

Abstract
The mechanisms underlying adaptive resistance of melanoma to targeted therapies remain unclear. By combining ChIP sequencing with microarray-based gene profiling, we determined that ERBB3 is upregulated by FOXD3, a transcription factor that promotes resistance to RAF inhibitors in melanoma. Enhanced ERBB3 signaling promoted resistance to RAF pathway inhibitors in cultured melanoma cell lines and in mouse xenograft models. ERBB3 signaling was dependent on ERBB2; targeting ERBB2 with lapatinib in combination with the RAF inhibitor PLX4720 reduced tumor burden and extended latency of tumor regrowth in vivo versus PLX4720 alone. These results suggest that enhanced ERBB3 signaling may serve as a mechanism of adaptive resistance to RAF and MEK inhibitors in melanoma and that cotargeting this pathway may enhance the clinical efficacy and extend the therapeutic duration of RAF inhibitors.
Introduction
Hyperactivation of the RAS/RAF/MEK/ERK1/2 pathway is a driving force in many tumor types. This is particularly evident in malignant melanoma, an aggressive form of skin cancer, which is hallmarked by rapid progression, poor responsiveness to conventional chemotherapies, and low survival rates in patients with metastatic disease. ERK1/2 signaling is enhanced in melanoma through several mutually exclusive mechanisms. These include increased growth factor signaling (1), activating mutations in NRAS and KRAS (2), and, most prevalently, activating mutations in the serine/threonine kinase BRAF (3). Oncogenic BRAF mutations (in particular BRAFV600E) are found in 40%–50% of cutaneous melanomas, and targeting BRAF or its downstream targets, MEK1/2, elicits potent antiproliferative and proapoptotic effects (4–9).
Targeting oncogenic BRAF and/or MEK1/2 has been extensively pursued in the clinical arena, and the RAF inhibitor vemurafenib (PLX4032; marketed as Zelboraf) has gained approval from the Food and Drug Administration (FDA) for the treatment of mutant V600 BRAF melanoma. Compared with dacarbazine, the previous standard of treatment for melanoma, vemurafenib shows a remarkable response rate (48% in phase III trial) and improved progression-free and overall survival (10). However, despite these impressive results, approximately 15% of mutant BRAF melanoma patients progress on vemurafenib, and overall, approximately 50% of patients experience a loss of responsiveness after 6–7 months (10). These findings underscore the need to understand compensatory mechanisms that bypass the requirement for active BRAF in melanoma. Acquired resistance to RAF inhibitors has been associated with multiple mechanisms including the following: amplification of cyclin D1 (11); increased expression of kinases such as RAF1 (C-RAF) (12), MAP3K8 (COT1) (13), PDGFRB (14), and IGF1R (15); loss of PTEN/activation of AKT (16–18); splice variants of BRAF (19); mutations in MEK1 (20, 21); and oncogenic mutation of NRAS (14). Many of these alterations appear to be stable events either acquired after treatment with RAF inhibitors or selected for out of the general tumor cell population. In contrast, little is known about short-term, adaptive mechanisms that may protect melanoma cells from RAF inhibitors.
Recently, we identified stem cell/pluripotency transcription factor forkhead box D3 (FOXD3) as a protein induced upon BRAF/MEK pathway inhibition selectively in mutant BRAF melanomas (22). Furthermore, depletion of FOXD3 by RNAi enhanced PLX4032/4720-mediated apoptosis, while overexpression of FOXD3 was protective (23). The possibility of FOXD3 functioning as an adaptive mediator of the response to RAF inhibitors led us to explore the FOXD3 transcriptome to identify potentially druggable targets. Using microarray analysis and ChIP coupled to next-generation sequencing (ChIP-seq), we identified v-erb-b2 erythroblastic leukemia viral oncogene homolog 3/human epidermal receptor 3 (ERBB3 or HER3) as a direct transcriptional target of FOXD3. RAF or MEK inhibition and FOXD3 overexpression caused an increase in ERBB3 at the protein and mRNA level in a panel of melanoma cell lines, culminating in a marked enhancement in responsiveness to the ERBB3 ligand neuregulin-1 (NRG1). ERBB3 signaling in concert with ERBB2 promoted AKT signaling and cell viability. Finally, combined treatment of mutant BRAF melanoma cells with PLX4720 and the ERBB2/EGFR inhibitor lapatinib abolished NRG1/ERBB3 signaling in vitro and reduced tumor burden in vivo when compared with either treatment alone. These results suggest that mutant BRAF melanoma adaptively shifts to an ERBB3-dependent pathway in response to RAF/MEK inhibitors and that targeting this pathway in conjunction with RAF inhibitors may provide therapeutic benefit in the clinic.
Results
Identifying the FOXD3 transcriptome in melanoma.
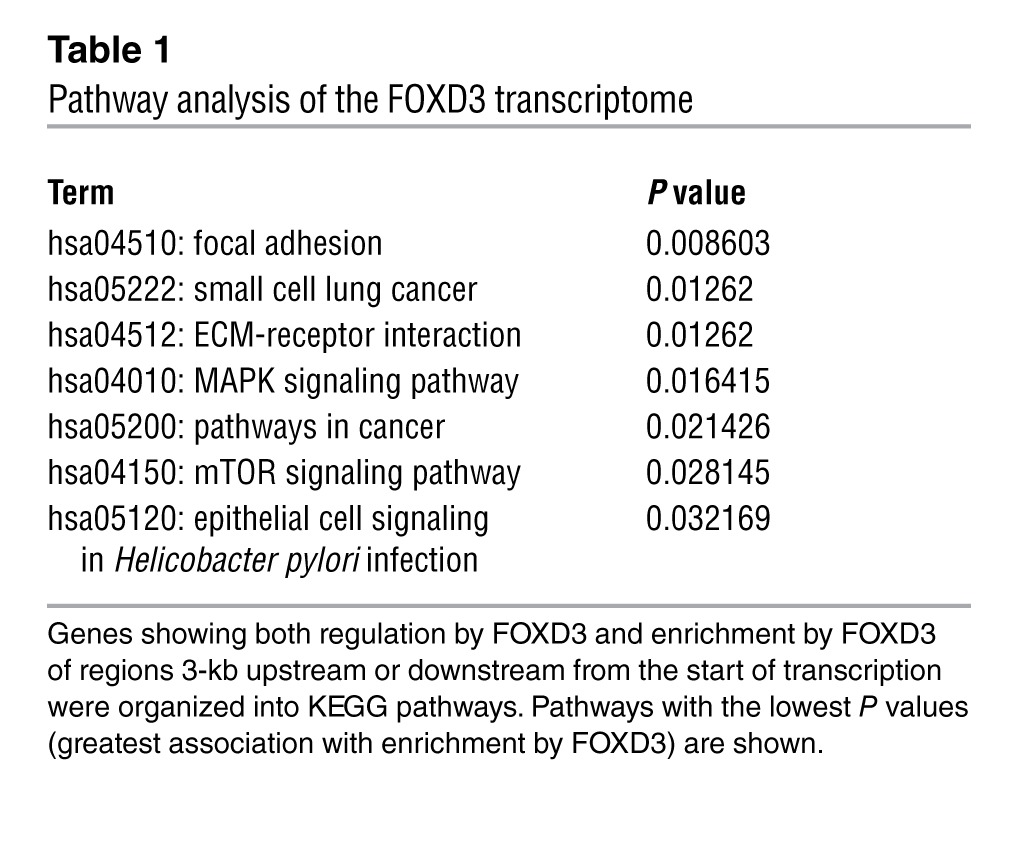
To understand the transcriptional impact of FOXD3 in melanoma cells, we utilized a microarray approach. We collected RNA from 3 unrelated mutant BRAF melanoma cell lines (WM115, WM793, and A375) that were engineered to inducibly express FOXD3 or the control gene β-galactosidase (LacZ) after 5 days of transgene induction (Figure 1A). This time point was chosen based on maximal phenotypic changes previously observed (22). Comparison of gene signatures among the 3 cell lines produced approximately 2,600 common genes differentially regulated by FOXD3-expressing cells compared with the LacZ controls (Figure 1B and Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI65780DS1). Since a large number of altered genes may represent secondary targets of FOXD3, we sought to narrow the scope of FOXD3-regulated genes to direct transcriptional targets. We performed ChIP-seq on V5-tagged FOXD3 IP from WM115TR-FOXD3. The results showed specific, reproducible enrichment foci across the genome with a preference for promoter regions (Figure 1C) and bidirectional promoters (data not shown). Analysis of genes located proximal to FOXD3 enrichment sites and showing regulation by FOXD3 indicated a preference for genes involved in focal adhesions, ECM-receptor interactions, MAPK and mTOR signaling, and other processes involved in cancer (Table 1 and Supplemental Figure 1), suggesting that FOXD3 is able to act as a major orchestrator of transcription in melanoma.
Figure 1. Microarray and ChIP-seq analysis of FOXD3 target genes.

(A) A375TR, WM115TR, and WM793TR cells expressing Dox-inducible FOXD3 were treated with or without 100 ng/ml Dox overnight. Induced V5-tagged FOXD3 was detected by immunoblotting for V5 and ERK1/2 as a loading control. WB, Western blot. (B) Heat map of common target genes downregulated (blue) or upregulated (red) by expression of FOXD3 compared with cells expressing LacZ. (C) Pie chart representation of the distribution of FOXD3 enrichment foci from ChIP-seq across the genome of WM115TR cells. Data represent 3 independent ChIP experiments compared with a pooled input control.
Table 1.
Pathway analysis of the FOXD3 transcriptome
ERBB3 is a direct transcriptional target of FOXD3.
Based on our previous data showing that FOXD3 promotes resistance to BRAF inhibition (23), we focused on genes that were druggable, given the translational nature of the study. We identified ERBB3 as a target upregulated by FOXD3 in the expression arrays and strongly enriched by FOXD3 in the ChIP-seq analysis (Figure 2A and Supplemental Table 1). ERBB3 expression is increased in response to targeted therapies such as lapatinib in breast cancer and gefitinib in lung cancer (24–27) and is also important for melanoma survival and proliferation (28, 29). ChIP-seq analysis showed that the first intron of ERBB3 was enriched by FOXD3. This region is well conserved between species and functions as an enhancer region for ERBB3 (30–32). Quantitative PCR (qPCR) showed dramatic enrichment of intron 1 over normal IgG only following FOXD3 expression (Figure 2B). Importantly, the V5 antibody did not enrich the promoter of an irrelevant gene, β-actin (ACTB), in a doxycycline-dependent (Dox-dependent) manner, verifying the specificity of FOXD3 enrichment. Enhanced expression on our microarrays coupled with binding of FOXD3 to the enhancer region suggests that FOXD3 directly upregulates the transcription of ERBB3. In support of this, IP of RNA polymerase II phosphoserine 2 (RNA pol II pSer2), a marker for transcriptional elongation (33, 34), significantly enriched ERBB3 intron 1 in cells expressing FOXD3 (Figure 2C). In addition we found that FOXD3 increased the expression of ERBB3 at both the mRNA (Figure 2D) and protein (Figure 2E) levels in WM115TR-FOXD3 cells. Similarly, induction of FOXD3 consistently enhanced the expression of ERBB3 in a panel of melanoma cells while consistently having no effect on the expression of other receptor tyrosine kinases (RTKs) known to convey resistance to targeted therapies (refs. 13–15, 25, and Figure 2F).
Figure 2. ERBB3 is a direct transcriptional target of FOXD3.

(A) Map of the ERBB3 locus showing read coverage for IP and input; aligned reads were visualized using the Integrated Genomics Viewer 2.0 (57). Relative signal of merged ChIP experiments is represented by red peaks, while the signal of the pooled inputs is represented with light gray peaks. The intron 1 enhancer region is underlined. (B and C) WM115TR/FOXD3-V5 cells were treated with 100 ng/ml Dox (+ Dox) or without (– Dox) for 24 hours. Cells were lysed, DNA was sheared, and protein/chromatin complexes were IP with normal IgG (B and C), anti-V5 antibody (B), or anti-RNA pol II pSer2 (C). Enrichment of ERBB3 intron 1 was validated by qPCR. Enrichment of the β-actin promoter is included as a control for specificity. Results represent the mean ± SEM (n = 4). P values are indicated. (D) WM115TR/FOXD3-V5 cells were treated with or without Dox for 24 hours. qRT-PCR was performed following RNA extraction. Fold change in ERBB3 transcript was normalized to housekeeping gene EEF1A1. Results represent mean ± SEM (n = 3). P value is indicated. (E) WM115TR/FOXD3-V5 cells were treated with or without Dox (100 ng/ml) for 24, 48, or 72 hours, and then lysed and immunoblotted as indicated. (F) Lysates from WM793TR, 1205LuTR, SK-MEL-28TR, and A375TR expressing FOXD3 for 24 hours were blotted as indicated.
ERBB3 expression is enhanced by RAF/MEK inhibition in melanoma.
Previous studies showed that FOXD3 is upregulated in response to BRAF/MEK inhibition in mutant BRAF melanoma (22). We sought to determine whether inhibition of BRAF or MEK1/2 could recapitulate the effects on ERBB3 seen by the ectopic expression of FOXD3. Knockdown of BRAF by siRNA resulted in an increase in ERBB3 protein in WM115 cells (Figure 3A). Similarly, inhibition of BRAF or MEK with PLX4032 or AZD6244, respectively, induced both FOXD3 and ERBB3 in WM115 and 1205Lu cells (Figure 3B). This observation was reinforced by microarray data showing upregulation of ERBB3 in response to BRAF knockdown (Supplemental Figure 2A). Similarly, increased ERBB3 mRNA expression was also observed in 1205Lu cells treated with PLX4032 or AZD6244 (Figure 3C). In both WM115 and 1205Lu cells, the ERBB3 signal on microarrays was also reduced by FOXD3 targeting siRNA, both alone or in combination with BRAF siRNA or PLX4720 (Supplemental Figure 2, A and B). Another cell line, A375, showed enhanced surface expression of ERBB3 (Figure 3D) as well as a concomitant upregulation of ERBB3 mRNA in response to either PLX4032 or AZD6244 (data not shown). These data indicate that BRAF/MEK inhibition, like FOXD3 overexpression, positively regulates ERBB3 expression levels.
Figure 3. Inhibition of mutant BRAF and MEK1/2 enhances ERBB3 expression in melanoma cells.

(A) WM115 cells were transfected with reagent alone (–), a nontargeting control siRNA (Ctl), or BRAF-targeting siRNA alone (BRAF) for 96 hours. Cells were lysed and immunoblotted as indicated. (B) WM115 and 1205Lu cells were treated overnight with DMSO (–), 1 μM PLX4032 (PLX), or 3.3 μM AZD6244 (AZD), lysed, and immunoblotted as indicated. (C) qRT-PCR analysis of ERBB3 mRNA levels in 1205Lu cells treated with DMSO, PLX4032, or AZD6244 overnight. Housekeeping gene EEF1A1 transcript was used for normalization. Results represent mean ± SEM (n = 4). P values are indicated. (D) Flow cytometry analysis of ERBB3 surface expression in A375 cells treated with DMSO, PLX4032, or AZD6244 overnight.
NRG1/ERBB3 signaling to AKT is enhanced by RAF/MEK inhibition in a FOXD3-dependent manner.
To assess the impact of FOXD3 expression on ligand-induced ERBB3 signaling, we treated WM115TR-FOXD3 cells with increasing concentrations of NRG1β, a potent ERBB3 ligand (35), in either the presence or absence of FOXD3 induction. Upregulation of ERBB3 by FOXD3 was associated with an enhanced sensitivity to NRG1β at all doses analyzed, as assessed by phosphorylation of ERBB3 (Figure 4A). Phosphorylated YXXM motifs in ERBB3 recruit PI3K, leading to activation of AKT (36). Consistent with enhanced ERBB3 signaling, FOXD3-expressing cells displayed enhanced NRG1β-dependent phosphorylation of AKT (Figure 4A).
Figure 4. FOXD3 and RAF/MEK inhibition enhance responsiveness to NRG1β.

(A and B) WM115TR/FOXD3-V5 (A) or WM115 (B) were treated with or without Dox (100 ng/ml) or PLX4032 (1 μM), respectively, for 24 hours followed by treatment with the indicated concentration of NRG1β for 1 hour. Cell lysates were immunoblotted as indicated. (C) WM115 cells were treated overnight with DMSO, PLX4032 (1 μM), or AZD6244 (3.3 μM), followed by 1 additional hour with or without NRG1β (10 ng/ml). Cells were lysed and lysates immunoblotted as indicated. (D) WM115 cells were pretreated with PLX4032 for 0, 2, 4, 6, and 16 hours and then stimulated with NRG1β (10 ng/ml) for 30 minutes. Cell lysates were immunoblotted as indicated. (E) WM115 cells were transfected with either control siRNA or 2 distinct FOXD3-targeting siRNAs for 72 hours. Cells were then treated for an additional 24 hours with PLX4032 (1 μM) or DMSO, after which NRG1β (10 ng/ml) was added for an additional hour to activate ERBB3. Cell lysates were immunoblotted as indicated.
To determine whether inhibition of BRAF could elicit a similar result in melanoma cells, WM115 cells were treated overnight with PLX4032 to induce endogenous FOXD3 and ERBB3, or with vehicle DMSO. PLX4032 treatment increased the sensitivity of ERBB3 to NRG1β and also enhanced AKT phosphorylation in WM115 (Figure 4B) and A375 cells (Supplemental Figure 3A). PLX4032 not only enhanced the intensity of response to NRG1β stimulation (Figure 4B), but also the duration of downstream AKT phosphorylation (Supplemental Figure 3B). A transient increase in ERK1/2 phosphorylation was observed in PLX4032-treated cells after stimulation with NRG1β, but this was largely dissipated within 1 hour (Supplemental Figure 3B). Similar to PLX4032, treatment of cells with AZD6244 enhanced both ERBB3 and AKT phosphorylation in response to NRG1β stimulation (Figure 4C). The enhancement of NRG1β/ERBB3 signaling was seen in multiple cell lines in response to either PLX4032 or AZD6244 pretreatment (Figure 4C, Supplemental Figure 3, C and D, and data not shown). Of note, phosphorylation of AKT was potently induced in melanoma cells regardless of PTEN status, as A375 cells are PTEN competent, while WM115 and 1205Lu cells are PTEN deficient. Importantly, phosphorylation of p70/p85 S6-kinase and S6 ribosomal protein were inhibited by treatment with PLX4032 or AZD6244, but restored by treatment with NRG1β (Figure 4C and Supplemental Figure 3C), indicating a restoration of translational activity by NRG1β/ERBB3 signaling. In addition to NRG1β, enhanced ERBB3 and AKT activation in PLX4032-treated cells was also observed following stimulation with NRG1α and neuroglycan (Supplemental Figure 4).
We next examined the temporal relationship among RAF inhibition, FOXD3 induction, and enhanced NRG1β/ERBB3 signaling. Induction of FOXD3 could be seen as early as 2 hours after treatment with PLX4032 and steadily increased up until 16 hours. Enhanced NRG1β/ERBB3 signaling could be observed after 4 hours of PLX4032 treatment, gradually increasing through 16 hours (Figure 4D). These data suggest that FOXD3 upregulation precedes enhancement of NRG1β/ERBB3 signaling. Importantly, depletion of FOXD3 by siRNA ablated ERBB3 protein expression, both basal and PLX4032 induced, and prevented responsiveness to NRG1β stimulation in both WM115 and 1205Lu cells (Figure 4E and Supplemental Figure 3E).
RAF inhibitors enhance ERBB3 phosphorylation in vivo.
We extended our analysis of RAF inhibitors on ERBB3 phosphorylation to the in vivo setting. First, we administered PLX4720 to nude mice with intradermal A375 xenografts for 5 days. PLX4720 is the nonclinical analog for vemurafenib. Analysis of the harvested tumors by immunohistochemistry (IHC) showed a statistically significant (P < 0.05) increase in the proportion of cells with high levels of membrane-associated staining for phosphorylated ERBB3 (phospho-ERBB3) in PLX4720-treated tumors compared with controls (Figure 5A). These findings indicate that increased ERBB3 sensitivity following RAF inhibition in melanoma cells occurs in vivo as well as in vitro.
Figure 5. Increased ERBB3 phosphorylation following RAF inhibitor treatment in vivo.

(A) A375 xenografts taken from animals fed vehicle (n = 5) or PLX4720-laced chow (n = 4) for 5 days analyzed by IHC for phospho-ERBB3 (Y1289). Representative images are shown. Original magnification, ×20. The graph shows quantitation of phospho-ERBB3 intensity. Cells were scored by intensity of membrane-associated staining from 0 (no staining) to 3 (strong staining). *P = 0.016. (B) Biopsies from patient taken prior to vemurafenib treatment, on-treatment, or upon disease progression were stained for phospho-ERBB3. Representative images are shown from patient 1 (Pt_1). The graph shows quantitation of cellular staining. Tumor cells in each slide were scored in a blinded manner, and statistical differences among the 3 conditions were analyzed using the cumulative link model (i.e., proportional odds model). The level of phospho-ERBB3 in the on-treatment and progression samples is statistically different from the pretreatment sample (*P < 0.001). The on-treatment biopsies for patient 1 and melanoma patient_503 (MP_503) were taken after 15 days and 16 months, respectively. Original magnification, ×200. (C) ERBB3 phosphorylation was analyzed by immunohistochemical staining of paired pretreatment and progression samples. MP_20 and MP_6 progressed after 6 and 9 months, respectively, on RAF/MEK inhibitor. MP_47 progressed on RAF inhibitor after 7.5 months. Representative images are shown for MP_6. Graphs show quantitation of phospho-ERBB3 intensity staining on scale of 0–3. *P < 0.001. Original magnification, ×200. (D) Statistical analysis across samples from all 9 patients that displayed staining for phospho-ERBB3. Analysis was performed using an ordered logistic regression model with random intercept for each patient.
Next, to analyze whether enhanced ERBB3 phosphorylation occurs in patients receiving vemurafenib, IHC was performed using biopsies taken before vemurafenib treatment, 15 days on-treatment, and following disease progression. In 2 patients analyzed, we observed low ERBB3 phosphorylation prior to treatment. A statistically significant increase in ERBB3 phosphorylation was observed in 1 of the 2 patients following treatment with vemurafenib and persisting through relapse (Figure 5B). An additional biopsy from a long-term on-treatment patient, who had not yet progressed, also showed upregulation of phospho-ERBB3 staining (Figure 5B). This suggests that ERBB3 phosphorylation can be enhanced in patients undergoing vemurafenib treatment.
We extended our analysis to a larger set for which pretreatment and progression samples were available. This set of 9 paired samples came from mutant BRAF melanoma patients who had received either RAF inhibitor or combined RAF/MEK inhibitor. The latter combination has been shown to provide increased progression-free survival in mutant BRAF melanoma patients compared with RAF inhibitor alone (37). Three out of the 9 progression samples showed a statistically significant increase in ERBB3 phosphorylation compared with the match pretreatment sample (Figure 5C). Statistical analysis across samples using an ordered logistic regression model with random intercept for each patient showed that progression samples have 2.16 times (95% CI, P < 0.001) higher odds of having greater scores compared with pretreatment and that on-treatment samples have 3.30 times (95% CI, P < 0.001) higher odds of having greater scores compared with pretreatment (Figure 5D). These findings suggest that upregulation of ERBB3 is maintained in some cases of chronic vemurafenib treatment.
ERBB3 activation promotes resistance to RAF/MEK inhibitors.
Increased expression and activation of RTKs has been associated with acquired resistance to PLX4032 in both patients and cultured melanoma cells (14, 15). To determine whether the rapid sensitization of cells to NRG1β stimulation could provide a form of adaptive resistance to PLX4032 and AZD6244, we plated A375 cells at low density in the presence of DMSO, PLX4032, or AZD6244 with or without NRG1β. DMSO-treated cells rapidly grew to confluency regardless of NRG1β stimulation (Figure 6A). As expected, treatment of A375 cells with either PLX4032 or AZD6244 potently blocked the growth of colonies, while addition of NRG1β to PLX4032- or AZD6244 treated cells promoted colony growth (Figure 6, A and B). Additionally, NRG1β enhanced the viability of WM115, WM266-4, and WM239A cells treated with PLX4032 or AZD6244 for 72 hours, but did not enhance the viability of DMSO-treated cells (Figure 6C). These data indicate that NRG1β is able to partially restore viability and colony growth in RAF/MEK inhibitor–treated cells.
Figure 6. NRG1β/ERBB3 signaling promotes resistance to RAF/MEK inhibitors.

(A) A375 cells were plated at clonal density and treated with either DMSO, DMSO with NRG1β (10 ng/ml), PLX4032 (1 μM), PLX4032 with NRG1β, AZD6244 (3.3 μM), or AZD6244 with NRG1β. Medium and drugs were replenished every 3 days for 7 days, after which cells were fixed and stained with crystal violet. (B) Magnification of colonies in A (×40). (C) WM115, WM239A, and WM266-4 cells were treated with DMSO, PLX4032 (1 μM), or AZD6244 (3.3 μM) with or without NRG1β (10 ng/ml) for 72 hours, after which AlamarBlue was added to medium for 2 hours. Reduced AlamarBlue as analyzed by spectrophotometer to determine cell viability. DMSO-treated cell groups were set to 100% viable, and all other groups were normalized to these groups. Mean ± SEM (n = 4) and P values are shown. (D) 1205LuTR cells stably expressing Dox-inducible LacZ-targeting (LacZ 2.1) or 2 distinct ERBB3-targeting shRNAs were treated with or without Dox for 5 days, followed by treatment with PLX4032 (+) or DMSO (–) for 24 hours, and finally stimulated with 10 ng/ml NRG1β for 1 hour prior to lysis. Lysates were immunoblotted as indicated. (E) Mean fold change of tumor volume in 1205LuTR xenografts (n = 16 per condition) in nude mice fed either PLX4720 or vehicle-laced chow, expressing either LacZ-targeting or ERBB3-targeting shRNAs. Statistically significant comparisons of the LacZ-targeting and ERBB3-targeting shRNA xenografts are indicated by blue P values (LacZ vs. ERBB3 shRNA#10) or green P values (LacZ vs. ERBB3 shRNA#12).
To test the requirement for ERBB3 in responsiveness to NRG1β, 1205LuTR cells stably expressing control (LacZ-targeting) shRNA or ERBB3-targeting shRNA were created. Depletion of ERBB3 with 2 independent shRNAs effectively inhibited AKT phosphorylation in response to NRG1β stimulation in vitro (Figure 6D). To determine whether ERBB3 was important for resistance to RAF inhibitors in vivo, 1205LuTR xenografts harboring LacZ- or ERBB3-targeting shRNAs were established in nude mice, and the animals were subsequently fed vehicle or PLX4720-laden chow. 1205Lu cells were utilized, given that they displayed a high level of intrinsic resistance to PLX4720 in our previous studies (23). ERBB3-knockdown cells did not significantly alter the growth of xenografts in the vehicle group (Figure 6E). In contrast, ERBB3-knockdown cells showed a marked reduction in tumor growth in the PLX4720 treatment group (Figure 6E). These data indicate that ERBB3 signaling is important in the response to RAF inhibitors both in vitro and in vivo.
NRG1β/ERBB3 signaling requires ERBB2 in melanoma.
ERBB3 is deficient in intrinsic kinase activity and relies upon other ERBB family members to phosphorylate it in response to ligand binding (38). As such, we sought to identify the kinase responsible for ERBB3 phosphorylation. Concomitant with ERBB3 phosphorylation in cells, enhanced ERBB2 (also known as HER2) phosphorylation in response to NRG1β was observed (Figure 4, B and D, and Supplemental Figure 3, A–D). We also observed a statistically significant (P < 0.05) increase in cells expressing high levels of membrane-associated phospho-ERBB2 (Y1221/Y1222) in A375 xenografts fed PLX4720 chow for 5 days (Figure 7, A and B). To determine whether ERBB2 was responsible for phosphorylating ERBB3, WM115 cells were depleted of ERBB2 by RNA interference. Knockdown of ERBB2 abolished NRG1β/ERBB3 signaling (Figure 7C). Additionally, treatment of cells with increasing doses of lapatinib (Tykerb/Tyverb), a clinical ERBB2/EGFR inhibitor, effectively inhibited NRG1β-stimulated ERBB3 and AKT phosphorylation in a dose-dependent manner in both A375 and WM115 cells (Figure 7D and Supplemental Figure 5A). EGFR-specific inhibitors gefitinib (Iressa) and erlotinib (Tarceva) failed to inhibit NRG1β/ERBB3 signaling in WM115 cells (Supplemental Figure 5B), indicating EGFR is not the kinase responsible for ERBB3 phosphorylation. ERBB4, which is also a receptor for NRG1β, is mutated in a subset of melanomas (39) and can be inhibited by lapatinib (39, 40). However, ERBB4 was poorly detected in the cells used in this study and depletion of ERBB4 with siRNA did not inhibit NRG1β/ERBB3 signaling in WM115 cells (Supplemental Figure 5C), arguing against ERBB4 phosphorylation of ERBB3. These data indicate that ERBB2 is the coreceptor for ERBB3 when cells are challenged with BRAF/MEK inhibitors and is responsible for its phosphorylation.
Figure 7. ERBB2 is required for NRG1β/ERBB3 signaling in melanoma.

(A) Representative images of A375 xenografts taken from animals fed vehicle or PLX4720-laced chow for 5 days analyzed by IHC for phospho-ERBB2 (Y1221/Y1222). Original magnification, ×100. (B) Quantitation of phospho-ERBB2 intensity of tumor cells from vehicle (n = 5) or PLX4720-treated A375 xenografts (n = 5). *P = 0.001. (C) WM115 cells were transfected with control or ERBB2-targeting siRNA for 72 hours, then treated with PLX4720 or DMSO for an additional 24 hours followed by treatment with or without NRG1β (10 ng/ml) for 1 hour, lysed, and immunoblotted as indicated. (D) A375 cells were pretreated for 24 hours with PLX4032 (1 μM) and then treated with or without NRG1β and a dose range of lapatinib for 1 hour, lysed, and immunoblotted as indicated.
Combining RAF/MEK inhibitors with lapatinib provides a therapeutic benefit in vitro and in vivo.
To determine whether lapatinib prevents NRG1β/ERBB3-mediated resistance to PLX4032, A375 cells were plated at low density in the presence of PLX4032 and treated with either NRG1β alone, lapatinib alone, or both in combination. After 10 days, PLX4032-treated cells formed sizeable colonies in the presence of NRG1β alone, but failed to do so in the presence of lapatinib (Figure 8, A and B). Of note, lapatinib alone did not prevent the growth of A375 cells (Supplemental Figure 6A). Lapatinib could also ablate cell viability promoted by NRG1β in the presence of PLX4032 or AZD6244 in WM115 and 1205Lu cells (Supplemental Figure 6, B and C). To test the combination of lapatinib with BRAF inhibitors in vivo, we treated nude mice carrying 1205Lu or A375 xenografts with or without lapatinib in combination with PLX4720 or placebo. 1205Lu tumors showed a modest but statistically significant (P < 0.05) inhibition of tumor growth when treated with lapatinib alone (Figure 8C). In contrast, A375 tumors rapidly progressed in both vehicle and lapatinib-treated animals and showed no statistical difference in tumor burden (Figure 8D). PLX4720-treated animals showed a long latency in tumor progression, with both cell lines followed by steady tumor growth after about 14–15 days (Figure 8, C and D). Nearly half of the 1205Lu and A375 xenografts treated with PLX4720 alone reached a sacrificial threshold by 28 and 26 days, respectively (Figure 8, E and F). Remarkably, the combination of PLX4720 with lapatinib almost completely abolished 1205Lu tumor growth, with no mice reaching the sacrificial threshold (Figure 8, C and E). Similarly, A375 tumors in PLX4720/lapatinib-treated animals showed a longer latency period followed by slower tumor growth than PLX4720 alone, with only 1 out of 16 animals reaching a tumor volume necessitating animal sacrifice (Figure 8, E and F). These results indicate that lapatinib enhances the efficacy of PLX4720 and impairs the regrowth of PLX4720-resistant tumors.
Figure 8. Inhibition of ERBB2 ablates NRG1β/ERBB3-mediated growth in vitro and reduces tumor burden in vivo.

(A) A375 cells were plated in the presence of PLX4032 (1 μM) alone or with lapatinib (1 μM), NRG1β (10 ng/ml), or NRG1β combined with lapatinib. Medium and additives were replaced every 3 days, with cells fixed and stained with crystal violet after 7 days. (B) Magnification of colonies in A (×40). (C) Mean fold change ± SEM of tumor volume in 1205Lu xenografts (n = 16 per condition) in nude mice fed either PLX4720 or vehicle chow with or without daily lapatinib (100 mg/kg) by oral gavage. Statistically significant comparisons of the vehicle and lapatinib monotherapy groups are indicated by blue P values, whereas statistically significant comparisons of the PLX4720 monotherapy and PLX4720/lapatinib (PLX + Lap) combined therapy groups are indicated by red P values. (D) Mean fold change ± SEM of tumor volume in A375 xenografts (n = 16 per condition) in nude mice fed either PLX4720 or vehicle-laced chow with or without daily lapatinib (100 mg/kg) by oral gavage. Statistically significant comparisons of the PLX4720 monotherapy and PLX4720/lapatinib combined therapy groups are indicated by their respective P values. (E) Kaplan-Meier plot showing time to 3-fold increase in initial tumor volume of 1205Lu xenografts following treatment with PLX4720 chow alone or with lapatinib (100 mg/kg). P value is indicated. (F) Kaplan-Meier plot showing time to 10-fold increase in initial tumor volume of A375 xenografts following treatment with PLX4720 chow alone or with lapatinib (100 mg/kg). P value is indicated.
Discussion
In this study, we report that NRG1/ERBB3 signaling is dramatically enhanced in V600 BRAF harboring melanoma cells treated with RAF and MEK inhibitors and diminishes inhibitor effects on cell viability and tumor growth. Central to the enhanced ERBB3 signaling by PLX4032/AZD6244 is FOXD3, a transcription factor that is induced by RAF/MEK inhibition and can protect cells from PLX4032-mediated death. ERBB3 partners with ERBB2 and the enhanced signaling from ERBB3/ERBB2 complexes can be overcome by combining BRAF inhibitors with the ERBB2/EGFR inhibitor lapatinib. These data suggest that this combination, as well as others that target ERBB3/ERBB2 signaling, may have therapeutic value in the clinic to improve the efficacy of BRAF inhibitors and prolong duration of response.
Our data provide evidence that upregulation of ERBB3 through FOXD3 is a form of adaptive resistance to RAF/MEK inhibitors in mutant BRAF melanoma. We previously showed that FOXD3 was induced upon disruption of mutant BRAF signaling in melanoma and was capable of promoting survival of cells treated with PLX4032 (vemurafenib)/PLX4720 (22, 23). Here, we identify ERBB3 as a direct transcriptional target of FOXD3. This links the regulation of ERBB3 to the mutant BRAF/MEK/ERK pathway for what we believe is the first time. Regulation of ERBB3 by other forkhead box transcription factors has been previously reported. FOXO3a and FOXO1 promote the upregulation of ERBB3 in breast cancer cells treated with lapatinib via effective inhibition of PI3K/AKT signaling (26, 27). While we did not observe upregulation of ERBB3 by lapatinib or PI3K inhibitors in melanoma cells (data not shown), this compensatory feedback mechanism has a number of parallels to the model that we propose. Additionally, FOXA1 was shown to bind to the ERBB3 intronic enhancer region in androgen receptor–driven (AR-driven) breast cancer. In response to androgen stimulation, FOXA1 and AR were recruited to intron 1, where they promoted ERBB3 transcription (31). We found that FOXD3 strongly enriched the intronic enhancer region of ERBB3. While it is unclear whether FOXD3 occupies the same binding sites as FOXA1, FOXD3 is a pioneering factor for FOXA1 at certain loci during development (41). It would be interesting to know whether FOXD3 target genes in melanoma are also known targets of FOXA1.
RAF/MEK inhibitors sensitize V600 mutant BRAF melanoma cells to NRG1β, resulting in a dramatic increase in AKT phosphorylation. Increased PI3K/AKT signaling is one previously identified mechanism of resistance to BRAF inhibition (15, 17, 42). In our experiments, activation of AKT was seen regardless of PTEN status, which has been shown to be one determinant of responsiveness to BRAF inhibition (17, 43–45). Consistent with the importance of AKT signaling in response to RAF inhibitors, we found that directly inhibiting AKT with MK2206 was able to enhance the efficacy of PLX4032 and ablate the protective effects of NRG1β on 1205Lu and WM115 cells (Supplemental Figure 7, A–C). These data also indicate that AKT is one of the main effectors of ERBB3-mediated resistance to PLX4032. Interestingly, inhibition of either BRAF or MEK1/2 led to the decreased phosphorylation of S6 ribosomal protein. but treatment with NRG1β restored S6 ribosomal protein phosphorylation, indicating a shift of translational control from ERK1/2 to AKT signaling. This restoration of protein translation as well as the actions of AKT on apoptotic and cell-cycle proteins may contribute to the enhanced cell viability.
Prior reports have highlighted the upregulation of RTKs, such as IGF1R or PDGFRβ, in melanoma as possible mechanisms of resistance to RAF inhibitors (14, 15). We did not detect enhanced signaling from either RTK in response to their respective ligands when cells were pretreated with PLX4032 for 24 hours. This would suggest that these receptors become overexpressed or hyperactivated later in the development of resistance. Indeed, the adaptive mechanism we propose likely allows cells to persist until they acquire a permanent mechanism of resistance. Consistent with this notion, ERBB3 shows enhanced signaling within a few hours of drug treatment. We also observed a marked increase in phospho-ERBB3 in xenografts after 5-day treatment with PLX4720, indicating in vivo relevance. Increased ERBB3 phosphorylation was also detected in 2 out of 3 on-treatment patient samples available to us. Interestingly, vemurafenib-associated increased ERBB3 phosphorylation was also detected in 4 out of 11 progressing patients (counting samples from Figure 5, B and C), and thus, it may be associated with acquired resistance in some cases. Basal ERBB3 expression was variable across cell lines (Supplemental Figure 8A), and it is therefore likely that the upregulation of ERBB3, as opposed to its basal expression, modulates the response to RAF inhibitor. Additionally, endogenous NRG1 was expressed at very low levels in melanoma cells (Supplemental Figure 8A) and was not enhanced following treatment with RAF inhibitor (Supplemental Figure 8B). The notion that paracrine stimulation of ERBB3 occurs is supported by evidence that production of NRG1 from dermal fibroblasts influences melanocyte biology (46).
Despite lacking the strong kinase activity of its ERBB family members, ERBB3 boasts numerous PI3K-recruiting YXXM motifs and thus serves as a powerful signaling partner for its fellow family members. Furthermore, ERBB3 is upregulated in response to targeted therapies in breast cancer and non–small cell lung carcinoma (24, 25). Unlike melanoma, these cancers are often driven by oncogenic ERBB signaling, either through ERBB2 amplification in the case of breast cancer or EGFR amplification and/or mutation in lung cancer. In acquired resistance to ERBB2 and EGFR inhibitors, signaling through ERBB3 is restored by either ERBB3 upregulation or compensatory phosphorylation by amplified MET (24–27). Our findings add what we believe to be a novel twist to ERBB3 and drug resistance in which ERBB3 signaling is augmented to overcome inhibition of the mutant BRAF/MEK/ERK pathway. A recent study attributed resistance to PLX4032 in mutant BRAF colorectal cancer cells to enhanced EGFR phosphorylation (47). In colorectal cancer cells, inhibition of EGFR in combination with BRAF was able to ablate cell growth and tumorigenesis but melanoma cells did not show this dependence on EGFR. It is possible that EGFR and ERBB3 are governed by similar feedback loops in colorectal cancer and melanoma cells, respectively. Furthermore, we cannot exclude the possibility of RAF-dependent, but FOXD3-independent, mechanisms that contribute to enhanced ERBB3 sensitivity to NRG1 in melanoma.
Targeted therapies are rapidly displacing conventional chemotherapies for cancers with defined driver mutations. For these therapies to show persistent benefits in the clinic, compensatory mechanisms need to be identified and targeted in concert. We demonstrate that treatment of melanoma cells with lapatinib effectively ablated ERBB3 phosphorylation and NRG1β-mediated growth in vitro and enhanced the antitumor activity of PLX4720 in vivo. Although lapatinib does not target ERBB3 directly, it does effectively inhibit all other members of the ERBB family (40, 48) and therefore may prevent ERBB3 phosphorylation in response to other ERBB family ligands in vivo. As both vemurafenib and lapatinib are FDA approved, combinatorial treatment in the clinic is likely feasible and could potentially enhance the efficacy and duration of response to vemurafenib and other mutant BRAF inhibitors. It is noted that diarrhea and skin rash are common adverse effects associated with lapatinib treatment (49), and upregulation of ERBB3 may limit the antitumor actions of lapatinib (27). Monoclonal antibodies targeting ERBB3 have proven efficacious in lung carcinoma and breast and other nonmelanoma tumor models (27, 50, 51) and are now entering clinical trials (e.g., NCT00994123; ClinicalTrials.gov). Our in vivo depletion experiments provide the basis for directly targeting ERBB3 in combination with vemurafenib in mutant BRAF melanoma. Ongoing efforts are focused on utilizing clinical grade anti-ERBB3 monoclonal antibodies in combination with RAF inhibitors to more specifically target the ERBB3 adaptive response pathway in melanoma preclinical models.
Methods
Cell culture.
Human melanoma cell lines WM793, WM115, 1205Lu, WM266-4, and WM239A were donated by Meenhard Herlyn (Wistar Institute, Philadelphia, Pennsylvania, USA). SK-MEL-28 and A375 cells were purchased from ATCC. Tetracycline repressor–expressing (TR-expressing) sublines WM793TR, WM115TR, A375TR, and SK-MEL-28TR cells expressing Dox-inducible FOXD3 or LacZ have been previously described (22). 1205LuTR cells expressing Dox-inducible FOXD3 were generated in the same manner. A375 and A375TR were cultured in DMEM with 10% FBS and nonessential amino acids. All other cells except A375 and A375TR were cultured in MCDB 153 medium containing 20% Leibovitz L-15 medium, 2% fetal bovine serum, 0.2% sodium bicarbonate, and 5 μg/ml insulin.
Inhibitors, growth factors, and function-blocking antibodies.
AZD6244 and lapatinib for in vitro use were purchased from Selleck Chemicals. Lapatinib for in vivo use was provided by the Thomas Jefferson University Hospital pharmacy. PLX4032, PLX4720, and PLX4720 rodent chow were provided by Gideon Bollag at Plexxikon. Recombinant human NRG1β was purchased from Cell Signaling Technology. Gefitinib and erlotinib were provided by Ulrich Rodeck (Thomas Jefferson University).
RNA interference.
1205Lu and WM115 cells were transfected for 5 hours with chemically synthesized siRNAs (Dharmacon) at a final concentration of 25 nM using Lipofectamine RNAiMAX (Invitrogen). For in vivo experiments, 1205LuTR cells stably expressing Dox-inducible shRNAs were generated by lentiviral transduction. Sequences for siRNA and shRNA and lentivirus information can be found in the Supplemental Methods.
Microarray analysis.
Total cellular RNA was extracted using the PerfectPure RNA Cultured Cell Kit (5 Prime). For FOXD3 overexpression experiments, RNA was collected after 5 days of either FOXD3 or LacZ induction. Microarrays were performed by MOgene LC using Agilent-014850 Whole Human Genome Microarrays, and analysis was performed by Kimmel Cancer Center Genomics facility. False discovery rates were estimated using the procedure introduced by Storey (52). Genes with an absolute fold change of at least 1.5 and false discovery rate of less than 25% were considered significant. Microarray data were deposited in the GEO database (GSE43962).
ChIP and ChIP-seq.
WM115TR/FOXD3-V5 cells were induced with Dox for 24 hours and then fixed with 1% formaldehyde for 10 minutes. ChIP was performed using the EZ-ChIP kit and protocol (Millipore). Precleared lysates were incubated overnight with protein G Dynabeads (Invitrogen); beads were washed and eluted overnight at 65°C in ChIP elution buffer (25 mM Tris, pH 7.5, 10 mM EDTA, 0.5% SDS, and 200 mM NaCl). Eluate was treated with RNase A and proteinase K followed by removal of beads and purification of DNA. Antibodies used were normal IgG (Cell Signaling Technology), V5 (Invitrogen), and anti-RNA pol II CTD repeat YSPTSPS (phospho S2) antibody (Abcam). Purified DNA was analyzed by qPCR using iQ SYBR Green Supermix (Bio-Rad), 0.8 μM oligonucleotide primers, and 5 μl ChIP product. The primers used are listed in Supplemental Methods. Primer specificity was confirmed by melt curve analysis and TAE gel electrophoresis. Reaction conditions were as follows: denaturation at 94°C for 30 seconds, annealing at 50°C for 30 seconds, and elongation at 72°C for 30 seconds, with 50 cycles in total. PCR was performed on an iCycler with MyiQ version 1.0 software (Bio-Rad). Relative DNA enrichment levels were calculated using the Comparative Ct method (ΔCt) (53). For ChIP-seq, cells were treated with Dox for 48 hours prior to ChIP. Next generation sequencing and analysis were performed on V5-IP and input DNA by the Kimmel Cancer Center Genomics facility.
ChIP-seq read-mapping, peak-finding, and annotation.
Alignment of ChIP-seq reads to the human hg19 genome was performed using Applied Biosystems Bioscope 1.3 software ChIP-seq analysis pipeline, with default settings. Model-based Analysis of ChIP-Seq (MACS) software version 1.4.1 (54, 55) was used to predict ChIP-binding peaks, comparing the IP samples against total chromatin input. Default peak-calling parameters were used, except the P value cutoff for peak detection was set to a more stringent value of 1 × 10–12. The resulting set of predicted ChIP-binding peaks was analyzed for enrichment of genomic features, including introns, exons, promoter, and intergenic regions, using Cis-regulatory Element Annotation System software, version 1.0.2 (56). Promoter occupancy rates were estimated in regions 3 kb upstream and downstream of transcription start sites.
Western blotting.
Cells were lysed and analyzed by Western blotting, as previously described (16). A list of antibodies can be found in the Supplemental Methods. Chemiluminescence was visualized on a VersaDoc MultiImager and quantitated using Quantity-One software (Bio-Rad).
qRT-PCR.
Total cellular RNA was extracted using the PerfectPure RNA Cultured Cell Kit. cDNA was made using the iScript cDNA Synthesis Kit (Bio-Rad). qPCR and analysis, including statistics, was performed as with ChIP experiments. The primers used are listed in Supplemental Methods.
Flow cytometry.
Detached cells were incubated in PBS with 2% BSA and 50 μl PE-conjugated anti-ERBB3 antibody (R&D Systems) on ice for 45 minutes. Washed cells were analyzed by flow cytometry on a BD FACSCalibur flow cytometer (BD Biosciences). Data were analyzed by FlowJo software (Tree Star Inc.).
Cell viability assays.
Cells (2 × 105) were plated in complete medium in the presence/absence of 10 ng/ml NRG1β and treated with either DMSO, PLX4032 (1 μM), AZD6244 (3.3 μM), lapatinib (1 μM), or combinations of lapatinib with either PLX4032 or AZD6244. Cells were cultured for 72 hours, at which time medium was replaced with complete medium containing 1× AlamarBlue (Invitrogen) with respective inhibitors/NRG1β added. Cells were allowed to reduce AlamarBlue for approximately 2 hours. Medium was collected in triplicate from each condition, and the absorbances of oxidized and reduced AlamarBlue were measured at wavelengths 600 nM and 570 nM, respectively, in a Multiskan Spectrum spectrophotometer (Thermo Fisher Scientific). The change in viability was calculated from the resulting absorbances using the manufacturer’s guidelines. All conditions were normalized to the DMSO control.
Colony formation assays.
A375 (1 × 103) cells were plated per 10-cm dish in complete medium with inhibitors or NRG1β, which were replenished every 3 days. After 7 days, cells were stained with crystal violet in formalin, plates were imaged by scanner, and colonies were imaged on a Nikon Eclipse Ti inverted microscope (Nikon) with NIS-Elements AR 3.00 software (Nikon). The percentage plate coverage is indicated as determined from 5 independent areas using ImageJ software ( http://rsbweb.nih.gov/ij/).
In vivo growth and survival assays.
Melanoma cells (1 × 106) were injected intradermally into female athymic mice (NU/J: Jackson) and allowed to grow for 10–14 days to reach appropriate volume (∼40 mm3 for A375, ∼200 mm3 for 1205Lu, and ∼100 mm3 for 1205LuTR). Mice were fed either AIN-76A chow or AIN-76A with 417 mg/kg PLX4720 chow. For lapatinib experiments, mice received either vehicle (0.5% methylcellulose, 0.1% Tween 80 in sterile PBS) or 100 mg/kg lapatinib suspended in vehicle by oral gavage daily (except on days 16, 17, 23, and 24 for A375 experiments). For shRNA experiments, mice were exposed to 2 mg/ml Dox in drinking water beginning 3 days prior to chow treatment. Measurements of tumor size were taken every 3–4 days using digital calipers, and tumor volume was determined by the following formula: volume = (length × width2) × 0.52. Time-to-event (survival) was determined by a 10-fold increase in baseline volume for the A375 experiment and a 3-fold increase in baseline volume for the 1205Lu experiment. The maximum allowable tumor size for 1205Lu and 1205LuTR cells was limited by the development of skin necrosis requiring euthanasia.
IHC.
Tissue samples from A375 intradermal xenografts were obtained from mice that were fed either control or PLX4720 chow for 5 days. Tissue was fixed in formalin and paraffin embedded. Sections were stained with anti–phospho-ERBB3 Y1289 (21D3) and phospho-ERBB2 Y1221/Y1222 (6B12) antibodies (Cell Signaling Technology) and scored in a blinded manner for staining intensity utilizing a digital Aperio ScanScope GL system and ImageScope software. Statistical analysis of staining quantitation was determined separately for each antibody using a proportional odds mixed model accounting for random effects to adjust for sample variation (R Statistical Software).
Patient samples.
Samples were formalin fixed and paraffin embedded immediately following isolation. IHC was performed using anti–phospho-ERBB3 Y1289 (21D3). Staining was scored in a blinded manner, as above.
Statistics.
For statistical analysis of qPCR and cell viability assays, 2-tailed t tests assuming unequal variances were performed using Excel (Microsoft). Statistical analysis for tumor growth data was conducted using a mixed-effects model and Tukey’s corrected pairwise comparisons of mean fold change in volume between treatment groups (SAS statistical software). Statistical analysis for time-to-event (survival) was conducted using log-rank comparison of Kaplan-Meier curves (SAS statistical software), and α for all experiments was 0.05. Additionally, analysis was performed across samples from all 9 patients that displayed staining for phospho-ERBB3 (3 sets of samples were uniformly negative and were not included). We utilized an ordered logistic regression model (i.e., proportional odds model) with random intercept for each patient. The ordered logistic regression model assumes that the odds of receiving a score greater than or equal to k is odds ratio (OR) times higher for progression than pretreatment, where the number OR is a constant for k = 1 or 2. We used the package ordinal of software R. For all analyses, P values of less than 0.05 were considered statistically significant.
Study approval.
All animal experiments were approved by the IACUC and performed in a facility at Thomas Jefferson University accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC). Patient samples were collected under a protocol approved by the IRB at the The University of Pennsylvania. All patients gave informed consent.
Supplementary Material
Acknowledgments
The work was supported by the American Cancer Society (RSG-08-03-01-CSM), the NIH (R01-CA160495, R01-GM067893, and R01-CA125103), the Department of Defense (W81XWH-11-1-0448), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to A.E. Aplin). E.V. Abel and K. Basile were supported in part by the Joanna M. Nicolay Melanoma Foundation. The Genomics and Flow Core Facilities in the Kimmel Cancer Center are supported by National Cancer Institute support grant 1P30CA56036. We are grateful to Gideon E. Bollag (Plexxikon) for providing PLX4032, PLX4720, and PLX4720 chow; Meenhard Herlyn (Wistar Institute) for WM melanoma cell lines; and Ulrich Rodeck (Thomas Jefferson University/Kimmel Cancer Center) for gefitinib and erlotinib. We thank Mark Pawlowski for performing IHC; Franco Capozza for advice on in vivo experiments; and Michele Weiss for scientific advice. We also acknowledge the help of Terry Hyslop and Tingting Zhan in the Division of Biostatistics, Department of Pharmacology and Experimental Therapeutics, Thomas Jefferson University.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(5):2155–2168. doi:10.1172/JCI65780.
References
- 1.Satyamoorthy K, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63(4):756–759. [PubMed] [Google Scholar]
- 2.Reifenberger J, et al. Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int J Cancer. 2004;109(3):377–384. doi: 10.1002/ijc.11722. [DOI] [PubMed] [Google Scholar]
- 3.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Sumimoto H, et al. Inhibition of growth and invasive ability of melanoma by inactivation of mutated BRAF with lentivirus-mediated RNA interference. Oncogene. 2004;23(36):6031–6039. doi: 10.1038/sj.onc.1207812. [DOI] [PubMed] [Google Scholar]
- 5.Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, Marais R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS One. 2008;3(7):e2734. doi: 10.1371/journal.pone.0002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13(16):4934–4942. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- 7.Boisvert-Adamo K, Aplin AE. B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene. 2006;25(35):4848–4856. doi: 10.1038/sj.onc.1209493. [DOI] [PubMed] [Google Scholar]
- 8.Bhatt KV, Spofford LS, Aram G, McMullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;24(21):3459–3471. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 9.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smalley KSM, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E–mutated melanomas. Mol Cancer Ther. 2008;7(9):2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montagut C, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johannessen CM, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468(7326):968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spofford LS, Abel EV, Boisvert-Adamo K, Aplin AE. Cyclin D3 expression in melanoma cells is regulated by adhesion-dependent phosphatidylinositol 3-kinase signaling and contributes to G1-S progression. J Biol Chem. 2006;281(35):25644–25651. doi: 10.1074/jbc.M600197200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paraiso KHT, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–2760. doi: 10.1158/0008-5472.CAN-10-2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shao Y, Aplin AE. Akt3-mediated resistance to apoptosis in B-RAF–targeted melanoma cells. Cancer Res. 2010;70(16):6670–6681. doi: 10.1158/0008-5472.CAN-09-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emery CM, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagle N, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–3096. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abel EV, Aplin AE. FOXD3 is a mutant B-RAF-regulated inhibitor of G1-S progression in melanoma cells. Cancer Res. 2010;70(7):2891–2900. doi: 10.1158/0008-5472.CAN-09-3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basile KJ, Abel EV, Aplin AE. Adaptive upregulation of FOXD3 and resistance to PLX4032/4720-induced cell death in mutant B-RAF melanoma cells. Oncogene. 2012;31(19):2471–2479. doi: 10.1038/onc.2011.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sergina NV, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445(7126):437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 26.Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci U S A. 2011;109(8):5021–5026. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrett JT, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci U S A. 2011;108(12):5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reschke M, et al. HER3 is a determinant for poor prognosis in melanoma. Clin Cancer Res. 2008;14(16):5188–5197. doi: 10.1158/1078-0432.CCR-08-0186. [DOI] [PubMed] [Google Scholar]
- 29.Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ. NRG1 / ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res. 2009;22(6):773–784. doi: 10.1111/j.1755-148X.2009.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad M, et al. SOX10 directly modulates ERBB3 transcription via an intronic neural crest enhancer. BMC Dev Biol. 2011;11:40. doi: 10.1186/1471-213X-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni M, et al. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell. 2011;20(1):119–131. doi: 10.1016/j.ccr.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera-Feliciano J, et al. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. 2006;133(18):3607–3618. doi: 10.1242/dev.02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell. 2004;13(1):67–76. doi: 10.1016/S1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- 34.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20(21):2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 35.Pinkas-Kramarski R, Shelly M, Glathe S, Ratzkin BJ, Yarden Y. Neu differentiation factor/neuregulin isoforms activate distinct receptor combinations. J Biol Chem. 1996;271(32):19029–19032. doi: 10.1074/jbc.271.32.19029. [DOI] [PubMed] [Google Scholar]
- 36.Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16(5):1373–1383. doi: 10.1158/1078-0432.CCR-09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sithanandam G, Anderson LM. The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther. 2008;15(7):413–448. doi: 10.1038/cgt.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prickett TD, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41(10):1127–1132. doi: 10.1038/ng.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu C, et al. Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure. 2008;16(3):460–467. doi: 10.1016/j.str.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, et al. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23(24):2824–2838. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi H, Kong X, Ribas A, Lo RS. Combinatorial treatments that overcome PDGFRβ-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71(15):5067–5074. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xing F, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring V600EBRAF. Oncogene. 2012;31(4):446–457. doi: 10.1038/onc.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng W, Yennu-Nanda VG, Scott A, Chen G, Woodman SE, Davies MA. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res. 2011;25(2):248–258. doi: 10.1111/j.1755-148X.2011.00950.x. [DOI] [PubMed] [Google Scholar]
- 45.Gopal YNV, et al. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi W, et al. The fibroblast-derived paracrine factor neuregulin-1 has a novel role in regulating the constitutive color and melanocyte function in human skin. J Cell Sci. 2010;123(pt 18):3102–3111. doi: 10.1242/jcs.064774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 48.Nelson MH, Dolder CR. Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Ann Pharmacother. 2006;40(2):261–269. doi: 10.1345/aph.1G387. [DOI] [PubMed] [Google Scholar]
- 49.Burris HA 3rd. Dual kinase inhibition in the treatment of breast cancer: initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist. 2004;9(suppl 3):10–15. doi: 10.1634/theoncologist.9-suppl_3-10. [DOI] [PubMed] [Google Scholar]
- 50.Schoeberl B, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70(6):2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schoeberl B, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Sig. 2009;2(77):ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 52.Storey JD. A direct approach to false discovery rates. J R Stat Soc Ser B Stat Methodol. 2002;64(pt 3):479–498. doi: 10.1111/1467-9868.00346. [DOI] [Google Scholar]
- 53.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Feng J, Liu T, Zhang Y. Using MACS to identify peaks from ChIP-Seq data. Curr Protoc Bioinformatics. 2011. chapter 2: unit 2.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25(19):2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 57.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.