

Abstract
NOD1 {nucleotide-binding oligomerization domain 1; NLRC [NOD-LRR (leucine-rich repeat) family with CARD (caspase recruitment domain) 1]} and NOD2 (NLRC2) are among the most prominent members of the NLR (NOD-LRR) family –proteins that contain nucleotide-binding NACHT domains and receptor-like LRR domains. With over 20 members identified in humans, NLRs represent important components of the mammalian innate immune system, serving as intracellular receptors for pathogens and for endogenous molecules elaborated by tissue injury. NOD1 and NOD2 proteins operate as microbial sensors through the recognition of specific PG (peptidoglycan) constituents of bacteria. Upon activation, these NLR family members initiate signal transduction mechanisms that include stimulation of NF-κB (nuclear factor-κB), stress kinases, IRFs (interferon regulatory factors) and autophagy. Hereditary polymorphisms in the genes encoding NOD1 and NOD2 have been associated with an increasing number of chronic inflammatory diseases. In fact, potential roles for NOD1 and NOD2 in inflammatory disorders have been revealed by investigations using a series of animal models. In the present review, we describe recent experimental findings associating NOD1 and NOD2 with various autoimmune and chronic inflammatory disorders, and we discuss prospects for development of novel therapeutics targeting these NLR family proteins.
Keywords: inflammation, nuclear factor-κB (NF-κB), nucleotide-binding oligomerization domain 1 (NOD1), nucleotide-binding oligomerization domain (NOD) leucine-rich repeat (NLR), nucleotide-binding oligomerization domain 2 (NOD2)
Abbreviations: AP-1, activator protein 1; BMT, bone marrow transplant; β-TrCP, β-transducin repeat-containing protein; CARD, caspase recruitment domain; DAP, L-Ala-γ-D-Glu-m-diaminopimelic acid; DC, dendritic cell; EAE, experimental autoimmune encephalomyelitis; ERK, extracellular-signal-regulated kinase; HFD, high-fat diet; IAP, inhibitor of apoptosis protein; BIR, baculovirus IAP repeat; c-IAP1, cellular IAP1; IBD, inflammatory bowel disease; IKK, inhibitory κB kinase; IL-6, interleukin 6; IFN, interferon; IRF, interferon regulatory factor; JNK, c-Jun N-terminal kinase; KO, knockout; LRR, leucine-rich repeat; LUBAC, linear ubiquitin chain assembly complex; MAPK, mitogen-activated protein kinase; MAVS, mitochondrial antiviral signalling; MDP, muramyl dipeptide; MKK, MAPK kinase; NEMO, NF-κB essential modulator; NOD, nucleotide-binding oligomerization domain; NLR, NOD-LRR; NLRC, NOD-LRR family with CARD; PG, peptidoglycan; RIP2, receptor-interacting protein 2; SCF, stem cell factor; TAB, TGF (transforming growth factor)-β-activated kinase-binding protein; TRAF, tumour-necrosis-factor-receptor-associated factor; TBK1, TRAF-associated nuclear factor-κB activator-binding kinase 1; TLR, Toll-like receptor; XIAP, X-linked IAP
INTRODUCTION
Humans are constantly challenged by an enormous diversity of microbes and viruses, relying on the innate immune system for initial responses to infectious pathogens. A complex network of innate immunity genes regulates a delicate balance between stimulation and suppression of host immune responses to foreign substances, ensuring appropriate homoeostasis in the gut, skin, lung and various body surfaces. Imbalances in these innate immunity systems contribute to serious infectious diseases and to chronic inflammatory and autoimmune diseases. The success of this immune response relies on the recognition of conserved molecular structures that are commonly present in microbes (and other types of pathogens), but not in the host, by ‘pattern recognition’ molecules that act as microbial sensors. In this context, NLRs [NACHT and LRR (leucine-rich repeat) domain-containing proteins] constitute a major subfamily of innate immunity proteins [1,2], acting primarily as intracellular PRRs (pattern recognition receptors) involved in the detection of cytoplasmic PAMPs (pathogen-associated molecular patterns) and endogenous products of tissue injury [DAMPs (danger- associated molecular patterns)] [3]. The recognition of the signals initiates a variety of host defence pathways through the activation of NF-κB, stress kinases, IRFs (interferon regulatory factors), inflammatory caspases and autophagy [4–7].
The NLR protein family comprises 22 human members [8–11]. Numerous orthologues and paralogues are also present in both vertebrate (e.g., rodents, fish) and invertebrate marine species (e.g. chordates) but not in insects (Drosophila) or nematodes (Caenorhabditis elegans) [8,12]. Moreover, the NLR family shares structural similarity with a subset of plant disease-resistance (R) proteins involved in the hypersensitive response induced by plant pathogens [13]. The typical architecture of NLR proteins includes a C-terminal region comprising variable numbers of LRR domains that are involved in recognition of conserved microbial patterns or other ligands; a centrally located nucleotide-binding NACHT domain that mediates self-oligomerization and is essential for NLR activation; and a N-terminal effector domain, which is responsible for the interaction with adaptor molecules or downstream effector proteins that mediate the signal transduction functions of NLRs. Based on the nature of their N-terminal domains, the NLRs have been divided into three subgroups, which possess (i) CARDs (caspase recruitment domains), (ii) PYDs (pyrin domains), or (iii) other domains, such as BIRs [baculovirus IAP (inhibitor of apoptosis protein) repeat domains] [1,4,9,14].
CARD-CARRYING NLR PROTEINS
The human genome encodes at least five NLRs that contain CARDs, a homophilic protein interaction motif typically comprised of a bundle of six to seven antiparallel α-helices [10,15]. The CARD-carrying NLRs include NOD1 [NLRC1 (NOD-LRR family with CARD 1), also termed as CARD4 or CLR7.1], NOD2 (NLRC2, also termed as CARD15; BLAU; IBD1; PSORAS1; CLR16.3), NLRP1 (also termed NALP1; NAC; CARD7), NLRC4 (also termed IPAF; CLAN; CARD12), and the class II MHC complex transactivator CIITA type I (also termed NLRA) [10,15]. NOD1 is widely expressed in many cell types and tissues in vivo, whereas NOD2 has been found in macrophages, DCs (dendritic cells), Paneth cells, keratinocytes, intestinal epithelium, lung, oral cavity and osteoblasts [16–25]. Nevertheless, both proteins are activated by recognition of specific motifs (mostly, muropeptides) present in bacterial PG (peptidoglycan). In contrast with TLRs (Toll-like receptors), which are cell-surface pathogen receptors found in the plasma membrane, NOD1 and NOD2 are cytoplasmic proteins. Interestingly, NOD1 and NOD2 dynamically traffic to intracellular membranes upon detection of PG derivatives [26,27].
PG is a major component of the Gram-positive bacterial cell wall, while in Gram-negative bacteria it is found as a thin layer in the periplasmic space. NOD2 detects and directly binds MDP (muramyl dipeptide) [28,29], a motif that is present in the PGs of both Gram-positive and Gram-negative bacteria, which is also a major component of many immunoadjuvants. In contrast, the recognition of bacterial PG by NOD1 is dependent on the presence of the m-DAP (L-Ala-γ-D-Glu-m-diaminopimelic acid), an amino acid characteristic of most Gram-negative and some Gram-positive bacteria, such as Listeria monocytogenes and Bacillus spp. [7,30,31]. The minimal structure detected by NOD1 is the dipeptide tri-DAP [30,32]. It was biochemically demonstrated that NOD1-activating tri-DAP directly interacts with the LRR domain of NOD1, suggesting that it is a direct ligand (agonist) of NOD1 [33].
Activation of NOD1 and NOD2 involves oligomerization mediated by the nucleotide-binding NACHT domains, thus creating a platform for activation of signalling molecules. The CARDs of NOD1 and NOD2 bind the CARD domain of RIP2 (receptor-interacting protein 2) {RICK [RIP-like interacting CLARP (caspase-like apoptosis-regulatory protein) kinase]}/Ripk2 (receptor-interacting serine/threonine protein kinase 2)/CARDIAK [CARD (caspase recruitment domain)-containing ICE (interleukin-1β-converting enzyme)-associated kinase] through homophilic CARD–CARD interactions (Figure 1). Gene KO (knockout) studies indicate that RIP2 is a critical mediator of NOD1 and NOD2 signallings [34–36], though precise details are unclear.
Figure 1. Major NOD-dependent signalling pathways.
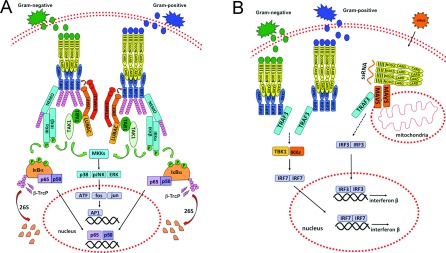
(A) NF-κB and AP-1 pathways. Bacterial PG-derived peptides γ-D-glutamyl-m-diaminopimelic acid (iE-DAP) and MDP are recognized by the cytosolic receptors NOD1 and NOD2. These ligands bind to NOD1 or NOD2 through the LRR domain of these molecules. This interaction initiates the activation of NOD1 and NOD2 due to the induction of a complex conformational change that results in protein oligomerization and further interaction with downstream effectors. NOD1 or NOD2 assembly recruits RIP2 through CARD–CARD interactions, resulting in RIP2 ubiquitination by IAPs and recruitment of LUBAC complex by XIAP, with further binding of the TAB1/TAK1 complex. It is believed that TAK1 gets activated through autophosphorylation and stimulates downstream IKK complex, including Lys63-linked polyubiquitination of NEMO (IKKγ), the regulatory subunit of the IKK complex, which also consists of the catalytic subunits IKK1 (IKKα) and IKK2 (IKKβ). This event is followed by IKK2 phosphorylation, which further phosphorylates the NF-κB inhibitor IκBα. IκBα is then ubiquitinated by the SCF/β-TrCP complex and further degraded by 26S proteasome. The degradation of IκBα releases NF-κB dimers to translocate into the nucleus, where they up-regulate target genes involved in host defence and apoptosis. NOD oligomerization and further RIP2 activation also recruits TAB/TAK1 complexes to mediate the phosphorylation of MAPKs, such as JNK, ERK and p38 MAPK, through the upstream activation of MKKs. These kinases translocate to the nucleus and then phosphorylate AP-1 transcription factors (c-fos, c-Jun, ATF and JDP family members) to mediate expression of target genes containing a TRE (TPA DNA-response element). (B) MAVS/IRF pathway. Activation of both NOD1 and NOD2 by bacterial products induces receptor oligomerization and RIP2 recruitment, which in turn binds TRAF3 and induces TBK1/IKKϵ activation through a mechanism that is not completely understood. This is followed by IRF transcription factor dimerization and activation, resulting in binding to and induction of type I IFN genes. Similarly, virus-derived single-stranded RNA binds NOD2 and induces its association with mitochondrial receptor MAVS, resulting in the activation of IRF3 transcription factors and induction of type I IFNs. TRAF3 also directly binds MAVS but its precise role requires further investigation.
RIP2 contains an N-terminal domain with homology with serine/threonine kinases and a C-terminal CARD. The exact role of the kinase activity of RIP2 is not fully understood, but studies using RIP2-KO and -KD (kinase dead) knockin mice indicate that the intact kinase activity is essential for RIP2 protein stability and proper innate immune responses triggered by NOD1 and NOD2 [37]. Also, the kinase domain of RIP2 binds IAPs, including XIAP (X-linked IAP), c-IAP1 (cellular IAP 1) and c-IAP2 [38,39]. Gene ablation studies have shown that IAPs can be required for NOD1 signalling, depending on the cellular type [38,39].
The RIP2-binding IAPs are E3 ligases, which are capable of partnering with different types of ubiquitin-conjugating enzymes (E2s), including those that catalyse the formation of Lys63-linked polyubiquitin chains. Lys63-linked chains of ubiquitin are not substrates for the proteasome, but rather represent a post-translational modification involved in protein activation and protein subcellular localization [40–42]. It has been reported that RIP2 is conjugated with Lys63-linked polyubiquitin chains at Lys209 upon NOD1 or NOD2 activation [43]. Additionally, XIAP was recently shown to recruit the LUBAC (linear ubiquitin chain assembly complex) E3 ligase complex to NOD protein complexes [44]. Altogether, these polyubiquitin chains attached to RIP2 attract the adapter protein TAB {TAK [TGF (transforming growth factor)-β-activated kinase]-binding protein} and its associated kinase TAK [45,46]. This complex also directly binds the BIR1 domain of XIAP, independently of Lys63-linked ubiquitination. TAK is an upstream activator of the IKK (inhibitory κB kinase) complex, as well as an activator of stress kinase cascades [through MKK (MAPK kinase) 6 phosphorylation] that result in JNK (c-Jun N-terminal kinase) and p38 MAPK (mitogen-activated protein kinase) activation [45] (Figure 1A). Indeed, treatment of macrophages with NOD1 agonists rapidly induces activation of p38, ERK (extracellular-signal-regulated kinase) and JNK [47]. Additionally, RIP2 interacts with the IKK subunit IKKγ [also called NEMO (NF-κB essential modulator)], promoting its modification with Lys63-linked polyubiquitin chains and resulting in activation of the IKK complex. The IKK complex then phosphorylates the NF-κB inhibitor IκBα, targeting it for Lys48-linked polyubiquitination and proteasome-dependent degradation [43,48] (Figure 1A). Upon phosphorylation at Ser32 and Ser36, IκBα binds the F-box ubiquitin ligase complex SCF (stem cell factor)/β-TrCP (β transducin repeat-containing protein), which is responsible for its ubiquitination [49]. After IκBα is degraded, free NF-κB translocates into the nucleus, where it drives the transcription of κB-containing genes [50,51] (Figure 1A).
In addition to NF-κB and stress kinases, NOD1 and NOD2 activate other innate immunity mechanisms. For example, the NACHT and LRR regions of NOD2 are reportedly required for the association with the mitochondrial outer membrane protein MAVS (mitochondrial antiviral signalling) [IPS-1 (IFN-β promoter stimulator protein 1), VISA (virus-induced signalling adaptor), Cardif] [52]. MAVS is an adapter protein originally implicated in innate immune responses to RNA viruses, which interacts with the RNA-binding proteins RIG-I (retinoic acid-inducible gene 1) and Helicard/MDA (melanoma differentiation-associated protein)-5 [3]. The MAVS complex stimulates IRF activation and induces the type I IFN (interferon) response [53]. The interaction of NOD2 with MAVS induces the activation of IRF3 and production of IFNβ (Figure 1B). Activation of NOD1 and NOD2 have also been reported to induce formation of a protein complex containing RIP2 and TRAF (tumour-necrosis-factor-receptor-associated factor) 3 (an E3 ligase that mediates Lys63-linked ubiquitination), leading to activation of IRF7 and induction of IFNβ, through a mechanism dependent on TBK1 (TRAF-associated nuclear factor-κB activator-binding kinase 1) and IKKϵ [54] (Figure 1B). In fact, infection by Helicobacter pylori activates a type I IFN response through a NOD1- and NOD2-dependent mechanism, leading to Stat1 (signal transducer and activation of transcription 1) activation and further clearance of bacterial burden [54].
NOD1 and NOD2 also enhance autophagy, an important process for removal of intracellular microbes via lysosome-mediated destruction [55–57]. The role of autophagy in innate immunity is becoming increasingly recognized [58,59]. NOD1 and NOD2 agonists are able to induce autophagy both in vitro and in vivo [55,56]. Indeed, both NOD proteins interact with and co-localize at the plasma membrane with ATG16L1 (autophagy related 16-like 1), an essential component of the ubiquitin-like system required for autophagosome formation [56]. Thus, NOD1 limits the bacterial burden by autophagy. After infection with Shigella flexneri, NOD1-deficient cells show higher amounts of intracellular bacteria compared with the wild-type cells [56]. Interestingly, like the MAVS-mediated IFN response, RIP2 is dispensable for the NOD-dependent autophagic response [56,57].
Recently, the pro-apoptotic protein Bid was identified as a functionally important component of the NOD1 and NOD2 signalling complexes [60]. Bid protein directly binds to NOD1 in vitro [61]. Also, Bid−/− macrophages are defective in cytokine production in response to NOD activation [60]. Precisely how Bid collaborates with NOD1 and NOD2 is unclear, but the finding extends the theme of robust connections between the cellular apoptosis machinery and innate immunity mechanisms. For example, NLRP1 is reportedly suppressed by anti-apoptotic proteins Bcl-2 and Bcl-XL [62,63]. The requirement of Bid for NOD1/NOD2 signalling raises questions about how the complex network of interactions of Bcl-2 family proteins (n>26 in humans) might impact innate immune responses. Bid, for example, is capable of binding most of the anti-apoptotic Bcl-2 family members [64,65], but the implications of those protein interactions for NOD1/NOD2 signalling remain to be defined.
NOD1 AND NOD2 IN HOST DEFENCE
NOD1 has been linked to host defence against various pathogenic bacteria, including L. monocytogenes [66], Pseudomonas aeruginosa [67] and H. pylori [54,68], being also required for IFNγ-mediated elimination of the parasitic protozoon Trypanosoma cruzi [69]. The PG component meso-DAP (NOD1 agonist) can activate human epithelial cells through NOD1 to secrete antibacterial factors and cytokines [23]. Similarly, NOD2 has been described as a major modulator of host immunity by detecting pathogens ranging from extracellular bacteria such as Staphylo-coccus aureus [70] to intracellular protozoans such as Toxoplasma gondii [71].
A number of in vivo studies have demonstrated a role for NOD1 and NOD2 in host defence. For instance, both NOD1 and NOD2 promote clearance of the intracellular pathogen Chlamydophila pneumoniae from the lungs [72]. Nod1−/− and Nod2−/− mice show delayed pulmonary bacterial clearance and evidence of defective iNOS (inducible nitric oxide synthase) expression and NO (nitric oxide) production (as observed using macrophages in vitro), suggesting that C. pneumoniae is recognized by both of these intracellular receptors [72]. Similarly, NOD1 and NOD2 regulate pulmonary innate immunity upon infection with Legionella pneumophila [73]. In this case, Nod1−/− and Nod2−/− mice show altered pulmonary inflammatory cell infiltration and impairment of cytokine response at different time points, suggesting that NOD1 and NOD2 distinctively modulate the pulmonary immune response after Legionella infection [73]. More recently, it was determined that NOD1 and NOD2 have also a crucial role for the IL-6 (interleukin 6)-dependent induction of mucosal TH17 responses at early stages of infection with Citrobacter rodentium and Salmonella typhimurium during colitis progression [74]. The TH17 response is an important component of mucosal immunity against bacterial pathogens in the lung and intestine, and it is dependent on the secretion of the cytokines IL-17 and IL-22 [74].
Many studies have demonstrated that recognition of pathogenic bacteria in intestinal cells lacking TLRs relies on the NOD1 activity [75–77]. In contrast with NOD2, NOD1 signalling is required as a ‘backup mechanism’ for activating NF-κB in human intestinal epithelial cells infected with Gram-negative enteric bacteria that can bypass TLR activation [76]. NOD1 is also a back- up to NOD2 in other respects. For example, in NOD2-deficient macrophages, which are tolerant to MDP and LPS (lipopolysaccharide), NOD1 agonists are fully capable of stimulating secretion of cytokines such as TNFα and IL-6 and of inducing NF-κB and MAPK activation [47].
The dual activation of both NF-κB and AP-1 (activator protein 1) signalling by NOD1 is responsible by a rapid induction of pro-inflammatory response upon H. pylori infection [78]. NOD1-deficient mice exhibit increased bacterial loads in the stomach when compared with wild-type counterparts after oral administration of H. pylori [68]. Moreover, aggregates of H. pylori products co-localize with NOD1 and some polyubiquitinated proteins in colonized gastric epithelium, which suggests a role for NOD1 in proximal sensing of H. pylori [79].
NOD1 has been implicated in IKK1 and NF-κB activation in the context of S. flexneri infection [80]. Infection of HEK (human embryonic kidney)-293 cells with S. flexneri activates JNK in a NOD1-dependent manner [75]. NOD1 was also shown to be essential for p38 MAPK phosphorylation in endothelial cells during L. monocytogenes infection [66]. Similarly, NOD2 has been implicated in the modulation of NF-κB and MAPK signalling in the bacterial pneumonia model. Specifically, reduced activation of NF-κB and MAPKs (and associated impairment of expression of cell adhesion molecules) was detected in the lungs of Nod2−/− mice challenged with Escherichia coli [81]. An enhanced bacterial burden and reduced neutrophil and cytokine/chemokine levels were observed in the lungs of Nod2−/− mice following E. coli infection [81]. Altogether, these results support a role for NOD1 and NOD2 in the innate immune responses against a variety of bacterial pathogens.
More recently, NOD2 signalling was involved in the activation of human monocytes and further differentiation into DCs upon Mycobacterium leprae infection, the aetiological agent of leprosy. Both live M. leprae and NOD2 inducer MDP, but not TLR ligands, stimulate IL-32 production, which is required for monocyte differentiation into DCs [82]. In fact, IL-32 is sufficient to induce the differentiation of DCs with potent MHC class I antigen-presenting function, which is apparently relevant to the progress of leprosy [82]. The identification of MDP as a mycobacterial ligand that triggers IL-32 production provides insight into mechanisms by which bacterial derivatives function as powerful adjuvants for vaccines.
NOD1 AND NOD2 IN INFLAMMATORY DISEASES
Dysregulation of NLR function has been described in a variety of maladies, including chronic inflammation, autoimmunity and cancer predisposition [11,83]. For instance, polymorphisms associated with the human NOD1 and NOD2 genes have been correlated with elevated cancer risk. For NOD1, polymorphisms [detected as SNPs (small nucleotide polymorphisms)] may be associated with several types of malignancy, including gastric, ovarian, prostate and lung cancer as well as lymphoma [84]. NOD2 polymorphisms have been associated with multiple human inflammatory disorders, including Crohn's disease, Blau syndrome, early-onset sarcoidosis, and atopic diseases, which cause NF-κB constitutive activation [11,85–87]. Intestinal macrophages isolated from Crohn's disease patients harbouring disease-associated alleles of NOD2 produce elevated levels of NF-κB targets, including the pro-inflammatory cytokines TNFα (tumour necrosis factor α) and IL-1β and IL-6 [87,88]. The three most common mutations (A702T, G908A and Leu1007fs) account for approximately 80% of NOD2 variants associated with Crohn's disease [89]. These NOD2 variant alleles might be found in up to one-third of Crohn's disease patients [90]. The presence of one variant allele increases the risk of developing Crohn's disease from 1.5- to 4.3-fold, and the presence of two copies has been proposed to increase the risk by 20- to 40-fold [91–93].
The functional analysis of Crohn's disease-associated NOD2 variants was achieved by the generation of knockin mice (Nod22939iC) for the most prevalent Crohn's disease susceptibility Nod2 allele in humans, 3020insC (corresponding to Leu1007fs mutation), which drives the formation of a truncated protein lacking 33 amino acids on its C-terminus. It was observed that NF-κB DNA-binding activity, expression of NF-κB target genes (including various cytokines) and IL-1β secretion are largely increased in MDP-stimulated macrophages derived from Nod22939iC mice when compared with wild-type counterparts [87]. Knockin mice treated with dextran sodium sulfate, an agent that disrupts intestinal mucosal barrier function and permits bacterial invasion, show severe and extensive inflammatory lesions of the colon as well as increased mortality [87]. Since Nod2−/− mice do not show increased inflammation [94], these results suggested that Nod22939iC is a gain-of-function allele whose product induces inflammation through elevated IKK and caspase 1 activation in response to MDP. Nevertheless, this ‘gain-of-function’ proposal is controversial, since it stands in contrast with a variety of studies that illustrate NOD2-deficient phenotypes, including (for instance) decreased PG-responsiveness for NOD2 mutations associated with Crohn's disease, with reduced cytokine production by monocytes, and reduced antimicrobial activity of intestinal epithelial cells [95].
Linkages of NOD1 gene variants to human disease have not been as commonly observed as for NOD2, despite the homology in protein structure and downstream signalling involving NOD1 and NOD2. In asthma and IBD (inflammatory bowel disease), an insertion-deletion polymorphism within intron 9 of NOD1 has been correlated with increased susceptibility to the disease, although no subsequent population studies have supported a linkage with IBD [11,96,97]. This mutation apparently contributes to differences in the expression of certain spliced mRNAs encoding NOD1 protein isoforms lacking repeats within the LRR domain. Since these isoforms lack proper ligand binding, it has been suggested that these variants might interfere with the activity of the full-length NOD1 [11]. Alternatively, since experimental deletion of the LRRs from NLRs renders these proteins constitutively active [16,17,98], it could be that these isoforms have ligand-independent activity. In diseases such as asthma or IBD, the expression of these isoforms appears to be altered relative to the common forms of NOD1 protein [11].
NOD1 has been implicated in the PG-dependent activation of infiltrating DCs that migrate into the CNS (central nervous system), an essential event associated with progression of multiple sclerosis [99]. In an animal model for multiple sclerosis, EAE (experimental autoimmune encephalomyelitis), Nod1−/− and Nod2−/− mice show a marked decrease in hind-limb paralysis, diminished inflammatory cells infiltrates into brain and spinal cord tissues and decreased axon demyelination [99]. Nevertheless, this phenotype was not exclusive to Nod1−/− or Nod2−/− mice. In fact, protection from disease progression was more profound in Rip2−/− mice than in Nod1−/− or Nod2−/− mice, which is consistent with the likely redundant roles of NOD1 and NOD2. Moreover, Rip2−/− mice were protected from disease progression caused by PG injection, indirectly implicating NOD1 and NOD2 given that they are upstream activators of RIP2.
Activation of NOD1 signalling has been recently correlated with the progression of cardiovascular inflammation. Nishio et al. [100] have shown that NOD1 activation enhances cytokine production by human coronary artery endothelial cells in vitro, and administration of NOD1 agonists in mice induces coronary arteritis with dense cellular infiltrates (i.e. neutrophils and macrophages), similar to the acute phase of Kawasaki disease, a rare childhood disease characterized by inflammation of blood vessels (vasculitis). In contrast, subcutaneous injection of MDP or other bacterial components (zymosan, OK432) did not induce arteritis [100], suggesting a specific role for NOD1 activity in vascular inflammation.
NOD1 activity also promotes ocular inflammation in a dose- and time-dependent manner [101]. NOD1 is expressed within the eye and its activation results in uveitis (inflammation of the middle layer of the eye) via an IL-1β-dependent mechanism. IL-1β processing and the respective mediators caspase 1 and IL-1R1 (interleukin-1 receptor 1) are essential for this pathological process. IL-1β can be produced within the eye upon treatment with NOD1 or NOD2 ligands (i.e. DAP or MDP respectively) in a caspase 1-dependent manner. However, caspase 1 and IL-1β are not essential for ocular inflammation mediated by NOD2, though they were involved in systemic inflammatory responses triggered by MDP [101]. Interestingly, certain strains of Chlamydia trachomatis infect the ocular tissue, a leading cause of preventable blindness, and primarily induce an inflammatory response by activating NF-κB through NOD1. Overexpression of a dominant-negative form of NOD1 or its depletion by RNA interference partially inhibits NF-κB activation during chlamydial infection in vitro [102]. These results suggest that NOD1 is a potential sensor for chlamydia.
It was recently reported that NOD1 has a role in fetal inflammation [103], which has been implicated in pre-term labour – a major cause of neonatal mortality. Fetal inflammation is possibly associated with innate immune responses in the context of intrauterine bacterial infection. Human term placental tissues and isolated term trophoblast (from third trimester gestation) express NOD1, but not NOD2. In vitro treatment of term trophoblast with NOD1 ligand (DAP), but not with NOD2 ligand (MDP), induces IL-6 secretion. High doses of DAP administered to pregnant mice cause pre-term birth within 24 h of injection and are accompanied by striking fetal pro-inflammatory responses, characterized by elevated levels of a wide variety of pro-inflammatory cytokines [103]. Thus, NOD1 apparently participates in modulating infection-associated pregnancy complications, which might result in premature births.
Insulin resistance in the context of Type 2 diabetes can be associated with chronic low-level inflammation, with innate immunity constituents implicated in this pathobiology. In this regard, NLR family proteins have been identified as critical factors involved in HFD (high-fat diet)-induced inflammation and insulin intolerance [104]. Nod1/Nod2-double-KO mice are protected from HFD-induced insulin intolerance, lipid accumulation and inflammation in adipose tissue and liver. Conversely, NOD1 activation alone (by injection of NOD1 ligand FK565) stimulates insulin resistance in vivo, involving changes in both glucose clearance and glucose production. In contrast, NOD2 activation only modestly reduces glucose clearance [104], possibly due to a more limited tissue distribution of NOD2 compared with NOD1. Indeed, NOD1 is expressed in the tissues most important for glucose homoeostasis, such as liver, muscle and adipose tissue [16]. Treatment with doses of NOD1 ligand that elicit only minor changes in circulating pro-inflammatory mediators, can cause adipose tissue inflammation and insulin resistance. Ex vivo, NOD1 activation was reported to cause pro-inflammatory cytokine secretion and to impair insulin-stimulated glucose uptake directly in cultured adipocytes [104]. Treatment of 3T3-L1 adipocytes with NOD1 ligand (Tri-DAP) impairs IRS-1 (insulin receptor substrate-1) tyrosine phosphorylation (a major substrate of the insulin receptor protein tyrosine kinase), leading to impaired downstream phosphorylation of Akt [also known as PKB (protein kinase B)] and GSK (glycogen synthase kinase), resulting in decreased insulin-dependent glucose uptake [105]. This phenotype was specifically reverted by using siRNAs (small interfering RNAs) for NOD1, resulting in decreased cytokine expression and restoring insulin-induced glucose uptake. Taken together, these results imply a possible role for NOD1 in systemic insulin resistance. It remains to be determined which NOD1-related pathway is prominently involved in type 2 diabetes encountered in humans.
CHEMICAL INHIBITORS OF NOD1 AND NOD2
The modulation of innate immune response targets is one of the major goals in the development of novel therapeutics for human autoimmune and chronic inflammatory diseases. The innate system resides at the intersection of the pathways of microbial recognition (host-defence), inflammation and cell death, thereby offering various therapeutic targets [106]. In this context, NOD1 and NOD2 are of particular interest, since they recognize distinct structures derived from bacterial PGs and directly activate multiple inflammatory pathways that result in cytokine production (NF-κB, stress kinases, IFN response, etc.). Theoretically, chemical antagonists of NOD1 or NOD2 could have applications in several acute and chronic diseases, in which dampening the pro-inflammatory response of the innate immune system might be beneficial. Table 1 provides a list of potential in vivo experimental models for testing the utility of NOD1 inhibitors.
Table 1. Experimental animal models implicating NOD1 in diseases.
A current list of potential NOD1-related diseases is shown, which includes experimental procedures to pursue tests using in vivo models, time of analysis and specific comments about their applications. The respective references are also listed. ED, embryonic day; KO, knockout; LPS, lipopolysaccharide, MOG, myelin oligodendrocyte glycoprotein.
| Disease | End points | Time | Comments | Reference |
|---|---|---|---|---|
| Colitis | Salmonella infection with further caecum collection for scoring of intestinal inflammation. Cytokine and bacterial load quantification. | 24–72 h post-infection | Reduced overall pathology and cytokine production is only evident in NOD1/NOD2 double KO mice | [112] |
| Insulin resistance (Type 2 diabetes) | Hyperinsulinaemic–euglycaemic clamps in mice injected with agonists. Systemic and tissue-specific inflammation evaluation in vivo. Analysis of cytokine secretion, glucose uptake and insulin signalling in adipocytes and primary hepatocytes in vitro. | HFD for 16 weeks | NOD1 agonist causes inflammation and insulin resistance directly in primary hepatocytes from wild-type, but not NOD1 KO mice. | [104] |
| Preterm delivery | Injection of pregnant mice on ED 14.5 with agonists and further monitoring of preterm delivery (as evidenced by the delivery of one or more pups within 24 h) and measurement of fetal weight. Cytokine-chemokine profiling from isolated organs. | 24–48 h post-injection | Cytokine–chemokine profiling was performed with fetal tissue and placenta and decidua from pregnant mice at ED 16.5. | [103] |
| Multiple sclerosis (EAE) | Mice immunization by subcutaneous injection of a MOG peptide and accompanied by pertussis toxin (co-injection with agonists and/or inhibitors), and further clinical scoring. | Up to 30 days after injection | Histology of paraffin sections of mice spinal cords were accessed to evaluate inflammatory infiltration and axon demyelination. | [99] |
| Ocular inflammation (uveitis) | Intravitreal injection of agonists and/or inhibitors. Intravital video microscopy, histology, and immunohistochemistry. | 5–72 h post-injection | Inflammation is dependent on IL-1β production. | [101] |
| Vascular inflammation | Oral or subcutaneous administration of agonists (and/or inhibitors) intraperitoneally primed with or without LPS. | 1 week after treatment. | Use of FK565 as a NOD1 agonist due to higher plasma stability. | [100] |
It seems likely that NOD proteins could be targeted by small- molecule drugs, given that they rely on nucleotide triphosphate binding for their activation. Indeed, Bielig et al. [107] identified, for the first time that certain arene-Cr(CO)3 complexes decrease inflammatory responses and diminish NOD2-mediated NF-κB signalling in vitro. These compounds appear to be selective for NOD-mediated inflammatory pathways, inasmuch as they are ineffective at inhibiting NF-κB activation induced by TLR2, TLR4 or TNF receptors. These compounds were touted to be the first organochromium complexes with a defined biological activity on human cells [107]. Nevertheless, due to the limited number of analogues tested to better define compound specificity [limited SAR (structure–activity relationship) analysis] and the non-drug-like properties of organochromium compounds (which might limit their pharmacological attributes), an in-depth chemical and biological profiling is necessary before pursuing further in vivo applications for these compounds as putative NOD2 inhibitors. Table 2 provides a list of some presumptive in vivo experimental models to test potential NOD2 inhibitors.
Table 2. Experimental animal models implicating NOD2 in diseases.
A current list of potential NOD2-related diseases is shown, which includes experimental procedures to pursue tests using in vivo models, time of analysis and specific comments about their applications. The respective references are also listed. BAL, bronchoalveolar lavage; BMT, bone marrow transplant; DSS, dextran sodium sulfate; GVHD, graft versus host disease; OVA, ovalbumin; ZIA, zymosan-induced arthritis.
| Disease | End points | Time | Comments | Reference |
|---|---|---|---|---|
| Colitis | 4% DSS supplied in drinking water for 6 days with or without co-administration of MDP followed by measurement of body weight, serum amyloid A and histological grading. | 6 days after the DSS introduction | NOD2 suppresses DSS-induced colitis through cross-talk with multiple TLR pathways. | [113] |
| GVHD (graft versus host disease) | 90 days following irradiation mice were given allo-BMT and target organs were scored for GVHD, T-cell infiltration as well as DC activation and animal survival. | 7–21 days after the allo-BMT | NOD2 deficiency in allo-BMT recipients exacerbates systemic and organ GVHD. | [114] |
| Allergy and asthma | Intranasal administration of OVA for 3 days followed by immunization and challenge with OVA 14 days later for 4 days. Lung inflammation was assessed by histology for cell infiltration and by ELISA for BAL fluid cytokine content. | 4–24 h after last OVA dose | NOD2, but not NOD1 stimulation prevents induction of tolerance. | [115] |
| Arthritis | Injection of zymosan in the knee joint followed by protease activity measured by NIR substrate as well as cell infiltration detected with histological analysis. | 3 days post intra-arterial injection | Differential role of NOD1 and NOD2 observed in ZIA. | [116] |
| Tuberculosis | Aerosol delivery of M. tuberculosis and assessment of CFU, cytokines in BAL fluid and survival. | From 4 weeks to 6 months after infection | NOD2 involved in both innate and adaptive immunity in resistance to Mycobacterium tuberculosis | [117] |
| Lyme disease | Subcutaneous injection of Borrelia burgdorferi followed by measuring joint inflammation by joint thickness, histology grading and IL-6 mRNA. | 4 weeks following subcutaneous injection | NOD2, but not NOD1 is involved in initiation of inflammation and later tolerance. | [118] |
NOD1 inhibitors have been identified by using a HTS (high throughput screening) approach, which employed a cell-based NF-κB reporter gene assay. From an ~ 290000 library of diverse, drug-like compounds, a class of 2-aminobenzimidazoles was reported as the first selective NOD1 inhibitors [108,109]. These compounds were shown to suppress NF-κB signalling stimulated by NOD1 ligands and NOD1 overexpression, but not by various other NF-κB inducers, including activators of NOD2, TLRs, TNF-family receptors, RNA receptors (MAVS activators), DNA-damaging agents and PKC (protein kinase C) activators [108]. The mechanism of action behind the prototypical inhibitor, dubbed as Noditinib-1, is based on the modulation of the NOD1 protein conformation and its subcellular localization, and does not involve competitive displacement of ATP from the NOD1 protein [108]. This class of NOD1 inhibitors also suppresses activation of the IFN pathway, in addition to NF-κB. Further improvements on the potency and pharmacological profile (including in vivo stability) of Noditinib-1 will yield a promising asset for testing the efficacy and safety of NOD1 inhibitors in preclinical models of disease. From the phenotype of NOD1-KO mice, we might predict that chemical antagonists of NOD1 would predispose to adverse events related to infection risk [110,111]. However, unlike gene KOs, pharmacological agents only reduce but rarely completely ablate the activity of their targets, providing optimism that a NOD1 inhibitor strategy could be safe.
CONCLUSIONS AND FUTURE DIRECTIONS
To date, NOD1 and NOD2 are among the best studied NLR family proteins. Experimental investigations of these proteins have provided insights into the diverse roles of this class of innate immunity proteins in host defence and inflammatory diseases. The full spectrum of cellular mechanisms responsible for regulating NOD1/NOD2 activity remain to be determined, as well as the downstream events that connect NOD signalling complexes to activation of NF-κB, stress kinases, IFN responses, autophagy and other processes. Importantly, therapeutic opportunities for pharmacological modulation of NOD1 and NOD2 are largely unexplored. A critical issue concerns the question of whether NOD1 or NOD2 activity can be safely suppressed without causing undue detriment to host defences against endogenous microflora and pathogenic micro-organisms. For inflammatory and autoimmune diseases where the severity of NOD1- or NOD2-mediated inflammation warrants, it seems plausible that cycles of brief therapy with NOD1 or NOD2 inhibitors (applied under the cover of antibiotic therapy) could potentially quiet inflammation, produce remissions, and thus provide an opportunity for re-establishing immune homoeostasis. Much work lies ahead as these and other questions are addressed en route to harnessing the expanding base of knowledge about NOD1 and NOD2 for clinical benefit.
References
- 1.Kanneganti T. D., Lamkanfi M., Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Ting J. P., Lovering R. C., Alnemri E. S., Bertin J., Boss J. M., Davis B. K., Flavell R. A., Girardin S. E., Godzik A., Harton J. A., et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Creagh E. M., O’Neill L. A. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Murray P. J. NOD proteins: an intracellular pathogen- recognition system or signal transduction modifiers? Curr. Opin. Immunol. 2005;17:352–358. doi: 10.1016/j.coi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F., Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 6.Petrilli V., Dostert C., Muruve D. A., Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Tattoli I., Travassos L. H., Carneiro L. A., Magalhaes J. G., Girardin S. E. The nodosome: NOD1 and NOD2 control bacterial infections and inflammation. Semin. Immunopathol. 2007;29:289–301. doi: 10.1007/s00281-007-0083-2. [DOI] [PubMed] [Google Scholar]
- 8.Harton J. A., Linhoff M. W., Zhang J., Ting J. P. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 2002;169:4088–4093. doi: 10.4049/jimmunol.169.8.4088. [DOI] [PubMed] [Google Scholar]
- 9.Inohara Chamaillard, McDonald C., Nunez G. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 10.Proell M., Riedl S. J., Fritz J. H., Rojas A. M., Schwarzenbacher R. The NOD-like receptor (NLR) family: a tale of similarities and differences. PLoS ONE. 2008;3:e2119. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carneiro L., Magalhaes J., Tattoli I., Philpott D., Travassos L. NOD-like proteins in inflammation and disease. J. Pathol. 2008;214:136–148. doi: 10.1002/path.2271. [DOI] [PubMed] [Google Scholar]
- 12.Messier-Solek C., Buckley K. M., Rast J. P. Highly diversified innate receptor systems and new forms of animal immunity. Semin. Immunol. 2010;22:39–47. doi: 10.1016/j.smim.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Ausubel F. M. Are innate immune signaling pathways in plants and animals conserved? Nat. Immunol. 2005;6:973–979. doi: 10.1038/ni1253. [DOI] [PubMed] [Google Scholar]
- 14.Carneiro L. A., Travassos L. H., Girardin S. E. NOD-like receptors in innate immunity and inflammatory diseases. Ann. Med. 2007;39:581–593. doi: 10.1080/07853890701576172. [DOI] [PubMed] [Google Scholar]
- 15.Chamaillard M., Girardin S. E., Viala J., Philpott D. J. NODs, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell. Microbiol. 2003;5:581–592. doi: 10.1046/j.1462-5822.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 16.Inohara N., Koseki T., del Peso L., Hu Y., Yee C., Chen S., Carrio R., Merino J., Liu D., Ni J., et al. NOD1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J. Biol. Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 17.Ogura Y., Inohara N., Benito A., Chen F. F., Yamaoka S., Nunez G. NOD2, a NOD1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez O., Pipaon C., Inohara N., Fontalba A., Ogura Y., Prosper F., Nunez G., Fernandez-Luna J. L. Induction of NOD2 in myelomonocytic and intestinal epithelial cells via nuclear factor-κB activation. J. Biol. Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 19.Hisamatsu T., Suzuki M., Reinecker H. C., Nadeau W. J., McCormick B. A., Podolsky D. K. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 20.Tada H., Aiba S., Shibata K., Ohteki T., Takada H. Synergistic effect of NOD1 and NOD2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect. Immun. 2005;73:7967–7976. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogura Y., Lala S., Xin W., Smith E., Dowds T. A., Chen F. F., Zimmermann E., Tretiakova M., Cho J. H., Hart J., et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003;52:1591–1597. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voss E., Wehkamp J., Wehkamp K., Stange E. F., Schroder J. M., Harder J. NOD2/CARD15 mediates induction of the antimicrobial peptide human β-defensin-2. J. Biol. Chem. 2006;281:2005–2011. doi: 10.1074/jbc.M511044200. [DOI] [PubMed] [Google Scholar]
- 23.Uehara A., Fujimoto Y., Fukase K., Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 2007;44:3100–3111. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Uehara A., Sugawara Y., Kurata S., Fujimoto Y., Fukase K., Kusumoto S., Satta Y., Sasano T., Sugawara S., Takada H. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell. Microbiol. 2005;7:675–686. doi: 10.1111/j.1462-5822.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 25.Marriott I., Rati D. M., McCall S. H., Tranguch S. L. Induction of NOD1 and Nod2 intracellular pattern recognition receptors in murine osteoblasts following bacterial challenge. Infect. Immun. 2005;73:2967–2973. doi: 10.1128/IAI.73.5.2967-2973.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lecine P., Esmiol S., Metais J. Y., Nicoletti C., Nourry C., McDonald C., Nunez G., Hugot J. P., Borg J. P., Ollendorff V. The NOD2–RICK complex signals from the plasma membrane. J. Biol. Chem. 2007;282:15197–15207. doi: 10.1074/jbc.M606242200. [DOI] [PubMed] [Google Scholar]
- 27.Kufer T. A., Kremmer E., Adam A. C., Philpott D. J., Sansonetti P. J. The pattern-recognition molecule NOD1 is localized at the plasma membrane at sites of bacterial interaction. Cell. Microbiol. 2008;10:477–486. doi: 10.1111/j.1462-5822.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- 28.Girardin S. E., Boneca I. G., Viala J., Chamaillard M., Labigne A., Thomas G., Philpott D. J., Sansonetti P. J. NOD2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 29.Mo J., Boyle J. P., Howard C. B., Monie T. P., Davis B. K., Duncan J. A. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J. Biol. Chem. 2012;287:23057–23067. doi: 10.1074/jbc.M112.344283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Girardin S. E., Travassos L. H., Herve M., Blanot D., Boneca I. G., Philpott D. J., Sansonetti P. J., Mengin-Lecreulx D. Peptidoglycan molecular requirements allowing detection by NOD1 and NOD2. J. Biol. Chem. 2003;278:41702–41708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 31.Viala J., Sansonetti P., Philpott D. J. NODs and ‘intracellular’ innate immunity. C. R. Biol. 2004;327:551–555. doi: 10.1016/j.crvi.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 33.Laroui H., Yan Y., Narui Y., Ingersoll S. A., Ayyadurai S., Charania M. A., Zhou F., Wang B., Salaita K., Sitaraman S. V., et al. L-Ala-γ-D-Glu-meso-diaminopimelic acid (DAP) interacts directly with leucine-rich region domain of nucleotide-binding oligomerization domain 1, increasing phosphorylation activity of receptor-interacting serine/threonine-protein kinase 2 and its interaction with nucleotide-binding oligomerization domain 1. J. Biol. Chem. 2011;286:31003–31013. doi: 10.1074/jbc.M111.257501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chin A. I., Dempsey P. W., Bruhn K., Miller J. F., Xu Y., Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature. 2002;416:190–194. doi: 10.1038/416190a. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi K., Inohara N., Hernandez L. D., Galan J. E., Nunez G., Janeway C. A., Medzhitov R., Flavell R. A. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- 36.Fairhead T., Lian D., McCully M. L., Garcia B., Zhong R., Madrenas J. RIP2 is required for NOD signaling but not for Th1 cell differentiation and cellular allograft rejection. Am. J. Transplant. 2008;8:1143–1150. doi: 10.1111/j.1600-6143.2008.02236.x. [DOI] [PubMed] [Google Scholar]
- 37.Nembrini C., Kisielow J., Shamshiev A. T., Tortola L., Coyle A. J., Kopf M., Marsland B. J. The kinase activity of Rip2 determines its stability and consequently NOD1- and NOD2-mediated immune responses. J. Biol. Chem. 2009;284:19183–19188. doi: 10.1074/jbc.M109.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertrand M. J., Doiron K., Labbe K., Korneluk R. G., Barker P. A., Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009;30:789–801. doi: 10.1016/j.immuni.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Krieg A., Correa R. G., Garrison J. B., Le Negrate G., Welsh K., Huang Z., Knoefel W. T., Reed J. C. XIAP mediates NOD signaling via interaction with RIP2. Proc. Natl. Acad. Sci. U.S.A. 2009;106:14524–14529. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pickart C. M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 41.Lauwers E., Jacob C., Andre B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol. 2009;185:493–502. doi: 10.1083/jcb.200810114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurley J. H., Stenmark H. Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu. Rev. Biophys. 2011;40:119–142. doi: 10.1146/annurev-biophys-042910-155404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasegawa M., Fujimoto Y., Lucas P. C., Nakano H., Fukase K., Nunez G., Inohara N. A critical role of RICK/RIP2 polyubiquitination in NOD-induced NF-κB activation. EMBO J. 2008;27:373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damgaard R. B., Nachbur U., Yabal M., Wong W. W., Fiil B. K., Kastirr M., Rieser E., Rickard J. A., Bankovacki A., Peschel C., et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell. 2012;46:746–758. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 46.Chen F., Bhatia D., Chang Q., Castranova V. Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ. 2006;13:1835–1838. doi: 10.1038/sj.cdd.4402014. [DOI] [PubMed] [Google Scholar]
- 47.Kim Y. G., Park J. H., Daignault S., Fukase K., Nunez G. Cross-tolerization between NOD1 and NOD2 signaling results in reduced refractoriness to bacterial infection in NOD2-deficient macrophages. J. Immunol. 2008;181:4340–4346. doi: 10.4049/jimmunol.181.6.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbott D. W., Wilkins A., Asara J. M., Cantley L. C. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004;14:2217–2227. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 49.Spencer E., Jiang J., Chen Z. J. Signal-induced ubiquitination of IκBα by the F-box protein Slimb/β-TrCP. Genes Dev. 1999;13:284–294. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Q., Verma I. M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 51.Perkins N. D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 52.Sabbah A., Chang T. H., Harnack R., Frohlich V., Tominaga K., Dube P. H., Xiang Y., Bose S. Activation of innate immune antiviral responses by NOD2. Nat. Immunol. 2009;10:1073–1080. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seth R. B., Sun L., Chen Z. J. Antiviral innate immunity pathways. Cell Res. 2006;16:141–147. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 54.Watanabe T., Asano N., Fichtner-Feigl S., Gorelick P. L., Tsuji Y., Matsumoto Y., Chiba T., Fuss I. J., Kitani A., Strober W. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest. 2010;120:1645–1662. doi: 10.1172/JCI39481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cooney R., Baker J., Brain O., Danis B., Pichulik T., Allan P., Ferguson D. J., Campbell B. J., Jewell D., Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 56.Travassos L. H., Carneiro L. A., Ramjeet M., Hussey S., Kim Y. G., Magalhaes J. G., Yuan L., Soares F., Chea E., Le Bourhis L., et al. NOD1 and NOD2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 57.Travassos L. H., Carneiro L. A., Girardin S., Philpott D. J. NOD proteins link bacterial sensing and autophagy. Autophagy. 2010;6:409–411. doi: 10.4161/auto.6.3.11305. [DOI] [PubMed] [Google Scholar]
- 58.Levine B., Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeretssian G., Correa R. G., Doiron K., Fitzgerald P., Dillon C. P., Green D. R., Reed J. C., Saleh M. Non-apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96–99. doi: 10.1038/nature09982. [DOI] [PubMed] [Google Scholar]
- 61.Askari N., Correa R. G., Zhai D., Reed J. C. Expression, purification, and characterization of recombinant NOD1 (NLRC1): a NLR family member. J. Biotechnol. 2012;157:75–81. doi: 10.1016/j.jbiotec.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruey J. M., Bruey-Sedano N., Luciano F., Zhai D., Balpai R., Xu C., Kress C. L., Bailly-Maitre B., Li X., Osterman A., et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. doi: 10.1016/j.cell.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 63.Faustin B., Chen Y., Zhai D., Le Negrate G., Lartigue L., Satterthwait A., Reed J. C. Mechanism of Bcl-2 and Bcl-XL inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proc. Natl. Acad. Sci. U.S.A. 2009;106:3935–3940. doi: 10.1073/pnas.0809414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang K., Yin X. M., Chao D. T., Milliman C. L., Korsmeyer S. J. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- 65.Clohessy J. G., Zhuang J., de Boer J., Gil-Gomez G., Brady H. J. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J. Biol. Chem. 2006;281:5750–5759. doi: 10.1074/jbc.M505688200. [DOI] [PubMed] [Google Scholar]
- 66.Opitz B., Puschel A., Beermann W., Hocke A. C., Forster S., Schmeck B., van Laak V., Chakraborty T., Suttorp N., Hippenstiel S. Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J. Immunol. 2006;176:484–490. doi: 10.4049/jimmunol.176.1.484. [DOI] [PubMed] [Google Scholar]
- 67.Travassos L. H., Carneiro L. A., Girardin S. E., Boneca I. G., Lemos R., Bozza M. T., Domingues R. C., Coyle A. J., Bertin J., Philpott D. J., et al. NOD1 participates in the innate immune response to Pseudomonas aeruginosa. J. Biol. Chem. 2005;280:36714–36718. doi: 10.1074/jbc.M501649200. [DOI] [PubMed] [Google Scholar]
- 68.Viala J., Chaput C., Boneca I. G., Cardona A., Girardin S. E., Moran A. P., Athman R., Memet S., Huerre M. R., et al. NOD1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 69.Silva G. K., Gutierrez F. R., Guedes P. M., Horta C. V., Cunha L. D., Mineo T. W., Santiago-Silva J., Kobayashi K. S., Flavell R. A., Silva J. S., et al. Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J. Immunol. 2010;184:1148–1152. doi: 10.4049/jimmunol.0902254. [DOI] [PubMed] [Google Scholar]
- 70.Hruz P., Zinkernagel A. S., Jenikova G., Botwin G. J., Hugot J. P., Karin M., Nizet V., Eckmann L. NOD2 contributes to cutaneous defense against Staphylococcus aureus through α-toxin-dependent innate immune activation. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12873–12878. doi: 10.1073/pnas.0904958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shaw M. H., Reimer T., Sanchez-Valdepenas C., Warner N., Kim Y. G., Fresno M., Nunez G. T cell-intrinsic role of NOD2 in promoting type 1 immunity to Toxoplasma gondii. Nat. Immunol. 2009;10:1267–1274. doi: 10.1038/ni.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimada K., Chen S., Dempsey P. W., Sorrentino R., Alsabeh R., Slepenkin A. V., Peterson E., Doherty T. M., Underhill D., Crother T. R., et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berrington W. R., Iyer R., Wells R. D., Smith K. D., Skerrett S. J., Hawn T. R. NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur. J. Immunol. 2010;40:3519–3527. doi: 10.1002/eji.201040518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geddes K., Rubino S. J., Magalhaes J. G., Streutker C., Le Bourhis L., Cho J. H., Robertson S. J., Kim C. J., Kaul R., Philpott D. J., et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 2011;17:837–844. doi: 10.1038/nm.2391. [DOI] [PubMed] [Google Scholar]
- 75.Girardin S. E., Tournebize R., Mavris M., Page A. L., Li X., Stark G. R., Bertin J., DiStefano P. S., Yaniv M., Sansonetti P. J., et al. CARD4/NOD1 mediates NF-κB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2:736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim J. G., Lee S. J., Kagnoff M. F. NOD1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by Toll-like receptors. Infect. Immun. 2004;72:1487–1495. doi: 10.1128/IAI.72.3.1487-1495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zilbauer M., Dorrell N., Elmi A., Lindley K. J., Schuller S., Jones H. E., Klein N. J., Nunez G., Wren B. W., Bajaj-Elliott M. A major role for intestinal epithelial nucleotide oligomerization domain 1 (NOD1) in eliciting host bactericidal immune responses to Campylobacter jejuni. Cell. Microbiol. 2007;9:2404–2416. doi: 10.1111/j.1462-5822.2007.00969.x. [DOI] [PubMed] [Google Scholar]
- 78.Allison C. C., Kufer T. A., Kremmer E., Kaparakis M., Ferrero R. L. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J. Immunol. 2009;183:8099–8109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- 79.Necchi V., Sommi P., Ricci V., Solcia E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS ONE. 2010;5:e9716. doi: 10.1371/journal.pone.0009716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim M. L., Jeong H. G., Kasper C. A., Arrieumerlou C. IKKα contributes to canonical NF-κB activation downstream of NOD1-mediated peptidoglycan recognition. PLoS ONE. 2010;5:e15371. doi: 10.1371/journal.pone.0015371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Theivanthiran B., Batra S., Balamayooran G., Cai S., Kobayashi K., Flavell R. A., Jeyaseelan S. NOD2 Signaling contributes to host defense in the lungs against Escherichia coli infection. Infect. Immun. 2012;80:2558–2569. doi: 10.1128/IAI.06230-11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Schenk M., Krutzik S. R., Sieling P. A., Lee D. J., Teles R. M., Ochoa M. T., Komisopoulou E., Sarno E. N., Rea T. H., Graeber T. G., et al. NOD2 triggers an interleukin-32-dependent human dendritic cell program in leprosy. Nat. Med. 2012;18:555–563. doi: 10.1038/nm.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis B. K., Wen H., Ting J. P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kutikhin A. G. Role of NOD1/CARD4 and NOD2/CARD15 gene polymorphisms in cancer etiology. Hum. Immunol. 2011;72:955–968. doi: 10.1016/j.humimm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 85.Franchi L., Park J. H., Shaw M. H., Marina-Garcia N., Chen G., Kim Y. G., Nunez G. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 2008;10:1–8. doi: 10.1111/j.1462-5822.2007.01059.x. [DOI] [PubMed] [Google Scholar]
- 86.Hugot J. P., Chamaillard M., Zouali H., Lesage S., Cezard J. P., Belaiche J., Almer S., Tysk C., O’Morain C. A., Gassull M., et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 87.Maeda S., Hsu L. C., Liu H., Bankston L. A., Iimura M., Kagnoff M. F., Eckmann L., Karin M. NOD2 mutation in Crohn's disease potentiates NF-κB activity and IL-1β processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 88.Lala S., Ogura Y., Osborne C., Hor S. Y., Bromfield A., Davies S., Ogunbiyi O., Nunez G., Keshav S. Crohn's disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 89.Rescigno M., Nieuwenhuis E. E. The role of altered microbial signaling via mutant NODs in intestinal inflammation. Curr. Opin. Gastroenterol. 2007;23:21–26. doi: 10.1097/MOG.0b013e32801182b0. [DOI] [PubMed] [Google Scholar]
- 90.Niess J. H., Klaus J., Stephani J., Pfluger C., Degenkolb N., Spaniol U., Mayer B., Lahr G., von Boyen G. B. NOD2 polymorphism predicts response to treatment in Crohn's disease – first steps to a personalized therapy. Dig. Dis. Sci. 2012;57:879–886. doi: 10.1007/s10620-011-1977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ahmad T., Armuzzi A., Bunce M., Mulcahy-Hawes K., Marshall S. E., Orchard T. R., Crawshaw J., Large O., de Silva A., Cook J. T., et al. The molecular classification of the clinical manifestations of Crohn's disease. Gastroenterology. 2002;122:854–866. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 92.Brant S. R., Picco M. F., Achkar J. P., Bayless T. M., Kane S. V., Brzezinski A., Nouvet F. J., Bonen D., Karban A., Dassopoulos T., et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn's disease phenotypes. Inflamm. Bowel Dis. 2003;9:281–289. doi: 10.1097/00054725-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 93.Lesage S., Zouali H., Cezard J. P., Colombel J. F., Belaiche J., Almer S., Tysk C., O’Morain C., Gassull M., Binder V., et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am. J. Hum. Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pauleau A. L., Murray P. J. Role of NOD2 in the response of macrophages to Toll-like receptor agonists. Mol. Cell. Biol. 2003;23:7531–7539. doi: 10.1128/MCB.23.21.7531-7539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kufer T. A., Fritz J. H., Philpott D. J. NACHT-LRR proteins (NLRs) in bacterial infection and immunity. Trends Microbiol. 2005;13:381–388. doi: 10.1016/j.tim.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 96.Hysi P., Kabesch M., Moffatt M. F., Schedel M., Carr D., Zhang Y., Boardman B., von Mutius E., Weiland S. K., Leupold W., et al. NOD1 variation, immunoglobulin E and asthma. Hum. Mol. Genet. 2005;14:935–941. doi: 10.1093/hmg/ddi087. [DOI] [PubMed] [Google Scholar]
- 97.McGovern D. P., Hysi P., Ahmad T., van Heel D. A., Moffatt M. F., Carey A., Cookson W. O., Jewell D. P. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 2005;14:1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 98.Bertin J., Nir W. J., Fischer C. M., Tayber O. V., Errada P. R., Grant J. R., Keilty J. J., Gosselin M. L., Robison K. E., Wong G. H., et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-κB. J. Biol. Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 99.Shaw P. J., Barr M. J., Lukens J. R., McGargill M. A., Chi H., Mak T. W., Kanneganti T. D. Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity. 2011;34:75–84. doi: 10.1016/j.immuni.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nishio H., Kanno S., Onoyama S., Ikeda K., Tanaka T., Kusuhara K., Fujimoto Y., Fukase K., Sueishi K., Hara T. NOD1 ligands induce site-specific vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 2011;31:1093–1099. doi: 10.1161/ATVBAHA.110.216325. [DOI] [PubMed] [Google Scholar]
- 101.Rosenzweig H. L., Galster K. T., Planck S. R., Rosenbaum J. T. NOD1 expression in the eye and functional contribution to IL-1β-dependent ocular inflammation in mice. Invest. Ophthalmol. Visual Sci. 2009;50:1746–1753. doi: 10.1167/iovs.08-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Welter-Stahl L., Ojcius D. M., Viala J., Girardin S., Liu W., Delarbre C., Philpott D., Kelly K. A., Darville T. Stimulation of the cytosolic receptor for peptidoglycan, NOD1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell. Microbiol. 2006;8:1047–1057. doi: 10.1111/j.1462-5822.2006.00686.x. [DOI] [PubMed] [Google Scholar]
- 103.Cardenas I., Mulla M. J., Myrtolli K., Sfakianaki A. K., Norwitz E. R., Tadesse S., Guller S., Abrahams V. M. NOD1 activation by bacterial iE-DAP induces maternal-fetal inflammation and preterm labor. J. Immunol. 2011;187:980–986. doi: 10.4049/jimmunol.1100578. [DOI] [PubMed] [Google Scholar]
- 104.Schertzer J. D., Tamrakar A. K., Magalhaes J. G., Pereira S., Bilan P. J., Fullerton M. D., Liu Z., Steinberg G. R., Giacca A., Philpott D. J., et al. NOD1 activators link innate immunity to insulin resistance. Diabetes. 2011;60:2206–2215. doi: 10.2337/db11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao L., Hu P., Zhou Y., Purohit J., Hwang D. NOD1 activation induces proinflammatory gene expression and insulin resistance in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 2011;301:E587–E598. doi: 10.1152/ajpendo.00709.2010. [DOI] [PubMed] [Google Scholar]
- 106.Ulevitch R. J. Therapeutics targeting the innate immune system. Nat. Rev. Immunol. 2004;4:512–520. doi: 10.1038/nri1396. [DOI] [PubMed] [Google Scholar]
- 107.Bielig H., Velder J., Saiai A., Menning M., Meemboor S., Kalka-Moll W., Kronke M., Schmalz H. G., Kufer T. A. Anti-inflammatory arene–chromium complexes acting as specific inhibitors of NOD2 signalling. Chem. Med. Chem. 2010;5:2065–2071. doi: 10.1002/cmdc.201000320. [DOI] [PubMed] [Google Scholar]
- 108.Correa R. G., Khan P. M., Askari N., Zhai D., Gerlic M., Brown B., Magnuson G., Spreafico R., Albani S., Sergienko E., et al. Discovery and characterization of 2-aminobenzimidazole derivatives as selective NOD1 inhibitors. Chem. Biol. 2011;18:825–832. doi: 10.1016/j.chembiol.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khan P. M., Correa R. G., Divlianska D. B., Peddibhotla S., Sessions E. H., Magnuson G., Brown B., Suyama E., Yuan H., Mangravita-Novo A., et al. Identification of inhibitors of NOD1-induced nuclear factor-κB activation. ACS Med. Chem. Lett. 2011;2:780–785. doi: 10.1021/ml200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Le Bourhis L., Magalhaes J. G., Selvanantham T., Travassos L. H., Geddes K., Fritz J. H., Viala J., Tedin K., Girardin S. E., Philpott D. J. Role of NOD1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2009;77:4480–4486. doi: 10.1128/IAI.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Clarke T. B., Davis K. M., Lysenko E. S., Zhou A. Y., Yu Y., Weiser J. N. Recognition of peptidoglycan from the microbiota by NOD1 enhances systemic innate immunity. Nat. Med. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Geddes K., Rubino S., Streutker C., Cho J. H., Magalhaes J. G., Le Bourhis L., Selvanantham T., Girardin S. E., Philpott D. J. NOD1 and NOD2 regulation of inflammation in the Salmonella colitis model. Infect. Immun. 2010;78:5107–5115. doi: 10.1128/IAI.00759-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Watanabe T., Asano N., Murray P. J., Ozato K., Tailor P., Fuss I. J., Kitani A., Strober W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J. Clin. Invest. 2008;118:545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Penack O., Smith O. M., Cunningham-Bussel A., Liu X., Rao U., Yim N., Na I. K., Holland A. M., Ghosh A., Lu S. X., et al. NOD2 regulates hematopoietic cell function during graft-versus-host disease. J. Exp. Med. 2009;206:2101–2110. doi: 10.1084/jem.20090623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duan W., Mehta A. K., Magalhaes J. G., Ziegler S. F., Dong C., Philpott D. J., Croft M. Innate signals from NOD2 block respiratory tolerance and program TH2-driven allergic inflammation. J. Allergy Clin. Immunol. 2010;126:1284–1293. doi: 10.1016/j.jaci.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rosenzweig H. L., Clowers J. S., Nunez G., Rosenbaum J. T., Davey M. P. Dectin-1 and NOD2 mediate cathepsin activation in zymosan-induced arthritis in mice. Inflamm. Res. 2011;60:705–714. doi: 10.1007/s00011-011-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Divangahi M., Mostowy S., Coulombe F., Kozak R., Guillot L., Veyrier F., Kobayashi K. S., Flavell R. A., Gros P., Behr M. A. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J. Immunol. 2008;181:7157–7165. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 118.Petnicki-Ocwieja T., DeFrancesco A. S., Chung E., Darcy C. T., Bronson R. T., Kobayashi K. S., Hu L. T. NOD2 suppresses Borrelia burgdorferi mediated murine Lyme arthritis and carditis through the induction of tolerance. PLoS ONE. 2011;6:e17414. doi: 10.1371/journal.pone.0017414. [DOI] [PMC free article] [PubMed] [Google Scholar]