

Abstract
Background and Purpose
Modulation of Kv7/M channel function represents a relatively new strategy to treat neuronal excitability disorders such as epilepsy and neuropathic pain. We designed and synthesized a novel series of pyrazolo[1,5-a] pyrimidin-7(4H)-one compounds, which activate Kv7 channels. Here, we characterized the effects of the lead compound, QO-58, on Kv7 channels and investigated its mechanism of action.
Experimental Approach
A perforated whole-cell patch technique was used to record Kv7 currents expressed in mammalian cell lines and M-type currents from rat dorsal root ganglion neurons. The effects of QO-58 in a rat model of neuropathic pain, chronic constriction injury (CCI) of the sciatic nerve, were also examined.
Key Results
QO-58 increased the current amplitudes, shifted the voltage-dependent activation curve in a more negative direction and slowed the deactivation of Kv7.2/Kv7.3 currents. QO-58 activated Kv7.1, Kv7.2, Kv7.4 and Kv7.3/Kv7.5 channels with a more selective effect on Kv7.2 and Kv7.4, but little effect on Kv7.3. The mechanism of QO-58's activation of Kv7 channels was clearly distinct from that used by retigabine. A chain of amino acids, Val224Val225Tyr226, in Kv7.2 was important for QO-58 activation of this channel. QO-58 enhanced native neuronal M currents, resulting in depression of evoked action potentials. QO-58 also elevated the pain threshold of neuropathic pain in the sciatic nerve CCI model.
Conclusions and Implications
The results indicate that QO-58 is a potent modulator of Kv7 channels with a mechanism of action different from those of known Kv7 openers. Hence, QO-58 shows potential as a treatment for diseases associated with neuronal hyperexcitability.
Keywords: KCNQ/M-channel, PPOs, opener, neuropathic pain, QO-58, DRG
Introduction
The M-type K+ channel plays an important role in controlling neuronal excitability (Hu et al., 2007). It is now well established that the KCNQ gene family (Kv7) underlies the molecular basis of M currents (Wang et al., 1998). Spontaneous mutations in Kv7 subunits cause epilepsy in humans and mice. In addition, Kv7 channels are expressed in the sensory system, such as the trigeminal ganglion neurons (Yoshida and Matsumoto, 2005) and dorsal root ganglion (DRG) (Linley et al., 2008), and are possibly involved in migraine and neuropathic pain. Therefore, modulation of Kv7.2/Kv7.3 channels represents a new strategy for treating neuronal excitability disorders such as migraine, epilepsy and neuropathic pain (Dedek et al., 2001; Munro and Dalby-Brown, 2007; Wuttke et al., 2007).
Over the past few years, multiple compounds have been reported to activate Kv7 channels (Miceli et al., 2008; Xiong et al., 2008). The prototype activator of Kv7 channels is retigabine (Kapetanovic et al., 1995). Retigabine activates Kv7.2, Kv7.3, Kv7.4 and Kv7.5 channels, but inhibits Kv7.1 channels (Tatulian et al., 2001). A crucial Trp236 residue in the cytoplasmic part of S5 is believed to be important for retigabine-induced activation of Kv7.2 channels (Wuttke et al., 2005). Zinc pyrithione (ZnPy) has been found to strongly potentiate all Kv7 channels except Kv7.3 (Xiong et al., 2007). The key determinants of this effect of ZnPy include a leucine residue in S5 (Leu249) and another one within the linker (Leu275) between S5 and the pore region; these are different from retigabine's activation sites (Xiong et al., 2008). Fenamates, including meclofenamic acid and diclofenac, currently used as non-steroidal anti-inflammatory drugs (NSAIDs), are another series of Kv7 channel activators (Peretz et al., 2005). Diclofenac activates Kv7.4 but blocks Kv7.5 channels (Brueggemann et al., 2011). Compounds NH6 and NH29 were synthesized based on the structural template of diclofenac (Peretz et al., 2007). NH29 acts as a gating modifier and is targeted at the voltage sensor of Kv7.2 channels (Peretz et al., 2010). Recently, we found that another NSAID drug, celecoxib, also interacts with the retigabine binding site to activate Kv7 channels (Du et al., 2011).
Activators of Kv7 channels have been shown to have great potential for clinical applications. Retigabine has recently been found to be an effective treatment of epileptic diseases clinically (Fattore and Perucca, 2011; Weisenberg and Wong, 2011). Noticeably, retigabine also has analgesic effects, especially in animal models of chronic inflammatory (Linley et al., 2008; Liu et al., 2008) and neuropathic pain (Blackburn-Munro and Jensen, 2003; Rose et al., 2011). In addition, flupirtine, an analogue of retigabine, is also a potent Kv7 channel activator. Encouragingly, flupirtine is already used as a centrally acting, non-opioid analgesic for the treatment of a variety of pain states (Devulder, 2010).
In an effort to find new chemical structures of Kv7/M-channel openers, we designed and synthesized a novel series of pyrazolo[1,5-a] pyrimidin-7(4H)-one compounds (PPOs), which have the ability to open Kv7 channels (Jia et al., 2011; Qi et al., 2011). We evaluated and analysed the effects of about 120 analogues of PPOs on Kv7.2/Kv7.3 channels to construct a structure-activity relationship (SAR). From this SAR study, we found that a trifluoromethyl group at the 2-position is required for this activity; at the 3-position, substitution with a phenyl or naphthyl group afforded best activity and electron withdrawing substitutes on the aromatic ring at the 5-position is the most important site for activity. The optimization of PPOs based on the SAR results produced the lead compound QO-58, which showed the best EC50 (0.06 ± 0.01 μM) for activation of Kv7.2/Kv7.3 (Qi et al., 2011). In the present study, we characterized the effects of QO-58 on Kv7 channels using whole-cell patch-clamp recording techniques (Figure 1A). Our results indicate that QO-58 is a potent activator of Kv7 channels with a mechanism of action different from those of known Kv7 openers. QO-58 also has the potential to be developed as a treatment for diseases associated with neuronal hyperexcitability.
Figure 1.
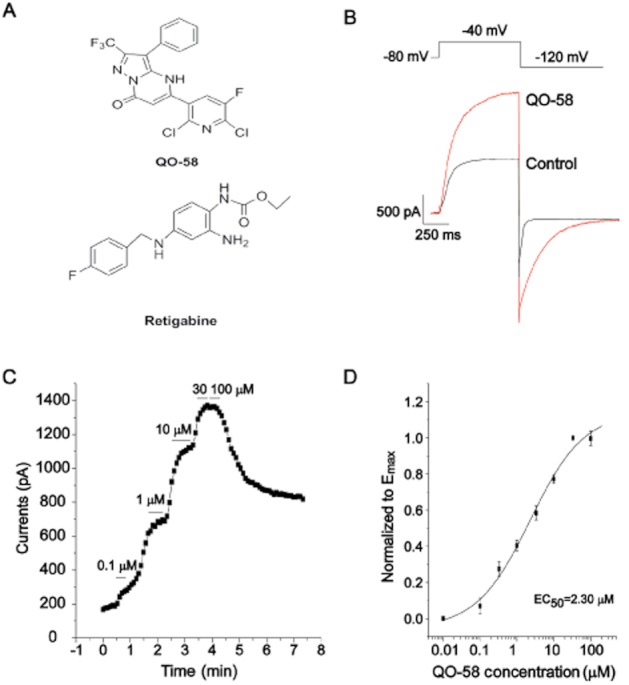
Compound QO-58 enhances Kv7.2/Kv7.3 channel currents. (A) The structure of compound QO-58 and retigabine. (B) Typical outward currents elicited by step depolarization to −40 mV from a holding potential of −80 mV in the absence and presence of 3 μM QO-58. (C) QO-58 concentration-dependently increased Kv7.2/Kv7.3 channel currents generated at −40 mV. (D) QO-58-induced outward currents were normalized to the maximal effect (Emax) value and fit to a logistic function; EC50 value was 2.3 ± 0.8 μM and the slope factor was 0.6 ± 0.1 (n = 6).
Methods
Compounds
QO-58 and retigabine (Purity >98% by HPLC-DAD) were synthesized in the Department of New Drugs Development, School of Pharmacy, Hebei Medical University, and their purity was verified by MS and NMR analysis (Qi et al., 2011). XE991 was purchased from Sigma (St Louis, MO, USA).
DNA constructs
Plasmids encoding human Kv7.1, human Kv7.2, rat Kv7.3, human Kv7.4 and human Kv7.5 (GenBank accession numbers: NM000218, AF110020, AF091247, AF105202 and AF249278, respectively) were kindly provided by Diomedes E. Logothetis (Virginia Commonwealth University, Richmond, VA, USA). Kv7.2 (R207W), Kv7.2 (L275A), Kv7.2 (Y284C), Kv7.2 (A306T) and Kv7.2 (W236L) mutants were kindly provided by Zhaobing Gao (Chinese Academy of Sciences, Shanghai, China). Kv7.2 (VVY224225226AIC) mutants were produced by Pfu DNA polymerase with a QuickChange kit (Stratagene, La Jolla, CA, USA). The structure of the mutants was confirmed with DNA sequencing.
The nomenclature of Kv7 potassium channels and other receptors and channels conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Cell culture
Stable CHO cells expressing Kv7.2/7.3 channels (a kind gift from Professor Wang Kewei, Peking University) were grown in DMEM supplemented with 10% fetal calf serum, 1× non-essential amino acids, 600 μg·mL−1 G418 and 600 μg·mL−1 hygromycin B.
HEK293 cells were cultured in DMEM supplemented with 10% fetal calf serum and antibiotics. For transfection of 8 wells of cells, a mixture of 4 μg Kv7, 4 μg RFP pcDNAs and 6 μL Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was prepared in 1.2 mL DMEM. The mixture was then applied to the cell culture wells and incubated for 4–6 h. Recordings were made 24 h after cell transfection, and the cells were used within 48 h.
Rat DRG neuron culture
DRGs were extracted from the intervertebral foramina of 7-day-old Sprague-Dawley rats (provided by Experimental Animal Center of Hebei Province). A total of 40 adult male Sprague-Dawley rats were used in our study. The ganglia were digested at 37°C with 1 mg·mL−1 collagenase for 30 min, followed by another 30 min digestion with 2.5 mg·mL−1 trypsin. They were subsequently suspended at least twice in DMEM plus 10% fetal calf serum to stop digestion. Thereafter, the ganglia were plated on poly-D-lysine-coated glass coverslips. The neurons were cultured for 4 days and used within 48 h.
The results of all studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Electrophysiology
For current measurements in the CHO cell, HEK293 cells and DRG neurons, recordings were performed using the perforated (amphotericin B, 250 μg·mL−1, Sigma) patch-clamp technique at room temperature. The signals were amplified using an HEAK EPC10 patch-clamp amplifier. The acquisition rate was 10 kHz and signals were filtered at 2.5 kHz. Patch electrodes were pulled with a micropipette puller (Sutter Instruments, Novato, CA, USA) and fire polished to a final resistance of 1–2 MΩ. Series resistances were compensated by 60–80%. The internal and external solution for the mammalian cell lines and rat DRG neuron recording was as follows (in mM): KCl 150, MgCl2 5, HEPES 10, pH 7.4 adjusted with KOH; NaCl 160, KCl 2.5, MgCl2 1, CaCl2 2, glucose 10, HEPES 20 and pH 7.4 adjusted with NaOH respectively.
Neuropathic pain model of sciatic nerve chronic constriction injury
Adult male Sprague-Dawley rats (weighing 160∼180 g, provided by Experimental Animal Center of Hebei Province) were used in this experiment. The use of animals in this study was approved by the Animal Care and Ethical Committee of Hebei Medical University (Shijiazhuang, China), under the IASP guidelines for animal use. The rats were kept in plastic cages in a room with a temperature of 22–25°C, under 12/12 h light/dark cycle (lights on from 7 h to 19 h). Food and water were available ad libitum. The operation procedures for establishing the neuropathic pain model with sciatic nerve chronic constriction injury (CCI) have been described previously (Sommer and Schafers, 1998). Briefly, the rats were anaesthetized with an i.p. injection of sodium pentobarbital (10–20 mg kg−1). The depth of anaesthesia was assessed by responses to the pinching of the rats legs; no response to the pinching was taking as the sign for further operation.
Four ligatures (chromic catgut 4-0) were placed around the nerve with a distance of 1 mm between each ligature. The ligatures were loosely tied until a short flick of the ipsilateral hind limb was observed. The rats with above operation were divided into five groups with eight animals in each group: control solvent (17% PEG400) group, 12.5 mg·kg−1 retigabine group, and three QO-58 groups with different doses of 12.5 mg·kg−1, 25 mg·kg−1 and 50 mg·kg−1. Drugs were given by i.p. injection twice a day. Mechanical and heat nociceptive threshold was assessed before and after performing surgery on days 1, 3, 5, 7, 9, 11 and 19.
Mechanical test
Response threshold to mechanical stimulus was assessed as described previously (Chaplan et al., 1994). Briefly, calibrated nylon filaments (Von Frey hair, Stoelting Co, Chicago, IL, USA) with different bending forces were applied to the mid-plantar surface of the right hind paw of the rats. The filaments were applied starting with the softest and continuing in ascending order of stiffness. A brisk withdrawal of the right hind limb was considered a positive response.
Radiant heat test
Response to heat stimulus was tested on the right hind paw of the rats using a radiant heat lamp source (Hargreaves et al., 1988). The intensity of the radiant heat stimulus was maintained at 25 ± 0.1°C. Response of right hind paw withdrawal threshold (elapse time) was noted.
Data analysis and statistics
The concentration-response curve was fitted by logistic equation: 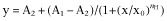 , where x is the drug concentration, and nH is the Hill coefficient. The current activation curves were generated by plotting the normalized tail current amplitudes against the step potentials and were fitted with a Boltzmann equation: y = A/{1 + exp[(Vh − Vm)/k]}, where A is the amplitude of relationship, Vh is the voltage for half-maximal activation, Vm is the test potential and k is the slope factor of the curve. The activation and deactivation traces were fitted to a single exponential function: I = A × [1 − exp(−t/τ)], where I is the current, A is amplitudes, t is time and τ is the time constant. Results are expressed as means ± SEM. Statistical analysis of differences between groups was carried out using Student's t-test or paired t-test. P-values ≤0.05 were considered significant.
, where x is the drug concentration, and nH is the Hill coefficient. The current activation curves were generated by plotting the normalized tail current amplitudes against the step potentials and were fitted with a Boltzmann equation: y = A/{1 + exp[(Vh − Vm)/k]}, where A is the amplitude of relationship, Vh is the voltage for half-maximal activation, Vm is the test potential and k is the slope factor of the curve. The activation and deactivation traces were fitted to a single exponential function: I = A × [1 − exp(−t/τ)], where I is the current, A is amplitudes, t is time and τ is the time constant. Results are expressed as means ± SEM. Statistical analysis of differences between groups was carried out using Student's t-test or paired t-test. P-values ≤0.05 were considered significant.
Results
The effects of compound QO-58 on expressed Kv7.2/Kv7.3 currents
We first studied the effect of compound QO-58 ( Figure 1A) on the activation of Kv7.2/Kv7.3 channels stably expressed in CHO cells. Superfusion of the cells with 3 μM QO-58 increased both the activated stead state current (−40 mV) and the deactivating tail current (−120 mV) amplitudes of Kv7.2/Kv7.3 channels (Figure 1B). Figure 1C shows the concentration-dependent effects of QO-58 on Kv7.2/Kv7.3 currents elicited by the depolarization potential of −40 mV. We then constructed a concentration-response relationship for the effects of QO-58 on Kv7.2/Kv7.3 currents (Figure 1D). QO-58 (100 μM) induced a maximal 6.15 ± 0.76-fold increase in the Kv7.2/Kv7.3 currents recoded at −40 mV. The current increase produced by 100 μM QO-58 was taken as the Emax, which was used to normalize the increase in Kv7.2/Kv7.3 currents produced by all concentrations of QO-58. The normalized concentration-response curves were fitted by a modified logistic equation, and the EC50 value was 2.3 ± 0.8 μM and the Hill coefficient was 0.6 ± 0.1 (Figure 1D) (n = 6).
Next, we studied the effect of compound QO-58 on voltage-dependent activation of Kv7.2/Kv7.3 currents. Superfusion of the cells with 0.3, 1 and 3 μM QO-58 increasingly shifted the threshold for channel activation to more hyperpolarized potentials (Figure 2A). Tail current amplitudes at −120 mV resulting from different test potentials were normalized and fitted by the Boltzmann function. QO-58 concentration-dependently shifted the V1/2 of Kv7.2/Kv7.3 currents to more negative potentials, the EC50 value was 1.2 ± 0.2 μM and the Hill coefficient was 1.2 ± 0.3 (n = 6) (Figure 2B,C).
Figure 2.

Compound QO-58 shifts the voltage-dependence of Kv7.2/Kv7.3 channel activation. (A) Series of outward currents elicited by depolarizing voltage steps (hold at −80 mV, in 10 mV incremental voltage steps from −130 to +30 mV) with increasing concentrations of QO-58. (B) The activation curves were generated in the absence of QO-58(▪), and in the presence of 0.1 μM, 0.3 μM, 1 μM, 3 μM, 10 μM QO-58. (C) The magnitude of the QO-58-induced shifts in V1/2 (ΔV1/2, towards more negative potentials) was calculated and plotted against QO-58 concentration; EC50 value was 1.2 ± 0.2 μM and a slope factor was 1.2 ± 0.3 (n = 6). (D) The effects of QO-58 on the activation and deactivation kinetics of Kv7.2/Kv7.3 currents. (E) Summary for results depicted in (D) (n = 5, *P < 0.05).
Then we studied the effects of QO-58 on the kinetics of Kv7.2/Kv7.3 currents. The activation and deactivation currents were both fitted to a single exponential function. Application of 10 μM QO-58 significantly slowed channel activation and deactivation kinetics (Figure 2D,E).
Selectivity of compound QO-58 on Kv7 channels
The Kv7 family of potassium channels consists of five subtypes, Kv7.1 to Kv7.5 (Ng et al., 2011). We studied the selectivity of QO-58 on Kv7 channels expressed in HEK293 cell. Kv7.5 did not produce significant currents when expressed alone, thus Kv7.5 was co-expressed with Kv7.3 and the effect of QO-58 was tested on the heterologous Kv7.3/Kv7.5 currents. When Kv7 currents were recorded at −40 mV, QO-58 (10 μM) significantly increased the Kv7.1, Kv7.2, Kv7.4 and Kv7.3/Kv7.5 currents, but only slightly increased Kv7.3 currents (Figure 3B).
Figure 3.
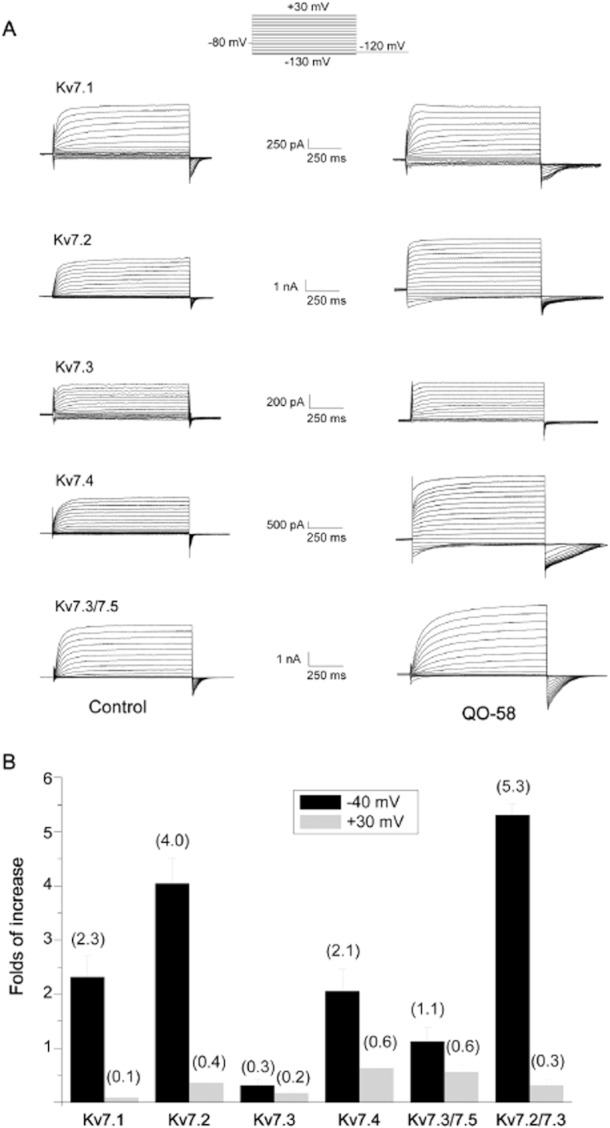
Compound QO-58 enhances Kv7 channel currents. (A) Whole-cell currents with Kv7 channel in the absence (left panels) and presence (right panels) of 10 μM QO-58. (B) Histogram plotting of the QO-58 effect on Kv7 channel currents generated by step depolarization at −40 mV and at +30 mV (n = 5–8).
QO-58 concentration-dependently increased Kv7.1, Kv7.2, Kv7.4 and Kv7.3/Kv7.5 channel currents recorded at −40 mV; the EC50 values for QO-58 were 7.0 ± 1.0, 1.3 ± 1.0, 0.6 ± 0.1 and 5.2 ± 2.2 μM for Kv7.1, Kv7.2, Kv7.4 and Kv7.3/Kv7.5 channels, respectively (Figure 4). Thus, QO-58 is more potent at activating Kv7.2 and Kv7.4 channels than the other channels. QO-58 (10 μM) produced a substantial leftward shift of the V1/2 of Kv7.1, Kv7.2, Kv7.4 and Kv7.3/Kv7.5 currents by 21.7 ± 1.1 mV (n = 7), 56.8 ± 5.4 mV (n = 6), 58.7 ± 2.9 mV (n = 6) and 47.4 ± 2.8 mV (n = 5), respectively; on the other hand, the V1/2 of Kv7.3 was shifted to the right by 2.7 ± 0.1 mV. (Figure 5 and Table 1) (n = 5).
Figure 4.
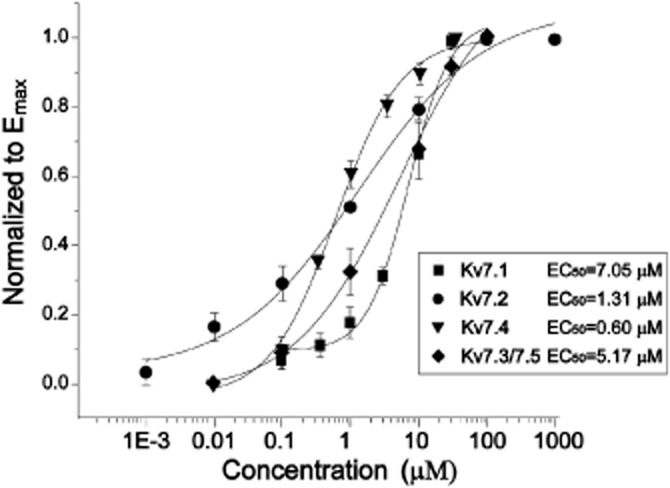
Concentration-dependent effects of QO-58 on Kv7 channel currents. The currents were measured at −40 mV. The concentration-response relationships were fitted with the logistic function (n = 5–8).
Figure 5.
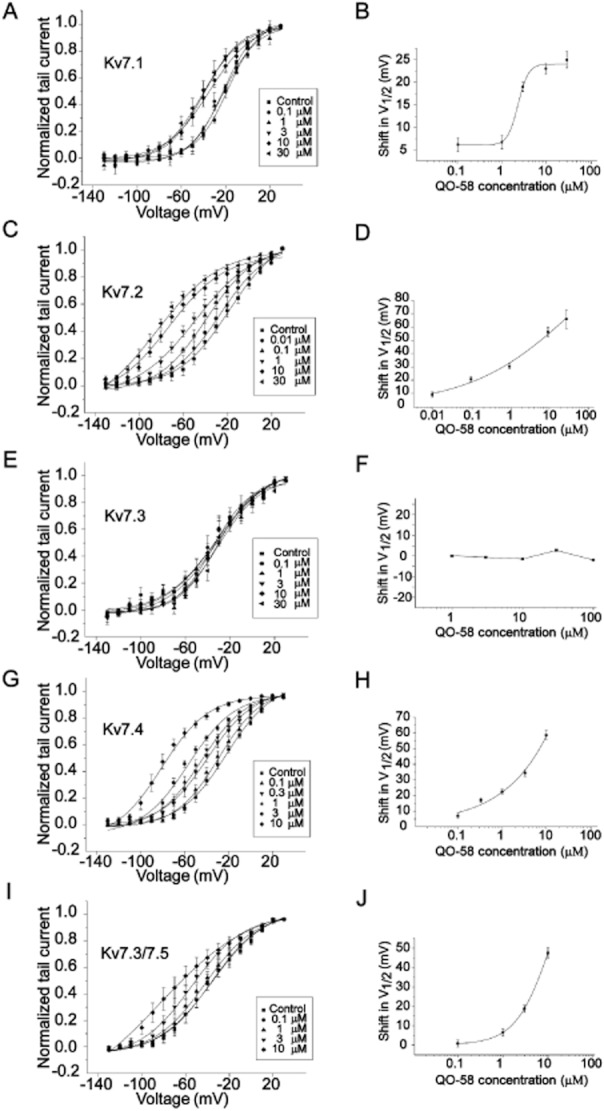
Concentration-dependent effects of QO-58 on the voltage-dependent activation of Kv7 channel currents. The left panels (A,C,E,G,I) show activation curves for Kv7 currents generated from tail currents with increasing concentrations of QO-58. The right panels (B,D,F,H,J) show the magnitude of the QO-58-induced shifts in V1/2 calculated and plotted against QO-58 concentration (n = 5–7).
Table 1.
The maximal changes of V1/2 of Kv7 subtype currents induced by QO-58 (10 μM)
| V1/2 (mV) (control) | V1/2 (mV) (QO-58) | ΔV1/2 (mV) | |
|---|---|---|---|
| Kv7.1 | −17.8 ± 0.8 | −39.5 ± 1.1 | 21.7 ± 1.1 |
| Kv7.2 | −18.1 ± 1.9 | −74.9 ± 5.4 | 56.8 ± 5.4 |
| Kv7.3 | −32.4 ± 3.1 | −35.1 ± 2.1 | 2.7 |
| Kv7.4 | −22.0 ± 1.4 | −80.7 ± 2.9 | 58.7 ± 2.9 |
| Kv7.2/Kv7.3 | −13.2 ± 1.1 | −58.3 ± 1.7 | 45.1 ± 1.7 |
| Kv7.3/Kv7.5 | −35.5 ± 2.4 | −51.7 ± 2.8 | 47.2 ± 2.8 |
All values are mean ± SEM (n = 5–7). ΔV1/2 means the difference between V1/2 of control and that of compound.
QO-58 (10 μM) significantly slowed the activation kinetics of Kv7.4 and Kv7.3/Kv7.5 currents, and slowed the deactivation kinetics of Kv7 currents. It was noteworthy that QO-58 largely increased the deactivation time constant of Kv7.2 by 6.2-fold, and greatly increased the deactivation time constant of Kv7.4 by 36.4-fold (Supporting Information Figure S1 and Table 2) (n = 5–7).
Table 2.
The effects of QO-58 on channels activation and deactivation kinetics of Kv7 subtypes
| Time constants (ms) | |||
|---|---|---|---|
| Control | QO-58 (10 μM) | ||
| Kv7.1 | Activation τ (−40 mV) | 122.5 ± 22.8 | 175 ± 35.9 |
| Deactivation τ (−120 mV) | 31.2 ± 2.3 | 84.2 ± 26.8 | |
| Kv7.2 | Activation τ (−40 mV) | 96.6 ± 12.0 | 137.6 ± 13.4 |
| Deactivation τ (−120 mV) | 15 ± 1.5 | 96.4 ± 12.5 | |
| Kv7.3 | Activation τ (−40 mV) | 166.3 ± 25.7 | 175 ± 18.9 |
| Deactivation τ (−120 mV) | 18.5 ± 3.0 | 29.3 ± 4.2 | |
| Kv7.4 | Activation τ (−40 mV) | 34.4 ± 4.4 | 100.5 ± 3.9 |
| Deactivation τ (−120 mV) | 8.7 ± 0.2 | 316.6 ± 125.8 | |
| Kv7.2/Kv7.3 | Activation τ (−40 mV) | 76.0 ± 2.5 | 179.5 ± 26.4 |
| Deactivation τ (−120 mV) | 28.3 ± 6.8 | 278.5 ± 57.3 | |
| Kv7.3/Kv7.5 | Activation τ (−40 mV) | 83 ± 16.6 | 137 ± 19.6 |
| Deactivation τ (−120 mV) | 32.1 ± 1.8 | 253.2 ± 61.9 | |
All values are mean ± SEM (n = 4–7).
In all, QO-58 was found to be a potent Kv7 channel opener with more selective effect on Kv7.2 and Kv7.4 channels, but has a minor effect on the Kv7.3 channel.
The mechanism for QO-58 activation of Kv7 currents
The Trp236 residue in Kv7.2 is believed to be a key amino acid in mediating the activation by retigabine, thus mutation of tryptophan to leucine [Kv7.2 (W236L)] renders the channel insensitive to retigabine (Wuttke et al., 2005). Hence, we studied the effect of QO-58 on this mutant Kv7.2 (W236L). As shown in Figure 6C,D, QO-58 (10 μM) still markedly enhanced the Kv7.2 (W236L) currents (the currents were increased by 1.6 ± 0.4-folds at −50 mV) in this mutant channel and induced a leftward shift of the activation curve, with V1/2 changed as effectively as in the wild-type Kv7.2 (n = 5, P < 0.05). These data suggest that QO-58 does not share the same mechanism of action as retigabine for activation of Kv7 currents (Wuttke et al., 2005).
Figure 6.

The effect of QO-58 on wild-type Kv7.2, Kv7.2 (W236L) and Kv7.2 (AIC) channel currents. (A) The Kv7.2 currents were elicited by a series of depolarizing voltage steps from −130 to +30 mV from a holding potential of −120 mV in the absence and presence of 10 μM QO-58. (B) QO-58 induced a leftward shift of the activation curve from V1/2 = −18.1 ± 1.9 mV to V1/2 = −74.9 ± 5.4 mV. (C,D) The effects of QO-58 and RTG on Kv7.2 (W236L) currents. QO-58 and RTG induced a leftward shift of the activation curve from V1/2 = −33.3 ± 1.7 mV to V1/2 = −92.8 ± 2.7 mV and −41.6 ± 2.0 mV respectively (n = 5). (E,F) The effects of QO-58 and RTG on Kv7.2 (AIC) currents. QO-58 and RTG induced a leftward shift of the activation curve from V1/2 = −81.0 ± 2.2 mV to V1/2 = −88.0 ± 2.1 mV and −95.5 ± 1.7 mV respectively (n = 5).
As it was found that QO-58 enhances all the Kv7 channel currents except Kv7.3 (Figure 3), we compared the sequences of these Kv7 channels and found that Val224Val225Tyr226 inside the S4 and S5 segments of the Kv7 were conserved in Kv7.1, Kv7.2, Kv7.4 and Kv7.5, while in Kv7.3 these residues were Ala-Ile-Cys. Thus, we made and tested a mutant Kv7.2 [VVY224,225,226AIC (Kv7.2 (AIC)]; compared with the wild-type Kv7.2 channel, Kv7.2 (AIC) had two major different features. Firstly, Kv7.2 (AIC) currents demonstrated a prominent inactivation (Figure 6E); secondly, the voltage-dependent activation of Kv7.2 (AIC) was greatly shifted to a more negative potential, and the V1/2 was now −81.0 ± 2.2 mV. QO-58 (10 μM) abolished the inactivation and did not affect the steady-state current amplitude of Kv7.2 (AIC). On the other hand, RTG (10 μM) also abolished the inactivation but more importantly increased greatly the steady-state current amplitude of Kv7.2 (AIC) (Figure 6E). Similarly, QO-58 was significantly less effective than RTG in further hyperpolarizing the voltage-dependent activation of Kv7.2 (AIC) (Figure 6F). These results indicate that Val224Val225Tyr226 in Kv7.2 are involved in QO-58 activation of Kv7 channels, and also further suggest that QO-58 and RTG activate Kv7.2 channel using a different mechanism.
We made further efforts to identify the amino acids in Kv7.2 possibly involved in the activity of QO-58. We constructed a docking simulation model for the interaction of QO-58 with residues within S5 and S6 of the Kv7.2 channel (Supporting Information Figure S2), a procedure we have used in our previous work (Du et al., 2011). Two residues, Aal306 and Leu275 in Kv7.2, were identified for possibly interacting with QO-58. We then tested the effects of QO-58 on Kv7.2 (A306T) and Kv7.2 (L275A) mutants (Supporting Information Figure S3). Two things are clear from these results. Firstly, the potentiating effects of QO-58 on the maximum current amplitude of Kv7.2 were converted to an inhibitory effect in Kv7.2 (A306T). Secondly, the effects of QO-58 on the voltage-dependent activation of Kv7.2 were greatly reduced; a change in V1/2 of 56.8 mV by QO-58 was seen for Kv7.2, and a change of 28.5 mV was seen for Kv7.2 (A306T) and 37.3 mV for Kv7.2 (L275A). These results suggest that alanine 306 and leucine 275 in Kv7 could also be important for mediating the activation of the Kv7 channels by QO-58.
Mutation on Kv7.2 and Kv7.3 channel can lead to benign familial neonatal convulsions (BFNCs) (Coppola et al., 2003; Zhou et al., 2006). We tested the effect of QO-58 on two of these mutants: Kv7.2 (R207W) and Kv7.2 (Y284C). Interestingly, QO-58 activated both Kv7.2 (R207W) and Kv7.2 (Y284C) mutants. These results indicate that QO-58 may be beneficial for treating benign familial neonatal convulsions (Supporting Information Figure S4).
QO-58 enhances the native M current in rat DRG neurons
We next examined the potential effect of QO-58 on native M-type K+ currents in rat DRG neurons. M currents were activated by a depolarizing voltage of −20 mV, and tail M currents were observed at −60 mV (Figure 7A). Both QO-58 and RTG increased M currents by approximately 25% at 10 μM (Figure 7B). QO-58 also induced significant hyperpolarization of the resting membrane potential (RMP) to about −18.4 mV. Application of an M-channel antagonist, XE991, completely abolished the membrane hyperpolarization induced by QO-58 (Figure 7C,D). A perfusion of QO-58 immediately abolished the repetitive firing spikes from a DRG neuron (Figure 7E).
Figure 7.
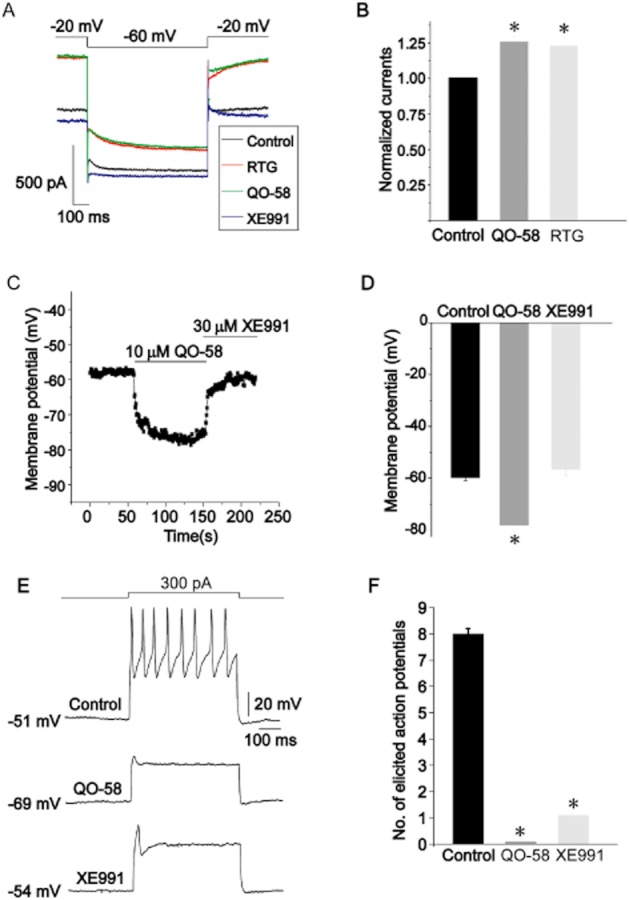
QO-58 enhances the M-current in DRG neurons. (A) M-current were activated at −20 mV and deactivated at −60 mV. QO-58 (10 μM) and RTG (10 μM) enhanced M-current similarly; XE991 inhibited M-current. (B) Normalized M-current amplitude in the presence of 10 μM QO-58 and 10 μM RTG (n = 5). (C) The resting membrane potential (RMP) of an isolated DRG neuron was monitored when 10 μM QO-58 was applied followed by the addition of 30 μM XE991. (D) Summary of results shown in (C): −59.7 ± 1.2 mV (control), −78.1 ± 2.3 mV (QO-58) and −56.6 ± 2.5 mV (XE991) respectively (n = 8–10). (E) Action potentials were evoked by application of a 300 pA depolarizing current. (F) Summary of results shown in (E) (n = 3). *P < 0.05.
QO-58 elevated the threshold of neuropathic pain in the CCI model
A CCI of the sciatic nerve model was successfully established. The rats operated on to create CCI of the sciatic nerve displayed significantly reduced withdrawal threshold for mechanical stimulus and shortened withdrawal latency to thermal stimulus, from day 1 after the operation (Figure 8A,B). QO-58 and retigabine significantly increased the threshold of the mechanical stimulus 1–19 days after the operation. QO-58 concentration-dependently prolonged the withdrawal latency for a response to the thermal stimulus, and in the 50 mg·kg−1 QO-58 groups the withdrawal latency was prolonged to its pre-operation level.
Figure 8.
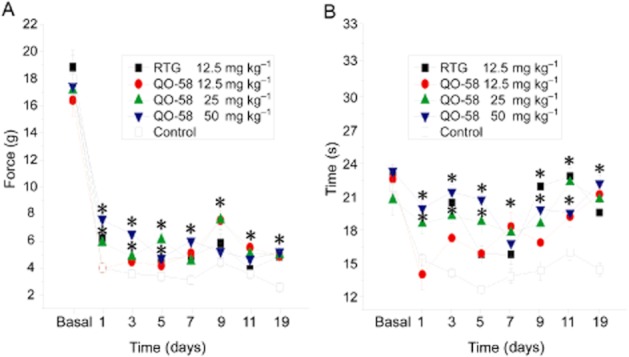
The effect of QO-58 on nociceptive behaviour induced by CCI of sciatic nerve. (A) Dose-dependent effect of QO-58 and RTG on withdrawal threshold to mechanical stimulus. (B) Dose-dependent effect of QO-58 and RTG on withdrawal time in response to radiant thermal stimulus. n = 6–8 rats per group, *P < 0.05.
Discussion and conclusions
Chemical modulators of Kv7 channels have become candidates for the treatment of diseases related to neuronal hyperexcitability. In addition, Kv7 channel modulators are also valuable probes for elucidating the channel gating mechanisms and their functional roles. In this study, we demonstrated that the compound QO-58, which we recently developed, is a potent modulator of the Kv7 channels. Importantly, QO-58 activated Kv7 channels with a mechanism clearly distinct from that used by retigabine.
Two outstanding features can be deduced from the observed effects of QO-58 on Kv7 currents. The first is a marked shift in the voltage-dependent activation of some Kv7 channels towards a more negative potential; the V1/2 for Kv7.2, Kv7.4, Kv7.2/Kv7.3, Kv7.3/Kv7.5 were shifted about −40∼–60 mV, whereas the V1/2 for Kv7.1 was shifted about −20 mV, whereas the V1/2 for Kv7.3 was not affected (Table 1). This effect of QO-58 has important implications. The negative shift of the voltage-dependent activation of Kv7 channel would lower the membrane potential threshold for Kv7 activation and thus would inevitably hyperpolarize the membrane potential, which would reduce the excitability of the cells expressing Kv7 channels. The above notion was well manifested in the effects of QO-58 action on DRG neurons: QO-58 significantly shifted the resting membrane potential of DRG neurons to a hyperpolarization potential and greatly reduced the firing spikes of these neurons (Figure 7). As Kv7.2/Kv7.3 (Wang et al., 1998) and Kv7.3/Kv7.5 (Shah et al., 2002) have been shown to underlie the neuronal M-type currents, these results corroborate the effects of QO-58.
The second outstanding feature of the effects of QO-58 on Kv7 is the significant slowing of the Kv7 deactivation kinetics. Although QO-58 also slowed the activation kinetics of Kv7, the effects on the deactivation kinetics were much stronger. Again, the efficacy of QO-58 on slowing the channel deactivation followed the same order as that found to shift the voltage-dependent activation of the different Kv7 subtypes; namely, the effects were greater on Kv7.2, Kv7.4, Kv7.2/Kv7.3, Kv7.3/Kv7.5 channels, but were less on Kv7.1, and were negligible on Kv7.3 (Table 2). As both the activation and the deactivation of Kv7 channel were slowed by QO-58, and slowing of channel activation would reduce the channel activity, it must be that the slowing of the channel deactivation by QO-58 contributed to the overall enhancement of the Kv7 channel. Thus the Kv7 channel became difficult to close in the presence of QO-58.
On the other hand, the effects of QO-58 on the amplitudes of Kv7 were less striking. Although QO-58 markedly enhanced the current amplitudes of Kv7 at negative membrane potentials (such as those at −40, −50 mV), it had much less effect on the maximal saturating current amplitudes recorded at more positive potentials; in fact, in the presence of QO-58, the current amplitudes at +30 mV were only increased by about 20–60% (Figure 3B).
It is clear that QO-58 uses a different mechanism from RTG to activate Kv7 channels. A tryptophan (W236) in Kv7.2 is the key determinant for the activation of Kv7.2 by RTG (Wuttke et al., 2005). Clearly, Trp236 in Kv7.2 did not mediate the activation of Kv7.2 by QO-58, as QO-58 activated Kv7.2 (W236L) as effectively as wild-type Kv7.2 (Figure 6). Also, in contrast to RTG (Tatulian et al., 2001) but similar to ZnPy (Xiong et al., 2007), QO-58 had little effect on Kv7.3. Val224Val225Tyr226 are conserved in all Kv7 except Kv7.3. Further, the mutant Kv7.2 (VVY224 225,226AIC) was not activated by QO-58 but still activated by RTG. These results suggest that Val224Val225Tyr226 in Kv7.2 play an important role in the activation of Kv7 channels by QO-58. However, these results should be interpreted with caution; the properties of Kv7.2 (AIC) currents greatly changed compared with those of Kv7.2 (Figure 6), as the channel currents became inactive and the voltage-dependent activation was greatly shifted to the more negative potentials. It is not clear whether these changes affect the effects of QO-58 in an allosteric manner. Nevertheless, the finding that this Kv7.2 (AIC) was still activated by RTG suggests that Val224Val225Tyr226 in Kv7.2 is indeed important for QO-58 activation of Kv7. With the help of docking modelling, we found that the mutation of Kv7.2 (A306T) and Kv7.2 (L275A) reduced the efficacy of QO-58 to activate Kv7.2. QO-58 may interact with regions involving Ala306 and Leu275 of Kv7.2 to activate Kv7 channels.
In some aspects, QO-58 affected Kv7 currents similarly to a recently reported opener of Kv7 channels, ZTZ240 (Gao et al., 2010). The major effects of ZTZ240 on Kv7 include hyperpolarization of voltage-dependent activation and a marked slowing of channel deactivation, which are the characteristics of QO-58's actions. Furthermore, like QO-58, ZTZ240 does not activate Kv7 through the sites used by RTG, and interestingly, Ala309 is important for the effects of ZTZ240 whereas Ala306 seems to be important for the effect of QO-58; Ala306 and Ala309 are neighbouring amino acids. However, there are several differences between QO-58 and ZTZ240 regarding their effects on Kv7 channels: firstly, QO-58 but not ZTZ240 activated the Kv7.1 channel. Secondly, compared with ZTZ240, QO-58 has a more selective effect on Kv7.4.
The CCI of a peripheral nerve is a widely used experimental model for neuropathic pain. In this study, we found that QO-58, as well as RTG, significantly reduced the nociceptive responses to mechanical and thermal stimuli in CCI rats, which suggests these K+ channel openers have the potential to be developed further for treatment of neuropathic pain.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (30730031 to H.L.Z., 30500112 to X.N.D.), the National Basic Research Program (2007CB512100) and a National 863 project (2006AA02Z183 to H.L.Z.). The Hebei Provincial Natural Science Foundation of China (C2011206021 to Y.M.) is also acknowledged. We thank Bo Qiu and Liman Huo for their kind help.
Glossary
- BFNC
benign familial neonatal convulsions
- CCI
chronic constriction injury
- DRG
dorsal root ganglion
- PPOs
pyrazolo[1,5-a] pyrimidin-7(4H)-ones
- RTG
retigabine
- SAR
structure-activity relationship
Conflict of interest
There are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 The effect of QO-58 on the activation and deactivation kinetics of Kv7 channel currents. The left panel shows the effects of QO-58 (10 μM) on the activation and deactivation kinetics of Kv7 currents. The right panel shows the summarized data of the activation and deactivation time constants of Kv7 currents (n = 4–6, *P < 0.05).
Figure S2 Docking results for the interaction of QO-58 with residues within S5 and S6 of the Kv7.2 channel. The open (A and B) and the closed (C) conformations of S5 and S6 segments with the pore loop were shown. (D) The 3D structure of compound QO-58. D shows the hydrogen bonds (blue dotted lines) between QO-58. Residues Leu272 and Leu275 are shown in red and yellow colours respectively.
Figure S3 The effects of QO-58 on wild-type Kv7.2, Kv7.2 (A306T), Kv7.2 (L275A) currents. (A,C,E) The currents were recorded using the voltage protocol shown in Figure 6 in the absence and presence of 10 μM QO-58. (B,D,F) The change of V1/2 from −46.3 ± 1.2 mV to −74.8 ± 2.8 mV was seen for Kv7.2 (A306) and from −30.2 ± 1.9 mV to −67.5 ± 3.5 mV for Kv7.2 (L275A) (n = 4–6, *P < 0.05).
Figure S4 The effects of QO-58 on BFNC mutant channel currents. (A) The Kv7.2 (R207W) and Kv7.2 (Y284C) channel currents were recorded in the absence and presence of 10 μM QO-58 using the voltage protocol holding at −80 mV, in 10 mV incremental voltage steps from −70 to +50 mV. (B) Histogram plotting of the QO-58 effect on Kv7.2 (R207W) and Kv7.2 (Y284C) channel currents generated by step depolarization at −40 mV and at +30 mV (n = 3–4, *P < 0.05).
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460:109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Martin JL, Cribbs LL, Byron KL. Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol Pharmacol. 2011;79:10–23. doi: 10.1124/mol.110.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Coppola G, Castaldo P, Miraglia del Giudice E, Bellini G, Galasso F, Soldovieri MV, et al. A novel KCNQ2 K+ channel mutation in benign neonatal convulsions and centrotemporal spikes. Neurology. 2003;61:131–134. doi: 10.1212/01.wnl.0000069465.53698.bd. [DOI] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98:12272–12277. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devulder J. Flupirtine in pain management: pharmacological properties and clinical use. CNS Drugs. 2010;24:867–881. doi: 10.2165/11536230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Du XN, Zhang X, Qi JL, An HL, Li JW, Wan YM, et al. Characteristics and molecular basis of celecoxib modulation on K(v)7 potassium channels. Br J Pharmacol. 2011;164:1722–1737. doi: 10.1111/j.1476-5381.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattore C, Perucca E. Novel medications for epilepsy. Drugs. 2011;71:2151–2178. doi: 10.2165/11594640-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang T, Wu M, Xiong Q, Sun H, Zhang Y, et al. Isoform-specific prolongation of Kv7 (KCNQ) potassium channel opening mediated by new molecular determinants for drug-channel interactions. J Biol Chem. 2010;285:28322–28332. doi: 10.1074/jbc.M110.116392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia C, Qi J, Zhang F, Mi Y, Zhang X, Chen X, et al. Activation of KCNQ2/3 potassium channels by novel pyrazolo[1,5-a]pyrimidin-7(4H)-one derivatives. Pharmacology. 2011;87:297–310. doi: 10.1159/000327384. [DOI] [PubMed] [Google Scholar]
- Kapetanovic IM, Yonekawa WD, Kupferberg HJ. The effects of D-23129, a new experimental anticonvulsant drug, on neurotransmitter amino acids in the rat hippocampus in vitro. Epilepsy Res. 1995;22:167–173. doi: 10.1016/0920-1211(95)00050-x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci. 2008;28:11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zhang X, Wang C, Zhang G, Zhang H. Antihistamine mepyramine directly inhibits KCNQ/M channel and depolarizes rat superior cervical ganglion neurons. Neuropharmacology. 2008;54:629–639. doi: 10.1016/j.neuropharm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Martire M, Taglialatela M. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol. 2008;8:65–74. doi: 10.1016/j.coph.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Munro G, Dalby-Brown W. Kv7 (KCNQ) channel modulators and neuropathic pain. J Med Chem. 2007;50:2576–2582. doi: 10.1021/jm060989l. [DOI] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A, et al. Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, et al. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol. 2007;97:283–295. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- Peretz A, Pell L, Gofman Y, Haitin Y, Shamgar L, Patrich E, et al. Targeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifier. Proc Natl Acad Sci U S A. 2010;107:15637–15642. doi: 10.1073/pnas.0911294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Zhang F, Mi Y, Fu Y, Xu W, Zhang D, et al. Design, synthesis and biological activity of pyrazolo[1,5-a]pyrimidin-7(4H)-ones as novel Kv7/KCNQ potassium channel activators. Eur J Med Chem. 2011;46:934–943. doi: 10.1016/j.ejmech.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Rose K, Ooi L, Dalle C, Robertson B, Wood IC, Gamper N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain. 2011;152:742–754. doi: 10.1016/j.pain.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544(Pt 1):29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Schafers M. Painful mononeuropathy in C57BL/Wld mice with delayed wallerian degeneration: differential effects of cytokine production and nerve regeneration on thermal and mechanical hypersensitivity. Brain Res. 1998;784:154–162. doi: 10.1016/s0006-8993(97)01327-9. [DOI] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Weisenberg JL, Wong M. Profile of ezogabine (retigabine) and its potential as an adjunctive treatment for patients with partial-onset seizures. Neuropsychiatr Dis Treat. 2011;7:409–414. doi: 10.2147/NDT.S14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Wuttke TV, Jurkat-Rott K, Paulus W, Garncarek M, Lehmann-Horn F, Lerche H. Peripheral nerve hyperexcitability due to dominant-negative KCNQ2 mutations. Neurology. 2007;69:2045–2053. doi: 10.1212/01.wnl.0000275523.95103.36. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H, Li M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol. 2007;3:287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008;29:99–107. doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Matsumoto S. Effects of alpha-dendrotoxin on K+ currents and action potentials in tetrodotoxin-resistant adult rat trigeminal ganglion neurons. J Pharmacol Exp Ther. 2005;314:437–445. doi: 10.1124/jpet.105.084988. [DOI] [PubMed] [Google Scholar]
- Zhou XH, Ma AQ, Liu XH, Huang C, Zhang YM, Shi RM. A novel mutation in KCNQ2 gene causes benign familial infantile convulsions (BFIC) in a Chinese family. Zhonghua Er Ke Za Zhi. 2006;44:487–491. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.