

Summary
Inflammasome assembly activates caspase-1 and initiates the inflammatory cell death program pyroptosis, which is protective against numerous pathogens. Consequently, several pathogens, including the plague causing bacterium Yersinia pestis, avoid activating this pathway to enhance their virulence. However, bacterial molecules that directly modulate the inflammasome have yet to be identified. Examining the contribution of Yersinia type III secretion effectors to caspase-1 activation, we identified the leucine-rich repeat effector YopM as a potent antagonist of both caspase-1 activity and activation. YopM directly binds caspase-1, which both inhibits caspase-1 activity and sequesters it to block formation of the mature inflammasome. Caspase-1 activation antagonizes Yersinia survival in vivo and consequently YopM inhibition of caspase-1 is required for Yersinia pathogenesis. Thus, a bacterium obstructs pyroptosis utilizing a direct mechanism of caspase-1 inhibition which is distinct from known viral or host inhibitors.
Introduction
Pyroptosis is an inherently inflammatory program of cell death directed by the cysteine protease caspase-1. Activation of caspase-1 is protective against numerous pathogens but aberrant activation or dysregulation is harmful to the host and involved in several inflammatory disorders (Bergsbaken et al., 2009). Correspondingly, this system is controlled by nucleotide-binding domain, leucine-rich-repeat- containing (NLR) family of proteins that detect pathogen-associated molecular patterns (PAMPs) and initiate a response to an invading pathogen by activating caspase-1. The resulting cytokine release and cell lysis creates an inflammatory environment directing immune responses to the site of infection. Accordingly, several pathogens antagonize this pathway to enhance their virulence (Ashida et al., 2011).
The etiologic agent of plague, Yersinia pestis, and the closely related pathogen, Y. pseudotuberculosis (Ypstb), initially replicate in the host without inducing inflammation (Bergsbaken and Cookson, 2007; Lathem et al., 2005; Price et al., 2012). Yersinia promote their survival by both evading detection and actively subverting immune signaling. Yersinia target several cell signaling pathways, through multiple type III secretion (T3S) system translocated effector proteins: YopO, YopE, YopM, YopH, and YopJ (Trosky et al., 2008). One consequence of T3S by pathogenic bacteria is the cytosolic delivery of microbial factors that act as PAMPs for the activation of caspase-1 (Lamkanfi and Dixit, 2009). While Yersinia can activate caspase-1, the specific factors detected as PAMPs have not yet been identified (Bergsbaken and Cookson, 2007; Brodsky et al., 2010).
One factor altering caspase-1 activation during Yersinia infection is YopK. This T3S regulator restricts effector translocation into host cells, likely limiting PAMP translocation, and consequently limiting caspase-1 activation as well (Brodsky et al., 2010; Dewoody et al., 2011). We examined Yersinia effectors individually for repression of caspase-1 activation, and identified YopM as a potent antagonist of both caspase-1 activity and activation. We demonstrate that activation of caspase-1 is detrimental to Yersinia survival in the host; cell death and inflammation limits bacterial replication in vivo and promotes host survival. Consequently, YopM inhibition of caspase-1 is a requirement of Yersinia pathogenesis. YopM acts by directly binding caspase-1 to block caspase-1 activity and this binding also sequesters caspase-1 and aborts inflammasome formation. The resulting “pre-inflammasome” contains NLR and the adapter protein ASC, but not caspase-1, and appears to be an intermediate step of normal inflammasome development. Together these data indicate that repression of inflammation by YopM is important for potentiating the virulence of the deadly pathogen Yersinia pestis.
Results
YopM prevents macrophage pyroptosis in response to Yersinia infection
During infection with Ypstb or Y. pestis, caspase-1 activation occurs predominantly in activated cells and either TLR2 or TLR4 stimuli are sufficient signals for this priming (Bergsbaken and Cookson, 2007). We observe caspase-1 activation is accelerated in macrophages infected with YersiniaΔ (Figure 1a, Fig S1), a Ypstb mutant competent for type III secretion but lacking effector proteins. We therefore hypothesized that one or more effector blocks caspase-1 activation. YopJ induces death by caspase-1-independent apoptosis (Bergsbaken and Cookson, 2007) and a YopJ allele unique to Y. pestis KIM activates caspase-1 (Lilo et al., 2008), however, YopJ deletion in Ypstb did not impact caspase-1 activation (Figure 1B). The effectors YopO, YopE, and YopH also did not alter caspase-1 activation (Figure 1B). YopK mutant bacteria activated greater caspase-1 (Figure 1B), likely due to its regulatory role in effector translocation (Brodsky et al., 2010; Dewoody et al., 2011). Significantly enhanced caspase-1 activation also occurred upon deletion of YopM (Figure 1B, Fig S1), a protein of enigmatic function, but nonetheless linked with anti-inflammatory activity (Leung et al., 1990; McCoy et al., 2010; McDonald et al., 2003). Complementation with YopM from Ypstb YPIII or Y. pestis CO92 (99.5% amino acid identity), depressed caspase-1 activation by YersiniaΔ (Figure 1C). Thus, YopM can act independently of other effectors and has conserved function between species, suggesting YopM is important for both pathogens.
Figure 1. see also Figure S1. YopM prevents caspase-1 induction of pyroptosis.
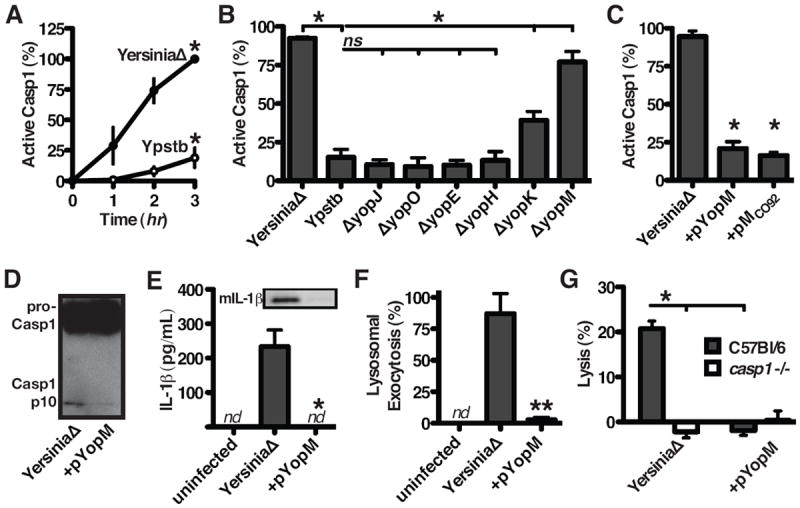
(A to E) Macrophages were infected with the Ypstb strains indicated (A to C) Cells with active caspase-1 were counted at the times indicated (A) or after 90 minutes of infection (B and C). (D) Caspase-1 immunoblot of infected cells indicates YopM prevents caspase-1 maturation. (E) ELISA shows release of IL-1β, confirmed by western blot for mature IL-1β, during YersiniaΔ infection but not during infection with YersiniaΔ expressing YopM in trans (+pYopM). (F) Lysosomal exocytosis, determined by immunofluorescence staining of surface LAMP1 on intact cells, was induced by infection and blocked by YopM. (G) Cellular lysis was measured from macrophages infected 2 hrs with Ypstb; lysis was caspase-1-dependent (indicating pyroptosis) and blocked by YopM. “ns”, p>0.05; “nd”, none detectable; data in A, B, C, E, F, and G are presented as mean +/ - SEM.
Caspase-1 activation is typically accompanied by processing of caspase-1 into p20 and p10 subunits, which is inhibited by YopM (Figure 1D). Active caspase-1 is required for activation of the pro-inflammatory cytokines IL-1β (Bergsbaken et al., 2009), and release of bioactive IL-1β is also prevented by YopM (Figure 1E). Additionally, lysosomal exocytosis, a caspase-1-dependent release of antimicrobial factors that can act on extracellular bacteria (Bergsbaken et al., 2011), is blocked by YopM (Figure 1F). The terminal cellular event directed by caspase-1 is lysis, where the release of inflammatory cellular contents amplifies local inflammatory responses (Bergsbaken et al., 2009; Lamkanfi et al., 2010) and deprives intracellular pathogens a replicative niche (Miao et al., 2010). YersiniaΔ induces lysis, prevented by YopM (Figure 1G). Infection with YersiniaΔ does not induce lysis of macrophages from casp1-/- mice (which are also casp11-/-, an upstream activator of caspase-1 in some circumstances (Kayagaki et al., 2011)) (Figure 1G). Together these data indicate YopM, delivered during Yersinia infection into relevant host target cells (Marketon et al., 2005), blocks important antimicrobial responses directed by caspase-1.
YopM promotes virulence by inhibiting caspase-1 in vivo
Timely inflammation is important in the host’s response to infection, suggesting that the ability of Yersinia to inhibit caspase-1 could impact bacterial virulence in vivo. YopM mutants are highly attenuated (Leung et al., 1990; McCoy et al., 2010; McPhee et al., 2010; Ye et al., 2009), and our in vitro observations (Figure 1) suggested attenuation may result from the inability to inhibit caspase-1. Two orders of magnitude more Ypstb were recovered from wild-type mice when the bacteria expressed YopM (Figure 2A), highlighting the requirement of YopM during infection. In caspase-1-/- mice, growth of ΔyopM Ypstb was restored and comparable to WT Ypstb (Figure 2A), indicating a major function of YopM in vivo is preventing caspase-1 activation. Wild-type and caspase-1-/- mice are both susceptible to WT (YopM+) Ypstb (Figure 2B). Wild-type (caspase-1+/+) mice survive infection with ΔyopM Ypstb but there is attenuation reversal of ΔyopM Ypstb in caspase-1-/- mice (Figure 2C). These results demonstrate that Ypstb is sensitive to the potent anti-microbial responses directed by caspase-1, thus YopM inhibition of caspase-1 in vivo is required for virulence.
Figure 2. YopM is required in vivo for inhibition of caspase-1.
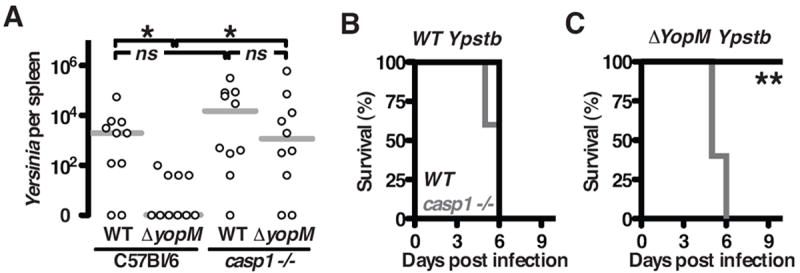
(A) Four days post intra-peritoneal inoculation with 1000 CFU of Ypstb, few bacteria were recovered from the spleens of C57BL/ 6 infected with ΔyopM Ypstb, while this strain colonized casp1-/- mice similar to fully virulent Ypstb; each point represents a single mouse. (B) WT Ypstb is lethal to both wild-type C57BL/ 6 mice (black) and casp1-/- mice (gray). (C) However, only casp1-/- mice are susceptible to ΔyopM Ypstb. Results are from combined experiments; asterisks indicate statistical differences by nonparametric Mann–Whitney U-test (titers, A) or Wilcoxon test (survival curves, B and C), *P < 0.05, **P < 0.005.
YopM binds caspase-1 to inhibit activity
Caspase-1 is activated in the inflammasome, a macromolecular complex formed by NLR family proteins in response to PAMP stimuli (Bauernfeind et al., 2009; Martinon et al., 2002). The NLR protein NLRC4 detects bacterial flagellin; NLRP3 activates caspase-1 in response to numerous stimuli including inorganic irritants, pore-forming toxins, and nigericin; and NLRP1 responds to Bacillus anthracis lethal toxin (LT). Transduced YopM protects macrophages from caspase-1 activation in response to all of these stimuli (Figure 3A), indicating YopM is sufficient to block caspase-1 activation triggered by multiple NLRs and diverse cognate PAMPs.
Figure 3. see also Figure S2. YopM is a pseudosubstrate inhibitor of caspase-1 activity.
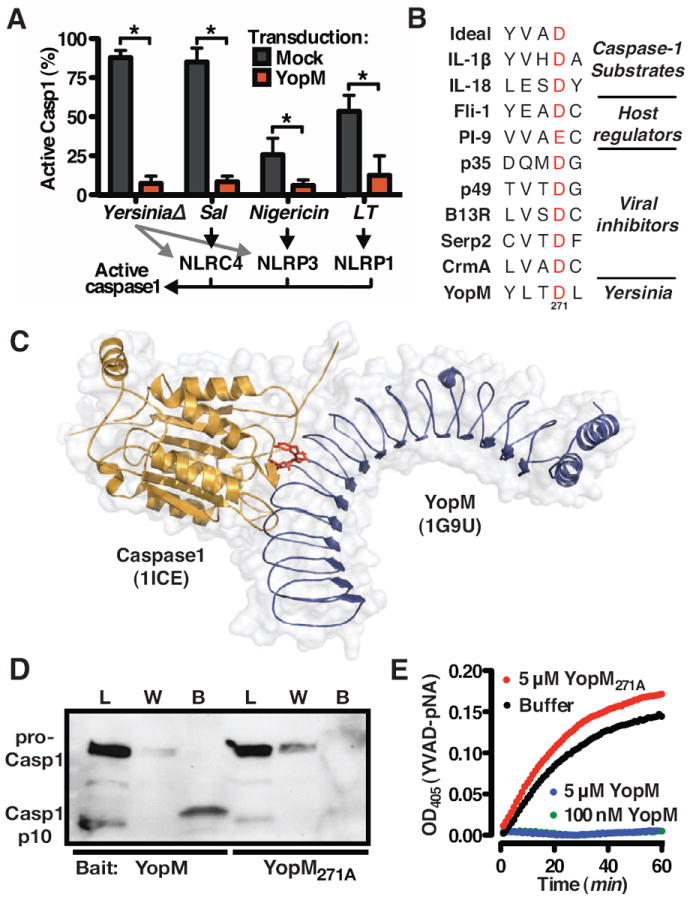
(A) Macrophages were challenged or treated with YersiniaΔ, Salmonella typhimurium, nigericin, or anthrax LT (10); retroviral transduction of YopM blocks caspase-1 activation in response to each of these stimuli; data are presented as mean +/ - SEM. (B) The conserved substrate recognition site bound by caspase-1 for selected known substrates and inhibitors, aligned to the similar sequence identified in YopM. The highly conserved aspartic acid is highlighted in red. (C) Model of YopM (blue, pdb: 1G9U) docking Caspase-1 (yellow, pdb: 1BMQ) via pseudosubstrate site (red). (D) Lysates from macrophages undergoing pyroptosis (infected with YersiniaΔ, lacking YopM) were incubated with 6xHis-YopM or 6xHis-YopM271A as bait for pull-down and immunoblot detection of caspase-1. The sample loaded (L) contains both the 45kDa procaspase-1 as well as the cleaved p10 subunit indicative of mature caspase-1. Caspase-1 was detected in the wash volumes (W) with YopM271A but not YopM; the bound fraction (B) contained caspase-1 only when YopM was used as bait. Enrichment of p10 over procaspase-1 in the YopM-bound (B) fraction compared with the loading (L) indicates YopM may have a higher affinity for mature caspase-1. (E) YopM, but not YopM271A, blocks proteolysis of caspase-1 substrate YVAD-pNA.
Avoiding caspase-1 activation is a beneficial strategy for many pathogenic bacteria (Bergsbaken et al., 2009; Miao et al., 2010), although bacterial inhibitors specifically antagonizing caspase-1 have yet to be described. For viruses, which similarly benefit from avoiding or delaying inflammatory responses, several proteins structurally resembling endogenous host regulators block caspase-1 activity (Lamkanfi and Dixit, 2009; Nathaniel et al., 2004). We identified a four amino acid sequence in a exposed loop of YopM similar to caspase-1 substrate YVAD and to endogenous and virus-encoded inhibitors of caspase-1 (Figure 3B). Immature and active caspase-1 both bind substrates with equal affinity (Elliott et al., 2009); pseudosubstrate inhibitors can block either the activation or activity of caspase-1 (Li et al., 2008), suggesting YopM may similarly bind and inactivate caspase-1 (Figure 3C).
Using recombinant YopM as bait to pull-down proteins from lysates of YersiniaΔ-infected macrophages undergoing pyroptosis, confirms YopM binds endogenous caspase-1 (Figure 3D). Caspase-1 substrates and pseudosubstrate inhibitors contain an aspartic acid residue critical for the protein:protein interaction (Li et al., 2008; Thornberry et al., 1997); altering this amino acid of YopM (Figure 3B) renders the protein (YopM271A) incapable of binding caspase-1 (Figure 3D). Since YopM preferentially binds cleaved, active caspase-1 (Figure 3D), we determined whether substrate hydrolysis by caspase-1 is blocked by YopM binding. YopM, but not YopM271A, directly blocks substrate cleavage by purified active caspase-1 (Figure 3E) without being cleaved itself (Fig S2), indicating that YopM and caspase-1 directly interact, that YopM occupies the substrate site of caspase-1, and that a pseudosubstrate sequence of YopM is required for binding caspase-1.
YopM blocks formation of mature inflammasomes
During pyroptosis, NLRs complex with the adapter ASC to form a structure where pro-caspase-1 is cleaved into an active state (Bauernfeind et al., 2009; Martinon et al., 2002; Stehlik et al., 2003). In vivo, these inflammasome structures are visualized as a macromolecular complex containing active caspase-1 (Fink et al., 2008). We hypothesized that YopM binding of caspase-1 might alter the formation or content of natural inflammasomes formed in response to infection. Ypstb induces formation of NLRP3 foci, a process unaltered by YopM (Figure 4A and 4B). NLRP3 requires the adapter protein ASC to bind and activate caspase-1; formation of ASC-containing pyroptosomes (Fernandes-Alnemri et al., 2007) were similarly unaltered by YopM (Figure 4C and 4D). Despite assembly of this NLR-ASC ‘pre-inflammasome’ scaffold during infection (Fig S3A), caspase-1 is not recruited into foci in the presence of YopM delivered by Ypstb (Figure 4E and 4F). Caspase-1 was recruited to the inflammasome in the presence of YopM271A, the mutant that cannot bind caspase-1 (Figure 4F). Further, caspase-1 activation was blocked in cells where it could not be recruited to form inflammasomes (Figure 4G and 4H) and caspase-1 colocalized with YopM (Figure 4I, Fig S3B). Consistent with these observations, YopM, but not YopM271A, blocks caspase-1 activation in response to PAMPs that activate NLRP1-, NLRP3- or NLRC4-inflammasomes (Fig S3C). Together these results indicate naturally delivered YopM preempts caspase-1 activation by binding and sequestering caspase-1, resulting in the formation of developmentally arrested pre-inflammasomes lacking caspase-1 (Figure 4G, 4H). Thus, YopM blocks pyroptosis by a unique bifunctional mechanism: the penultimate step of inflammasome formation is interrupted and substrate processing is directly inhibited.
Figure 4. see also Figure S3. YopM inhibits inflammasome formation and caspase-1 activation.
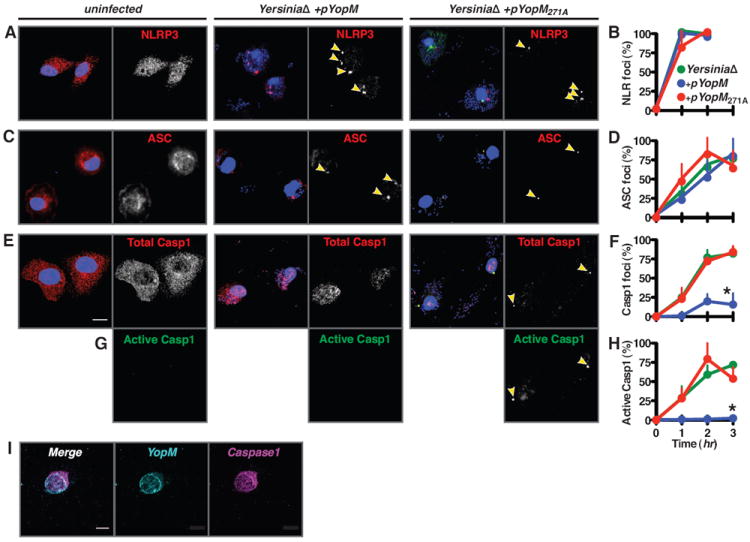
Wild-type or casp1-/- macrophages infected with Ypstb were fixed and protein localization visualized (A, C, E, G, I) and quantified (B, D, F, H) by immunofluorescence microscopy. Both NLRP3 (A and B) and the inflammasome adaptor protein ASC (C and D) rapidly form foci (arrowheads) in response to infection by Ypstb whether expressing YopM (+pYopM), inactive YopM (+pYopM271A), or no YopM at all (YersiniaΔ) (blue, red, and green lines, respectively, in B, D, F, H). (E and F) In contrast, recruitment of caspase-1 to foci is blocked by YopM but not YopM271A. (G and H) Caspase-1 is not activated in cells in which YopM can bind caspase-1. (I) In YopM-transduced, infected macrophages, YopM and caspase-1 colocalize. Representative confocal images are shown with macrophage nuclei visible by DNA staining (blue) and foci indicated with arrowheads; scale bars are 10 μm; data in B, D, F, and H are presented as mean +/ - SEM.
Discussion
Timely induction of inflammation is important for immunity to many pathogens. Inflammation during Yersinia infection is biphasic; an initial non-inflammatory phase of bacterial replication is followed by inflammation (Bergsbaken and Cookson, 2007; Lathem et al., 2005; Price et al., 2012). We have demonstrated that Ypstb not only evades innate immune detection, as previously presumed, but more specifically targets immune cells with an effector to bind and disable caspase-1. These results provide insight into the exceptional ability of Yersinia to initially replicate in the host without eliciting inflammation.
Our results explain a virulence requirement for YopM that has previously been enigmatic. Yersinia primarily target macrophages for T3S translocation during infection (Marketon et al., 2005). Using relevant host-derived cells and physiologic quantities of T3S-delivered YopM, we have identified a critical target of YopM. Several reports have shown that ΔyopM Yersinia are hyper-inflammatory and attenuated in vivo; however previously identified proteins that can bind YopM, α-thrombin, α1-antitrypsin, Rsk1, and Prk2, do not account this phenotype (Leung et al., 1990; McDonald et al., 2003; McPhee et al., 2010). Thus, YopM appears to have multiple targets and functions in vivo, like many other toxins and effectors, and these activities may be interconnected. The caspase-1 binding site of YopM is in a distinct central region of the protein not required for Rsk1 binding (McCoy et al., 2010; McPhee et al., 2010) or nuclear translocation (Trosky et al., 2008), but previously shown to be important for virulence (Skrzypek et al., 2003). Mutation of the Rsk1 binding site of YopM also disrupts a nuclear localization signal, and may contribute to caspase-1 inhibition, as both caspase-1 and viral inhibitors of caspase-1 also translocate to the nucleus (Fankhauser et al., 2000; Rodriguez et al., 2003). Our observations that YopM targets caspase-1 provides a mechanism for the anti-inflammatory properties of YopM.
Disruption of inflammasome formation and caspase-1 activation is a successful strategy of Poxviruses, which use multiple inhibitors that either block caspase-1 proteolysis or inhibit inflammasome formation (Lamkanfi and Dixit, 2009). Remarkably, Yersinia has both activities in a single dual function virulence factor. The bacterial origin of YopM possibly explains the lack of structural homology to viral inhibitors, which appear to be coopted host regulators (Fink et al., 2008; Nathaniel et al., 2004). Based on structural similarity, YopM is a potent, but only partial molecular mimic of Flightless-1, an endogenous pseudosubstrate inhibitor containing a gelsolin-like domain and leucine-rich repeats (Fink et al., 2008). Furthermore, like YopM, Flightless-1 inhibits both the activation and activity of caspase-1. However, YopM also inhibits inflammasome formation, like CARD-containing inhibitors of caspase-1 activation. Together these observations suggest Yersinia acquired caspase-1 inhibitory activity through convergent evolution, accounting for its unique, multipronged mechanism for inhibiting caspase-1 and regulating inflammation.
These data highlight the importance of caspase-1 in combating microbial infection and indicate that Yersinia are highly susceptible to pyroptosis, but normally prevent caspase-1 activation with YopM. Multiple Yersinia factors contribute to minimizing inflammation by restricting availability of PAMPs; directly targeting caspase-1 and inflammasome formation with YopM ensures Yersinia can limit host inflammatory responses and thereby establish infection.
Experimental Procedures
Bacteria and plasmids
Bacterial strains and plasmids are described in Table S1.
Cell culture
Macrophages were generated from femur exudates of C57BL/ 6 (Jackson Laboratories) or Caspase1-/-11-/- (from C. Roy, Yale University) mice as described in the supplemental methods, activated with 100 ng/ ml LPS (List Biologicals) 18 hrs pre-infection.
Retroviral vectors (Table S1) were transfected into Ecotropic Phoenix cells (ATCC). The recombinant viruses were transduced into macrophages and transductants selected with puromycin.
Infection conditions
Yersinia grown overnight at 25°C were diluted 1:40 into LB with 20mM MgCl2 and 20mM Na2C2O4 and grown at 25°C for 1 hr then 37°C for 2 hrs. Bacteria with expression plasmids were grown in media containing 50 μg/ ml carbenicillin and 0.02% arabinose. Bacteria washed in cold PBS were added to a MOI of 10 and spun onto macrophages at 150 g for 3 min. NLRP1 was induced with 1 mg/ ml anthrax lethal toxin subunits PA and LF (List Biologicals); NLRP3 with 20 mM nigericin (Sigma-Aldrich); and NLRC4 with Salmonella grown in LB overnight at 37°C, diluted 1:40, and grown 3 hrs in LB containing 0.3 M NaCl. All infections and toxin treatments of macrophages was for 1.5 hrs unless otherwise stated. In Figure 3A, macrophages from LT-susceptible BALB/ c mice (Jackson Laboratories) were used. LDH release was determined by Cytotox 96 kit (Promega) 2 hrs post-infection. In all other experiments, infections were done in the presence of 5 mM glycine to protect against cell lysis (Fink and Cookson, 2006).
Immunofluorescence microscopy
Caspase-1 activation was determined by Fam-YVAD-FMK (Immunochemistry Technologies) staining of macrophages infected on glass coverslips. After PBS washing, cells were fixed with Cytofix (BD). Lysosomal exocytosis was visualized by incubation of macrophages with anti-LAMP1 antibody (553792, BD) pre-fixation, as previously (Bergsbaken et al., 2011). Immunofluorescent staining of other proteins was performed post-fixation in Cytoperm (BD) with antibodies specific to HA (32-6700, Invitrogen), NLRP3 (sc-66846, Santa Cruz), ASC (AL177, Enzo), or Caspase-1 (sc-514, Santa Cruz) with Alexa fluor secondary antibodies (Invitrogen). Coverslips mounted with ProLong (Molecular Probes) were examined by confocal microscope (Leica SL or Zeiss LSM 510) at the Wm. Keck Center for Advanced Studies in Neural Signaling. Caspase-1 activation or foci formation was enumerated by counting the fraction of positive cells in at least four separate fields.
Immunoblotting and cytokine analysis
Supernatants from infected macrophages were sterilized by 0.22-mm filter and IL-1β release was quantified by ELISA (R&D Systems) or concentrated using a 10,000 MWCO Centricon Plus-20 centrifugal filter device (Millipore), separated by SDS-PAGE, transferred to PVDF membrane, and immunoblotted with anti–IL-1β antibody (AF-401-NA, R&D Systems). Caspase-1 and YopM immunoblots were performed with antibody against HA (sc-805 or 32-6700, Invitrogen), 6xHis (37-2900, Invitrogen), or caspase-1 p10 (sc-514, Santa Cruz), from cells lysed in M-PER protein extraction reagent (Thermo) with sonication.
Protein interaction and functional studies
Pyroptosis was induced in 107 macrophages by infection with YersiniaΔ at MOI 10 for 2 hrs. Lysates were added to 10 μg of recombinant 6xHisYopM or 6xHisYopM271A bound to HisPur Cobalt Resin (Thermo Fisher) and incubated at 4°C for 2 hr. After four washes with binding buffer, bound proteins were eluted with SDS-PAGE sample buffer containing imidazole and analyzed by immunoblot.
10U recombinant caspase-1 (Enzo) was pre-incubated with 6xHisYopM or 6xHisYopM271A for 5 min at 37°C in assay buffer (Enzo). Caspase-1 substrate Ac-YVAD-pNA (Enzo) was added to 200 uM and cleavage detected by monitoring absorbance at 405 nm (Spectramax M3, Molecular Devices).
The suitability of the YLTD motif of YopM as a caspase-1 pseudosubstrate was confirmed with the software PeptideCutter (ExPASy). Model interaction between YopM and caspase-1 was created in PyMOL by superimposing the YLTD sequence of YopM (1G9U) with the YVAD bound to caspase-1 (1ICE).
Animal experiments
Yersinia overnight cultures enumerated by Coulter counter (Multisizer 4, Beckman Coulter) were diluted in PBS for intraperitoneal delivery of 1000 CFU in 100 μl to 6–8 wk female mice. Mice were monitored for survival (5 mice each group) or sacrificed on day 4 and tissue homogenized and plated for CFU. Mice were housed in specific pathogen-free conditions and experiments performed by University of Washington Institutional Animal Care and Use Committee guidelines.
Statistical analysis
Statistical significance was calculated by unpaired Student t test (*P < 0.05, **P < 0.005) using GraphPad Prism, unless otherwise indicated. Data are representative of at least three independent experiments.
Supplementary Material
Highlights.
Yersinia type III secreted effector YopM prevents caspase-1-dependent cell death
Caspase-1 inhibition by YopM is required for bacterial virulence in vivo
YopM blocks pyroptotic cell death by directly binding caspase-1
YopM sequestration of caspase-1 arrests inflammasome maturation
Acknowledgments
We thank L. Cummings, M. Johnson, and W. Loomis for assistance with mouse experiments and F. Fang, S. Miller, and members of the Cookson lab for helpful discussion and critical reading of the manuscript. This work was supported by HG02360 and AI057141 to BC and NIH training grant AI055396 to CL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. Cell death and infection: A double-edged sword for host and pathogen survival. J Cell Biol. 2011;195:931–942. doi: 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, et al. Cutting Edge: NF-kB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Cookson BT. Macrophage Activation Redirects Yersinia - Infected Host Cell Death from Apoptosis to Caspase-1-Dependent Pyroptosis. PLoS Pathog. 2007;3:e161. doi: 10.1371/journal.ppat.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Micro. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, den Hartigh AB, Loomis WP, Cookson BT. Coordinated Host Responses during Pyroptosis: Caspase-1-Dependent Lysosome Exocytosis and Inflammatory Cytokine Maturation. J Immunol. 2011;187:2748–2754. doi: 10.4049/jimmunol.1100477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia Effector Protein Promotes Virulence by Preventing Inflammasome Recognition of the Type III Secretion System. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewoody R, Merritt PM, Houppert AS, Marketon MM. YopK regulates the Yersinia pestis type III secretion system from within host cells. Mol Microbiol. 2011;79:1445–1461. doi: 10.1111/j.1365-2958.2011.07534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JM, Rouge L, Wiesmann C, Scheer JM. Crystal structure of procaspase-1 zymogen domain reveals insight into inflammatory caspase autoactivation. J Biol Chem. 2009;284:6546–6553. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu J, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci. 2008;105:4312–4317. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. The Inflammasomes. PLoS Pathog. 2009;5:e1000510. doi: 10.1371/journal.ppat.1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathem WW, Crosby SD, Miller VL, Goldman WE. Progression of primary pneumonic plague: A mouse model of infection, pathology, and bacterial transcriptional activity. Proc Natl Acad Sci. 2005;102:17786–17791. doi: 10.1073/pnas.0506840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KY, Reisner BS, Straley SC. YopM inhibits platelet aggregation and is necessary for virulence of Yersinia pestis in mice. Infect Immun. 1990;58:3262–3271. doi: 10.1128/iai.58.10.3262-3271.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yin HL, Yuan J. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J Cell Biol. 2008;181:321–333. doi: 10.1083/jcb.200711082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilo S, Zheng Y, Bliska JB. Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect Immun. 2008;76:3911–3923. doi: 10.1128/IAI.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. Plague Bacteria Target Immune Cells During Infection. Science. 2005;309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The Inflammasome∷ A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-[beta] Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- McCoy M, Marre ML, Lesser CF, Mecsas J. The C-Terminal Tail of Yersinia pseudotuberculosis YopM Is Critical for Interacting with RSK1 and for Virulence. Infect Immun. 2010;78:2584–2598. doi: 10.1128/IAI.00141-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Vacratsis P, Bliska J, Dixon J. The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem. 2003;278:18514–18523. doi: 10.1074/jbc.M301226200. [DOI] [PubMed] [Google Scholar]
- McPhee JB, Mena P, Bliska JB. Delineation of Regions of the Yersinia YopM Protein Required for Interaction with the RSK1 and PRK2 Host Kinases and Their Requirement for Interleukin-10 Production and Virulence. Infect Immun. 2010;78:3529–3539. doi: 10.1128/IAI.00269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathaniel R, MacNeill AL, Wang Y-X, Turner PC, Moyer RW. Cowpox virus CrmA, Myxoma virus SERP2 and baculovirus P35 are not functionally interchangeable caspase inhibitors in poxvirus infections. J Gen Virol. 2004;85:1267–1278. doi: 10.1099/vir.0.79905-0. [DOI] [PubMed] [Google Scholar]
- Price PA, Jin J, Goldman WE. Pulmonary infection by Yersinia pestis rapidly establishes a permissive environment for microbial proliferation. Proc Natl Acad Sci. 2012;109:3083–3088. doi: 10.1073/pnas.1112729109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypek E, Myers-Morales T, Whiteheart S, Straley S. Application of a Saccharomyces cerevisiae Model To Study Requirements for Trafficking of Yersinia pestis YopM in Eucaryotic Cells. Infect Immun. 2003;71:937–947. doi: 10.1128/IAI.71.2.937-947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of p rocaspase-1 activation. J Immunol. 2003;171:6154–6163. doi: 10.4049/jimmunol.171.11.6154. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. A Combinatorial Approach Defines Specificities of Members of the Caspase Family and Granzyme B. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Trosky JE, Liverman ADB, Orth K. Yersinia outer proteins: Yops. Cell Microbiol. 2008;10:557–565. doi: 10.1111/j.1462-5822.2007.01109.x. [DOI] [PubMed] [Google Scholar]
- Ye Z, Kerschen EJ, Cohen DA, Kaplan AM, Van Rooijen N, Straley SC. Gr1+ cells control growth of YopM-negative yersinia pestis during systemic plague. Infect Immun. 2009;77:3791–3806. doi: 10.1128/IAI.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.