kit), developed with a 3,3′-diaminobenzidine substrate-chromogen system (Vector Laboratories, Burlingame, CA), and counterstained with methyl green. Bright field microscopy was performed, and images were acquired with a ×40 objective (Nikon Eclipse E800). For quantification, ≥8 fields were imaged from ≥4 tibiae per group (control vehicle, control PTH, Ocy-PPRKO vehicle, and Ocy-PPRKO PTH). Subsequently, ImageJ (21) was used to quantify the RANKL-stained osteocyte area and normalized to total number of osteocytes per field. Statistical analysis was performed using two-way ANOVA and Tukey's HSD test. Similar staining protocol was followed for Periostin IHC on tibiae except the anti-Periostin antibody (AF2955, R&D Systems) was diluted in Can Get Signal immunostain solution B (Toyobo, Japan).
IHC for sclerostin expression was performed on deparaffinized vertebrae. Antigen retrieval was performed using proteinase K for 15 min at RT. The subsequent steps were similar to those described for RANKL IHC above, except no secondary antibody was used because we used the biotinylated anti-mouse sclerostin antibody (1:50 dilution; R&D Systems, Inc., Minneapolis, MN), which was diluted in Tris/NaCl blocking buffer. Bright field microscopy was performed, and images were acquired with a ×20 objective (Nikon Eclipse E800). For quantification, ≥12 fields were imaged from ≥5 vertebrae per group (control vehicle, control PTH, Ocy-PPRKO vehicle, and Ocy-PPRKO PTH), and sclerostin-positive osteocytes were counted and normalized to total number of osteocytes per field. Two-way ANOVA was performed, and the significance of p values was determined based on the Q scores obtained using Tukey's HSD test.
In Situ Hybridization
In situ hybridization was carried out as described previously (22). The antisense probe for Col1α1 has been reported (22).
Serology
Serum was isolated from the blood collected by retro-orbital bleeding. Serum mouse TRAP5b (mouse TRAP assay), PINP (rat/mouse PINP enzyme immunoassay), CTX (RatLaps enzyme immunoassay), and RANKL (Quantikine ELISA Kit, R&D Systems) were measured using ELISA (Immunodiagnostic Systems Ltd., UK). Serum total calcium was measured by calcium LiquiColor arsenazo kit (Stanbio Laboratory), and inorganic phosphate was quantified by phospho liquid-UV kit (Stanbio Laboratory) as per the manufacturer's instructions. Serum PTH was measured using mouse intact PTH ELISA kit (Immutopics) according to the manufacturer's protocol.
Anabolic and Catabolic PTH Treatment
Human PTH(1–34) (MGH Peptide Core Facility) was dissolved in 0.1% TFA, aliquoted, stored frozen at −80 °C, and subsequently diluted to the appropriate concentration in vehicle (2% heat-inactivated mouse serum, 0.1 n HCl, and 0.9% sodium chloride) immediately before administration. For anabolic treatment, control and Ocy-PPRKO female mice, randomly subdivided into sham or treatment groups, were injected with 100 μl of either vehicle or PTH peptide. We treated mice with 80 μg/kg PTH(1–34) anabolic dose s.c. 5 days/week for 4 consecutive weeks. For catabolic treatment, control and Ocy-PPRKO male mice, randomly subdivided into sham or treatment groups, were s.c. implanted with Alzet Micro-Osmotic Pump model 1002 (DURECT Corp.) to deliver 100 μl of either vehicle or PTH peptide (100 μg/kg/day) for 2 consecutive weeks. Upon sacrifice, left femurs and left tibiae were cleaned of soft tissue, fixed in 10% phosphate-buffered formalin, pH 7.2, for 48 h, and stored in 70% ethanol until further use. Left femur was used for microCT and histomorphometry analysis, and the left tibia was used for histology. Right femurs and right tibiae were used to isolate RNA.
Histomorphometric Analysis
Mice were intraperitoneally injected with 20 mg/kg calcein and 40 mg/kg demeclocycline on days 9 and 2 before sacrifice, respectively. The distal femur metaphyses were removed and fixed in 70% ethanol. They were then dehydrated, infiltrated, and embedded in methyl methacrylate. Undecalcified 4-μm-thick sections were cut using a microtome (Leica RM 2255, Heidelberg, Germany). Consecutive sections were toluidine blue-stained for static measurements. Histomorphometric parameters were measured using an Osteomeasure image analysis system (OsteoMetrics, Atlanta, GA) coupled to a photomicroscope and personal computer, and the results were expressed according to standardized nomenclature (23). A sampling site of 2 mm2 was established in the secondary spongiosa at 400 μm below the growth plate. Trabecular bone volume was assessed as a percentage of total tissue volume (BV/TV, %), and trabecular thickness (μm), Tb.N (1/mm), and Tb.Sp (μm) were calculated. The cancellous bone surfaces lined by osteoblasts and osteoclasts were measured. Osteoblasts were expressed per bone surface (Ob.S/BS, %), bone perimeter (N.Ob/B.Pm/mm), and tissue area (N.Ob/T.Ar/mm2). Osteoclasts were expressed per bone surface (Oc.S/BS, %), bone perimeter (N.Oc/B.Pm/mm), and tissue area (N.Oc/T.Ar/mm2). The data in Table 5 comparing catabolic response among four groups (control vehicle, control PTH, Ocy-PPRKO vehicle, and Ocy-PPRKO PTH) were analyzed using ANOVA with Tukey/Kramer.
TABLE 5.
Blunted catabolic response to continuous PTH administration in Ocy-PPRKO detected by histomorphometric analysis in femurs
Values are mean ± S.E., ANOVA with Tukey/Kramer.
| Parameter | Control vehicle, N = 9 | Control PTH, N = 8 | Ocy-PPRKO vehicle, N = 8–9 | Ocy-PPRKO PTH, N = 8 | ANOVA p value |
|---|---|---|---|---|---|
| BV/TV (%) | 11.17 ± 0.97 | 10.29 ± 1.23 | 15.39 ± 0.62a,b | 13.19 ± 1.24 | 0.02 |
| Tb.Th (μm) | 33.78 ± 1.70 | 34.05 ± 2.21 | 37.24 ± 1.29 | 36.85 ± 2.07 | 0.4 |
| Tb.N (mm) | 3.27 ± 0.16 | 3.17 ± 0.17 | 4.13 ± 0.10a,b | 3.53 ± 0.18c | 0.000 |
| Tb.Sp (μm) | 278.8 ± 18.1 | 287.8 ± 18.3 | 206 ± 6.5a,b | 252.1 ± 17.5 | 0.003 |
| Ob.S/BS (%) | 8.35 ± 1.83 | 24.94 ± 5.54a | 2.45 ± 1.19b | 7.37 ± 1.64b | 0.000 |
| N.Ob/T.Ar (mm2) | 41.40 ± 8.30 | 129.59 ± 32.59a | 15.22 ± 7.03b | 38.80 ± 8.49b | 0.000 |
| N.Ob/B.Pm (mm) | 6.51 ± 1.32 | 19.89 ± 4.60a | 1.80 ± 0.79b | 5.72 ± 1.27b | 0.000 |
| OV/TV (%) | 0.062 ± 0.013 | 0.230 ± 0.076a | 0.010 ± 0.003b | 0.036 ± 0.012b | 0.002 |
| OS/BS (%) | 3.30 ± 0.7 | 10.62 ± 3.5a | 0.47 ± 0.1b | 1.71 ± 0.5b | 0.002 |
| O.Th (μm) | 3.00 ± 0.20 | 3.32 ± 0.21 | 2.46 ± 0.48 | 2.94 ± 0.36 | 0.348 |
| Oc.S/BS (%) | 1.26 ± 0.28 | 3.43 ± 0.56a | 0.72 ± 0.23b | 1.27 ± 0.23b | 0.000 |
| N.Oc/T.Ar (mm2) | 2.72 ± 0.52 | 8.51 ± 1.68a | 2.42 ± 0.73b | 3.37 ± 0.67b | 0.000 |
| N.Oc/B.Pm (mm) | 0.44 ± 0.09 | 1.29 ± 0.21a | 0.30 ± 0.09b | 0.49 ± 0.10b | 0.000 |
| ES/BS (%) | 0.43 ± 0.10 | 1.26 ± 0.26a | 0.26 ± 0.12b | 0.57 ± 0.14b | 0.001 |
a p < 0.05 compared with control vehicle.
b p < 0.05 compared with control PTH.
c p < 0.05 compared with Ocy-PPRKO.
RNA Extraction and Purification
RNA was extracted from whole bones (femur and tibia) and from osteocyte-enriched calvaria. Calvariae and long bones from control and Ocy-PPRKO mice were subjected to sequential collagenase and EDTA digestions to remove endosteal and periosteal osteoblasts and bone marrow cells as detailed previously (4). For basal mRNA expression analysis, osteocyte-enriched calvariae were frozen in liquid nitrogen. RNA was extracted by homogenization in TRIzol (Invitrogen) as per the manufacturer's instructions. RNA quality and quantity were ascertained by UV spectrophotometry (NanoDrop 8000, Thermo Fisher).
Quantitative RT-PCR
Reverse transcription was performed on 0.5–1 μg of DNase-treated total RNA and oligo(dT) primers using Omniscript (Invitrogen) or QuantiTect (Qiagen) according to the manufacturer's instructions. Quantitative PCR for PPR, SOST, RANKL, OPG, and β-actin was performed using the QuantiTect SYBR Green PCR kit (Qiagen) on StepOnePlus (Applied Biosystems) as described previously (4). Primer sequences are available upon request. The data were normalized to β-actin, and statistical significance was determined using a t test in Excel, and p ≤ 0.05 was considered significant.
Cyclic AMP Measurement
Tibiae, femurs, and calvariae were isolated from 8–12-week-old mice. Briefly, tibiae were dissected and cleaned of adherent tissues. Distal and proximal epiphyses were removed, and the bone marrow was flushed out using 2–3 ml of α-minimal essential medium supplemented with 0.1% bovine serum albumin (BSA) and 25 mm HEPES, pH 7.4. The remaining diaphyseal enriched region of the bones was cut into three pieces and sequentially digested as described above for RNA isolation. Each piece was then placed in ice-cold cAMP assay buffer (Dulbecco's modified essential medium containing 10 mm HEPES, 0.1% heat-inactivated BSA, and 1 mm isobutylmethylxanthine). Bone pieces were then incubated in cAMP assay buffer with the appropriate treatment at 37 °C for 15 min. The three pieces of each tibia were incubated with vehicle alone (assay buffer), 100 nm human PTH(1–34), or 0.1 μm forskolin. At the end of the incubation, the reaction was terminated by quickly removing the bones and placing them in 0.3 ml of cold 90% 2-propanol in 0.5 m HCl. Bones were then incubated for 16–18 h at 4 °C. Propanol extraction was repeated, and the combined extracts were evaporated by vacuum centrifugation. The dried extracts were redissolved in acetate buffer (50 mm sodium acetate, 0.05% sodium azide, pH 6.2) for measurement of cAMP by a specific RIA, as described previously (4). Bones were washed twice with 0.5 ml of acetone and once with 0.5 ml of ether and were air-dried and weighed. The results were normalized for the bone weight, and the data were expressed as picomoles of cAMP produced per mg of dry bone. Each experiment was repeated at least three times.
Total Body Weight and Dual X-ray Absorptiometry (DXA)
Total body weight in grams was recorded at the ages mentioned under “Results.” Mouse BMD, fat tissue, and lean tissue were measured by DXA using a Lunar PIXImus II densitometer (GE Medical System Luna, Madison, WI). In brief, animals were briefly anesthetized by s.c. administration of tri-bromoethanol, placed on a tray, and total body mineral density was acquired using the manufacturer's protocols. Post-acquisition skeletal analysis was performed by excluding the mouse head from the final BMD (g/cm2) determination. Fat and lean tissue weight were normalized to total body weight and expressed as g/kg. For total body weight, fat tissue, lean tissue, and BMD, statistical significance was determined using a t test, and p ≤ 0.05 was considered significant.
MicroCT Analysis
Bone morphology and microarchitecture were analyzed using a desktop high resolution μCT (μCT40, Scanco Medical, Brüttisellen, Switzerland), as described previously (24). Briefly, the femoral midshaft and L5 vertebral body were scanned using an x-ray energy of 70 keV, integration time of 200 ms, and a 12-μm isotropic voxel size. Longitudinal sections were scanned using an x-ray energy of 70 keV, a current of 114 μA, integration time of 200 ms, and an 18-μm isotropic voxel size; subsequently, the half of total femur length was measured, and distal to mid-diaphysis in the femurs were used for measuring trabecular parameters. For the trabecular bone region BV/TV (%), Tb.N (mm), and Tb.Sp (mm), and for the cortical bone region Cort.Th (mm), Cort.A (mm2), medullary area (mm2), cortical porosity (%),cortical bone mean density (mg hydroxyapatite/cm), and pMOI (mm4) were assessed.
Ex Vivo Osteoclastogenesis
We followed the protocol described previously (19) with modifications. Briefly, nonadherent spleen cells isolated from ∼12-week-old control mice were cultured with 10 ng/ml mouse M-CSF (R&D Systems) for 3 days and were used as osteoclast precursors. Osteocyte-enriched long bones were isolated from ∼10-week-old Ocy-PPRKO and control, weighed, cut into small pieces using aseptic technique, added to 24-well plates in 500 μl of complete medium (α-minimal essential medium with l-glutamine, 10% FBS, and penicillin/streptomycin) containing 0.1 μm dexamethasone, and incubated overnight at 37°C in a humidified atmosphere with 5% CO2. The next day, 20,000 osteoclast precursors were added to each well of the 24-well plates and cultured with 100 nm hPTH(1–34), 10 nm 1,25-dihydroxyvitamin D3, and/or 100 ng/ml murine OPG/Fc chimera (R&D Systems) in the presence of dexamethasone and M-CSF for 13 days with fresh ingredients added every 3–4 days. Next, these were stained for TRAP, and multinucleate cells with two or more nuclei were scored as TRAP-positive cells using an inverted microscope. The number of TRAP-positive cells per well were normalized to the weight of the bone pieces added to that well. Statistical significance was determined using a t test in Microsoft Excel with p ≤ 0.05 as significant. Photographic evidence was recorded with a Nikon Eclipse E800 microscope at a ×10 objective.
RESULTS
Generation of Mice Lacking PPR Expression in Osteocytes
To generate mice lacking PPR expression primarily in osteocytes (Ocy-PPRKO), mice with 10-kb DMP1-Cre were mated with mice in which exon E1 of PPR was flanked by Lox-P sites (Fig. 1A). Receptor ablation was demonstrated by genomic recombination as assessed by PCR analysis of genomic DNA. Presence of the ∼600-bp band, expected with successful Cre-based recombination, confirmed PPR gene deletion in calvaria and the osteocytes from long bones but not in other tissues (Fig. 1B). PPR gene knock-out resulted in significantly reduced PPR mRNA expression in osteocyte-enriched long bones from Ocy-PPRKO as compared with littermate controls, as we have recently reported (25). In contrast, PPR mRNA expression in primary calvarial osteoblasts derived from control and Ocy-PPRKO was unchanged (control = 100 ± 6.9%, Ocy-PPRKO = 112.1 ± 6.9%, expression normalized for β-actin; p = 0.25, NS) confirming that receptor expression was not altered in osteoblasts. In addition, osteocalcin mRNA expression was also similar (control = 100 ± 22.1%, Ocy-PPRKO = 54.2 ± 13.0%, expression normalized for β-actin; p = 0.14, NS) confirming the osteoblastic nature of these cells. Functional ablation of the receptor was ascertained by assessing cAMP accumulation in response to PTH in osteocyte-enriched long bone explants (OEBE). OEBE were treated with 100 nm hPTH(1–34) or 10 μm forskolin. Ocy-PPRKO bone explants had a statistically significant reduction (86.5% reduction) in cAMP accumulation in response to PTH compared with controls (Ocy-PPRKO = 15.0 ± 4.7 pmol/mg bone versus controls = 110.8 ± 20.5 pmol/mg bone; p < 0.001), whereas both Ocy-PPRKO and control mice demonstrated a robust cAMP response to forskolin (Ocy-PPRKO = 94.2 ± 58.3 pmol/mg bone versus controls = 66.8 ± 10.6 pmol/mg bone; p = 0.359) (Fig. 1C) demonstrating that Ocy-PPRKO mice retain intact adenylate cyclase responsiveness but lack cAMP accumulation following PTH administration due to reduced PPR expression in osteocytes (Fig. 1C). Moreover, primary calvarial osteoblasts from Ocy-PPRKO and control mice showed robust cAMP production when treated with either PTH or forskolin (Fig. 1D), demonstrating intact PPR signaling in osteoblasts.
FIGURE 1.

Generation of Ocy-PPRKO mice. A, mating scheme. B, allele-specific DNA recombination PCR showing specific PPR deletion, ascertained by the presence of an ∼600-bp band, in long bone and calvaria. The presence of the larger band (∼1.5 kb) is due to contamination with other bone cells (i.e. bone marrow cells, osteoblasts, and osteoclasts) that were not removed during DNA preparation in long bone and calvaria. cAMP response in osteocyte-enriched long bones (C) and calvarial osteoblasts (D) is shown. Gray bars represent vehicle; white bars represent 100 nm hPTH(1–34), and black bars represent 10 μm forskolin, and values are mean ± S.E.
Ocy-PPRKO Mice Displayed Increased Trabecular and Cortical Bone
PTH is the most important hormonal regulator of calcium and phosphate homeostasis. It exerts its effects by binding PPR expressed on its target organs, namely bone and kidney. To investigate if receptor deletion in osteocytes altered mineral homeostasis, we measured serum calcium and phosphate concentration. Both serum calcium (control = 7.24 ± 0.35 mg/dl, Ocy-PPRKO = 6.88 ± 0.62 mg/dl; n = 5, NS) and phosphate (control = 9.3 ± 0.89 mg/dl, Ocy-PPRKO = 8.4 ± 0.41 mg/dl, n = 4–5, NS) were indistinguishable between Ocy-PPRKO and controls, suggesting that other PPR-expressing cells, likely osteoblasts or kidney cells, regulate serum calcium and phosphate concentrations. Moreover, intact circulating PTH was also similar between Ocy-PPRKO and control (control = 111.5 ± 25.1 pg/ml, Ocy-PPRKO = 104.6 ± 25.2 pg/ml; n = 5–7 NS), indicating that the skeletal phenotype observed in Ocy-PPRKO mice is not a consequence of an altered parathyroid state.
We then analyzed the bone phenotype of Ocy-PPRKO mice. Considering that bone weight, fat tissue, and lean tissue contribute toward total body weight, we measured it at different ages. Total body weight was comparable between Ocy-PPRKO and control at 4 and 8 weeks of age (Fig. 2A). However, significantly increased body weight was recorded at 12 weeks for Ocy-PPRKO as compared with control (Fig. 2A). Increased body weight was not present in control (PPRfl/fl mice) and DMP1-Cre-expressing mice (Fig. 2A), indicating that the higher weight observed in Ocy-PPRKO animals was associated with the loss of PPR expression in osteocytes. As assessed by DXA, fat tissue (control = 839.5 ± 37.6 g/kg, Ocy-PPRKO = 845.3 ± 27.4 g/kg; n ≥ 6, NS) and lean tissue (control = 160.6 ± 35.9 g/kg, Ocy-PPRKO = 154.1 ± 25.7 g/kg; n ≥ 6, NS) weights were similar between 12-week-old Ocy-PPRKO and control. BMD was similar between Ocy-PPRKO and control mice at 8 weeks in both females (Fig. 2B) and males (Fig. 2C). However, at 12–15 weeks, a statistically significant increase in BMD was observed in both sexes, and it persisted at 20 weeks (Fig. 2, B and C). DMP1-Cre-only mice at 12 weeks of age were indistinguishable from controls (Fig. 2B).
FIGURE 2.
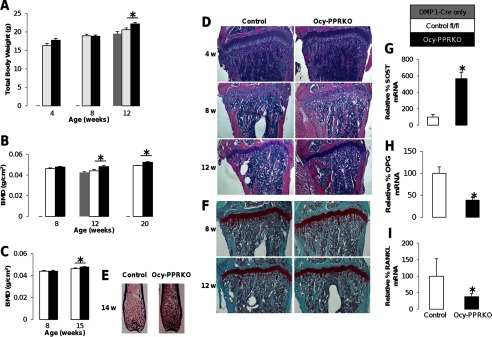
Increased body weight and BMD in Ocy-PPRKO. Control represents “PPR fl/fl” mice, and littermates were analyzed here and throughout. A, total body weight at 4 weeks (control, n = 5; Ocy-PPRKO, n = 6), 8–9 weeks (control, n = 12; Ocy-PPRKO, n = 11), and 12–14 weeks (DMP1-Cre only, n = 5; control, n = 13; Ocy-PPRKO, n = 15) in females. B, total BMD at 8 weeks (control, n = 8; Ocy-PPRKO, n = 9), 12–13 weeks (DMP1-Cre only, n = 7; control, n = 6; Ocy-PPRKO, n = 9), and 19–21 weeks (control, n = 3; Ocy-PPRKO, n = 3) in females. C, total BMD at 8 weeks (control, n = 7; Ocy-PPRKO, n = 6), and 15 weeks (control, n = 10; Ocy-PPRKO, n = 12) in males. D, H&E-stained tibiae from females, images acquired with a ×4 objective. w, weeks. E, Von Kossa stained femurs from males. F, Safranin O-stained tibiae from females, images acquired with a ×4 objective. Real time RT PCR for SOST (G), OPG (H), and RANKL (I) mRNA expression in osteocyte-enriched calvariae from 16-week-old control and Ocy-PPRKO males. Values are mean ± S.E., *, p ≤ 0.05.
H&E staining (Fig. 2D) of tibiae showed increased trabecular bone in Ocy-PPRKO as compared with control, starting at 8 weeks. Similarly, Von Kossa staining of femurs showed increased trabecular bone at 14 weeks (Fig. 2E).
To further assess the skeletal phenotype of these animals, we performed microCT analysis of both vertebrae (L5) and midshaft femurs in 5–6-week-old Ocy-PPRKO and controls. At this age, there were no statistically significant differences between Ocy-PPRKO and control mice, as shown in Table 1. MicroCT analysis at 12 weeks, however, showed a significant increase in trabecular bone volume fraction (BV/TV), Tb.N, and Tb.Th and a decrease in separation (Tb.Sp) in the vertebral body (L5) of Ocy-PPRKO as compared with control (Table 2). Analysis of femur midshafts showed a significant increase in cortical thickness (Cort.Th), area (Cort.A), and pMOI, in Ocy-PPRKO as compared with control (Table 2). Cortical density, medullary area, and cortical porosity were comparable in Ocy-PPRKO and control. (Table 2).
TABLE 1.
MicroCT analysis showed similar trabecular and cortical bone between 5- and 6-week-old Ocy-PPRKO and control females
Values are mean ± S.E., two-tailed t test assuming equal variance.
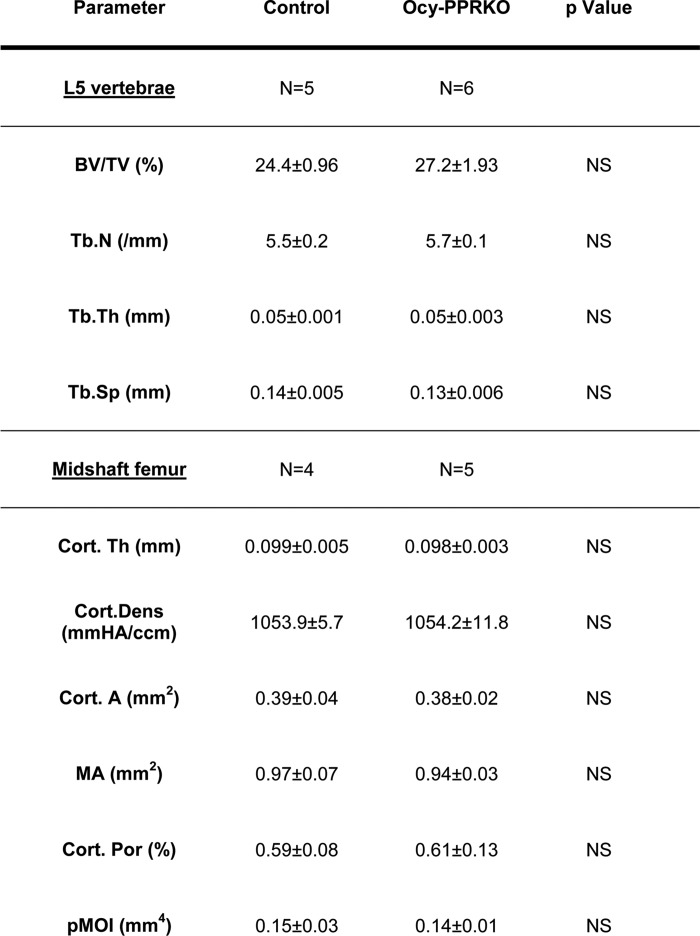
TABLE 2.
MicroCT analysis showed increased trabecular and cortical bone in 12-week-old Ocy-PPRKO females
Trabecular bone parameters were in L5 vertebrae. Cortical bone parameters were in the midshaft of femurs. Values are mean ± S.E., two-tailed t test assuming equal variance.
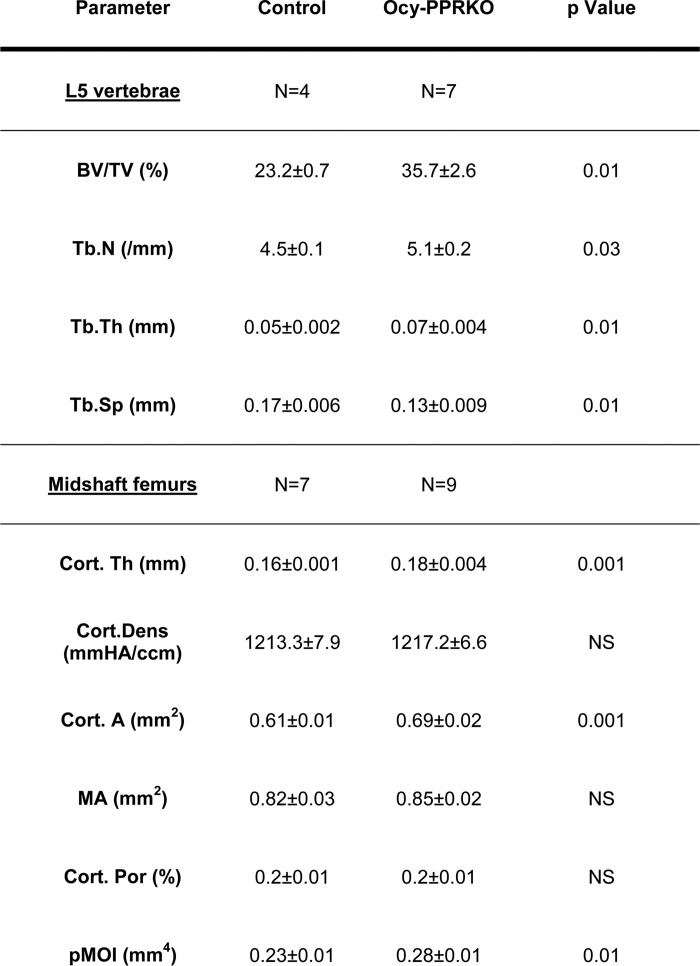
* p ≤ 0.05, and NS = not significant.
To investigate whether the increase in BMD was driven by reduced bone resorption or increased bone formation, or a combination of both, we measured, in these animals, serum markers of bone formation and bone resorption. PINP (control = 232.8 ± 26.8 ng/ml, Ocy-PPRKO = 185.1 ± 11.5 ng/ml; 12-week-old females, n ≥ 4, NS), CTX (control = 9.5 ± 1.9 ng/ml, Ocy-PPRKO = 7.0 ± 0.3 ng/ml; 12-week-old females, n ≥ 4, NS), and TRAP5b (control = 2.35 ± 0.31 units/liter, Ocy-PPRKO = 2.17 ± 0.18 units/liter; 12-week-old females, n ≥ 4, NS) were unchanged, as compared with controls.
Osteoblasts and osteoclasts number and function were reduced, although not significantly, in Ocy-PPRKO animals compared with controls (Table 5, vehicle treated animals), as demonstrated by a decrease in Ob.S/BS, N.Ob./T.Ar, N.Ob./B.Pm, osteoid surface (OS/BS), and osteoid volume (OV/TV), Oc.S/BS, N.Oc/T.Ar, N.Oc/B.Pm, and erosion surface. These findings were associated with a significant increase in both SOST mRNA expression (Fig. 2G) and in the number of Sclerostin-positive osteocytes (Fig. 3, E, top row, and F) in bones from these animals, suggesting that the suppression of the Wnt/β-catenin pathways might drive the reduced osteoblast number and activity. Reduced osteoblast function was also demonstrated by a dramatic reduction in Col1α1 mRNA expression, as shown by in situ hybridization in both trabecular and cortical bone (Fig. 3C), in Ocy-PPRKO animals compared with controls. Interestingly, a comparable number of Sclerostin-expressing osteocytes was observed in the cortical region of the vertebrae from Ocy-PPRKO and control, suggesting that cellular responses to PTH are site-specific (Fig. 3G).
FIGURE 3.
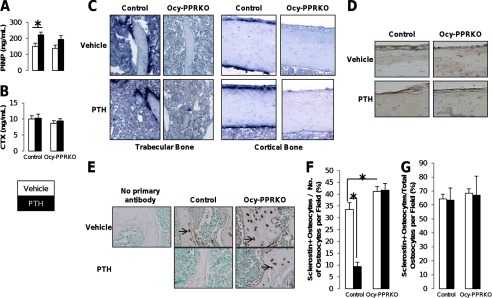
Defective osteoblast recruitment in response to intermittent PTH administration in Ocy-PPRKO. Anabolic response was analyzed in females injected with vehicle or 80 μg/kg/day hPTH(1–34). Serum levels of (A) PINP and (B) CTX (control, vehicle, n = 6; control, PTH, n = 7; Ocy-PPRKO, vehicle, n = 7; Ocy-PPRKO, PTH, n = 7). C, in situ for collagen type I α1 mRNA on tibiae. D, IHC for Periostin on tibiae. Images were acquired with a ×20 objective. E and F, lack of Sclerostin regulation in Ocy-PPRKO in response to PTH. E, representative images of sclerostin IHC in the trabecular bone in the vertebrae from females injected with vehicle or 50 nmol/kg hPTH(1–34) for 1.5 h. Arrows indicate sclerostin expression in osteocytes. Sclerostin-positive osteocytes per field in the trabecular region (F) and cortical ring (G) of the vertebrae were counted and normalized to total number of osteocytes per field. Images acquired with a ×20 objective. Values are mean ± S.E. *, p ≤ 0.05.
To examine cells of the osteoclast lineage, we analyzed RANKL and OPG expression. There was a significant decrease in both OPG (Fig. 2H) and RANKL (Fig. 2I) mRNA expression in osteocyte-enriched calvariae from Ocy-PPRKO compared with control, whereas the ratio RANKL/OPG remained unchanged. A significant 85% decrease in RANKL protein expression was also observed in osteocytes from Ocy-PPRKO as compared with control (Fig. 4, E, top row, and F).
FIGURE 4.

Blunted catabolic response in Ocy-PPRKO upon continuous PTH administration. Catabolic response was analyzed in males injected with vehicle or 100 μg/kg/day hPTH(1–34). Serum levels of PINP (A) and CTX (B) are shown (control, vehicle, n = 7; control, PTH, n = 7; Ocy-PPRKO, vehicle, n = 7–8; Ocy-PPRKO, PTH, n = 4). *, p ≤ 0.05. C, representative microCT images of mid-diaphysis to epiphysis of femurs. D, representative images of Von Kossa-stained femurs. E, representative images of RANKL IHC from tibiae. Arrows indicate osteocytes immunostained for RANKL. Images were acquired with a ×40 objective. F, RANKL-stained osteocyte area was quantified by ImageJ and normalized to total number of osteocytes per field. Values are mean ± S.E. NS, not significant. *, p ≤ 0.01.
To investigate whether the decrease in osteoclast activity was associated with the presence of unresorbed calcified cartilage, as recently reported for chondrocyte and osteoblast-specific RANKL knock-out mice (20), we performed safranin O staining on tibiae from Ocy-PPRKO and control. Growth plates in Ocy-PPRKO were indistinguishable from controls (Fig. 2F), indicating that PPR activation in osteocytes is not required for proper bone growth and cartilage resorption. Moreover, the presence of a normal growth plate demonstrates normal PPR expression and signaling in other bone cells, such as chondrocytes and osteoblasts. Altogether, these findings indicate that in the absence of PPR signaling in osteocytes there is an age-dependent increase in BMD, and in trabecular and cortical bone parameters that is associated with a decrease in osteoblast and osteoclast functions but not number.
Blunted Skeletal Response to Anabolic and Catabolic PTH Administration in Ocy-PPRKO
Ocy-PPRKO and control were then subjected to intermittent or continuous PTH treatment. As expected, intermittent hPTH(1–34) at a dose of 80 μg/kg/day induced a significant increase in vertebral BV/TV and Tb.N and a decrease in Tb.Sp (Table 3) in control. Analysis of midshaft femurs showed a significant increase in Cort.Th and Cort.A and pMOI in PTH-treated controls compared with vehicle treated animals (Table 3). In contrast, these trabecular and cortical parameters remained unchanged between vehicle- and PTH-treated Ocy-PPRKO (Table 3). No significant changes were observed in the other bone parameters between vehicle- and PTH-treated control or Ocy-PPRKO mice (Table 3). These data demonstrate that osteocyte-mediated signals are required for anabolic skeletal responses to PTH.
TABLE 3.
MicroCT analysis showed lack of anabolic response upon intermittent PTH administration in Ocy-PPRKO
Anabolic responses were analyzed in females injected with vehicle or 80 μg/kg/day hPTH(1–34). Trabecular bone parameters were in L5 vertebrae. Cortical bone parameters were in midshaft of femurs. Values are mean ± S.E., two-tailed t test assuming equal variance was performed to compare vehicle versus PTH-treated controls, and vehicle versus and PTH-treated Ocy-PPRKO.
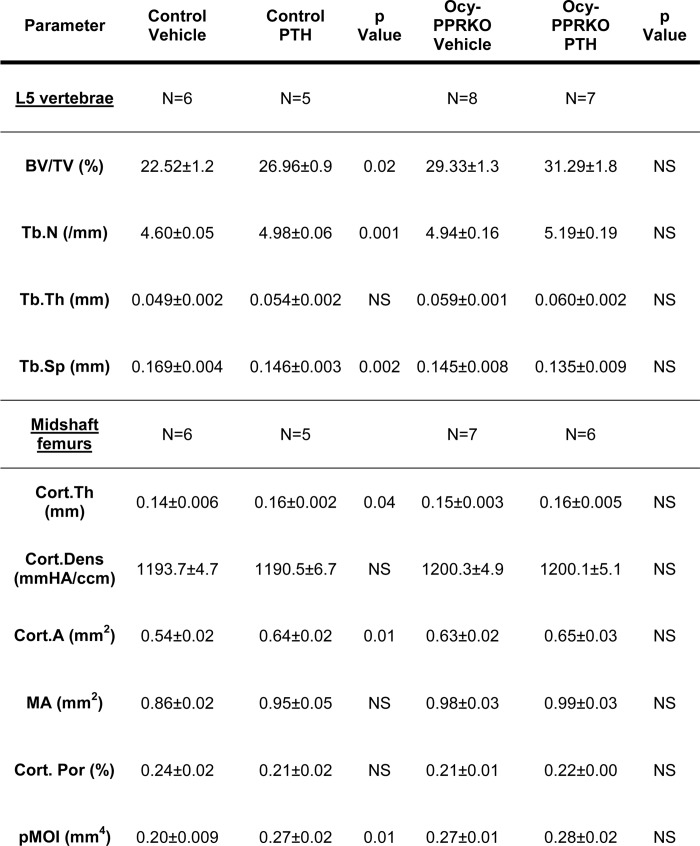
* p ≤ 0.05, and NS is not significant.
To evaluate the role of PPR signaling in osteocytes in modulating osteoblast and osteoclast activities during anabolic PTH treatment, PINP and CTX serum levels were measured. Intermittent administration of PTH significantly increased circulating PINP, a marker of osteoblast activity, in control but had no effect in Ocy-PPRKO (Fig. 3A). Serum CTX, a marker of osteoclast activity, remained unchanged in both groups (Fig. 3B). In Ocy-PPRKO, intermittent PTH failed to stimulate Collα1 mRNA expression (Fig. 3C). Periostin, a regulator of SOST/Sclerostin expression (26), was unchanged upon intermittent PTH administration both in control and Ocy-PPRKO mice (Fig. 3D). SOST/Sclerostin expression has been reported to return to base line by 24 h after PTH treatment (27), which limited our ability to analyze Sclerostin expression in intermittent PTH-treated bone samples because, as per our anabolic protocol, these were harvested 24 h after the last PTH injection. Therefore, to study Sclerostin regulation, we treated control and Ocy-PPRKO with vehicle or 50 nmol/kg hPTH(1–34) for 1.5 h. Although PTH induced a decrease in Sclerostin expression in the osteocytes from control, it failed to suppress Sclerostin expression in Ocy-PPRKO (Fig. 3, E and F). Interestingly, a similar sclerostin expression was observed in the cortical bone of vertebrae from both vehicle- and PTH-treated Ocy-PPRKO and control (Fig. 3G), suggesting a differential PPR-mediated regulation of sclerostin expression in trabecular and cortical sites.
Next, we examined the ability of Ocy-PPRKO to respond to continuous PTH administration. Mice were subjected to 2 weeks of continuous administration of 100 μg/kg/day of hPTH(1–34). MicroCT (Fig. 4C and Table 4) and Von Kossa staining (Fig. 4D) showed a catabolic response upon PTH treatment in control but not Ocy-PPRKO. PTH treatment induced a significant decrease in Tb.N and an increase in Tb.Sp in control mice, as shown by microCT analysis (Table 4). In contrast, these parameters remained unchanged between vehicle- and PTH-treated Ocy-PPRKO (Table 4), demonstrating that mice lacking PPR expression in osteocytes are resistant to continuous infusion of PTH. No significant changes were observed in other parameters between vehicle- and PTH-treated control or Ocy-PPRKO mice (Table 4).
TABLE 4.
MicroCT analysis showed lack of catabolic response to continuous PTH administration in Ocy-PPRKO
Catabolic response was analyzed in males treated with vehicle or 100 μg/kg/day of hPTH(1–34). Trabecular bone parameters were in mid-to-distal-diaphysis of femurs. Cortical bone parameters in midshaft of femurs. Values are mean ± S.E., two-way ANOVA with Tukey HSD.
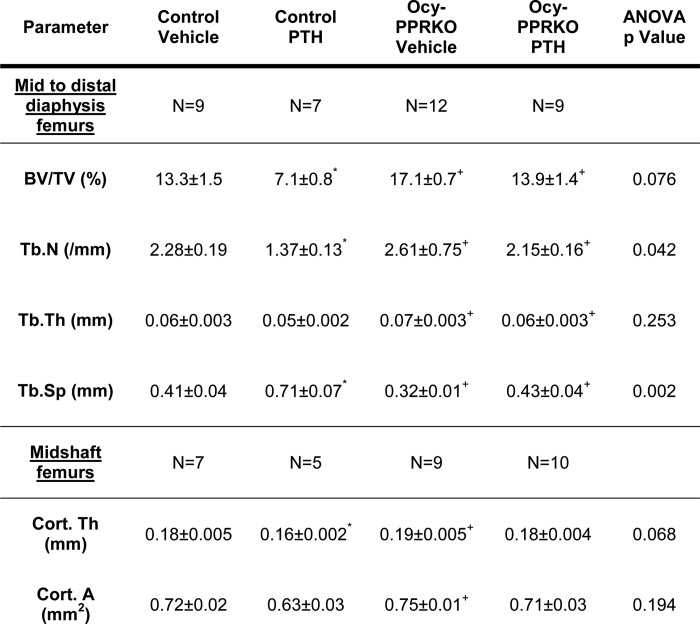
* p < 0.01 compared with control vehicle.
+ p < 0.01 compared with control PTH.
# p < 0.01 compared with Ocy-PPRKO.
Histomorphometric analysis demonstrated a significant increase in both osteoblast and osteoclast number and function, upon PTH treatment in controls, as demonstrated by a significant increase in Ob.S/BS, N.Ob./T.Ar, N.Ob./B.Pm, osteoid surface (OS/BS), osteoid volume (OV/TV), Oc.S/BS, N.Oc/T.Ar, N.Oc/B.Pm, and erosion surface (Table 5). None of these parameters were significantly changed in Ocy-PPRKO animals upon PTH administration (Table 5), indicating that deletion of PPR in osteocytes blunts the skeletal response to continuous PTH. Interestingly, PTH did significantly decrease Tb.N in Ocy-PPRKO mice and not in controls; however, this change was not associated with a decrease (or change) in any other bone parameter (Table 5). Moreover, histomorphometry further confirmed the increased BV/TV and Tb.N and the decreased Tb.Sp in Ocy-PPRKO compared with controls (Table 5).
Serum marker of osteoblast activity, PINP, was significantly elevated in PTH-treated control but remained unchanged in Ocy-PPRKO, further confirming the lack of PTH responsiveness in Ocy-PPRKO (Fig. 4A). Interestingly, serum levels of CTX, a marker of bone resorption, were significantly increased in Ocy-PPRKO (p < 0.05), and in controls the increase (167%) did not reach significance (p = 0.0508) (Fig. 4B). These results suggest that PTH-induced CTX elevation is independent of PPR signaling in osteocytes and most likely reflect a osteoblast-mediated, PTH-dependent mechanism(s).
To investigate if the lack of response to continuous PTH was due to an osteoclast defect, we analyzed the osteoclast-forming ability of osteocytes from Ocy-PPRKO and control in vitro. OEBE from Ocy-PPRKO and control were co-cultured with osteoclast precursors derived from wild-type spleens. Upon PTH treatment, multinucleate osteoclasts (MNC) were observed in both cultures (Fig. 5A). However, the number of MNC formed in Ocy-PPRKO cultures was significantly reduced (Fig. 5B; control MNC/mg OEBE = 4.6 ± 0.2, Ocy-PPRKO MNC/mg OEBE = 1.8 ± 0.2, p = 0.001). Importantly, OPG completely abrogated the osteoclastogenic ability of both Ocy-PPRKO and control OEBE (Fig. 5, A and B), suggesting a RANKL-dependent mechanism. The number of multinucleated osteoclasts formed upon 1,25-dihydroxyvitamin D treatment was similar in both Ocy-PPRKO and control cultures (Fig. 5), demonstrating reduced osteoclast formation in Ocy-PPRKO because of ablated PPR signaling.
FIGURE 5.

Reduced osteoclast formation in Ocy-PPRKO upon PTH stimulation in vitro. A, representative images of TRAP+ multinucleate osteoclasts induced by OEBE from Ocy-PPRKO and control when co-cultured with osteoclast precursors (Oc precursors) with or without hPTH(1–34), OPG, and vitamin D (vitD). Images were acquired with a ×10 objective. B, number of TRAP+ multinucleate osteoclasts induced by OEBE from Ocy-PPRKO and control. Values are mean ± S.E. NS, not significant. *, p ≤ 0.01.
To further investigate the reduced osteoclast activity in response to PTH observed by histomorphometry (Table 5), we analyzed RANKL expression in osteocytes upon catabolic PTH administration. ANOVA showed a significant difference in genotypes but not the PTH treatments or genotype and PTH treatment interaction. Importantly, Tukey HSD identified a significantly higher RANKL expression in both vehicle- and PTH-treated control osteocytes as compared with both vehicle- and PTH-treated Ocy-PPRKO osteocytes (Fig. 4, E and F). To further assess the role of RANKL in these animals, we measured its level in the serum. There was no difference in circulating RANKL between control and Ocy-PPRKO animals, and PTH treatment had no effect (control-veh = 129.7.8 ± 10.9 pg/ml, control-PTH = 101.78 ± 18.6 pg/ml, p = 0.19, NS; Ocy-PPRKO-veh = 98.8 ± 7.8 pg/ml, Ocy-PPRKO-PTH = 107.4 ± 20.5 pg/ml; p = 0.65, NS), suggesting that circulating RANKL might not reflect the local effects of this cytokine. RANKL levels in bone marrow supernatant of femurs and tibias of Ocy-PPRKO and controls were below the limit of detection of the assay used. Moreover, RANKL mRNA expression in RNA isolated from intact bones (including both bone marrow and osteoblasts) showed no changes in vehicle- or PTH-treated control and Ocy-PPRKO (relative % RANKL mRNA expression: control-veh = 100.0 ± 12.0%, control-PTH = 83.0 ± 2.9%, p = 0.198, NS; Ocy-PPRKO-veh = 53.4 ± 5.2%, Ocy-PPRKO-PTH = 86.2 ± 22.7%; p = 0.270, NS). These findings are in agreement with previous studies (20) and suggest that the contribution of RANKL produced by osteocytes is small relative to the total RANKL produced, yet its biological effects can be detected as lack of PTH catabolic actions. In the absence of PPR signaling in osteocytes, continuous PTH administration fails to increase both osteoblasts and osteoclasts and induce a skeletal effect.
DISCUSSION
It has been known for over a decade that PTH can exert its anabolic or catabolic effect on bone via activation of the PPR widely expressed on cells of the osteoblast lineage; however, the exact cellular target of its action on bone has remained elusive. Several mechanisms have been proposed, and they include recruitment and proliferation of osteoprogenitors cells, activation of bone lining cells (28, 29), inhibition of osteoblast and osteocyte apoptosis (30), and suppression of SOST/Sclerostin expression (27). Osteocytes, the most abundant cells in bone, have recently emerged as important modulators of bone modeling and remodeling (5). In this regard, the molecular roles of sclerostin and RANKL, two predominantly osteocytic skeletal factors, have been reported (5). Interestingly, both molecules are known targets of PTH actions with SOST/Sclerostin being suppressed and RANKL being increased upon receptor activation.
Here, using a murine model of PPR ablation in osteocytes, we provide, for the first time, evidence that receptor signaling in these cells is important to control both osteoblast and osteoclast functions via SOST/Sclerostin and RANKL-mediated mechanisms. Importantly, our studies demonstrated that PPR signaling in osteocytes is required for generating full skeletal responses to anabolic and catabolic PTH administration. During the PTH anabolic regimen, Sclerostin suppression is needed to allow a full hormonal effect and, in the absence of PPR signaling in osteocytes, PTH has little, if any, skeletal effects.
It has been previously reported that mice expressing a constitutively active PPR in osteocytes (DMP1-CaPTHR1) have increased trabecular and cortical bone (8, 31), suggesting that receptor signaling in osteocytes drives bone formation. Interestingly, mice lacking receptor expression in osteocytes, the Ocy-PPRKO mice, also displayed increased trabecular and cortical bone. The apparently similar phenotype, i.e. increase in trabecular and cortical bone parameters, observed in murine models of constitutive expression (DMP1-CaPTHR1) or deletion (Ocy-PPRKO) of PPR in osteocytes is driven by two distinct mechanisms. The increased trabecular and cortical bone in DMP1-CaPTHR1 is a result of increased osteoblast and osteoclast activities, increased bone formation rate, and high bone turnover. In contrast, the increased bone in the Ocy-PPRKO is the net result of reduced osteoblast and osteoclast function and low bone turnover state. Bone formation rate in Ocy-PPRKO mice is not changed, as is the mineral apposition rate (data not shown). Histomorphometric analysis showed a reduced, although not significant, number of both osteoblasts and osteoclast, and upon PTH administration, this cellular defect became evident (see Table 5). We can hypothesize that, under normal conditions, the lack of receptor expression in osteocytes does not significantly alter the number of osteoblasts and osteoclasts but reduces their function with a net increase in BMD, as demonstrated by both DXA, microCT, and histomorphometric analysis. However, when Ocy-PPRKO animals were subjected to a skeletal perturbation (such as intermittent or continuous PTH administration), they failed to properly increase both osteoblast and osteoclast numbers and activities as shown by histomorphometric analysis (Table 5), expression of Col1α1 (Fig. 3C), and Sclerostin (Fig. 3E). Interestingly, continuous administration of PTH failed to induce trabecular bone loss or increase osteoclast number in Ocy-PPRKO animals despite a significant increase in serum CTX. We can speculate that CTX might derive from skeletal sources other than the one analyzed or from the modest increase in osteoclast numbers detected by histomorphometry (Table 5).
We previously reported that mice with inducible PPR deletion (postnatally, Ocy-PPRcKO) (4) have mild osteopenia and homeostatic defects, whereas constitutive ablation of the receptor, as achieved in Ocy-PPRKO, induce an increase in BMD. We can speculate that the difference between Ocy-PPRcKO and Ocy-PPRKO could be ascribed mostly to the timing of receptor ablation (postnatally versus embryonic), different promoter activities, or tamoxifen effects independent of PPR activation. Moreover, in the conditional PPR knock-out, the severity of the phenotype might depend on the age of mice when the tamoxifen treatment was started, the dose of tamoxifen administered, and the duration of receptor ablation. To investigate whether a transient osteopenia was present in the Ocy-PPRKO as well, we performed microCT analysis on L5 vertebrae and femurs of mice at ∼5 weeks of age (similar age as for the inducible animals). As reported in Table 1, there were no detectable skeletal differences between controls and Ocy-PPRKO animals, suggesting that timing of receptor ablation, promoter activity, and tamoxifen effects might drive the osteopenic phenotype in the inducible model.
Increased osteoblast function in DMP1-CaPTHR1 was associated with decreased Sclerostin expression (31). Similarly, decreased osteoblast function in Ocy-PPRKO was associated with increased Sclerostin expression, supporting the concept that regulation of Sclerostin expression is an important effector of PPR signaling in osteocytes.
Osteocytic regulation of osteoclasts through osteoblast-mediated mechanism is well understood (5). Emerging literature, however, suggested that osteocytes could directly regulate osteoclasts. Apoptotic osteocytes release apoptotic bodies expressing RANKL, which, in turn, activate osteoclasts (6). Furthermore, targeted ablation of osteocytes using the 10-kb DMP1 promoter to drive the expression of diphtheria toxin receptor expression followed by a single injection of diphtheria toxin caused necrosis of 70% osteocytes in cortical bone and activated osteoclasts (6), demonstrating osteocytes mediated osteoclast regulation. Moreover, murine model of osteocyte-specific RANKL ablation showed osteopetrosis, demonstrating a critical role for osteocyte-derived RANKL in osteoclast function and ultimately bone remodeling (19, 20). In Ocy-PPRKO mice, we observed significantly reduced RANKL expression in osteocytes and reduced osteoclastogenesis in PTH-treated OEBEs from these animals, suggesting that the reduced osteoclast number upon continuous PTH administration and their activity is likely RANKL-dependent. Interestingly, upon continuous PTH administration, RANKL mRNA was not significantly increased in intact bones (including osteoblasts and bone marrow) of both controls and Ocy-PPRKO nor was it increased in osteocyte-enriched bones.
To investigate the role of PPR signaling in osteocytes in anabolic and catabolic skeletal effects, we subjected the Ocy-PPRKO mice to intermittent or continuous PTH regimens. Interestingly, both the anabolic and catabolic skeletal responses to PTH were blunted in Ocy-PPRKO mice, indicating an important role of PPR signaling in osteocytes in mediating the skeletal responses to the hormone. Immunohistochemical analysis of tibiae from Ocy-PPRKO mice revealed a lack of Sclerostin suppression or Col1α1 expression in response to PTH administration. We can hypothesize that in the absence of receptor signaling in osteocytes, PTH failed to suppress sclerostin and to increase osteoblast activity and, ultimately, induce an anabolic response. Moreover, lack of PPRs also significantly reduced RANKL expression in osteocytes and impaired osteoclast activities and ultimately bone remodeling (Fig. 6).
FIGURE 6.

PPR signaling in osteocytes directly regulates osteoblasts and osteoclasts. A, osteocytes have been reported to regulate osteoblasts, which in turn regulate osteoclasts via RANKL expression. Recent evidence suggests that RANKL in osteocytes regulates osteoclasts. B, in our murine model of constitutive PPR deletion in osteocytes, increased SOST mRNA and Sclerostin protein expression and decreased osteoblasts were observed. In addition, decreased RANKL mRNA and protein expression and decreased osteoclasts were observed.
In summary, we have shown here, for the first time, that PPR signaling in osteocytes is required for anabolic and catabolic responses to PTH.
Acknowledgments
We thank Dr. Henry M. Kronenberg, Endocrine Unit, Massachusetts General Hospital, for critical review of this manuscript, Drs. Tomoki Nakashima and Hiroshi Takayanagi from Tokyo Medical and Dental University for technical recommendation for the RANKL IHC, and Dr. Mary Bouxsein and Leeann Louis from Massachusetts General Hospital for suggestions on microCT analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant DK079161 from the NIDDK (to P. D. P.).
- PTH
- parathyroid hormone
- PTHrP
- PTH-related peptide
- PPR
- PTH/PTHrP type 1 receptor
- Ocy-PPRKO
- PPR deletion in osteocytes
- RANKL
- receptor activator of NF-κB ligand
- BMD
- bone mineral density
- ANOVA
- analysis of variance
- Tb.N
- trabecular number
- BS
- bone surface
- B.Pm
- bone perimeter
- T.Ar
- tissue area
- BV
- bone volume
- TV
- tissue volume
- Cort.Th
- cortical thickness
- Cort.A
- cortical area
- pMOI
- polar moment of inertia
- TRAP
- tartrate-resistant acid phosphatase
- veh
- vehicle
- OEBE
- osteocyte-enriched long bone explant
- IHC
- immunohistochemistry
- DXA
- dual x-ray absorptiometry
- Col1α1
- collagen type I α1
- NS
- not significant
- MNC
- multinucleate osteoclast
- PINP
- procollagen type 1 amino-terminal propeptide
- CTX
- carboxyl-terminal telopeptide, type 1
- microCT
- micro-computed tomography
- Tb.Sp
- trabecular separation
- OPG
- osteoprotegerin
- N.Ob.
- number of osteoblast
- Tb.Th
- trabecular thickness.
REFERENCES
- 1. Jüppner H., Abou-Samra A. B., Freeman M., Kong X. F., Schipani E., Richards J., Kolakowski L. F., Jr., Hock J., Potts J. T., Jr., Kronenberg H. M. (1991) A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 254, 1024–1026 [DOI] [PubMed] [Google Scholar]
- 2. Finkelstein J. S., Klibanski A., Schaefer E. H., Hornstein M. D., Schiff I., Neer R. M. (1994) Parathyroid hormone for the prevention of bone loss induced by estrogen deficiency. N. Engl. J. Med. 331, 1618–1623 [DOI] [PubMed] [Google Scholar]
- 3. Riancho J. A., Hernández J. L. (2012) Pharmacogenomics of osteoporosis: a pathway approach. Pharmacogenomics 13, 815–829 [DOI] [PubMed] [Google Scholar]
- 4. Powell W. F., Jr., Barry K. J., Tulum I., Kobayashi T., Harris S. E., Bringhurst F. R., Pajevic P. D. (2011) Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 209, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schaffler M. B., Kennedy O. D. (2012) Osteocyte signaling in bone. Curr. Osteoporos. Rep. 10, 118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonewald L. F. (2011) The amazing osteocyte. J. Bone Miner. Res. 26, 229–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qing H., Ardeshirpour L., Pajevic P. D., Dusevich V., Jähn K., Kato S., Wysolmerski J., Bonewald L. F. (2012) Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 27, 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rhee Y., Allen M. R., Condon K., Lezcano V., Ronda A. C., Galli C., Olivos N., Passeri G., O'Brien C. A., Bivi N., Plotkin L. I., Bellido T. (2011) PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 26, 1035–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leupin O., Kramer I., Collette N. M., Loots G. G., Natt F., Kneissel M., Keller H. (2007) Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone Miner. Res. 22, 1957–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kramer I., Baertschi S., Halleux C., Keller H., Kneissel M. (2012) Mef2c deletion in osteocytes results in increased bone mass. J. Bone Miner. Res. 27, 360–373 [DOI] [PubMed] [Google Scholar]
- 11. Genetos D. C., Toupadakis C. A., Raheja L. F., Wong A., Papanicolaou S. E., Fyhrie D. P., Loots G. G., Yellowley C. E. (2010) Hypoxia decreases sclerostin expression and increases Wnt signaling in osteoblasts. J. Cell. Biochem. 110, 457–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collette N. M., Genetos D. C., Economides A. N., Xie L., Shahnazari M., Yao W., Lane N. E., Harland R. M., Loots G. G. (2012) Targeted deletion of SOST distal enhancer increases bone formation and bone mass. Proc. Natl. Acad. Sci. U.S.A. 109, 14092–14097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semënov M., Tamai K., He X. (2005) SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 280, 26770–26775 [DOI] [PubMed] [Google Scholar]
- 14. Li X., Zhang Y., Kang H., Liu W., Liu P., Zhang J., Harris S. E., Wu D. (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 280, 19883–19887 [DOI] [PubMed] [Google Scholar]
- 15. Semenov M. V., He X. (2006) LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J. Biol. Chem. 281, 38276–38284 [DOI] [PubMed] [Google Scholar]
- 16. Baron R., Rawadi G. (2007) Targeting the Wnt/β-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148, 2635–2643 [DOI] [PubMed] [Google Scholar]
- 17. Fu Q., Jilka R. L., Manolagas S. C., O'Brien C. A. (2002) Parathyroid hormone stimulates receptor activator of NFκB ligand and inhibits osteoprotegerin expression via protein kinase A activation of cAMP-response element-binding protein. J. Biol. Chem. 277, 48868–48875 [DOI] [PubMed] [Google Scholar]
- 18. Huang J. C., Sakata T., Pfleger L. L., Bencsik M., Halloran B. P., Bikle D. D., Nissenson R. A. (2004) PTH differentially regulates expression of RANKL and OPG. J. Bone Miner. Res. 19, 235–244 [DOI] [PubMed] [Google Scholar]
- 19. Nakashima T., Hayashi M., Fukunaga T., Kurata K., Oh-Hora M., Feng J. Q., Bonewald L. F., Kodama T., Wutz A., Wagner E. F., Penninger J. M., Takayanagi H. (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 17, 1231–1234 [DOI] [PubMed] [Google Scholar]
- 20. Xiong J., Onal M., Jilka R. L., Weinstein R. S., Manolagas S. C., O'Brien C. A. (2011) Matrix-embedded cells control osteoclast formation. Nat. Med. 17, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi T., Chung U. I., Schipani E., Starbuck M., Karsenty G., Katagiri T., Goad D. L., Lanske B., Kronenberg H. M. (2002) PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development 129, 2977–2986 [DOI] [PubMed] [Google Scholar]
- 23. Parfitt A. M. (1988) Bone histomorphometry: proposed system for standardization of nomenclature, symbols, and units. Calcif. Tissue Int. 42, 284–286 [DOI] [PubMed] [Google Scholar]
- 24. Bouxsein M. L., Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., Müller R. (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468–1486 [DOI] [PubMed] [Google Scholar]
- 25. Fulzele K., Krause D. S., Panaroni C., Saini V., Barry K. J., Liu X., Lotinun S., Baron R., Bonewald L., Feng J. Q., Chen M., Weinstein L. S., Wu J. Y., Kronenberg H. M., Scadden D. T., Divieti Pajevic P. (2013) Myelopoiesis is regulated by osteocytes through Gsα-dependent signaling. Blood 121, 930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonnet N., Conway S. J., Ferrari S. L. (2012) Regulation of β-catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc. Natl. Acad. Sci. U.S.A. 109, 15048–15053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bellido T., Ali A. A., Gubrij I., Plotkin L. I., Fu Q., O'Brien C. A., Manolagas S. C., Jilka R. L. (2005) Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146, 4577–4583 [DOI] [PubMed] [Google Scholar]
- 28. Kim S. W., Pajevic P. D., Selig M., Barry K. J., Yang J.-Y., Shin C. S., Baek W.-Y., Kim J.-E., Kronenberg H. M. (2012) Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 27, 2075–2084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobnig H., Turner R. T. (1995) Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology 136, 3632–3638 [DOI] [PubMed] [Google Scholar]
- 30. Jilka R. L., Weinstein R. S., Bellido T., Roberson P., Parfitt A. M., Manolagas S. C. (1999) Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 104, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Brien C. A., Plotkin L. I., Galli C., Goellner J. J., Gortazar A. R., Allen M. R., Robling A. G., Bouxsein M., Schipani E., Turner C. H., Jilka R. L., Weinstein R. S., Manolagas S. C., Bellido T. (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One 3, e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]