

Abstract
Recent clinical and experimental studies are redefining the cellular and molecular bases of pulmonary arterial hypertension (PAH). The genetic abnormalities first identified in association with the idiopathic form of PAH — together with a vast increase in our understanding of cell signaling, cell transformation, and cell-cell interactions; gene expression; microRNA processing; and mitochondrial and ion channel function — have helped explain the abnormal response of vascular cells to injury. Experimental and clinical studies now converge on the intersection and interactions between a genetic predisposition involving the BMPR2 signaling pathway and an impaired metabolic and chronic inflammatory state in the vessel wall. These deranged processes culminate in an exuberant proliferative response that occludes the pulmonary arterial (PA) lumen and obliterates the most distal intraacinar vessels. Here, we describe emerging therapies based on preclinical studies that address these converging pathways.
Pathological features of PAH
Pulmonary arterial hypertension (PAH) is diagnosed by an elevation in mean pulmonary arterial (PA) pressure above 25 mmHg at rest or 30 mmHg with exercise. Patients usually present with much higher levels of PA pressure, but only vague and insidious symptoms of increasing fatigue and dyspnea; some patients are diagnosed only after syncopal episodes, which can reflect suprasystemic levels of PA pressure and low cardiac output. The causes of PAH were reclassified according to a consensus at the Fourth World Symposium of Pulmonary Hypertension in Dana Point, California, USA, in 2008 and published by Simonneau and colleagues in 2009 (see Dana Point classification of pulmonary hypertension and ref. 1). While this review focuses on pulmonary hypertension category 1 (i.e., PAH), pathophysiologic insights can also be gained from understanding other causes of pulmonary hypertension. Hence, experimental studies in hypoxic animals are also discussed.
In neonates and in infants, PAH likely results from failure of the neonatal pulmonary vasculature to dilate at birth, and pathological changes in the blood vessels are evident in the first few days of life. Most prominent is abnormal muscularization of distal PAs at the alveolar duct and wall levels and a striking reduction in their number. In older infants and adults, there is also progressive intimal hyperplasia leading to occlusive changes in the PAs and plexiform lesions (Figure 1 and see below).
Figure 1. Vascular abnormalities associated with PAH.
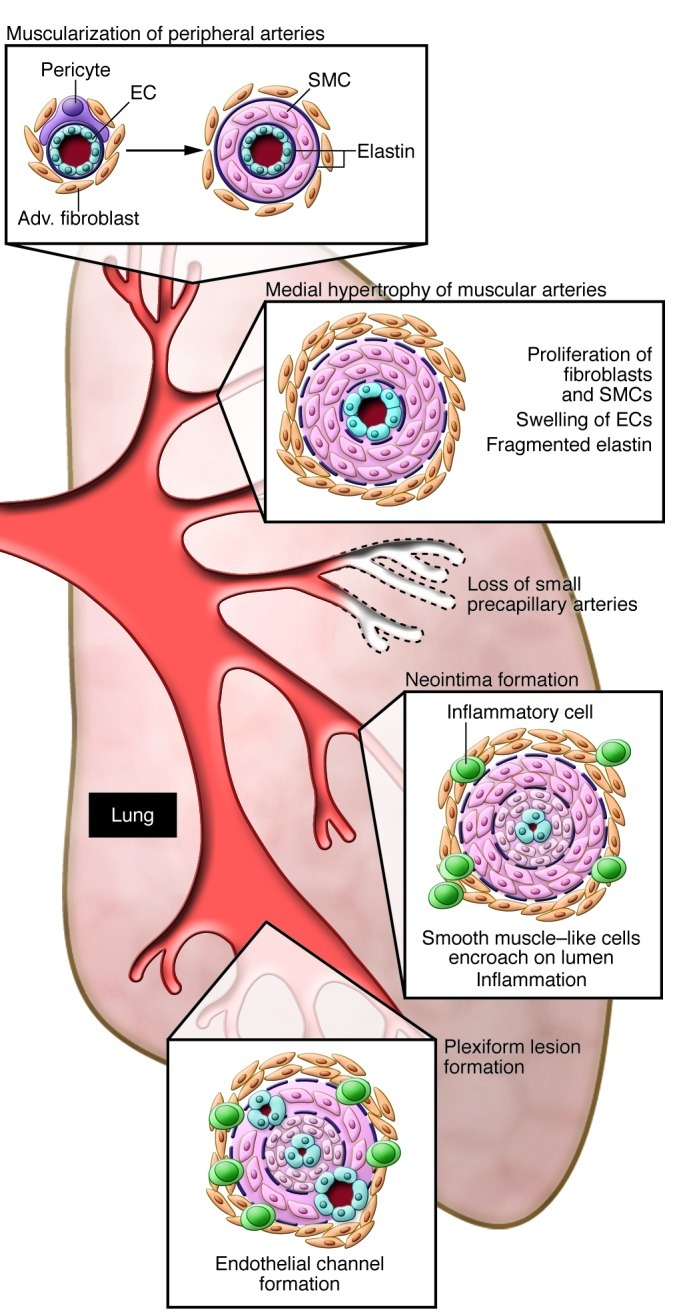
This schema depicts the abnormalities throughout the pulmonary circulation: abnormal muscularization of distal and medial precapillary arteries, loss of precapillary arteries, thickening of large PAs, and neointimal formation that is particularly occlusive in vessels less than 500–100 μM and in plexiform lesions therein.
PA EC alterations in the clinical (2) and experimental (3) setting precede muscularization of distal PAs. In PA ECs from patients with idiopathic PAH (IPAH), an increase in Tie2 receptor expression in ECs releases serotonin, mediating SMC proliferation (4). Dysfunctional ECs can either release factors that stimulate SMC proliferation, such as FGF-2 (5), or fail to produce agents that normally suppress proliferation of SMCs in response to growth factors, such as apelin (encoded by Apln) (6).

Muscularization of distal, normally nonmuscular, PAs at the alveolar duct and wall level is associated with differentiation of pericytes into SMCs that subsequently proliferate. The progressive thickening of the wall of more proximal intraacinar and preacinar muscular arteries and the obliteration associated with neointimal formation are attributed to increased proliferation and migration of cells considered to be SMCs because they are α-SMA positive (7). The origin of these cells is unclear, and the mechanism of their dysregulation is the subject of intense study. They may represent a specialized subpopulation of SMCs; they may originate as stem cells (8) or fibrocytes (9) or transform from ECs (10, 11). The loss of distal PAs could be caused by alterations in ECs and/or pericytes resulting in apoptosis (12).
Later in disease, there is dysregulated EC proliferation, producing aberrant channels in the otherwise obliterated lumen of the vessel and in the adventitia (i.e., the plexiform lesion). These channels may reflect clonal expansion of apoptosis-resistant ECs (13), or they may be derived from circulating endothelial progenitor cells (EPCs) that accumulate at sites of endothelial denudation or injury and expand locally (14). PA ECs from patients with IPAH produce decreased amounts of NO. Synthesized largely by eNOS in ECs of the pulmonary vasculature, NO is a vasodilator and suppressor of SMC proliferation. The reduction in NO may be related to high arginase levels (15), because l-arginine, the substrate of eNOS, is required to produce NO. ECs from patients with PAH may be highly proliferative in response to growth factors (14) and exhibit high rates of glycolysis (16) that reflect impaired mitochondrial metabolism. The dysfunction of these cells is revealed in their inability to form normal tubes in culture, consistent with the fact that they fail to restore occluded or lost precapillary vessels.
Strategies to improve NO delivery to tissues have been successful experimentally, and provision of nitrite can block both the sequelae of hypoxia and inflammation in PAH patients (see Emerging therapeutic strategies for pulmonary hypertension and ref. 17). Similarly, inhibiting asymmetric dimethyl arginine (ADMA) by activating dimethylarginine dimethyl hydrolase (DDAH) may be useful in allowing greater production of NO (18). Current therapies for PAH include targeting reduced prostacyclin and increased endothelin levels. A more complete discussion can be found in the supplemental material (available online with this article; doi: 10.1172/JCI60658DS1).

Additional features of PAH include thickening of the pulmonary adventitia and venous hypertrophy (19) and increased expression of TGF-β; matrix proteins such as elastin, fibronectin, and tenascin-C; and glycosaminoglycans (20). In addition to macrophages, B and T cells (21) abound in the perivascular space and are often seen invading the vessel wall, and there is heightened expression of inflammatory mediators such as S100A4 (22) and fractalkine (23).
Genetics of PAH: BMPR2 and other TGF-β family members
Genetic studies have demonstrated that 70% or more of patients with hereditary PAH (24, 25), and 10%–20% of patients with sporadic IPAH, are heterozygous for a mutation in bone morphogenetic protein (BMP) receptor type 2 (BMPR2). BMPR2 is a member of the TGF-β superfamily of growth factor receptors. Mutations can affect different functions of BMPR2, namely the ligand-binding domain, the signaling mechanism, and the interaction of the receptor with the cytoskeleton. BMPR2 is expressed ubiquitously and, in association with a coreceptor (usually BMPR1A), can signal through many different pathways, including pSmad1/5 (26), p-p38 (27), pERK, JNK, and Akt/PI3K (28, 29).
The penetrance of heritable PAH is low: 80% of family members carrying BMPR2 mutations will never develop PAH (30). The presence of a BMPR2 mutation is much lower (i.e., 6%–8%) in patients with PAH related to a congenital left-to-right shunt (31) and is rare in patients with PAH associated with appetite suppressants (32). The functional link between mutations in BMPR2 and PAH is reinforced by the fact that independent of a mutation in BMPR2, most IPAH patients have reduced BMPR2 protein expression, as do, to some extent, patients with PAH associated with other conditions (33). In addition, mutations in the effectors of the signaling pathway and in the TGF-β superfamily of receptors have also been described in patients with PAH and IPAH (34). For example, activin-like kinase type 1 (ALK1) and endoglin are mutated in patients with hereditary hemorrhagic telangiectasia and with PAH (35, 36). In fact, the ALK1-deficient mouse develops spontaneous pulmonary hypertension, whereas the BMPR2-heterozygous mouse model requires additional perturbations, such as both hypoxia and serotonin or inflammation, to elicit an exaggerated pulmonary hypertensive phenotype (37). In addition, somatic mutations in ECs from patients with pulmonary hypertension have been described (38).
New links have been established among BMPR2 signaling, Smad activation, and the processing of microRNAs (39), particularly miR-21, which can control functions critical to SMC contractility (40). Indeed, altered microRNA processing has been described in heritable PAH (41), and altered microRNA profiles are seen in experimental and clinical PAH (42).
Various strategies have been undertaken to rescue BMPR2 loss of function directly by gene therapy (43) and by improving trafficking of mutant receptors (44). Since loss of BMPR2 leads to an adverse response to TGF-β signaling (45), therapies that inhibit the TGF-β receptor ALK5 may also be efficacious in PAH (46).
BMPR2 and vascular cell dysfunction
When loss of BMPR2 is induced by RNA interference in PA ECs, the cells become susceptible to apoptosis (47). This vulnerability of ECs with loss of BMPR2 can explain the reduced number of peripheral alveolar duct and wall PAs. Our group has shown that BMPs, via BMPR2, activate the canonical Wnt signaling pathway to induce PA EC survival and proliferation and activate the noncanonical pathway to induce migration, critical features in angiogenesis and regeneration of damaged blood vessels (48). Further work by our group showed that a complex between β-catenin and PPARγ is essential in directing these functions and that a key downstream gene transcribed by this complex is apelin (6). Apelin has autocrine effects in promoting PA EC survival and migration and paracrine effects in suppressing aberrant PA SMC growth. Mice with PPARγ deleted in ECs develop spontaneous PAH (49) that is reversed by treatment with apelin (6). Our studies also showed the efficacy of using naturally occurring PPARγ adducts, such as nitro–fatty acids (NO2-FAs), to rescue BMPR2 dysfunction. Both NO2-FA and similarly reactive electrophilic keto-FAs display nanomolar affinities for PPARγ (50) and mediate distinctive patterns of PPARγ-dependent gene expression. This is in addition to their antioxidant properties, related to activation of Nrf2, and antiinflammatory properties, related to inhibition of NF-κB. Another transcription factor that has a beneficial effect in rescuing BMPR2 dysfunction in PA ECs appears to be Id1 (51).
Loss of BMPR2 causes proliferation of PA SMCs in response to TGF-β1 and BMP2, in contrast to the inhibition of SMC proliferation and increased susceptibility to apoptosis normally observed with these morphogens. Normal BMPR2 signaling negatively regulates PDGF (52) and likely other growth-promoting factors implicated in the pathobiology of PAH, such as EGF (53). Since BMP4 induces differentiation of fetal lung fibroblasts into SMCs and inhibits their proliferation (54), lack of BMP4/BMPR2 interaction might expand the myofibroblast population of cells, accounting for the adventitial and intimal thickening of the PAs in PAH.
In cultured PA SMCs, BMP2 regulates PPARγ transcriptional activity (52), and APOE is a key target gene. Mice with deletion of Apoe develop pulmonary hypertension on a high-fat diet (55), as do mice with deletion of Pparg in SMCs (52), even when fed a normal diet. In systemic arterial SMCs, apoE can repress proliferation by phosphorylating and internalizing the coreceptor of PDGF, LDL receptor–related protein 1 (LRP1) (56). Repression of the PDGF receptor by imatinib (Gleevec) can reverse experimental pulmonary hypertension in rats (57) and may improve outcome in patients with end-stage PAH (58). Moreover, dasatinib, a very potent inhibitor of the BCR/ABL tyrosine kinase, also appears promising as a future therapy (59).
In addition to apoE, another key target of PPARγ is adiponectin, which can sequester PDGF-BB (60) and repress PA SMC proliferation. Treatment of Apoe–/– mice with the PPARγ agonist rosiglitazone reverses PAH and raises adiponectin levels (55). However, rosiglitazone adversely affects PA EC survival in association with disruption of the PPARγ/β-catenin transcription factor complex (6). This side effect was not observed with naturally occurring adducts, such as NO2-FAs. Other sequelae of BMPR2 dysfunction implicated in proliferation of vascular SMCs include heightened production of osteoprotegrin (61) and tenascin-C (62). Osteoprotegerin is increased in pulmonary vascular lesions and in serum of patients with PAH and can increase PA SMC proliferation and migration. Tenascin-C can cluster integrins and activate growth factor receptors in SMC and induce their proliferation (63). Circulating osteopontin is also increased in PAH and indicates patients with more severe symptomatology (64). Given the functional significance of BMPR2 signaling, it is not surprising that penetrance of the mutation appears to depend on the level of production of BMPR2 from the normal allele (30). Conversely, haploinsufficiency of BMPR2 (65), or even expression of dominant-negative BMPR2 in SMCs (66), in mice requires addition of other stimuli to bring out a more severe PAH phenotype (67).
Drugs and toxins
The high incidence of PAH in patients taking anorexigens implicated serotonin-like compounds in the pathobiology of the disease (68). There are additive effects of serotonin and the anorexigen dexfenfluramine in causing PAH (69). There is also evidence in female PAH patients of an additive effect between seratonin and 17β-estradiol levels (70). Serotonin has structural and functional similarities to compounds such as cocaine and amphetamine. Abuse of these compounds is also associated with PAH (71). This suggests a relationship between heightened signaling via G protein–coupled serotonin and serotonin-like receptors and PAH. There is also evidence of interplay among HIV, cocaine abuse, and heightened activity of PDGF (72).
Serotonin receptors and transporters
Elevated serotonin levels and serotonin transport have been implicated in the pathology of experimental and clinical PAH. One study identified a gain-of-function polymorphism in the serotonin transporter in patients with PAH (73), but this modifier was not observed in different populations of PAH patients. Serotonin exaggerates vasoreactivity in the fawn-hooded rat (74), and there is attenuated severity of pulmonary vascular disease in mice lacking the serotonin transporter gene (75). In contrast, mice overexpressing the serotonin transporter globally (76) or specifically in SMCs (77) develop worsened hypoxia-induced PAH. PDGF-mediated proliferation of SMCs can be compounded by increased activity of the serotonin transporter, since this enhances PDGF receptor β–mediated signaling (78). Other studies have shown that serotonin-mediated stimulation of the serotonin transporter and the serotonin receptors induces expression of cyclins and c-fos (79), which are critical to the PA SMC proliferative response.
Serotonin stimulates an increase in S100A4, a member of the S100 family of calcium-binding proteins (80) that induces proliferation and migration of PA SMCs (80) and is increased in neointimal lesions from patients with IPAH and PAH associated with other conditions. Moreover, a mouse that overexpresses S100A4 can spontaneously (albeit rarely) develop pulmonary vascular pathology (22), and consistently does so when concomitantly infected with murine gamma herpes (MHV68, the murine homolog of HHV8; ref. 81). The latter study, coupled with the observation of increased incidence of PAH in patients with HIV, underscores the importance of immune mechanisms and inflammation in PAH pathobiology as well as the above-described interplay among serotonergic compounds, PDGF, and HIV (72).
Inflammation and immune mechanisms
Increasing attention is being focused on the proinflammatory state of the vessel wall in the progression of PAH. The development of PAH in a subset of patients with HIV infection may be a function of the patient’s HLA class II alleles (82). Moreover, a link was made between expression of HHV8 (associated with Kaposi sarcoma) and IPAH (83). It also appears that the Kaposi sarcoma virus can stimulate lysosome-mediated degradation of BMPR2 (84). Recent studies suggested a link between the HIV-nef gene and plexogenic pulmonary vascular lesions in PAH in HIV-infected patients and SIV-infected nonhuman primates (85).
There is a high incidence of PAH in areas of the world endemic for schistosomiasis. About 10% of patients with schistosomiasis will develop portal hypertension, and 10% of those will have PAH. Chronic infection of mice with high-dose cercariae results in severe but spotty lung vascular remodeling (86, 87), albeit with a relatively modest pulmonary hypertensive response. Treatment of the schistosomiasis in these mice induces regression of the pathology (86). Allergic responses to ovalbumin or to Aspergillus species in mice can also cause extensive spotty pulmonary vascular remodeling, without pulmonary hypertension (88). In this model, an IL-13–mediated increase in α-resistin is associated with SMC proliferation, but the functional significance of this molecule in scleroderma and PAH (89) and in experimental schistosomiasis (87) is not known. In mice lacking prostaglandin synthase, induction of allergic inflammation with the house dust mite induces intense pulmonary vascular remodeling, changes that are reversed by administration of prostaglandin E2 (90).
In the experimental setting, haploinsufficiency of BMPR2 is associated with an exaggerated pulmonary hypertensive response to an inflammatory stimulus (91). Other experimental models of chronic inflammation, such as repeated injections of endotoxin (92) or TNF-α (93), also cause pulmonary vascular changes. In the rodent model of pulmonary hypertension, depletion of T cell subsets worsens the pathology (94). This has been attributed to unbalanced B cell activity resulting from impaired Tregs (95). In the athymic rat given the VEGF receptor blocker, reconstitution with Tregs was sufficient to rescue PAH in association with elevated BMPR2 levels. In inflammatory PAH, complement appears to play an essential role (96).
Heightened circulating levels of cytokines and their receptors have been demonstrated in IPAH patients; these include fractalkine, stromal derived factor–1 (SDF-1), monocyte chemoattractant protein–1 (MCP-1), and granulocyte-monocyte colony-stimulating factor (reviewed in ref. 97). Loss of BMPR2 induces IL-6 (98), a cytokine that can cause severe pulmonary vascular disease in rodents (99) in association with SMC proliferation (100) mediated by the transcription factor Kruppel-like factor 5 (101). Regulation of epigenetic factors may reverse inflammatory processes. In hypoxia-associated PAH, activation of fibroblasts expressing elevated levels of cytokines was linked to increased histone deacetylase 1 (HDAC1), inhibition of which reversed both the fibroblast phenotype and PAH in experimental animals (102).
An increase in perivascular macrophages is essential to the development of hypoxia-induced pulmonary hypertension in experimental animals, and this phenomenon is observed in lung tissue from patients with IPAH (103). Mononuclear fibrocytes, cells that have characteristics of both fibroblasts and leukocytes (104), have been identified as key contributors to the remodeling of the pulmonary vasculature. These cells may migrate into the vessel wall through the angiomata located in the expanding adventitia (9). At least some of the cells in the neointimal lesions may have originated as invading fibrocytes; indeed, high levels of circulating fibrocytes are found in adults and children with pulmonary hypertension (105). Tertiary lymphoid tissue was described in patients with IPAH as evidence of altered immune regulation (106). Similarly, circulating autoantibodies are observed in patients with both autoimmune and idiopathic and other associated forms of pulmonary hypertension, but the role of these antibodies in the pathology is the subject of considerable investigation (107).
Previous studies suggested that heightened expression of the transcription factor nuclear factor of activated T cells c2 (NFATc2) (108), which is associated with activated inflammatory cells, may underlie PAH. Increased nuclear NFATc2 is observed in T cells from IPAH patients and in pulmonary vascular lesions, and this can lead to repression of voltage-gated K+ (Kv) 1.5 channel expression and influx of intracellular Ca+, causing contraction and proliferation of SMCs. NFATc2 nuclear translocation can be inhibited by cyclosporine as well as by tacrolimus (also known as FK-506). Recently, it was shown that miR-204 is reduced in PAH, and low miR-204 increases the level of phosphatase Shp2, which in turn activates NFATc2 (109).
Elastase activity
Ultrastructural studies of PAs from children with congenital heart defects and associated PAH suggested that elastolytic activity may be an early feature of this complication (2). Elevated serine elastase activity was subsequently documented in rodent and murine models of PAH (110), which led to the successful experimental use of elastase inhibitors to prevent pulmonary vascular pathology (Figure 2 and refs. 111, 112). The mechanism relating elastase activity to PAH is based on studies in cultured PA SMCs showing that serum and EC factors can mediate the cells’ production of serine elastase. This enzyme can release growth factors from the extracellular matrix (5) and induce production of matrix metalloproteinases and tenascin-C, a glycoprotein associated with activation of SMC growth factor receptors and survival pathways (Figure 2 and ref. 63). These studies led to work showing that elastase inhibitors not only prevent, but also reverse, experimental pulmonary hypertension by inducing apoptosis of SMCs (113). Regression of experimental pulmonary hypertension was also achieved by blocking a downstream effector of elastase, the EGF receptor (53). Blockade of the PDGF tyrosine kinase receptor by imatinib had a similar effect (57). Similarly, transfection of a dominant-negative survivin construct (114) blocked a downstream effector of growth factor signaling and was also effective in reversing PAH through SMC apoptosis. Recently, we found that the relevant PA SMC elastase in PAH is neutrophil elastase (115). PA SMCs from mice that overexpress S100A4 have elevated levels of neutrophil elastase mRNA and protein, as do PA SMCs from patients with IPAH. Inhibition of elastase with the naturally occurring serine elastase inhibitor elafin attenuated the development of neointimal lesions in the S100A4 mice that are infected with virus.
Figure 2. Factors that converge in the molecular pathogenesis of PAH.

This schema focuses on the interactions among inflammation, altered cellular metabolism, and genetic/epigenetic abnormalities in the pathogenesis of PAH.
An interesting link has been made between metabolism and inflammation by showing that the reduced adiponectin seen in insulin resistance (116) leads to recruitment of eosinophils that can play a profound role in the development of pulmonary vascular disease. Another study linked endothelin with the unfolded protein response, mitochondrial dysfunction, and inflammation (117).
Glycolytic metabolism and the Warburg effect
The fawn-hooded rat, which has defective serotonin metabolism and develops PAH in response to relative alveolar hypoxia at mile-high altitude, also has abnormal oxygen sensing in the mitochondria of SMCs, leading to reduced Kv channel function (118). Hyperpolarized mitochondria cause normoxic stimulation of HIF-1α (a phenomenon known as the Warburg effect), leading to reduced cytochrome c oxidase and SOD levels and impaired Kv channel expression and function (Figure 2). In the fawn-hooded rat, in association with hyperpolarized mitochondria, there is hypermethylation of the SOD2 promoter in PAs, but not aortic SMCs. Demethylating agents can reverse the apoptosis-resistant phenotype of the SMCs and the pulmonary hypertension in these rats (119).
Interestingly, BMP2-mediated BMPR2 signaling has been directly related to expression of Kv channels (120); conversely, serotonin can directly inhibit rat PA Kv channels (121). Moreover, transfer of Kv channels has been used as an experimental strategy in animal models to prevent and reverse PAH (122). Reversal of the mitochondrial abnormality and Kv channel dysfunction can be achieved through the pyruvate dehydrogenase kinase (PDK) inhibitor dicholoracetate (DCA) (118), in association with regression of pulmonary vascular remodeling. The link between metabolism and inflammation is reinforced by studies showing that the cytokine TNF-α can cause mitochondrial hyperpolarization that is also reversible by DCA (123).
Manipulation of the glycolytic pathway can also be achieved by suppression of malonyl conenzyme decarboxylase. This reduces FA oxidation and reverses chronic pulmonary hypertension (124). Interestingly, ER stress increases the distance between the ER and the mitchondria in PA, but not systemic SMCs, and this occurs through activation of ATF6 and transcription of Nogo-B. The increased distance results in inhibition of calcium-sensitive mitochondrial enzymes and decreases mitochondrial-dependent apoptosis (123), thus facilitating SMC proliferation.
It appears that the inability of cells to undergo autophagy (125) is linked to heightened oxidative stress, HIF-1α stabilization, increased intracellular Ca+, suppression of Kv channel function, and worsening of hypoxia-induced pulmonary hypertension (126). An increase in HIF-2α also promotes PAH, as is evident in von Hippel Lindau disease (127).
Recent reports of elevated transient receptor potential (TRP) calcium channels TRP3 and TRP6 in PA SMCs from IPAH patients indicated that inhibition of these channels can repress the heightened proliferation observed in SMCs (128). Inhibition of PKA or activation of cAMP seem to have a similar effect (129).
Gender and estrogen metabolism
Reduced expression of BMPR2 from the normal allele can distinguish unaffected from PAH-affected family members with a BMPR2 mutation (30), and this appears to be estrogen dependent. The female predisposition to IPAH may also be related to aberrant expression of a cytochrome, CYP1B1, that leads to a mitogenic estrogen metabolite (130). Experimental studies attribute the manifestation of pulmonary vascular pathology in S100A4-overexpressing female mice to 17β-estradiol induction of receptor for advanced glycosylation end-products (RAGE), the S100A4 receptor (131). In contrast, both estrogen and activation of the estrogen receptor β (ERβ) is protective against the development of experimental pulmonary hypertension (132, 133), so preferential engagement of the ERα may dictate the development of PAH in females. Also, the lack of insulin resistance and of PAH in Apoe–/– female mice fed a Western diet has been attributed to heightened levels of adiponectin (55); the low level of adiponectin in males is related to testosterone-mediated degradation. Metabolic syndrome, however, is increased in females with IPAH compared with the general population (134), so low adiponectin may place females at heightened risk for PAH.
Stem and progenitor cells
A major effort is being directed at understanding the mechanisms underlying recruitment of circulating EPCs (135) and endothelial-derived autocrine signals (136), with the view toward applying this knowledge to regenerating normal microvessels. EPCs engineered to express eNOS were successfully applied first to experimental PAH (137), and are currently being tested in the clinical setting. Mesenchymal cells also attenuate experimentally induced PAH and improve RV performance (138, 139).
Although the focus in understanding the mechanism of PAH has been on the small PAs (less than 500 μM), changes in impedance (140) resulting from stiffening of the more proximal as well as the distal PAs may also be a critical determinant of RV function (141). BMPR2 mutations associated with PAH may also influence the remodeling pathology of proximal PAs and of cardiac myocytes and fibroblasts. For example, a mosaic deletion of BMPR1A improves distal PA remodeling, but creates stiffer proximal PAs (142). Basic questions remain as to why the RV might fail under conditions such as scleroderma (143), when PA pressure and resistance are not highly elevated. However, a discussion of RV and large PA function in the context of PAH merits its own review.
New directions
This review attempts to show that the myriad molecules and pathways implicated in PAH pathobiology are all interconnected. There is a genetic predisposition that is linked either directly or indirectly to the BMPR2 signaling pathway in vascular and in inflammatory/immune cells. This affects the response of the tissue to an infectious or inflammatory stimulus. Inflammation and oxidative stress also have profound effects on mitochondrial metabolism and gene regulation (Figure 2).
Thus far, in clinical or in experimental studies, little attention has been given to the microbiome in pulmonary hypertension and to the induction of inflammasomes and their potential role in the remodeling associated with PAH (144). Next-generation sequencing combined with proteomics and metabolomics will further identify gene variants and epigenetic changes that underlie the pathobiology of PAH. The use of induced pluripotent stem cells differentiated into vascular cells could be useful (145) in better stratifying and personalizing newly emerging PAH therapies (see Emerging therapeutic strategies for pulmonary hypertension), if these cells reflect the genetic and epigenetic features causing the cellular dysfunction associated with PAH.
Supplementary Material
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(12):4306–4313. doi:10.1172/JCI60658.
References
- 1.Simonneau G, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 suppl):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch M, et al. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension: A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest. 1986;55(6):632–653. [PubMed] [Google Scholar]
- 3.Rosenberg HC, Rabinovitch M. Endothelial injury and vascular reactivity in monocrotaline pulmonary hypertension. Am J Physiol. 1988;255(6 pt 2):H1484–H1491. doi: 10.1152/ajpheart.1988.255.6.H1484. [DOI] [PubMed] [Google Scholar]
- 4.Dewachter L, et al. Angiopoietin/Tie2 pathway influences smooth muscle hyperplasia in idiopathic pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1025–1033. doi: 10.1164/rccm.200602-304OC. [DOI] [PubMed] [Google Scholar]
- 5.Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular-matrix bound basic fibroblast growth factor. J Cell Physiol. 1996;166(3):495–505. doi: 10.1002/(SICI)1097-4652(199603)166:3<495::AID-JCP4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 6.Alastalo TP, et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. 2011;121(9):3735–3746. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150(4):1349–1360. [PMC free article] [PubMed] [Google Scholar]
- 8.Passman JN, et al. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc Natl Acad Sci U S A. 2008;105(27):9349–9354. doi: 10.1073/pnas.0711382105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davie NJ, et al. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L668–L678. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 10.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ Res. 2002;90(11):1189–1196. doi: 10.1161/01.RES.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 11.Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009;10:95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyrick B, Clarke SW, Symons C, Woodgate DJ, Reid L. Primary pulmonary hypertension: a case report including electronmicroscopic study. Br J Dis Chest. 1974;68(1):11–20. doi: 10.1016/0007-0971(74)90003-5. [DOI] [PubMed] [Google Scholar]
- 13.Taraseviciene-Stewart L, Gera L, Hirth P, Voelkel NF, Tuder RM, Stewart JM. A bradykinin antagonist and a caspase inhibitor prevent severe pulmonary hypertension in a rat model. Can J Physiol Pharmacol. 2002;80(4):269–274. doi: 10.1139/y02-047. [DOI] [PubMed] [Google Scholar]
- 14.Masri FA, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 15.Xu W, et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18(14):1746–1748. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 16.Xu W, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104(4):1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuckerbraun BS, et al. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121(1):98–109. doi: 10.1161/CIRCULATIONAHA.109.891077. [DOI] [PubMed] [Google Scholar]
- 18.Leiper J, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13(2):198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 19.Chazova I, Loyd JE, Zhdanov VS, Newman JH, Belenkov Y, Meyrick B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol. 1995;146(2):389–397. [PMC free article] [PubMed] [Google Scholar]
- 20.Jones P, Cowan K, Rabinovitch M. Progressive pulmonary vascular disease is characterized by a proliferative response related to deposition of tenascin-C and is preceded by subendothelial accumulation of fibronectin. Am J Pathol. 1997;150(4):1349–1360. [PMC free article] [PubMed] [Google Scholar]
- 21.Liptay MJ, et al. Neointimal macrophages colocalize with extracellular matrix gene expression in human atherosclerotic pulmonary arteries. J Clin Invest. 1993;91(2):588–594. doi: 10.1172/JCI116238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenway S, et al. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164(1):253–262. doi: 10.1016/S0002-9440(10)63115-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balabanian K, et al. CX(3)C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(10):1419–1425. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- 24.Lane KB, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 25.Deng Z, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudarakanchana N, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11(13):1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 27.Nohe A, et al. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol Chem. 2002;277(7):5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- 28.Grijelmo C, et al. Proinvasive activity of BMP-7 through SMAD4/src-independent and ERK/Rac/JNK-dependent signaling pathways in colon cancer cells. Cell Signal. 2007;19(8):1722–1732. doi: 10.1016/j.cellsig.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 29.Tian Q, He XC, Hood L, Li L. Bridging the BMP and Wnt pathways by PI3 kinase/Akt and 14-3-3zeta. Cell Cycle. 2005;4(2):215–216. [PubMed] [Google Scholar]
- 30.Hamid R, Cogan JD, Hedges LK, Austin E, Phillips JA. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat. 2009;30(4):649–654. doi: 10.1002/humu.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts KE, et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J. 2004;24(3):371–374. doi: 10.1183/09031936.04.00018604. [DOI] [PubMed] [Google Scholar]
- 32.Humbert M, et al. BMPR2 germline mutations in pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J. 2002;20(3):518–523. doi: 10.1183/09031936.02.01762002. [DOI] [PubMed] [Google Scholar]
- 33.Atkinson C, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–1678. doi: 10.1161/01.CIR.0000012754.72951.3D. [DOI] [PubMed] [Google Scholar]
- 34.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88(1):E2–E11. doi: 10.1161/01.RES.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 35.Harrison RE, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet. 2003;40(12):865–871. doi: 10.1136/jmg.40.12.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaouat A, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–448. doi: 10.1136/thx.2003.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerkic M, et al. Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc Res. 2011;92(3):375–384. doi: 10.1093/cvr/cvr232. [DOI] [PubMed] [Google Scholar]
- 38.Aldred MA, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(9):1153–1160. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454(7200):56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang H, et al. Bone morphogenetic protein 4 promotes vascular smooth muscle contractility by activating microRNA-21 (miR-21), which down-regulates expression of family of dedicator of cytokinesis (DOCK) proteins. J Biol Chem. 2012;287(6):3976–3986. doi: 10.1074/jbc.M111.303156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drake KM, et al. Altered MicroRNA processing in heritable pulmonary arterial hypertension: an important role for Smad-8. Am J Respir Crit Care Med. 2011;184(12):1400–1408. doi: 10.1164/rccm.201106-1130OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caruso P, et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30(4):716–723. doi: 10.1161/ATVBAHA.109.202028. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds AM, et al. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1182–L1192. doi: 10.1152/ajplung.00020.2006. [DOI] [PubMed] [Google Scholar]
- 44.Yang J, et al. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: implications for familial pulmonary arterial hypertension. Circ Res. 2008;102(10):1212–1221. doi: 10.1161/CIRCRESAHA.108.173567. [DOI] [PubMed] [Google Scholar]
- 45.Morrell NW, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790–795. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 46.Long L, et al. Altered bone morphogenetic protein and transforming growth factor-beta signaling in rat models of pulmonary hypertension: potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation. 2009;119(4):566–576. doi: 10.1161/CIRCULATIONAHA.108.821504. [DOI] [PubMed] [Google Scholar]
- 47.Teichert-Kuliszewska K, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–217. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 48.de Jesus Perez VA, et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184(1):83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guignabert C, et al. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1082–L1090. doi: 10.1152/ajplung.00199.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schopfer FJ, et al. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. J Biol Chem. 2010;285(16):12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valdimarsdottir G, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106(17):2263–2270. doi: 10.1161/01.CIR.0000033830.36431.46. [DOI] [PubMed] [Google Scholar]
- 52.Hansmann G, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(5):1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 54.Jeffery TK, Upton PD, Trembath RC, Morrell NW. BMP4 inhibits proliferation and promotes myocyte differentiation of lung fibroblasts via Smad1 and JNK pathways. Am J Physiol Lung Cell Mol Physiol. 2005;288(2):L370–L378. doi: 10.1152/ajplung.00242.2004. [DOI] [PubMed] [Google Scholar]
- 55.Hansmann G, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115(10):1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 56.Takayama Y, May P, Anderson RG, Herz J. Low density lipoprotein receptor-related protein 1 (LRP1) controls endocytosis and c-CBL-mediated ubiquitination of the platelet-derived growth factor receptor beta (PDGFR beta). J Biol Chem. 2005;280(18):18504–18510. doi: 10.1074/jbc.M410265200. [DOI] [PubMed] [Google Scholar]
- 57.Schermuly RT, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. N Engl J Med. 2005;353(13):1412–1413. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 59.Montani D, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 60.Arita Y, et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation. 2002;105(24):2893–2898. doi: 10.1161/01.CIR.0000018622.84402.FF. [DOI] [PubMed] [Google Scholar]
- 61.Lawrie A, et al. Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am J Pathol. 2008;172(1):256–264. doi: 10.2353/ajpath.2008.070395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ihida-Stansbury K, et al. Tenascin-C is induced by mutated BMP type II receptors in familial forms of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;291(4):L694–L702. doi: 10.1152/ajplung.00119.2006. [DOI] [PubMed] [Google Scholar]
- 63.Jones P, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the αvβ3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139(1):279–293. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lorenzen JM, et al. Osteopontin in patients with idiopathic pulmonary hypertension. Chest. 2011;139(5):1010–1017. doi: 10.1378/chest.10-1146. [DOI] [PubMed] [Google Scholar]
- 65.Beppu H, et al. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1241–L1247. doi: 10.1152/ajplung.00239.2004. [DOI] [PubMed] [Google Scholar]
- 66.West J, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–1114. doi: 10.1161/01.RES.0000126047.82846.20. [DOI] [PubMed] [Google Scholar]
- 67.Long L, et al. Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res. 2006;98(6):818–827. doi: 10.1161/01.RES.0000215809.47923.fd. [DOI] [PubMed] [Google Scholar]
- 68.Abenhaim L, Humbert M. Pulmonary hypertension related to drugs and toxins. Curr Opin Cardiol. 1999;14(5):437–441. doi: 10.1097/00001573-199909000-00013. [DOI] [PubMed] [Google Scholar]
- 69.Dempsie Y, et al. Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice. Circulation. 2008;117(22):2928–2937. doi: 10.1161/CIRCULATIONAHA.108.767558. [DOI] [PubMed] [Google Scholar]
- 70.White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. 2011;90(2):373–382. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- 71.Chin KM, Channick RN, Rubin LJ. Is methamphetamine use associated with idiopathic pulmonary arterial hypertension? Chest. 2006;130(6):1657–1663. doi: 10.1378/chest.130.6.1657. [DOI] [PubMed] [Google Scholar]
- 72.Dhillon NK, et al. Effect of cocaine on human immunodeficiency virus-mediated pulmonary endothelial and smooth muscle dysfunction. Am J Respir Cell Mol Biol. 2011;45(1):40–52. doi: 10.1165/rcmb.2010-0097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eddahibi S, et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation. 2003;108(15):1839–1844. doi: 10.1161/01.CIR.0000091409.53101.E8. [DOI] [PubMed] [Google Scholar]
- 74.Le Cras TD, Kim DH, Markham NE, Abman AS. Early abnormalities of pulmonary vascular development in the Fawn-Hooded rat raised at Denver’s altitude. Am J Physiol Lung Cell Mol Physiol. 2000;279(2):L283–L291. doi: 10.1152/ajplung.2000.279.2.L283. [DOI] [PubMed] [Google Scholar]
- 75.Eddahibi S, et al. Attenuated hypoxic pulmonary hypertension in mice lacking the 5-hydroxytryptamine transporter gene. J Clin Invest. 2000;105(11):1555–1562. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacLean MR, et al. Overexpression of the 5-hydroxytryptamine transporter gene: effect on pulmonary hemodynamics and hypoxia-induced pulmonary hypertension. Circulation. 2004;109(17):2150–2155. doi: 10.1161/01.CIR.0000127375.56172.92. [DOI] [PubMed] [Google Scholar]
- 77.Guignabert C, et al. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res. 2006;98(10):1323–1330. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, Li M, Warburton RR, Hill NS, Fanburg BL. The 5-HT transporter transactivates the PDGF{beta} receptor in pulmonary artery smooth muscle cells. FASEB J. 2007;21(11):2725–2734. doi: 10.1096/fj.06-8058com. [DOI] [PubMed] [Google Scholar]
- 79.Simon AR, et al. 5-HT induction of c-fos gene expression requires reactive oxygen species and Rac1 and Ras GTPases. Cell Biochem Biophys. 2005;42(3):263–276. doi: 10.1385/CBB:42:3:263. [DOI] [PubMed] [Google Scholar]
- 80.Lawrie A, et al. Interdependent serotonin transporter and receptor pathways regulate S100A4/Mts1, a gene associated with pulmonary vascular disease. Circ Res. 2005;97(3):227–235. doi: 10.1161/01.RES.0000176025.57706.1e. [DOI] [PubMed] [Google Scholar]
- 81.Spiekerkoetter E, et al. Reactivation of gammaHV68 induces neointimal lesions in pulmonary arteries of S100A4/Mts1-overexpressing mice in association with degradation of elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294(2):L276–L289. doi: 10.1152/ajplung.00414.2007. [DOI] [PubMed] [Google Scholar]
- 82.Morse JH, et al. Primary pulmonary hypertension in HIV infection: an outcome determined by particular HLA class II alleles. Am J Respir Crit Care Med. 1996;153(4 pt 1):1299–1301. doi: 10.1164/ajrccm.153.4.8616557. [DOI] [PubMed] [Google Scholar]
- 83.Cool CD, et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003;349(12):1113–1122. doi: 10.1056/NEJMoa035115. [DOI] [PubMed] [Google Scholar]
- 84.Durrington HJ, et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J Biol Chem. 2010;285(48):37641–37649. doi: 10.1074/jbc.M110.132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marecki JC, et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am J Respir Crit Care Med. 2006;174(4):437–445. doi: 10.1164/rccm.200601-005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crosby A, et al. Praziquantel reverses pulmonary hypertension and vascular remodeling in murine schistosomiasis. Am J Respir Crit Care Med. 2011;184(4):467–473. doi: 10.1164/rccm.201101-0146OC. [DOI] [PubMed] [Google Scholar]
- 87.Graham BB, et al. Schistosomiasis-induced experimental pulmonary hypertension: role of interleukin-13 signaling. Am J Pathol. 2010;177(3):1549–1561. doi: 10.2353/ajpath.2010.100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Daley E, et al. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med. 2008;205(2):361–372. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Angelini DJ, et al. Resistin-like molecule-beta in scleroderma-associated pulmonary hypertension. Am J Respir Cell Mol Biol. 2009;41(5):553–561. doi: 10.1165/rcmb.2008-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lundequist A, et al. Prostaglandin E(2) exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. J Immunol. 2010;184(1):433–441. doi: 10.4049/jimmunol.0902835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song Y, Jones JE, Beppu H, Keaney JF, Jr, Loscalzo J, Zhang YY. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation. 2005;112(4):553–562. doi: 10.1161/CIRCULATIONAHA.104.492488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meyrick B, Brigham K. Repeated eschrerichia coli endotocin-induced pulmonary inflammation causes chronic pulmonary hypertension in sheep. Lab Invest. 1986;55(2):164–176. doi: 10.1111/1523-1747.ep12280663. [DOI] [PubMed] [Google Scholar]
- 93.Stevens T, Janssen PL, Tucker AD. Acute and long-term TNF-alpha administration increases pulmonary vascular reactivity in isolated rat lungs. J Appl Physiol. 1992;73(2):708–712. doi: 10.1152/jappl.1992.73.2.708. [DOI] [PubMed] [Google Scholar]
- 94.Taraseviciene-Stewart L, et al. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175(12):1280–1289. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tamosiuniene R, et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res. 2011;109(8):867–879. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bauer EM, Zheng H, Comhair S, Erzurum S, Billiar TR, Bauer PM. Complement C3 deficiency attenuates chronic hypoxia-induced pulmonary hypertension in mice. PLoS One. 2011;6(12):e28578. doi: 10.1371/journal.pone.0028578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schober A, Zernecke A. Chemokines in vascular remodeling. Thromb Haemost. 2007;97(5):730–737. [PubMed] [Google Scholar]
- 98.Brauner EV, et al. Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med. 2008;177(4):419–425. doi: 10.1164/rccm.200704-632OC. [DOI] [PubMed] [Google Scholar]
- 99.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104(2):236–244. doi: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perros F, et al. Fractalkine-induced smooth muscle cell proliferation in pulmonary hypertension. Eur Respir J. 2007;29(5):937–943. doi: 10.1183/09031936.00104706. [DOI] [PubMed] [Google Scholar]
- 101.Courboulin A, et al. Kruppel-like Factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res. 2011;12:128. doi: 10.1186/1465-9921-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li M, et al. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187(5):2711–2722. doi: 10.4049/jimmunol.1100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vergadi E, et al. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation. 2011;123(18):1986–1995. doi: 10.1161/CIRCULATIONAHA.110.978627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frid MG, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168(2):659–669. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yeager ME, et al. Circulating fibrocytes are increased in children and young adults with pulmonary hypertension. Eur Respir J. 2012;39(1):104–111. doi: 10.1183/09031936.00072311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perros F, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185(3):311–321. doi: 10.1164/rccm.201105-0927OC. [DOI] [PubMed] [Google Scholar]
- 107.Dib H, et al. Targets of anti-endothelial cell antibodies in pulmonary hypertension and scleroderma. Eur Respir J. 2012;39(6):1405–1414. doi: 10.1183/09031936.00181410. [DOI] [PubMed] [Google Scholar]
- 108.Bonnet S, et al. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc Natl Acad Sci U S A. 2007;104(27):11418–11423. doi: 10.1073/pnas.0610467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Courboulin A, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208(3):535–548. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Todorovich-Hunter L, Dodo H, Ye C, McCready L, Keeley FW, Rabinovitch M. Increased pulmonary artery elastolytic activity in adult rats with monocrotaline-induced progressive hypertensive pulmonary vascular disease compared with infant rats with nonprogressive disease. Am Rev Respir Dis. 1992;146(1):213–223. doi: 10.1164/ajrccm/146.1.213. [DOI] [PubMed] [Google Scholar]
- 111.Ye C, Rabinovitch M. Inhibition of elastolysis by SC-37698 reduces development and progression of monocrotaline pulmonary hypertension. Am J Physiol. 1991;261(4 pt 2):H1255–H1267. doi: 10.1152/ajpheart.1991.261.4.H1255. [DOI] [PubMed] [Google Scholar]
- 112.Maruyama K, et al. Chronic hypoxic pulmonary hypertension in rats and increased elastolytic activity. Am J Physiol. 1991;261(6 pt 2):H1716–H1726. doi: 10.1152/ajpheart.1991.261.6.H1716. [DOI] [PubMed] [Google Scholar]
- 113.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6(6):698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 114.McMurtry MS, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115(6):1479–1491. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim YM, et al. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol. 2011;179(3):1560–1572. doi: 10.1016/j.ajpath.2011.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lawrie A, et al. Paigen diet-fed apolipoprotein E knockout mice develop severe pulmonary hypertension in an interleukin-1-dependent manner. Am J Pathol. 2011;179(4):1693–1705. doi: 10.1016/j.ajpath.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yeager ME, Belchenko DD, Nguyen CM, Colvin KL, Ivy DD, Stenmark KR. Endothelin-1, the unfolded protein response, and persistent inflammation: role of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2012;46(1):14–22. doi: 10.1165/rcmb.2010-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 119.Archer SL, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fantozzi I, et al. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L993–L1004. doi: 10.1152/ajplung.00191.2005. [DOI] [PubMed] [Google Scholar]
- 121.Cogolludo A, et al. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: role of 5-HT2A receptors, caveolin-1, and KV1.5 channel internalization. Circ Res. 2006;98(7):931–938. doi: 10.1161/01.RES.0000216858.04599.e1. [DOI] [PubMed] [Google Scholar]
- 122.McMurtry MS, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95(8):830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 123.Sutendra G, et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3(88):88ra55. doi: 10.1126/scitranslmed.3002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sutendra G, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2(44):44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 125.Coller HA. Cell biology. The essence of quiescence. Science. 2011;334:1074–1075. doi: 10.1126/science.1216242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee SJ, et al. Autophagic protein LC3B confers resistance against hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med. 2011;183(5):649–658. doi: 10.1164/rccm.201005-0746OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hickey MM, et al. The von Hippel-Lindau Chuvash mutation promotes pulmonary hypertension and fibrosis in mice. J Clin Invest. 2010;120(3):827–839. doi: 10.1172/JCI36362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Abe K, et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ Res. 2004;94(3):385–393. doi: 10.1161/01.RES.0000111804.34509.94. [DOI] [PubMed] [Google Scholar]
- 129.Zhang S, et al. Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1202–L1210. doi: 10.1152/ajplung.00214.2006. [DOI] [PubMed] [Google Scholar]
- 130.Austin ED, et al. Alterations in estrogen metabolism: Implications for higher penetrance of FPAH in females. Eur Respir J. 2009;34(5):1093–1099. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dempsie Y, et al. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res. 2011;12:159. doi: 10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Umar S, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184(6):715–723. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lahm T, et al. 17β-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185(9):965–980. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zamanian RT, et al. Insulin resistance in pulmonary arterial hypertension. Eur Respir J. 2009;33(2):318–324. doi: 10.1183/09031936.00000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kumar PA, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147(3):525–538. doi: 10.1016/j.cell.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ding BS, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147(3):539–553. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96(4):442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 138.Deng W, St Hilaire RC, Chattergoon NN, Jeter JR, Jr, Kadowitz PJ. Inhibition of vascular smooth muscle cell proliferation in vitro by genetically engineered marrow stromal cells secreting calcitonin gene-related peptide. Life Sci. 2006;78(16):1830–1838. doi: 10.1016/j.lfs.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 139.Baber SR, et al. Intratracheal mesenchymal stem cell administration attenuates monocrotaline-induced pulmonary hypertension and endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2007;292(2):H1120–H1128. doi: 10.1152/ajpheart.00173.2006. [DOI] [PubMed] [Google Scholar]
- 140.Weinberg CE, et al. Extraction of pulmonary vascular compliance, pulmonary vascular resistance, and right ventricular work from single-pressure and Doppler flow measurements in children with pulmonary hypertension: a new method for evaluating reactivity: in vitro and clinical studies. Circulation. 2004;110(17):2609–2617. doi: 10.1161/01.CIR.0000146818.60588.40. [DOI] [PubMed] [Google Scholar]
- 141.Mahmud M, Champion HC. Right ventricular failure complicating heart failure: pathophysiology, significance, and management strategies. Curr Cardiol Rep. 2007;9(3):200–208. doi: 10.1007/BF02938351. [DOI] [PubMed] [Google Scholar]
- 142.Vanderpool RR, El-Bizri N, Rabinovitch M, Chesler NC. Patchy deletion of Bmpr1a potentiates proximal pulmonary artery remodeling in mice exposed to chronic hypoxia [published online ahead of print February 8, 2012]. Biomech Model Mechanobiol. doi: 10.1007/s10237-012-0379-6. doi: 10.1007/s10237-012-0379-6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chung L, et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest. 2010;138(6):1383–1394. doi: 10.1378/chest.10-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 145.Ikonomou L, et al. Programmatic change: lung disease research in the era of induced pluripotency. Am J Physiol Lung Cell Mol Physiol. 2011;301(6):L830–L835. doi: 10.1152/ajplung.00255.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.