

Abstract
Cell division cycle 25 A (Cdc25A), a dual-specificity protein phosphatase, is one of the most crucial cell cycle regulators, which removes the inhibitory phosphorylation in cyclin-dependent kinases (CDKs), such as CDK2, CDK4, and CDK6, and positively regulates the activities of CDKs that lead to cell cycle progression. In addition, Cdc25A also acts as a regulator of apoptosis. Overexpression of Cdc25A promotes tumorigenesis, and is frequently observed in various types of cancer. Here we briefly summarize current understanding of the role of Cdc25A in cell proliferation and apoptosis, as well as the impact of overexpression of Cdc25A on tumorigenesis.
Keywords: Apoptosis, cancer, Cdc25A, cell cycle, cell proliferation
1. Introduction
One of the most prominent features in cancer cells is deregulated growth [1, 2]. This is often due to mutations of genes that endow cancer cells with the ability of uncontrolled proliferation and resistance to apoptosis [1, 3]. The uncontrolled proliferation is mostly attributed to cell cycle deregulation [4, 5]. Cell cycle is a tightly controlled procedure, which needs a delicate signaling network to determine the proper progression [5, 6]. Cyclin-dependent kinases (CDKs) are foremost cell cycle regulators that phosphorylate and activate downstream players like retinoblastoma (Rb) protein to promote cell cycle progression [7]. Not surprisingly, the activities of CDKs are also precisely regulated by multiple events such as phosphorylation, dephosphorylation and protein-protein interaction [7], among which, the removal of inhibitory phosphorylation on CDKs by cell division cycle 25 (Cdc25) phosphatase, a dual-specificity protein phosphatase, is critical for activation of CDKs [7,8].
The Cdc25 dual phosphatase family has three members: Cdc25A, Cdc25B, and Cdc25C [9]. Although the catalytic domains of these phosphatases are well conserved, their regulatory domains, which decide their subcellular distribution and turnover, are greatly diverse [10]. While Cdc25B and Cdc25C promote G2/M progression by primarily dephosphorylating CDK1 at T14/Y15, two inhibitory phosphorylation sites [11], Cdc25A plays a more extensive role in assisting both G1/S and G2/M progression by dephosphorylating CDK4 at Y17 [12], CDK6 at Y24 [13], as well as CDK2 and CDK1 at T14/Y15 [11, 14]. More importantly, overexpression of Cdc25A has been frequently documented in multiple cancer cell lines, which is highly associated with the malignancy and poor prognosis in cancer patients [10, 15].
The human Cdc25A protein has 524 amino acid residues, with two distinct regions: N-terminal regulatory domain and C-terminal catalytic domain [9]. The regulatory domain of Cdc25A contains a series of phosphorylation sites that are involved in regulating protein stability and protein-protein interaction [16, 17]. Besides, this domain also contains nuclear localization sequence (NLS) and nuclear exportation sequence (NES), which determine the subcellular location of Cdc25A [18]. The catalytic domain of Cdc25A shares a common HCX5R motif with other protein tyrosine phosphatases, and has a shallow active pocket that is accessible for substrates containing either phospho-tyrosine (p-Y) or phospho-threonine (p-T) [19]. In this review, we focus on summarizing the role of Cdc25A in cell proliferation and apoptosis. In addition, we discuss the impact of Cdc25A overexpression on tumorigenesis.
2. The role of Cdc25A in regulating cell proliferation
2.1 G1/S entry
The proper cell cycle progression from G1 to S phase requires a series of genes related to DNA synthesis to be expressed timely. These genes encode proteins such as dihydrofolate reductase (DHFR), thymidine kinase (TK), and ribonucleotide reductase (RR), which commonly share similar E2F binding sites in their promoters [20]. The binding of E2F transcriptional activators to the promoters is associated with the acetylation and tri-methylation on these genes, inducing the expression of these genes and the G1/S cell cycle progression [21].
In the G0 and early G1 phase, hypophosphorylated Rb protein serves as a major inhibitor of E2Fs by directly binding to and inhibiting the activation domain of E2Fs [22], as well as recruiting co-repressors such as histone deacetylases (HDACs) to the target genes [22]. As cell cycle progresses from G1 to S, the phosphorylation level of Rb gradually increases, resulting in dissociation of E2F from Rb and activation of E2Fs [22] (Fig.(1)).
Fig. (1).
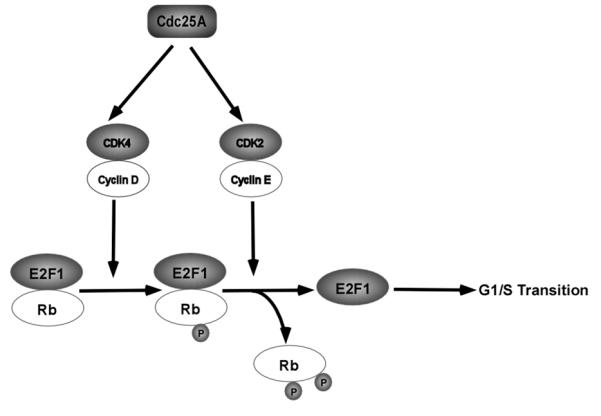
Promotion of G1/S transition by Cdc25A. Cdc25A activates cyclin D-CDK4 complex and cyclin E-CDK2 complex, which phosphorylate Rb and dissociate Rb from complex with E2F1, releasing inhibition on E2F1, whose activity is essential for G1/S transition.
Phosphorylation of Rb protein is at least partially mediated by cyclin D-CDK4/CDK6 complexes [23, 24], and this event is considered to be the rate limiting step during the G1/S cell cycle transition [23, 24]. Masatoshi et al. demonstrated that cyclin D-CDK4 complex is able to phosphorylate Rb at S780 in vivo and in vitro [25]. Although accumulation of p-S780 on Rb is not enough for dissociation of E2F from Rb [26], it is essential for further phosphorylation of Rb by cyclin E-CDK2 complex in the late G1 phase [26] (Fig. (1)).
The observation that microinjection of Cdc25A antibodies to the proliferating cells results in G1 cell cycle arrest suggests a significant role of Cdc25A in G1/S cell cycle progression [27]. Later, it was demonstrated that transforming growth factor beta (TGF-β)-induced downregulation of Cdc25A causes accumulation of inhibitory phosphorylation, p-Y17, on CDK4 and consequent G1 arrest [12]. Consistently, in vitro studies showed that GST-bound Cdc25A can remove inhibitory phosphorylation on CDK4 [28], confirming Cdc25A as a positive regulator of CDK4. In addition, inhibition of Cdc25A expression by TGF-β also results in accumulating inhibitory phosphorylation, p-Y24, on CDK6, decreasing CDK6 activity by at least two folds, and reducing Rb phosphorylation [13]. The results indicate that CDK6 is also positively regulated by Cdc25A (Fig. (1)).
Besides cyclin D-CDK4/6 complexes, Cdc25A is also able to dephosphorylate p-T14/p-Y15, two inhibitory phophorylation residues, on CDK2 in later G1 phase [14]. The activated cyclin E-CDK2 complex phosphorylates Rb at S567, eventually rendering the dissociation of Rb from E2F, releasing repression on E2F transcription activity, and promoting G1/S transition [26] (Fig. (1)). A study using ectopic expression of Cdc25A disclosed that overexpression of Cdc25A can accelerate G1/S transition by prematurely upregulating the CDK2 activity [29]. However, this study surprisingly showed that the activities of cyclin D-CDK4/6 complexes are not affected by overexpression of Cdc25A and not associated with Cdc25A-mediated acceleration of G1/S progression [29]. This contradictory observation might root from lack of inhibitory phophorylation on both CDK4 and CDK6, which needs induction by ultraviolet (UV) or TGF-β, in proliferating immortalized epithelial cell line (MCF-10A) and NRK cells [29].
As Cdc25A is important for cell cycle progression, its activity has to be timely and precisely regulated during the whole cell cycle. This can be achieved by multiple mechanisms including regulation of Cdc25A expression at transcriptional [30, 31], translational [32, 33], and post-translational level [16], as well as regulation of catalytic efficiency of Cdc25A by modulating phosphatase activity [32] and enzyme-substrate interaction [17, 34].
The promoter of Cdc25A gene has three putative E2F binding regions: E2F-A, E2F-B, and E2F-C, which locate around −60, 0, and −160 bps to the transcription start site individually [35]. Upon serum starvation, E2Fs were observed to bind to E2F-A region in the complex with Rb proteins, which inhibits the transcription of Cdc25A gene in NIH 3T3 cells [36]. After addition of serum, the E2F1 binds to the E2F-B region on Cdc25A gene and activates Cdc25A transcription, which is necessary for E2F1-induced G1 phase progression and G1/S transition in the originally quiescent Rat1 fibroblasts cells [30]. E2F1-dependent Cdc25A transcription might be a consequence of replacement of E2F-Rb complex on putative E2F sites by free E2F1, which is at least partially acetylated [37].
In addition to E2F1, E2F2 and E2F3 can also activate Cdc25A transcription [30]. However, the induction of Cdc25A transcription by E2F2 and E2F3 is not only less potent but also timely later than that by E2F1 [30]. A possible explanation of such observation is that E2F1 induces Cdc25A transcription by direct binding, whereas E2F2 and E2F3 do not [30]. Cycloheximide, which blocks de novo protein synthesis, inhibits E2F2- and E2F3-induced Cdc25A transcription [30], implying that other proteins are required for E2F2- and E2F3-induced Cdc25A transcription.
Besides E2Fs, signal transducer and activator of transcription 3 (STAT3) was demonstrated to mediate cytokine-induced G1/S transition by upregulating Cdc25A transcription and activities of CDKs [38]. Another study revealed that upon IL-6 treatment, STAT3 binds to the promoter of Cdc25A gene and activates Cdc25A transcription in serum-starved cells [39]. Such activation appears to be in a Myc-dependent manner, as knockdown of Myc neutralizes the effects of STAT3 on activation of Cdc25A transcription [39]. This is consistent with the finding that Cdc25A is a transcriptional target of Myc/Max heterodimer [31]. Likely, binding of STAT3 to Cdc25A promoter facilities recruitment of Myc, even though there is no direct interaction between STAT3 and Myc [39]. On the contrary, STAT3 can also inhibit Cdc25A transcription by recruiting Rb to the promoter under reactive oxygen species (ROS) stimulation [39]. The molecular mechanism by which recruitment of Rb by STAT3 inhibits Cdc25A transcription is still not known. Possibly, Rb represses STAT3-induced Cdc25A transcription by recruiting histone deacetylases or methytranferases.
After Cdc25A mRNA is translated, the de novo synthesized protein will undergo various post-translational modifications, among which, phosphorylation is the most prevalent one. The N-terminal domain of Cdc25A contains multiple phosphorylation residues, which modulate either Cdc25A stability or Cdc25A-CDK interaction to match cellular requirement [16, 17]. The most important post-translational modification of Cdc25A is cyclin E-CDK2 complex-dependent phosphorylation, which activates Cdc25A to promote G1/S progression [27]. This, together with the fact that cyclin E-CDK2 is positively regulated by Cdc25A, suggests that there is a feedforward loop between cyclin E-CDK2 complex and Cdc25A [27]. The kinetic data indicates that the feedforward loop between cyclin E-CDK2 complex and Cdc25A is critical for cells to overcome the restriction point (R-point or G1 checkpoint) [40], after which next round DNA replication is committed [41].
As keeping faithful transmission of genomic content from an individual cell to its daughter is the key to survival of organisms, a cell has to withdraw from DNA synthesis in response to genotoxic stress [42]. One feasible strategy for this goal is to downregulate cellular Cdc25A level, which can be achieved by promoting Cdc25A turnover upon DNA damage [43]. The genotoxic stress initiates DNA damage response (DDR) pathway by activating the serine/threonine (S/T) kinases ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related) [44]. Activated ATM or ATR phosphorylates a number of downstream signaling cascades, including cell cycle checkpoint proteins (Chk1 and Chk2) [45] (Fig. (2)).
Fig. (2).
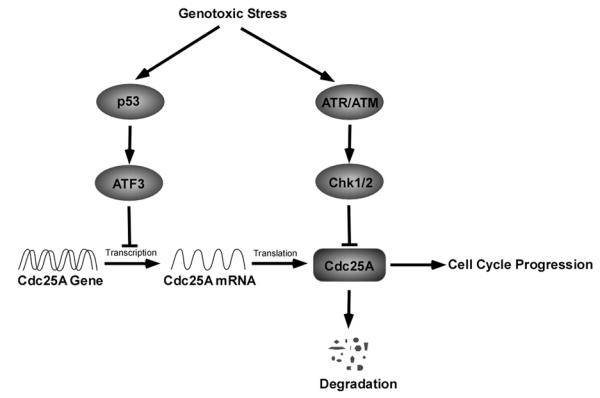
Regulation of Cdc25A in response of genotoxic stress. Upon DNA damage, p53 and ATR/ATM machinery are activated individually. Activated p53 inhibits Cdc25A transcription through ATF3 activation, and activated ATR/ATM promotes Cdc25A turnover through checkpoint activation.
Chk1 was originally considered to be activated by ATR in the presence of single-stranded DNA (ssDNA) [46], but a recent study undermined this point by providing evidence that Chk1 can be phosphorylated and activated by ATM in response to radiation [47]. Activated Chk1 phosphorylates multiple residues on Cdc25A, among which, phosphorylation at S76 is necessary for further phosphorylation at S79 and S82, residues locating in DSG motif (TDS82GFCLDS88PGPLD), by casein kinase 1α (CK1α) [48-50]. After phosphorylation at S82, an E3 ligases complex, SCF (Skp1/Cul1/F-box protein) binds to Cdc25A through interaction between DSG motif and β-TrCP (F-box protein) to facilitate Cdc25A ubiquitination and subsequent degradation [50, 51], which, in turn, causes inhibition of CDK2 activity [52] and G1/S cell cycle arrest [53] (Fig. (2)).
S76 residue of Cdc25A is also a phoshorylation target of glycogen synthase kinase 3 beta (GSK3β) [54], which has long been known to negatively affect G1 cell cycle progression by promoting cyclin D1 and c-Myc degradation [55, 57]. The GSK3β-induced phosphorylation at S76, which requires a prime phosphorylation at T80 by Polo-like kinase 3 (Plk-3) [54], is associated with SCF-mediated Cdc25A degradation and G1/S cell cycle arrest [54].
Chk2, another checkpoint protein, was reported to be phosphorylated and activated by ATM upon ionizing radiation (IR) [58], and activated Chk2 phosphorylates Cdc25A at S123, which promotes Cdc25A degradation and downregulates CDK2 activity resulting in blocking DNA synthesis and inducing S-phase checkpoint [58]. However, other studies argued that it is Chk1, but not Chk2, that plays an essential role in regulating Cdc25A level in response to DNA damage, because there is no much difference in kinetics of IR-induced Cdc25A degradation between Chk2−/− and Chk2+/+ HCT116 cells [59].
According to the two-wave model [14], the G1/S checkpoint is initiated and maintained by two unique pathways: one is ATR/ATM-Chk1/2-Cdc25A cascade, which is rapidly activated to arrest G1/S transition in response to DNA damage [14]; the other is p53-mediated cell cycle arrest, which appears later, but lasts longer [14]. p53-associated G1 arrest was demonstrated to be mediated by p21Cip1 [60, 61]. Interestingly, Rother et al. found that p53 expression is inversely correlated to the mRNA and protein level of Cdc25A [62]. Since p21Cip1 was found to bind to Cdc25A promoter and repress Cdc25A transcription upon DNA damage [63], p21Cip1 was considered to be necessary for p53-mediated repression of Cdc25A transcription. Nevertheless, the p53 homologues such as TAp63α and p73α can activate p21Cip1 transcription, but cannot repress Cdc25A transcription, indicating that p53-mediated suppression of Cdc25A may be independent of p21Cip1 [62].
Since there is no binding sequence for p53 in Cdc25A promoter, Demidova et al. analyzed the Cdc25A promoter sequence to look for binding sites for other regulators, and found that this region contains binding sequence for activating transcription factor 3 (ATF3), a direct target of p53 [64]. It has been demonstrated that ATF3 mediates p53-induced transcription repression of Cdc25A, which is responsible for maintaining long term cell cycle arrest in response to genotoxic stress [64] (Fig. (2)).
In addition to DNA damage, hypoxia was found to be capable of affecting Cdc25A expression in colon cancer cells [65]. Hypoxia-associated downregulation of Cdc25A is mediated by p21Cip1 and miR-21, but independent of p53 and DDR pathway [65]. Combining the finding that hypoxia-induced S phase accumulation also requires p21Cip1 and miR-21, it is highly suspected that hypoxia-induced S phase arrest is a consequence of hypoxia-mediated downregulation of Cdc25A [65]. A recent report revealed that the 3′-Untranslated Region (3′-UTR) of Cdc25A mRNA contains a putative binding site for miR-21, which is essential for interaction between miR-21 and Cdc25A mRNA [66]. This interaction downregulates Cdc25A expression, thereby arresting cells at G1/S entry [66].
Unlike transcriptional or post-translational regulation, the effect of translational regulation of Cdc25A on cell cycle is largely unexplored. Lin et al. reported that protein boule-like (BOLL protein) is another factor that utilizes the 3′-UTR region to regulate Cdc25A expression [32]. The interaction between 3′-UTR of Cdc25A and BOLL protein does not affect the stability of Cdc25A mRNA as miR-21 does, but promotes translation of Cdc25A mRNA [32]. Although BOLL protein is widely known as a meiotic regulator, whether BOLL protein-mediated Cdc25A translation contributes to cell proliferation remains to be determined.
Another currently known translation factor of Cdc25A is eukaryotic initiation factor 2 alpha (eIF2α), which plays a major role in regulating global translation upon cellular stress [67]. In response to cellular stress, phosphorylation of eIF2α at S51 increases its affinity to eukaryotic initiation factor 2B (eIF2B) to form a complex that inhibits the guanine nucleotide exchange in translation [68, 69]. Treatment of cells with salubrinal, an eIF2α inhibitor, decreases the expression of Cdc25A, implying that Cdc25A translation can be regulated by eIF2α [33]. However, it is unclear how eIF2α regulates Cdc25A translation.
The phosphatidylinositol 3-kinases (PI3K)-mammalian target of rapamycin (mTOR) cascade has long been known as an important signaling pathway that regulates translation initiation [70]. The observation that LY294002, a PI3K inhibitor, reduces the expression of Cdc25A indicates involvement of PI3K signaling in Cdc25A expression [71]. As mTOR is also a G1/S cell cycle regulator [72], it is possible that Cdc25A plays a role in mTOR-associated G1/S cell cycle progression. However, how PI3K-mTOR cascade regulates Cdc25A expression remains to be defined.
2.2 G2/M entry
The cyclin B-CDK1 complex was identified as a major component of maturation promoting factor that drives G2/M progression [73]. The activation of CDK1 at the G2/M boundary includes two independent events: one is CAK-mediated phosphorylation at T161 [74, 75], which is necessary for cyclin B-CDK1 complex formation [76], and the other is Cdc25-mediated dephosphorylation at T14/Y15 [11]. It was demonstrated that the dephosphorylation and activation of CDK1 by Cdc25A is a rate limiting step during the G2/M transition, and activated cyclin B-CDK1 complex phosphorylates Cdc25A at S17/S115, which stabilizes Cdc25A by uncoupling it from check point regulation [77]. Therefore, cyclin B-CDK1 complex forms a feedforward loop with Cdc25A to overcome the G2/M checkpoint [77]. Cdc25B was considered to initiate this feedforward loop between cyclin B-CDK1 complex and Cdc25A by dephosphorylating inhibitory phosphorylation on CDK1 [77]. Mitra et al. further substantiated that cyclin A-CDK2 complex positively regulates CDK1 activity by activating Cdc25B in S phase [78]. At the mitotic exit, Cdc25A is targeted by an E3 ligase complex, APC/C (anaphase promoting complex/cyclosome), which binds to Cdc25A through Ken-Box motif in a phosphorylation-independent manner and specifically ubiquitinates Cdc25A causing its degradation [79] (Fig. (3)).
Fig. (3).
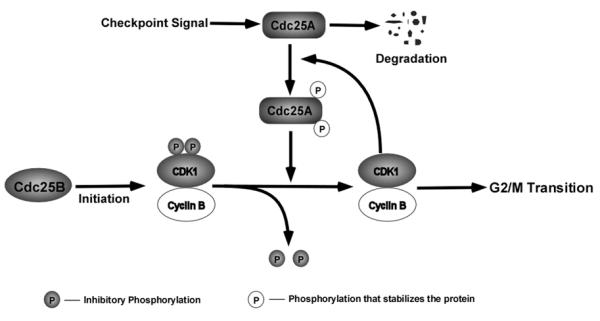
Regulation of G2/M transition by Cdc25A. Cdc25B initiates CDK1 activity by dephosphorylating p-T14/Y15 on CDK1. Activated CDK1 stabilizes Cdc25A by phosphorylating at S17/S115 on Cdc25A, which can feedforwardly elevate CDK1 activity by removing inhibitory phosphorylation on CDK1. The feedforward loop between CDK1 and Cdc25A is critical for G2/M transition.
Timofeev et al. reported their striking discovery that overexpression of Cdc25A accumulates p-Y15 on CDK1, whereas the activity of CDK1 is still high [80]. Moreover, this study also revealed that Cdc25A activates CDK7, which, in turn, phosphorylates CDK1 at T161 and promotes the cyclin B-CDK1 complex formation [80]. Therefore, Cdc25A, in addition to Cdc25B, might play a role in initiating feedforward loop between cyclin B-CDK1 and Cdc25A.
In order to avoid entering mitosis with damaged DNA that threatens cell survival, a cell has to stop prematurely initiated mitosis at G2/M entry [81]. Like G1/S entry, DDR pathway plays a critical role in controlling G2/M entry by regulating Cdc25A abundance [82]. It was revealed that activated Chk1 phosphorylates NIMA (never in mitosis gene A)-related kinase 11 (NEK11) at S273 and causes NEK11 activation [83]. The activated NEK11, in turn, phosphorylates Cdc25A at S82, which requires a prime phosphorylation of Cdc25A at S76 by Chk1 [83]. Phosphorylation of DSG motif (p-S82) thus provides a docking site for β-TrCP-mediated SCF complex binding, which promotes ubiquitination and proteasome-mediated degradation of Cdc25A [83]. A new model points out that dimerized 14-3-3λ serves as a scaffold to form a Cdc25A-Chk1-14-3-3λ ternary complex, which is essential to phosphorylate Cdc25A at S76 [84].
Besides proteasome-mediated degradation, in vitro studies showed that checkpoint signaling also accumulates phosphorylation on Cdc25A at S123, which decreases the stability of Cdc25A and arrests cells at G2/M transition [58, 85]. However, in vivo results disclosed that although S123A knock-in mutants have a longer half-life than that in normal cells upon IR, the G2/M cell cycle checkpoint is not perturbed in the mutants [86], implying that the IR-induced G2/M cell cycle arrest does not really require Cdc25A degradation. Meanwhile, the authors also showed that Cdc25A S123AKI/KI accumulates at centrosome and promotes centrosome duplication by increasing CDK1 activity upon IR [86]. This, together with G2/M cell cycle arrested by IR, causes uncoupling of centrosome duplication and DNA replication, which eventually leads to supernumerary centrosomes [86].
The elongated half-life of Cdc25A S123AKI/KI does not disturb the IR-induced G2/M checkpoint, suggesting that other post-translational modifications of Cdc25A affect G2/M cell cycle progression in response to IR-induced checkpoint activation. Chen et al. reported that phosphorylation of Cdc25A at T507 or S178 provides a docking site for 14-3-3 binding [17]. The interaction between 14-3-3 and Cdc25A prevents Cdc25A from binding to cyclin B-CDK1 complex; thus, phosphorylation of Cdc25A at T507 inactivates cyclin B-CDK1 complex and inhibits associated mitotic entry [17]. This is supported by another study that Chk1 phosphorylates Xenopus Cdc25A (Xe-Cdc25A) at T504 (corresponding to human T507), which prevents Cdc25A from interacting with Cdk1-cyclin A, Cdk1-cyclin B, or Cdk2-cyclin E and consequently inactivates these cyclin-CDK complexes [34]. Therefore, phosphorylation of Cdc25A at T507, rather than Cdc25A degradation, is exactly responsible for activating mitotic checkpoint [34]. Strikingly, p-T507-triggered inhibition of CDK1 activity does not require 14-3-3 protein, implying that 14-3-3 has other role such as preventing dephosphorylation of Cdc25A at T507, rather than inhibiting activation of cyclin-CDK complexes [34].
Recently, a member of Cdc14 dual-specificity phosphatase family, Cdc14A, was found to be able to negatively regulate G2/M progression and CDK1 activity by downregulating phosphatase activity, but not stability, of Cdc25A through an unknown mechanism [87].
3. The role of Cdc25A in apoptosis
Apoptosis is a precisely programmed procedure of cell death, which is controlled by multiple complicated signaling networks [88]. Apoptosis signal-regulating kinase 1 (ASK1) is one of the most important stress kinases regulating dual specificity mitogen-activated protein kinase kinase 4 (MKK4)-c-Jun N-terminal kinases (JNK) cascade [89-90], and p38 mitogen-activated protein kinase (MAPK) [90]. The activation of JNK pathway affects a serious of apoptosis-related proteins such as p53, Bax, and c-Jun [91, 92], and leads to apoptosis [93]. Similarly, activated p38 causes apoptosis by regulating various factors such as NFκB, p53, and BimEL, a Bcl-2 family member [93-95].
Generally, ASK1 is activated by dissociating from the complex with thioredoxin in response to oxidative stress [96]. Zou et al. found that cytoplasmic Cdc25A inhibits ASK1 activity and increases the resistance to oxidative stress-induced apoptosis [89]. The inhibitory effect of Cdc25A on ASK1 activity was demonstrated to be independent of its phosphatase activity, but dependent of interaction between Cdc25A and ASK1, which disrupts homo-oligomer formation of ASK1 [89]. Another report showed that nitrosative stress inhibits Cdc25A expression causing reduced interaction between Cdc25A and ASK1, which activates ASK1 [32] (Fig. (4)). Consistently, in vivo studies showed that a human cancer-predisposing polymorphism, Cdc25A S88F, causes mouse embryonic lethality and induces apoptosis due to being incapable of binding to and inhibiting ASK1 [97], implying that the DSG motif might be critical for interaction between Cdc25A and ASK1. Furthermore, ectopic expression of Cdc25A was found to result in suppressing serum starvation-induced apoptosis by activating AKT [98].
Fig. (4).
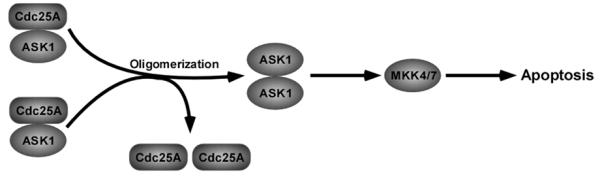
Inhibition of ASK1 activation and apoptosis by Cdc25A. Cdc25A inhibits ASK1 activity by binding to ASK1 and interrupting ASK1 oligomer formation, thus preventing cell apoptosis.
On the contrary, Cdc25A was also found to be required for Myc-dependent apoptosis [31]. This striking finding is supported by another report that Cdc25A mediates Myc-dependent apoptosis in vascular smooth muscle cells [99]. Cdc25A-associated apoptosis was thought to be related to deregulation of Cdc25A, which facilitates cells with damaged DNA to bypass the G2/M checkpoint leading to cell apoptosis [64]. Recent studies have also revealed that caspase-mediated cleavage of Cdc25A produces a C-terminal fragment, which has elevated phosphatase activity and nuclear localization sequence, but has no nuclear exportation sequence [100]. Consequently, this fragment is restricted exclusively in nuclei, where it activates CDK2 and induces apoptosis [100]. The role of C-terminal Cdc25A in apoptosis is further supported by the finding that C-terminal Cdc25A promotes apoptosis by activating CDK1 [101].
The discrepancy of the role of Cdc25A in apoptosis is associated with the subcellular distribution of Cdc25A [102]. Cdc25A functions as an apoptosis suppressor only when it is in the cytoplasm, whereas nuclear-accumulation of Cdc25A leads to cell apoptosis [102]. However, the observation that Cdc25A is predominantly settled in the cytoplasm [102] suggests that the major role of Cdc25A in apoptosis can be considered as a suppressor.
4. Effect of overexpression of Cdc25A on tumorigenesis
As Cdc25A is critical for both cell proliferation and apoptosis, it is not surprising that overexpression of Cdc25A has been observed frequently in various types of cancer, and found to be correlated to poor prognosis in patients [10, 15, 103]. The mechanisms of Cdc25A overexpression are complicated and need to be further investigated. However, some previous studies have provided valuable clues, indicating that overexpression of Cdc25A in cancer cells is at least partially due to transcriptional and post-translational deregulation [10].
One of the major causes for deregulation of Cdc25A transcription is dysfunction of some transcription repressors, particularly p53 [64]. Studies have revealed that mutation in p53 gene occurs frequently in a variety of cancers, including lung [104], breast [105], colorectal [106], head and neck [107], and gastric cancer [108], in which Cdc25A is also deregulated [109-113], indicating a close correlation between p53 mutation and Cdc25A overexpression in cancer cells. Demidova et al. found that p53-induced transcription repression of Cdc25A is mediated by ATF3 [64], suggesting that cancer-associated overexpression of Cdc25A might be a consequence of p53 gene mutation. Another cause for cancer-associated elevation of Cdc25A transcription is disruption of E2F1-Rb complex by human papillomavirus type 16 E7 oncoprotein [35], resulting in cell cycle deregulation [114]. As E7 oncoprotein is known for its ability to promote cancer development [115, 116], it is possible that overexpression of Cdc25A mediates E7-induced transformation and immortalization.
The discovery that aberrantly elongated half life of Cdc25A contributes to high cellular level of Cdc25A in breast cancer provides important evidence that links overexpression of Cdc25A to post-translational regulation [117]. Recent studies have revealed that ubiquitin hydrolase Dub3 can reduce Cdc25A turnover by removing ubiquitin chains from Cdc25A protein [118]. Consequently, overexpression of Cdc25A was observed to be correlated with high level of Dub3 in 9 of 12 breast cancer cell lines [118]. Besides, dysfunction of β-TRCP was also found to be associated with overexpression of Cdc25A in lung cancer [119].
The in vivo studies provided more evidence on how overexpression of Cdc25A affects cancer development. Ray et al. demonstrated that double transgenic expression of Cdc25A and ras/neu under mouse mammary tumor virus (MMTV) promoter causes mammary tumorigensis in mice much faster than single transgenic expression of MMTV-H-ras/neu [120]. More importantly, cells from double transgenic mice are much more capable of proliferation and invasion than from MMTV-H-ras/neu single transgenic mice [120]. However, transgenic expression of Cdc25A alone is not sufficient to induce tumorigensis, suggesting that overexpression of Cdc25A assists ras/neu-induced mammary tumorigensis in vivo [120]. Transgenic overexpression of Cdc25A causes damage of telomeric region of chromosome 4 [120], implying that overexpression of Cdc25A promotes tumorigensis partially by inducing certain DNA damage. Consistently, further studies revealed that the mouse with heterozygous Cdc25A+/− is more resistant to ras/neu-induced tumorigensis than that with homozygous Cdc25A+/+, which is a consequence of difference in proliferation capability between the cells from heterozygous and homozygous Cdc25A tissue [121]. Therefore, Cd25A plays a rate-limiting role in ras/neu-induced tumorigensis [122].
Since overexpression of Cdc25A is involved in tumor initiation and progression, and correlated with poor prognosis, Cdc25A has been considered as a potential target for cancer therapy [123-125]. Several categories of compounds have been explored for this purpose, including natural products, lipophilic acids, vitamin K analogues, electrophiles, and phosphate mimics [123]. Moreover, as mild heat shock (42°C) was found to destabilize Cdc25A, hyperthermia therapy is thought to have great potential to treat cancer patients [126].
Acknowledgments
This work was supported in part by NIH (CA115414 to S.H.), American Cancer Society (RSG-08-135-01-CNE to S.H.) and Carroll Feist Predoctoral Fellowship (to T.S.) from the Feist-Weiller Cancer Center, Louisiana State University Health Sciences Center, Shreveport, LA.
References
- [1].Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- [2].Evan G, Vousden K. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- [3].Croce C. Oncogenes and cancer. N. Engl. J. Med. 2008;358:502–511. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- [4].Golias C, Charalabopoulos A, Charalabopoulos K. Cell proliferation and cell cycle control: a mini review. Int. J. Clin. Pract. 2004;58:1134–1141. doi: 10.1111/j.1742-1241.2004.00284.x. [DOI] [PubMed] [Google Scholar]
- [5].Vermeulen K, Van Bockstaele D, Berneman Z. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Park M, Lee S. Cell cycle and cancer. J. Biochem. Mol. Biol. 2003;36:60–65. doi: 10.5483/bmbrep.2003.36.1.060. [DOI] [PubMed] [Google Scholar]
- [7].Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- [8].Ekholm S, Reed S. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 2000;12:676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- [9].Karlsson-Rosentha C, Millar J. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- [10].Boutros R, Lobjois V, Ducommun B. Cdc25 phosphatases in cancer cells: key players? Good targets? Nat. Rev. Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- [11].Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Terada Y, Tatsuka M, Jinno S, Okayama H. Requirement for tyrosine phosphorylation of CDK4 in G1 arrest induced by ultraviolet irradiation. Nature. 1995;376:358–362. doi: 10.1038/376358a0. [DOI] [PubMed] [Google Scholar]
- [13].Iavarone A, Massague J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature. 1997;387:417–422. doi: 10.1038/387417a0. [DOI] [PubMed] [Google Scholar]
- [14].Bartek J, Lukas J. Mammalian G1-and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell Biol. 2001;13:738–747. doi: 10.1016/s0955-0674(00)00280-5. [DOI] [PubMed] [Google Scholar]
- [15].Xu X, Yamamoto H, Sakon M, Yasui M, Ngan CY, Fukunaga H, Morita T, Ogawa M, Nagano H, Nakamori S, Sekimoto M, Matsuura N, Monden M. Overexpression of Cdc25A phosphatase is associated with hypergrowth activity and poor prognosis of human hepatocellular carcinomas. Clin. Cancer Res. 2003;9:1764–1772. [PubMed] [Google Scholar]
- [16].Busino L, Chiesa M, Draetta G, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–2056. doi: 10.1038/sj.onc.1207394. [DOI] [PubMed] [Google Scholar]
- [17].Chen M-S, Ryan CE, Piwnica-Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding. Mol. Cell Biol. 2003;23:7488–7497. doi: 10.1128/MCB.23.21.7488-7497.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kallstrom H, Lindqvist A, Pospisil V, Lundgren A, Rosenthal C. Cdc25A localisation and shuttling: characterisation of sequences mediating nuclear export and import. Exp. Cell Res. 2005;303:89–100. doi: 10.1016/j.yexcr.2004.09.012. [DOI] [PubMed] [Google Scholar]
- [19].Rudolph J. Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry. 2007;46:3595–3604. doi: 10.1021/bi700026j. [DOI] [PubMed] [Google Scholar]
- [20].DeGregori J, Kowalik T, Nevins J. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Blais A, Dynlacht B. E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- [23].Resnitzky D, Reed S. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol. Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kitagawa M, Higashi H, Jung H, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-CDK4 is different from that for phosphorylation by cyclin A/E-CDK2. EMBO. J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- [26].Harbour J, Luo R, Dei Santi A, Postigo A, Dean D. CDK phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- [27].Hoffmann I, Draetta G, Karsenti E. Activation of the phosphatase activity of human Cdc25A by a CDK2-cyclin E dependent phosphorylation at the G1/S transition. EMBO. J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Z, Southwick EC, Wang M, Kar S, Rosi KS, Wilcox CS, Lazo JS, Carr BI. Involvement of Cdc25A phosphatase in Hep3B hepatoma cell growth inhibition induced by novel K vitamin analogs. Cancer Res. 2001;61:7211–7216. [PubMed] [Google Scholar]
- [29].Blomberg I, Hoffmann I. Ectopic expression of Cdc25A accelerates the G1/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol. Cell Biol. 1999;19:6183–6194. doi: 10.1128/mcb.19.9.6183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. Cdc25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol. Cell Biol. 1999;19:6379–6395. doi: 10.1128/mcb.19.9.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature. 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- [32].Lin YM, Chung CL, Cheng YS. Posttranscriptional regulation of Cdc25A by BOLL is a conserved fertility mechanism essential for human spermatogenesis. J. Clin. Endocrinol. Metab. 2009;94:2650–2657. doi: 10.1210/jc.2009-0108. [DOI] [PubMed] [Google Scholar]
- [33].Tomko RJ, Jr., Lazo JS. Multimodal control of Cdc25A by nitrosative stress. Cancer Res. 2008;68:7457–7465. doi: 10.1158/0008-5472.CAN-08-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Uto K, Inoue D, Shimuta K, Nakajo N, Sagata N. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO. J. 2004;23:3386–3396. doi: 10.1038/sj.emboj.7600328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Katich S, Zerfass-Thome K, Hoffmann I. Regulation of the Cdc25A gene by the human papillomavirus type 16 E7 oncogene. Oncogene. 2001;20:543–550. doi: 10.1038/sj.onc.1204130. [DOI] [PubMed] [Google Scholar]
- [36].Chen X, Prywes R. Serum-induced expression of the cdc25A gene by relief of E2F- mediated repression. Mol. Cell Biol. 1999;19:4695–4702. doi: 10.1128/mcb.19.7.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Martinez-Balbas M, Bauer U, Nielsen S, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO. J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fukada T, Ohtani T, Yoshida Y, Shirogane T, Nishida K, Nakajima K, Hibi M, Hirano T. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell- cycle transition. EMBO. J. 1998;17:6670–6677. doi: 10.1093/emboj/17.22.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barre B, Vigneron A, Coqueret O. The STAT3 transcription factor is a target for the Myc and riboblastoma proteins on the Cdc25A promoter. J. Biol. Chem. 2005;280:15673–15681. doi: 10.1074/jbc.M413203200. [DOI] [PubMed] [Google Scholar]
- [40].Aguda B, Tang Y. The kinetic origins of the restriction point in the mammalian cell cycle. Cell Prolif. 1999;32:321–335. doi: 10.1046/j.1365-2184.1999.3250321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Trimarchi J, Lees J. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- [42].Zhou B, Elledge S. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- [43].Mailand N, Falck J, Lukas C, Sylju, aring, sen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- [44].Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- [45].Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- [46].Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for ATR in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou B-B, Bartek J, Khanna KK. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to Ionizing radiation. J. Biol. Chem. 2003;278:14806–14811. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- [48].Hassepass I, Voit R, Hoffmann I. Phosphorylation at serine 75 is required for UV- mediated degradation of human Cdc25A phosphatase at the S-phase checkpoint. J. Biol. Chem. 2003;278:29824–29829. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- [49].Donzelli M, Busino L, Chiesa M, Ganoth D, Hershko A, Draetta G. Hierarchical order of phosphorylation events commits Cdc25A to betaTrCP-dependent degradation. Cell Cycle. 2004;3:469–471. [PubMed] [Google Scholar]
- [50].Honaker Y, Piwnica-Worms H. Casein kinase 1 functions as both penultimate and ultimate kinase in regulating Cdc25A destruction. Oncogene. 2010;29:3324–3334. doi: 10.1038/onc.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello N, Hershko A, Pagano M, Draetta G. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- [52].Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta G. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO. Rep. 2000;1:71–79. doi: 10.1093/embo-reports/kvd018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA- damaging agents. J. Biol. Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- [54].Kang T;, Wei Y, Honaker Y, Yamaguchi H, Appella E, Hung M, Piwnica-Worms H. GSK-3β targets Cdc25A for ubiquitin-mediated proteolysis, and GSK-3β inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13:36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-Myc proteolysis and subnuclear localization. J. Biol. Chem. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- [56].Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB, Hung M-C, Meric-Bernstam F. Role of glycogen synthase kinase 3β in rapamycin-mediated cell cycle regulation and chemosensitivity. Cancer Res. 2005;65:1961–1972. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]
- [58].Falck J, Mailand N, Syljuasen R, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- [59].Jin J, Ang XL, Ye X, Livingstone M, Harper JW. Differential roles for checkpoint kinases in DNA damage-dependent degradation of the Cdc25A protein phosphatase. J. Biol. Chem. 2008;283:19322–19328. doi: 10.1074/jbc.M802474200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Narayanan B, Geoffroy O, Willingham M, Re G, Nixon D. p53/p21(WAF1/CIP1) expression and its possible role in G1 arrest and apoptosis in ellagic acid treated cancer cells. Cancer Lett. 1999;136:215–221. doi: 10.1016/s0304-3835(98)00323-1. [DOI] [PubMed] [Google Scholar]
- [61].Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- [62].Rother K, Kirschner R, Sanger K, Bohlig L, Mossner J, Engeland K. p53 downregulates expression of the G1/S cell cycle phosphatase Cdc25A. Oncogene. 2007;26:1949–1953. doi: 10.1038/sj.onc.1209989. [DOI] [PubMed] [Google Scholar]
- [63].Vigneron A, Cherier J, Barre B, Gamelin E, Coqueret O. The cell cycle inhibitor p21waf1 binds to the myc and cdc25A promoters upon DNA damage and induces transcriptional repression. J. Biol. Chem. 2006;281:34742–34750. doi: 10.1074/jbc.M602492200. [DOI] [PubMed] [Google Scholar]
- [64].Demidova AR, Aau MY, Zhuang L, Yu Q. Dual regulation of Cdc25A by Chk1 and p53-ATF3 in DNA replication checkpoint control. J. Biol. Chem. 2009;284:4132–4139. doi: 10.1074/jbc.M808118200. [DOI] [PubMed] [Google Scholar]
- [65].de Oliveira P, Zhang L, Wang Z, Lazo J. Hypoxia-mediated regulation of Cdc25A phosphatase by p21 and miR-21. Cell Cycle. 2009;8:3157–3164. doi: 10.4161/cc.8.19.9704. [DOI] [PubMed] [Google Scholar]
- [66].Wang P, Zou F, Zhang X, Li H, Dulak A, Tomko RJ, Jr., Lazo JS, Wang Z, Zhang L, Yu J. microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res. 2009;69:8157–8165. doi: 10.1158/0008-5472.CAN-09-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- [68].Wek R, Jiang H, Anthony T. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- [69].Sudhakar A, Ramachandran A, Ghosh S, Hasnain S, Kaufman R, Ramaiah K. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2α) promotes complex formation between eIF2α (P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry. 2000;39:12929–12938. doi: 10.1021/bi0008682. [DOI] [PubMed] [Google Scholar]
- [70].Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–6422. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- [71].Gao N, Flynn DC, Zhang Z, Zhong X-S, Walker V, Liu KJ, Shi X, Jiang B-H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am. J. Physiol. Cell Physiol. 2004;287:C281–291. doi: 10.1152/ajpcell.00422.2003. [DOI] [PubMed] [Google Scholar]
- [72].Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol. Res. 2004;50:545–549. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- [73].Stark G, Taylor W. Control of the G2/M transition. Mol. Biotechnol. 2006;32:227–248. doi: 10.1385/MB:32:3:227. [DOI] [PubMed] [Google Scholar]
- [74].Solomon M, Lee T, Kirschner M. Role of phosphorylation in p34cdc2 activation: identification of an activating kinase. Mol. Biol. Cell. 1992;3:13–27. doi: 10.1091/mbc.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Fesquet D, Labbe J, Derancourt J, Capony J, Galas S, Girard F, Lorca T, Shuttleworth J, Doree M, Cavadore J. The MO15 gene encodes the catalytic subunit of a protein kinase that activates Cdc2 and other cyclin-dependent kinases (CDKs) through phosphorylation of Thr161 and its homologues. EMBO. J. 1993;12:3111–3121. doi: 10.1002/j.1460-2075.1993.tb05980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ducommun B, Brambilla P, Felix M, Franza B, Karsenti E, Draetta G. Cdc2 phosphorylation is required for its interaction with cyclin. EMBO. J. 1991;10:3311–3319. doi: 10.1002/j.1460-2075.1991.tb04895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mailand N, Podtelejnikov A, Groth A, Mann M, Bartek J, Lukas J. Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO. J. 2002;21:5911–5920. doi: 10.1093/emboj/cdf567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mitra J, Enders G. Cyclin A/CDK2 complexes regulate activation of CDK1 and Cdc25 phosphatases in human cells. Oncogene. 2004;23:3361–3367. doi: 10.1038/sj.onc.1207446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta G. Dual mode of degradation of Cdc25 A phosphatase. EMBO. J. 2002;21:4875–4884. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Timofeev O, Cizmecioglu O, Settele F, Kempf T, Hoffmann I. Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J. Biol. Chem. 2010;285:16978–16990. doi: 10.1074/jbc.M109.096552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Cuddihy A, O’Connell M. Cell-cycle responses to DNA damage in G2. Int. Rev. Cytol. 2003;222:99–140. doi: 10.1016/s0074-7696(02)22013-6. [DOI] [PubMed] [Google Scholar]
- [82].Sorensen C, Melixetian M, Klein D, Helin K. NEK11: linking Chk1 and Cdc25A in DNA damage checkpoint signaling. Cell Cycle. 2010;9:450–455. doi: 10.4161/cc.9.3.10513. [DOI] [PubMed] [Google Scholar]
- [83].Melixetian M, Klein D, Sorensen C, Helin K. NEK11 regulates Cdc25A degradation and the IR-induced G2/M checkpoint. Nat. Cell Biol. 2009;11:1247–1253. doi: 10.1038/ncb1969. [DOI] [PubMed] [Google Scholar]
- [84].Kasahara K, Goto H, Enomoto M, Tomono Y, Kiyono T, Inagaki M. 14-3-3γ mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO. J. 2010;29:2802–2812. doi: 10.1038/emboj.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. U. S. A. 2002;99:14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shreeram S, Hee WK, Bulavin DV. Cdc25A serine 123 phosphorylation couples centrosome duplication with DNA replication and regulates tumorigenesis. Mol. Cell Biol. 2008;28:7442–7450. doi: 10.1128/MCB.00138-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Vazquez-Novelle MD, Mailand N, Ovejero S, Bueno A, Sacristan MP. Human Cdc14A phosphatase modulates the G2/M transition through Cdc25A and Cdc25B. J. Biol. Chem. 2010;285:40544–40553. doi: 10.1074/jbc.M110.133009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Elmore S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zou X, Tsutsui T, Ray D, Blomquist JF, Ichijo H, Ucker DS, Kiyokawa H. The cell cycle-regulatory Cdc25A phosphatase inhibits apoptosis signal-regulating kinase 1. Mol. Cell Biol. 2001;21:4818–4828. doi: 10.1128/MCB.21.14.4818-4828.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Matsukawa J, Matsuzawa A, Takeda K, Ichijo H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem. 2004;136:261–265. doi: 10.1093/jb/mvh134. [DOI] [PubMed] [Google Scholar]
- [91].Behrens A, Sibilia M, Wagner E. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- [92].Leppa S, Bohmann D. Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene. 1999;18:6158–6162. doi: 10.1038/sj.onc.1203173. [DOI] [PubMed] [Google Scholar]
- [93].Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- [94].Kim S-J, Hwang S-G, Shin DY, Kang S-S, Chun J-S. p38 kinase regulates nitric oxide-induced apoptosis of articular chondrocytes by accumulating p53 via NFκB- dependent transcription and stabilization by serine 15 phosphorylation. J. Biol. Chem. 2002;277:33501–33508. doi: 10.1074/jbc.M202862200. [DOI] [PubMed] [Google Scholar]
- [95].Cai B, Chang SH, Becker EBE, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- [96].Takeda K, Noguchi T, Naguro I, Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 2008;48:199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- [97].Bahassi e. M., Yin M, Robbins S, Li Y, Conrady D, Yuan Z, Kovall R, Herr A, Stambrook P. A human cancer-predisposing polymorphism in Cdc25A is embryonic lethal in the mouse and promotes ASK-1 mediated apoptosis. Cell Div. 2011;6:4. doi: 10.1186/1747-1028-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Fuhrmann G, Leisser C, Rosenberger G, Grusch M, Huettenbrenner S, Halama T, Mosberger I, Sasgary S, Cerni C, Krupitza G. Cdc25A phosphatase suppresses apoptosis induced by serum deprivation. Oncogene. 2001;20:4542–4553. doi: 10.1038/sj.onc.1204499. [DOI] [PubMed] [Google Scholar]
- [99].Macdonald K, Bennett MR. Cdc25A is necessary but not sufficient for optimal c-myc- induced apoptosis and cell proliferation of vascular smooth muscle cells. Circ. Res. 1999;84:820–830. doi: 10.1161/01.res.84.7.820. [DOI] [PubMed] [Google Scholar]
- [100].Mazars A, Fernandez-Vidal A, Mondesert O, Lorenzo C, Prevost G, Ducommun B, Payrastre B, Racaud-Sultan C, Manenti S. A caspase-dependent cleavage of Cdc25A generates an active fragment activating cyclin-dependent kinase 2 during apoptosis. Cell Death Differ. 2009;16:208–218. doi: 10.1038/cdd.2008.142. [DOI] [PubMed] [Google Scholar]
- [101].Chou S-T, Yen Y-C, Lee C-M, Chen M-S. Pro-apoptotic role of Cdc25A: activation of cyclin B1/Cdc2 by the Cdc25A C-terminal domain. J. Biol. Chem. 2010;285:17833–17845. doi: 10.1074/jbc.M109.078386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Leisser C, Rosenberger G, Maier S, Fuhrmann G, Grusch M, Strasser S, Huettenbrenner S, Fassl S, Polgar D, Krieger S, Cerni C, Hofer-Warbinek R, deMartin R, Krupitza G. Subcellular localisation of Cdc25A determines cell fate. Cell Death Differ. 2004;11:80–89. doi: 10.1038/sj.cdd.4401318. [DOI] [PubMed] [Google Scholar]
- [103].Kristjansdottir K, Rudolph J. Cdc25 phosphatases and cancer. Chem. Biol. 2004;11:1043–1051. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- [104].Chiba I, Takahashi T, Nau M, D’Amico D, Curiel D, Mitsudomi T, Buchhagen D, Carbone D, Piantadosi S, Koga H. Mutations in the p53 gene are frequent in primary, resected non-small cell lung cancer. Oncogene. 1990;5:1603–1610. [PubMed] [Google Scholar]
- [105].Sjalander A, Birgander R, Hallmans G, Cajander S, Lenner P, Athlin L, Beckman G, Beckman L. p53 polymorphisms and haplotypes in breast cancer. Carcinogenesis. 1996;17:1313–1316. doi: 10.1093/carcin/17.6.1313. [DOI] [PubMed] [Google Scholar]
- [106].Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV, Lane DP. p53 mutations in colorectal cancer. Proc. Natl. Acad. Sci. U. S. A. 1990;87:7555–7559. doi: 10.1073/pnas.87.19.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Olshan A, Weissler M, Pei H, Conway K. p53 mutations in head and neck cancer: new data and evaluation of mutational spectra. Cancer Epidemiol. Biomarkers Prev. 1997;6:499–504. [PubMed] [Google Scholar]
- [108].Uchino S, Noguchi M, Ochiai A, Saito T, Kobayashi M, Hirohashi S. p53 mutation in gastric cancer: a genetic model for carcinogenesis is common to gastric and colorectal cancer. Int. J. Cancer. 1993;54:759–764. doi: 10.1002/ijc.2910540509. [DOI] [PubMed] [Google Scholar]
- [109].Dixon D, Moyana T, King M. Elevated expression of the Cdc25A protein phosphatase in colon cancer. Exp. Cell Res. 1998;240:236–243. doi: 10.1006/excr.1998.3940. [DOI] [PubMed] [Google Scholar]
- [110].Cangi M, Cukor B, Soung P, Signoretti S, Moreira G, Ranashinge M, Cady B, Pagano M, Loda M. Role of the Cdc25A phosphatase in human breast cancer. J. Clin. Invest. 2000;106:753–761. doi: 10.1172/JCI9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Wu W, Fan Y-H, Kemp BL, Walsh G, Mao L. Overexpression of Cdc25A and Cdc25B is frequent in primary non-small cell lung cancer but is not associated with overexpression of c-myc. Cancer Res. 1998;58:4082–4085. [PubMed] [Google Scholar]
- [112].Gasparotto D, Maestro R, Piccinin S, Vukosavljevic T, Barzan L, Sulfaro S, Boiocchi M. Overexpression of Cdc25A and Cdc25B in head and neck cancers. Cancer Res. 1997;57:2366–2368. [PubMed] [Google Scholar]
- [113].Xing X, Chen J, Chen M. Expression of Cdc25 phosphatases in human gastric cancer. Dig. Dis. Sci. 2008;53:949–953. doi: 10.1007/s10620-007-9964-4. [DOI] [PubMed] [Google Scholar]
- [114].Nguyen DX, Westbrook TF, McCance DJ. Human papillomavirus type 16 E7 maintains elevated levels of the Cdc25A tyrosine phosphatase during deregulation of cell cycle arrest. J. Virol. 2002;76:619–632. doi: 10.1128/JVI.76.2.619-632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989;63:4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Loffler H, Syljuasen R, Bartkova J, Worm J, Lukas J, Bartek J. Distinct modes of deregulation of the proto-oncogenic Cdc25A phosphatase in human breast cancer cell lines. Oncogene. 2003;22:8063–8071. doi: 10.1038/sj.onc.1206976. [DOI] [PubMed] [Google Scholar]
- [118].Pereg Y, Liu B, O’Rourke K, Sagolla M, Dey A, Komuves L, French D, Dixit V. Ubiquitin hydrolase Dub3 promotes oncogenic transformation by stabilizing Cdc25A. Nat. Cell Biol. 2010;12:400–406. doi: 10.1038/ncb2041. [DOI] [PubMed] [Google Scholar]
- [119].He N, Li C, Zhang X, Sheng T, Chi S, Chen K, Wang Q, Vertrees R, Logrono R, Xie J. Regulation of lung cancer cell growth and invasiveness by β-TRCP. Mol. Carcinog. 2005;42:18–28. doi: 10.1002/mc.20063. [DOI] [PubMed] [Google Scholar]
- [120].Ray D, Terao Y, Fuhrken PG, Ma Z-Q, DeMayo FJ, Christov K, Heerema NA, Franks R, Tsai SY, Papoutsakis ET, Kiyokawa H. Deregulated Cdc25A expression promotes mammary tumorigenesis with genomic instability. Cancer Res. 2007;67:984–991. doi: 10.1158/0008-5472.CAN-06-3927. [DOI] [PubMed] [Google Scholar]
- [121].Ray D, Terao Y, Nimbalkar D, Hirai H, Osmundson EC, Zou X, Franks R, Christov K, Kiyokawa H. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 2007;67:6605–6611. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- [122].Ray D, Kiyokawa H. Cdc25A phosphatase: a rate-limiting oncogene that determines genomic stability. Cancer Res. 2008;68:1251–1253. doi: 10.1158/0008-5472.CAN-07-5983. [DOI] [PubMed] [Google Scholar]
- [123].Wang Z, Kar S, Carr B. Cdc25A protein phosphatase: a therapeutic target for liver cancer therapies. Anticancer Agents Med. Chem. 2008;8:863–871. doi: 10.2174/187152008786847675. [DOI] [PubMed] [Google Scholar]
- [124].Lazo J, Wipf P. Is Cdc25 a druggable target? Anticancer Agents Med. Chem. 2008;8:837–842. doi: 10.2174/187152008786847738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Lavecchia A, Di Giovanni C, Novellino E. Cdc25A and B dual-specificity phosphatase inhibitors: potential agents for cancer therapy. Curr. Med. Chem. 2009;16:1831–1849. doi: 10.2174/092986709788186084. [DOI] [PubMed] [Google Scholar]
- [126].Madlener S, Rosner M, Krieger S, Giessrigl B, Gridling M, Vo TPN, Leisser C, Lackner A, Raab I, Grusch M, Hengstschlager M, Dolznig H, Krupitza G. Short 42°C heat shock induces phosphorylation and degradation of Cdc25A which depends on p38MAPK, Chk2 and 14.3.3. Hum. Mol. Genet. 2009;18:1990–2000. doi: 10.1093/hmg/ddp123. [DOI] [PubMed] [Google Scholar]