

Background: Self-aggregation of β-amyloid plays an important role in the pathogenesis of Alzheimer disease.
Results: Small molecule inhibitors of β-amyloid fibril formation reduce β-amyloid mediated cell toxicity.
Conclusion: Rational design led to the successful development of small molecule inhibitors of β-amyloid oligomerization and toxicity.
Significance: Small molecules targeting β-amyloid misfolding may provide new treatments for Alzheimer disease.
Keywords: Alzheimer Disease, Medicinal Chemistry, Organic Chemistry, Protein Aggregation, Small Molecules, β-Amyloid Fibrillization
Abstract
Increasing evidence implicates Aβ peptides self-assembly and fibril formation as crucial events in the pathogenesis of Alzheimer disease. Thus, inhibiting Aβ aggregation, among others, has emerged as a potential therapeutic intervention for this disorder. Herein, we employed 3-aminopyrazole as a key fragment in our design of non-dye compounds capable of interacting with Aβ42 via a donor-acceptor-donor hydrogen bond pattern complementary to that of the β-sheet conformation of Aβ42. The initial design of the compounds was based on connecting two 3-aminopyrazole moieties via a linker to identify suitable scaffold molecules. Additional aryl substitutions on the two 3-aminopyrazole moieties were also explored to enhance π-π stacking/hydrophobic interactions with amino acids of Aβ42. The efficacy of these compounds on inhibiting Aβ fibril formation and toxicity in vitro was assessed using a combination of biophysical techniques and viability assays. Using structure activity relationship data from the in vitro assays, we identified compounds capable of preventing pathological self-assembly of Aβ42 leading to decreased cell toxicity.
Introduction
Alzheimer disease (AD)2 is the most common age-related neurodegenerative disorder affecting nearly 25 million patients (1). It is characterized by progressive cognitive decline and eventually a debilitating dementia (2). Currently available pharmacologic interventions are limited to compounds enhancing cholinergic function (acetylcholinesterase inhibitors) or acting as N-methyl-d-aspartic acid receptor antagonist, but these drugs only provide symptomatic relief without halting the progression of the disease (3–5). Thus, there is an enormous medical need for novel disease-modifying therapies that target the underlying neuropathological mechanisms involved in the development of AD.
The two pathological hallmarks of AD are senile amyloid-β (Aβ) plaques (6) and neurofibrillary tangles made of aggregated Tau (7). According to the amyloid cascade hypothesis, the aggregation of Aβ is the primary cause of the disease (8–11). Thus, removal of toxic amyloid deposits is a central therapeutic aim in AD (12). The Aβ1–42 peptide (Aβ42) is the more neurotoxic form of Aβ, as Aβ42 has more pronounced oligomerization and aggregation properties (13–15).
The majority of studies on Aβ toxicity suggest that low molecular weight soluble oligomers or high molecular weight prefibrillar intermediates account for its neurotoxicity (16–20), synapse loss, and cognitive impairment (21). Aβ fibrils and high molecular weight oligomers are rich in β-sheet structures, whereas low molecular weight oligomers (dimers, trimers, tetramers) do not adopt stable secondary structures. The different transient Aβ species most likely exist in equilibrium with each other (22–23).
The amyloid cascade hypothesis offers several strategies for therapeutic intervention, including inhibition of Aβ production (β- and/or γ-secretase inhibitors) or inhibition of Aβ aggregation and toxicity. Preventing Aβ aggregation is therapeutically attractive because this might be an exclusively pathological process and would not interfere with the physiological function of the amyloid precursor protein (20).
Thus, a potential method of treating AD is to administer small molecules capable of preventing Aβ oligomerization, fibrillization, and/or plaque formation (24–26). The majority of non-peptidic anti Aβ-aggregation inhibitors identified by in vitro screening are metal chelators (27), dyes (28, 29), and polyphenolic natural products (30–34).
An alternative approach is based on a rational design utilizing acylated 3-aminopyrazoles with a donor-acceptor-donor hydrogen bond pattern complementary to that of the β-sheet of Aβ42 (35, 36) (Fig. 1A). These compounds bearing 3-aminopyrazoles are either dimeric compounds, where the 3-aminopyrazole moieties are connected by a rigid oxalyl-linker, or oligomeric compounds, where the 3-aminopyrazole moieties are directly linked to each other by amide bonds (35, 36).
FIGURE 1.

A, Shown is the structure of the Ampox (N1,N2-bis(5-methyl-1H-pyrazol-3-yl)oxalamide); Ref. 35) compound, the numbering of the pyrazole moiety, the linker unit, and the possible donor-acceptor-donor interactions of the 3-amino-pyrazole moiety with Aβ42 peptide aggregates having cross-β-sheet conformation. B, structures of the small molecule inhibitors 1–14 of Aβ42 fibrillization containing different aromatic substituents at the 4- or 5-position of the 3-aminopyrazole moiety.
Highly ordered π-stacking interactions between aromatic ring systems play important roles in β-sheet formation and assembly of complex biological and chemical supramolecular structures (37). Previous studies suggested that the anti-aggregation properties of polyphenols result from their ability to interfere with π-stacking interactions between aromatic side chains in amyloid (38). Thus, we hypothesized that in addition to the already existing donor-acceptor-donor contacts, aromatic substituents attached at the 4- or 5-position of the 3-aminopyrazole ring should increase the anti-aggregation potency of our compounds. Herein, we demonstrate that rationally designed small molecules inhibit Aβ oligomerization, fibril formation, and protect against Aβ-induced toxicity.
EXPERIMENTAL PROCEDURES
Compound Synthesis
The synthesis of compounds 1-14 (Fig. 1B) from commercially available starting materials is described in supplemental Fig. S5–S9.
Thioflavin T (ThT) Fluorescence Assays
Aβ42 lyophilized powder (Bachem) was reconstituted in hexafluoroisopropanol to 1 mm. The peptide solution was sonicated for 15 min at room temperature and agitated overnight, and aliquots were made into non-siliconized microcentrifuge tubes. The hexafluoroisopropanol was then evaporated under a stream of argon. The resulting peptide film was dried under a vacuum for 10 min, tightly sealed, and stored at −80 °C until used.
For the ThT assay, a PBS solution in non-siliconized incubation tubes was prepared with final concentrations of 330 μm inhibitors, 33 μm Aβ42, and 10 μm ThT (Sigma). The final concentration of DMSO was 12.8%. Therefore, the final molar ratio of test compound to Aβ42 was 10:1. A solution containing Aβ42 and 10 μm ThT only was used to measure the maximal relative fluorescence unit (defined as 100% relative fluorescence units). A negative control without Aβ42 defined as 0% relative fluorescence unit was also prepared for each compound to exclude compound-derived fluorescence. The assay was run for 24 h at 37 °C, and the spectrofluorescence (excitation, 440 nm; emission, 485 nm) was read in 6 replicates in black 384-well assay plates (PerkinElmer Life Sciences) in a microplate reader (Tecan). The ThT-assay IC50 determination was performed as described above with the exception that the following 7 concentrations of test compound were used: 330, 82.5, 20.63, 5.16, 1.29, 0.32, and 0.08 μm. The IC50 values were determined from the percent inhibition of aggregation values obtained at the end of the measurement. Thereafter these values were plotted against the log of the inhibitor concentration. By fitting the data with a sigmoidal function in the Prism software (GraphPad Software), the IC50 values were obtained.
Aβ42 protofibrils were prepared essentially as described (20, 39). Briefly, 1 mg of lyophilized Aβ42 was solubilized in 50 μl of 100% anhydrous DMSO in a 1.5-ml sterile microtube. Then, 800 μl of high purity water was immediately added, and the pH of the resulting solution was adjusted to ∼7.6 by adding 10 μl of 2 m Tris-base, pH 7.6. The solution was centrifuged (16,000 × g; 4 °C; 10 min), and the supernatant was injected into a Superdex 75 column previously equilibrated with 50 ml of 10 mm Tris-HCl, pH 7.4. Aβ42 monomers were prepared as described above, except that 1 mg of lyophilized Aβ42 was solubilized in 6 m guanidine hydrochloride (1 ml), and the solution was directly injected into the Superdex 75 column (20, 39). Aβ42 was eluted at a flow rate of 0.5 ml/min, and 1-ml fractions were collected in 1.5-ml sterile microtubes (supplemental Fig. S10). The elution was monitored at UV absorbance A210, A254, and A280. Aβ42 concentration was determined from the A280 using the theoretical molar extinction coefficient 1490 m−1cm−1 (39, 40). In Jan et al. (39) we validated the reliability of this method.
Aβ42 monomers and protofibrils (20 μm) were separately co-incubated with the test compounds at the following molar ratios (Aβ42 test compounds): 1:0.5, 1:1, and 1:2 in 1.5-ml sterile microtubes (500 μl/tube, in duplicates). For this purpose, dilutions of the test compounds were prepared from stock solutions in 100% anhydrous DMSO in such a manner that each tube containing Aβ42 monomers or protofibrils received an identical volume of the test compound stock solutions. As controls, the equal volume of 100% anhydrous DMSO was separately added to Aβ42 monomers and protofibrils. For validation experiments, purified Aβ42 monomers and protofibrils were co-incubated with selected test compounds at a molar ratio of 1:4 (10 μm Aβ42, 40 μm compound).
The samples were incubated at 37 °C, and fibril formation was monitored by the ThT binding assay and transmission electron microscopy (TEM). ThT fluorescence was determined every 24 h up to 72 h of incubation. For this purpose, 80 μl of Aβ42 monomers or protofibrils with and without test compounds were mixed with 20 μl of ThT (100 μm) and 10 μl of glycine-NaOH, pH 8.5 (500 mm), in a Nunc 384-well fluorescence plate (100 μl/well). ThT fluorescence of each sample was measured in an Analyst AD fluorometer (Molecular Devices) at excitation and emission wavelengths of 450 and 485 nm, respectively.
Aβ42 fibrils were prepared as described (20, 39). Briefly, a concentrated solution (1 mg/ml) of the Aβ42 preparation to obtain protofibrils, containing monomers, protofibrils, and a small amount of fibrils, was incubated at 37 °C, pH 7.8, under mild agitation for 48 h. Aβ42 fibrils (100 μm) were incubated at 37 °C with either DMSO (40 μm) or the test compounds (40 μm) in 1.5-ml sterile microtubes (600 μl/tube, in duplicates), and fibril disaggregation was monitored by the ThT binding assay and TEM. ThT fluorescence was determined at 0 and 48 h before adding the test compounds to monitor Aβ42 fibril formation. After the addition of DMSO or the test compounds, ThT fluorescence was determined at 24 and 48 h.
Analysis of Soluble Aβ42 by SDS-PAGE
After 48 h of incubation at 37 °C, Aβ42 samples (monomers or protofibrils) with or without test compounds were centrifuged (16,000 × g, 15 min, 4 °C). In contrast, Aβ42 fibril samples with or without test compounds were ultracentrifuged (100,000 × g, 30 min, 4 °C). Then, 10–12 μl of the supernatant (monomers or protofibrils or fibrils) was mixed with SDS loading buffer (41) and subjected to electrophoresis on NuPAGE 4–12% Bis-tris SDS gels (Invitrogen). The supernatant from Aβ42 fibrils was also filtered through 0.22-μm filters previously equilibrated with 10 mm Tris-HCl, pH 7.4, by centrifugation (14,000 rpm, 10 min). The protein bands were visualized by silver staining using a commercial kit (Invitrogen).
TEM
A 5–10-μl droplet of sample containing Aβ42 was deposited on a 200-mesh Formvar-coated TEM grid (EM Sciences) and allowed to settle for 60 s. Then the excess solution was wicked away by gently applying a piece of blotting paper to the edge of the grid. Then a 10-μl droplet of 2% uranyl acetate was deposited on the grid and allowed to settle for 60 s. The excess solution was removed as above. Then the grid was vacuum-dried by gently applying the vacuum probe on a grid side. Image acquisition was carried out using a Phillips CM10 microscope operated at an acceleration voltage of 80 kV.
Fluorescence Correlation Spectroscopy (FCS)
Measurements were performed using a ConfoCor I (Zeiss, Evotec) instrument equipped with an argon ion laser. The pinhole diameter was 40 μm. The instrument was calibrated with the dye rhodamine 6G with a known diffusion coefficient in water at 20 °C of 2.8·10−6 cm2/s. The Aβ42 peptide was labeled N-terminally with the fluorescence dye Oregon green as described (36). Compounds were tested using preformed aggregates that were prepared freshly by diluting low concentrated (500 nm) DMSO-stock solutions of Oregon green-labeled Aβ42 1:1 with deionized water. For each measurement a new aliquot, stored at −70 °C, was thawed and sterile-filtered through a nylon filter (0.2 μm) to remove large aggregates. Final concentration of Oregon green-labeled Aβ42 peptide in the assay was 5 nm in PBS and 6% DMSO.
The compounds were dissolved in 100% anhydrous DMSO at 50-fold concentration of the desired assay concentration, so that a constant amount of 1 μl of ligand stock solution was always added to the test solution. This latter contained 10-fold concentrated PBS, water, 8 μm mercapto-ethylamine and was distributed as 48-μl aliquots to 12 standard reaction vials. Mercaptoethylamine was added to the assay mixtures to reduce the rather high triplett fraction of the dye Oregon green as a so called triplett quencher (42).
After the calibration of the FCS instrument, the assay was started by adding 1 μl of Aβ42-Oregon green-labeled peptide in DMSO, and the aggregation process was analyzed over a period of 5 h. Samples were prepared at least as duplicates. Data evaluation was carried out by averaging the fluorescence fluctuations for every well and counting data points that deviated more than 5-fold from the mean fluorescence signal, i.e. every peak at a Z-score equal or higher than +5 was counted as an Aβ42 aggregate (36). Because the intensity of the fluorescence peaks is related to the number of dye molecules present in Aβ42 aggregates, not only the number of peaks but also the product of number and height of peaks was evaluated. The results of every measurement were normalized to the values measured for the control samples (36).
Cell Viability Assay
To evaluate cell viability of SH-SY5Y neuroblastoma cells, a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was employed according to the manufacturer's instructions (Promega). Crude Aβ42 was prepared as described previously (20, 39). Briefly, 1 mg of lyophilized Aβ42 was solubilized in 20 μl of 100% anhydrous DMSO in a 1.5-ml sterile microtube. Then 800 μl of high purity water was immediately added, and the pH of the resulting solution was adjusted to ∼7.6 by adding 10 μl of 2 m Tris base, pH 7.6. The solution was always freshly prepared and used immediately. Crude Aβ42 with or without compound was incubated for 30 min in serum-free culture medium complemented with insulin and then added onto the cells (plated in 96-wellplates) for 24 h. The MTT-dye solution was added for the last 3 h of incubation. Then the cells were incubated for 1 h in a solubilization solution, and the blue formazan product was measured at 570 nm using 690 nm as a reference wavelength in a microplate reader (Tecan). The signal was expressed as percentage of the A570–690 count from the vehicle-treated cells.
Internalization Assay
To evaluate the internalization of crude Aβ42, SH-SY5Y cells were plated in 6-well plates at a density of 5 × 105 cells/well. Crude Aβ42 (3 μm) was preincubated for 1 h with compound (3 μm) in a 1:1 molar ratio. The SH-SY5Y cells were then incubated for 2 h with the crude Aβ42/compound mixture, washed with ice-cold PBS, trypsinized, and centrifuged in ice-cold PBS at 1500 rpm for 5 min. The cells were resuspended in 80 μl of cell lysis buffer (Cell Signaling Technology) for 5 min on ice and then briefly sonicated. The lysate was centrifuged for 5 min at 14,000 × g, and the protein concentration of supernatants was determined using a microbicinchoninic acid protein assay kit (Thermo Scientific). Internalized Aβ42 was detected with a human Aβ42 ELISA high sensitivity detection kit (Millipore) following the manufacturer instructions, and the data were normalized by the total protein concentration of the samples.
Nuclear Magnetic Resonance Experiments
One-dimensional and two-dimensional NMR experiments were performed at 5 °C on a 700-MHz spectrometer (Bruker) equipped with a cryogenic probe. Commercially available 15N-labeled Aβ42 (rPeptide) was solubilized in 100 mm NaOH at a concentration of 2 mg/ml. Compounds were dissolved in deuterated DMSO at 100 mm concentration. In the titration experiments the initial sample contained 50 μm 15N-labeled Aβ42 in 50 mm phosphate buffer (pH 7.5, adjusted after the addition of the labeled peptide and kept constant along the titration). The titration series contained compounds 6, 7, or 10 in increasing compound/peptide ratios up to a ratio of ∼30. At this ratio, the DMSO concentration was ∼1.4% (v/v). The reference experiment was conducted using DMSO at 1.4% concentration and the same pH but without any added compound. Chemical shift referencing and intensity normalization was performed on the basis of an internal 4,4-dimethyl-4-silapentane-1-sulfonic acid reference.
Saturation transfer difference (STD) experiments were conducted at the specified irradiation frequencies, with irradiation at 60 ppm used as the reference spectrum. The difference between the saturated and reference spectra (saturated-reference) is shown as the STD spectra. A saturation block of 5 s and a recycle delay of 7 s were used for STD experiments. The samples contained 0.4 mm compound with or without the added peptide (at compound/peptide ratio of 16:1). The sample without the added peptide did not show any STD effect at the used frequencies. Two preparations of Aβ42 were used for the STD experiments; the first was the normally solubilized Aβ42 supposed to be rich of the monomeric Aβ42 peptide, and the second was incubated at 37 °C for 24 h (without stirring) to be enriched in oligomeric species.
RESULTS
Screening for Inhibitors of Fibril Formation Using ThT Assays
We first investigated the effect of our compounds (Fig. 1B) on the aggregation and fibril formation of Aβ42 by the mean of different ThT fluorescent assays (43). Because this dye binds to amyloidogenic cross-β-sheet structures, ThT assays are widely used for the identification and quantification of amyloid fibrils and to monitor fibril formation kinetics (44). For this purpose a high concentration of ThT was added to Aβ42 fibrillization samples in order to be in excess compared with the number of potential ThT-fibril binding sites (44).
Aβ42 was prepared for the first screening assay as a resuspended peptide film containing monomeric and heterogenic mixtures of low molecular weight Aβ42 oligomers (<16 kDa), as determined by Western blot and centrifugation (Fig. 2A). We have shown that such crude Aβ42 preparations are toxic to cells (20). The incubation time was 24 h, as preliminary experiments have shown that within this time the aggregation process was completed (ThT signal reached a steady state). In general, a compound was considered active in this assay when at least 70% inhibition of Aβ42 aggregation was observed. The Trimer inhibitor (36) of Aβ42 aggregation was used as positive control in this assay and displayed ≥70% inhibition (data not shown).
FIGURE 2.
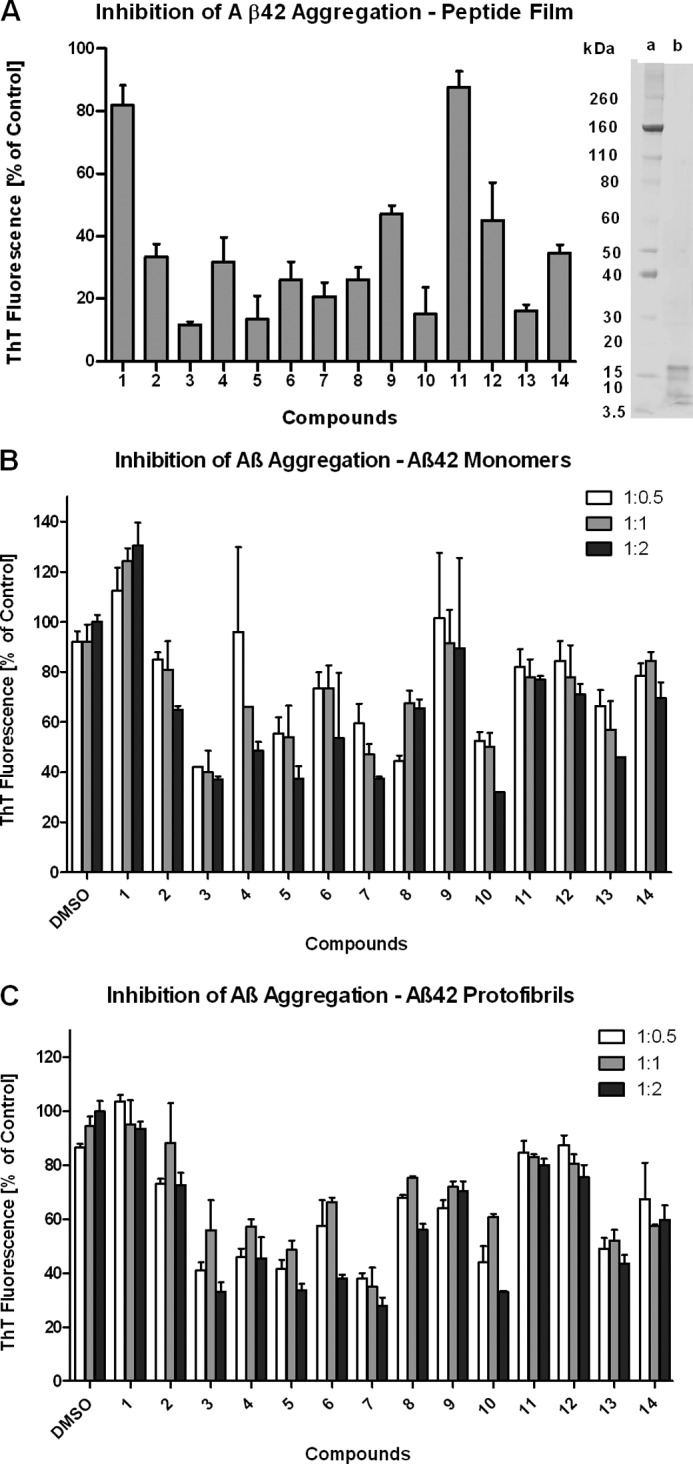
In vitro screening assays using Aβ42 peptide film (A), Aβ42 monomers (B), and Aβ42 protofibrils (C). A, the concentration of Aβ42 peptide film was 33 μm. The test concentration for compounds 1–14 was 330 μm, and the incubation time was 24 h. The data are expressed as the percentage (mean ± S.D.) of control conditions: Aβ42 aggregation with DMSO only. Freshly prepared Aβ42 peptide film (4 μg) was analyzed by SDS-PAGE to confirm the presence of oligomeric Aβ42 present (a, molecular weight marker; b, Aβ42 peptide film). B, the concentration of Aβ42 monomers was 20 μm. C, the concentration of Aβ42 protofibrils was 20 μm. Compounds 1–14 at 10 μm (1:0.5; 1% DMSO), 20 μm (1:1; 2% DMSO), and 40 μm (1:2; 4% DMSO) were co-incubated with Aβ42 monomers or protofibrils for 72 h. The data are expressed as the percentage (mean ± S.D.) of the ThT fluorescence of the 4% DMSO control.
Attaching bulky aromatic substituents at the 4-position of 3-aminopyrazole as for compound 1 (Fig. 1B) did not yield an active compound (Fig. 2A). This may be due to the fact that the rigid (-C(O)C(O)-) linker prevented additional interactions of the aromatic substituents with Aβ42. The incorporation of a more flexible (-CH2CH2-) linker in compound 2 increased its activity compared with 1. However, the inhibition of Aβ42 aggregation was still <70%, indicating that compounds with substituents at the 4-position of 3-aminopyrazole were suboptimal.
By employing a flexible (-CH2CH2-) linker and attaching aromatic substituents at the 5-position of the 3-aminopyrazole ring compounds 3–8, 10, and 13 (Fig. 1B) were obtained. Compounds having strong electron-donating groups (p-OCH3 for compound 4 or p-N(CH3)2 for compound 7), weak electron-donating groups (p-CH3 for compound 3 or p-phenyl for compound 8), or strong electron-withdrawing groups (p-Cl for compound 5 or p-F for compound 6) attached to the 5-phenyl-substituents, all displayed >70% of inhibition of Aβ42 aggregation in this assay (Fig. 2A). These data, however, showed no clear preference for the different phenyl substituents at the 5-position of 3-aminopyrazole. Furthermore, compounds 10 and 13 with a non-symmetrical substitution pattern at the 5-position of the 3-aminopyrazole moiety displayed equal activity. Thus, the heteroaromatic 2-thienyl moiety in compound 10 retained the inhibition of fibrillization activity. However, compound 9 bearing a heteroaromatic 2-furanyl moiety displayed a decrease in activity with <70% inhibition of Aβ42 aggregation. Compound 13 containing the flexible (-CH2CH2-) linker showed superior inhibition of Aβ42 aggregation when compared with the structurally related compounds 11 and 12 containing a partially reduced (-C(O)CH2-) linker. Compound 14 containing a shorter (-CH2-) linker was inferior to the otherwise identical compound 3 having a (-CH2CH2-) linker. However, the shorter but more flexible (-CH2-) linker was superior to the partially reduced (-C(O)CH2-) linker (compare compounds 11, 12, and 14; Fig. 2A). In summary, compounds containing a flexible (-CH2CH2-) linker unit and having an aromatic substituent attached to the 5-position of 3-aminopyrazole (Fig. 1B) demonstrated the strongest inhibition of Aβ42 fibrillization.
Investigating the Inhibition of Fibril Formation Using Purified Aβ42 Monomers and Protofibrils
To elucidate which Aβ42 species interact with our compounds and also to confirm the screening results obtained with the Aβ42 peptide film, we next investigated the potency of compounds 1–14 on inhibiting fibril formation of purified Aβ42 monomers or protofibrils, i.e. interference with Aβ42 nucleation or elongation. For this purpose Aβ42 monomers or Aβ42 protofibrils of defined size were prepared by size exclusion chromatography (supplemental Fig. S10) and characterized as previously described (39).
Testing the compounds by incubating them with Aβ42 monomers or protofibrils (Aβ42:compound molar ratios of 0.5, 1, and 2; 72-h incubation time) revealed that compounds 3–7, 10, and 13 led to a ≥50% inhibition of fibril maturation starting from Aβ42 monomers (Fig. 2B), and a Aβ42:compound ratio of 1:2 inhibited the elongation of Aβ42 protofibrils into mature fibrils (Fig. 2C). At substoichiometric concentrations (0.5 molar ratio), the inhibition of fibril formation was generally weaker for most of the compounds tested (Fig. 2, B and C).
In contrast, compounds 1, 2, 8, 9, 11, 12, and 14 exhibited low to moderate activity, i.e. ≤50% against the fibrillization of Aβ42 monomers (Fig. 2B) or protofibrils (Fig. 2C). Interestingly, compound 1 appeared to enhance the fibrillization of monomeric Aβ42 when compared with the control. The data showed that compounds 1 and 2 with an aromatic moiety at the 4-position of the 3-aminopyrazole moiety were less effective in inhibiting the fibrillization of both Aβ42 monomers and protofibrils when compared with compounds 3–7, 10, and 13 having an aromatic moiety at the 5-position of the 3-aminopyrazole. Unlike the results obtained with the Aβ42 peptide film, compounds 11 and 12 showed similar inhibition of Aβ42 fibril formation starting from both Aβ42 monomers and protofibrils. Again, compound 9, having the 2-furanyl moiety, displayed a weaker effect on inhibition of fibril formation (<50%) in both assays. In contrast to the results obtained with the crude Aβ42 peptide film, compound 8 was less potent when Aβ42 monomers or protofibrils were used, displaying <50% inhibition of Aβ42 fibril formation.
Taken together, the results from our in vitro screening assays (Fig. 2, A–C) were in good agreement. Thus, compounds 3, 5, 6, 7, 10, and 13 containing a flexible (-CH2CH2-) linker and different aromatic substituents at the 5-position of 3-aminopyrazole displayed potent inhibition of both Aβ42 nucleation and Aβ42 protofibril elongation.
Validation of Representative Hit Compounds and Elucidation of Their Mode of Action
Compound 1 was selected as a negative control because it did not show any activity in the previous in vitro screening assays. In contrast, compounds 5, 6, 7, and 10 displayed good inhibition of Aβ42 aggregation in the previous screening assays. These compounds were again tested in the ThT assay using the Aβ42 peptide film preparation to determine their IC50 values. For compound 1 the IC50 could not be determined (data not shown). The IC50 values for compounds 5, 6, 7, and 10 were 8.6, 40.5, 7.5, and 29 μm, respectively (Fig. 3 and supplemental Fig. S1). In summary, compound 7 displayed the most potent inhibition of Aβ42 fibrillization in the ThT assay followed by compounds 5, 10, and 6.
FIGURE 3.

IC50 determination assay using Aβ42 peptide film. The concentration of Aβ42 peptide film was 33 μm. The test concentration for compounds 5, 6, 7, and 10 were 330, 82.5, 20.63, 5.16, 1.29, 0.32, and 0.08 μm with an incubation time of 24 h. The IC50 values were determined from the fluorescence values obtained. The ThT IC50-data are expressed as the mean ± S.D.
To explore in more details the mechanism(s) by which compounds 1, 5, 6, 7, and 10 inhibited the fibrillization of Aβ42, we investigated their effect on the kinetic of Aβ42 fibril formation starting with purified Aβ42 monomer preparations. Fibril formation kinetic of Aβ42 monomers was studied over a period of 48 h with and without compounds (Fig. 4A) using a ThT readout (45). Incubation of Aβ42 monomers in the presence of compounds 5, 6, 7, and 10 resulted in an initial rise of the ThT signal after 24 h similar to Aβ42 monomers incubated without compounds. However, after 48 h the ThT signal was significantly lower as compared with the control (Fig. 4A). Compound 1 had no effect on Aβ42 monomer fibrillization as the increase in ThT signal over 48 h was comparable to the DMSO control.
FIGURE 4.

Inhibition of Aβ42 aggregation kinetics for compounds 1, 5, 6, 7, and 10 (40 μm) using 10 μm Aβ42 monomers. The total incubation time was 48 h, after which the samples were analyzed with three different assays: ThT fluorescence (A), SDS-PAGE (B), and TEM (C). The ThT-data are expressed as the mean ± S.D.; scale bar for TEM images, 100 nm. a.u., absorbance units.
Testing compounds in a fluorescent-based assay has an inherent risk of readout artifacts. As an example, compounds with absorption at ∼440 nm (the excitation wavelength of ThT) may quench or interfere with the fluorescence readout (44, 46). So, we employed an orthogonal, non-fluorescence-based assay based on the different sedimentation properties of soluble and aggregated Aβ42. For this, samples were centrifuged, and supernatants were analyzed for soluble Aβ42 protein (monomers, oligomers, and protofibrils) by SDS-PAGE (Fig. 4B). In agreement with the ThT data, SDS-PAGE analysis after 48 h of incubation at 37 °C revealed a substantial reduction in the content of soluble Aβ42 in the control and compound 1-treated sample, suggesting that almost all soluble Aβ42 was converted into insoluble fibrils. This was also in agreement with the TEM images (Fig. 4C) of the control and compound 1-treated sample showing bundles of extensive mature fibrils.
The samples treated with compounds 5, 6, 7, and 10 showed that significant amounts of soluble Aβ42 (monomers and protofibrils) remained in the supernatant, which was also in agreement with the ThT data. This suggests an interaction of compounds 5, 6, 7, and 10 with Aβ42 oligomers/protofibrils and subsequent prevention of their maturation into insoluble species. In agreement with the ThT and SDS-PAGE data, samples containing compounds 5, 6, 7, and 10 did not show mature fibrils in TEM but resulted in the formation of non-fibrillar clusters of curvilinear protofibrils (Fig. 4C). The curvilinear, irregular morphology of Aβ42 protofibrils is quite different from mature amyloid fibrils (47, 48).
Thus, compounds 5, 6, 7, and 10 did not sequester Aβ42 monomers and, hence, did not interfere with Aβ42 monomer oligomerization and seed formation. Instead, compounds 5, 6, 7, and 10 appeared to target prefibrillar Aβ42 oligomers to prevent fibrillization.
Next, we performed a kinetic analysis of the fibrillization of preformed Aβ42 protofibrils to probe the ability of compounds 1, 5, 6, 7, and 10 to interact with these structures. In the presence of DMSO alone, Aβ42 protofibrils displayed a ThT signal already at 0 h, confirming the presence of oligomeric aggregates with high β-sheet content (Fig. 5A). Then, over the first 24 h, the ThT fluorescence increased with time, consistent with the conversion of protofibrils into mature fibrils. The fibrillization process then slowed down over the next 24 h of incubation because the conversion of protofibrils into mature fibrils requires the presence of small, soluble Aβ42 species. The rise of the ThT signal for samples treated with compound 1 suggested a somehow promoted Aβ42 protofibril fibrillization process (Fig. 5A). This is in agreement with the TEM image (Fig. 5C) where the control and compound 1 sample displayed elongated fibrils among clusters of protofibrils.
FIGURE 5.

Inhibition of Aβ42 aggregation kinetics for compounds 1, 5, 6, 7, and 10 (40 μm) using 10 μm Aβ42 protofibrils. The total incubation time was 48 h after which the samples were analyzed with three different assays: ThT fluorescence (A), SDS-PAGE (B), and TEM (C). The ThT data are expressed as the mean ± S.D.; scale bar for TEM images, 100 nm. a.u., absorbance units.
In contrast, compounds 5, 6, 7, and 10 appeared to interact readily with Aβ42 protofibrils as evident by the significant decrease in ThT signal after adding the compounds to Aβ42 protofibrils at 0 h. The TEM images of samples containing compounds 5, 6, 7, and 10 were in agreement with the ThT data as they showed clusters of elongated curvilinear protofibrils (Fig. 5C). Thus, compounds 5, 6, 7, and 10 did not interfere with the oligomerization of Aβ42 protofibrils but blocked their fibrillization.
SDS-PAGE analysis was performed to quantify the remaining amount of soluble Aβ42 species (monomers, oligomers, and protofibrils). The Aβ42 protofibrils generated with our protocol were SDS-sensitive, and a band similar to monomeric Aβ42 was detected at 0 h of incubation (Fig. 5B). A significant decrease in the amount of soluble Aβ42 was observed after 48 h of incubation for samples containing compounds 1, 5, 6, and 10. Thus, in agreement with the TEM images, compounds 5, 6, and 10 were not able to disaggregate Aβ42 protofibrils. Interestingly, a larger amount of soluble Aβ42 was detected for samples containing compound 7 or the control after 48 h of incubation (Fig. 5B) as evident by the Aβ42 band intensity.
Thus, the most plausible mechanism of action for compounds 5, 6, 7, and 10 is an interaction with Aβ42 protofibrils that results in the formation of clusters of elongated curvilinear protofibrils with low β-sheet content. In addition, compound 7 caused the formation of soluble Aβ42 aggregates similar to SDS-sensitive Aβ42 protofibrils.
To test the ability of compounds 6, 7, and 10 to disaggregate amyloid fibrils, they were incubated with preformed mature Aβ42 fibrils, and the disaggregation was monitored by SDS-PAGE, TEM, and ThT fluorescence (Fig. 6). The crude Aβ42 preparation already displayed a strong ThT signal at 0 h that increased considerably over the next 48 h of Aβ42 fibril maturation (Fig. 6D). The addition of DMSO resulted in a slight decrease of the ThT signal after 24 of incubation, suggesting a minor interference with the ThT fluorescence. However, the increase of the ThT signal after 48 h of incubation suggested that DMSO did not interfere with the reassociation of mature fibrils. In contrast, samples containing compounds 6, 7, and 10 showed a significant decrease of the ThT signal after 24 and 48 h of incubation, indicating a dissociation of preformed Aβ42 fibrils. The ThT signal for samples containing compounds 6 and 10 after 24 and 48 h of incubation was similar to the initial crude Aβ42 preparation. In contrast, the ThT signal for samples containing compound 7 after 24 and 48 h of incubation was reduced when compared with the initial crude Aβ42 preparation (Fig. 6D).
FIGURE 6.

Disaggregation of Aβ42 fibrils by DMSO (40 μm) and compounds 6, 7, and 10 (40 μm) using 100 μm Aβ42 fibrils. The incubation time to form Aβ42 fibrils was 0 h (0hBC) and 48 h before adding DMSO and compounds 6, 7, and 10 (48hBC). Samples were then analyzed after 24 h (24hAC) and 48 h (48hAC) of incubation with DMSO and compounds 6, 7, and 10 using three different assays: SDS-PAGE (A), SDS-PAGE with filtration of the supernatant (B), TEM (C), and ThT fluorescence (D). The ThT data are expressed as the mean ± S.D. Scale bar for TEM images, 100 nm. a.u., absorbance units.
To verify any disaggregation of mature fibrils by DMSO and compounds 6, 7, and 10, the amount of soluble Aβ42 species (monomers, oligomers, and protofibrils) after 24 and 48 h of incubation was determined by SDS-PAGE analysis of the supernatant (Fig. 6A). Overall, the amount of soluble Aβ42 was not significantly different between the preformed Aβ42 fibrils and the samples treated with DMSO or compounds 6, 7, and 10, suggesting that there was no disaggregation of preformed Aβ42 fibrils. To determine the amount of Aβ42 monomers, the supernatant was additionally filtered and analyzed by SDS-PAGE (Fig. 6B). In general, the Aβ42 monomer bands were much weaker in intensity than the corresponding soluble Aβ42 bands (compare Fig. 6, A and B), suggesting that the majority of soluble Aβ42 species were oligomers and protofibrils. Neither DMSO nor compounds 6, 7, and 10 increased the amount of monomeric Aβ42, confirming that compounds 6, 7, and 10 did not disaggregate preformed Aβ42 fibrils. These findings were in agreement with the TEM images, where the DMSO sample as well as the samples treated with compounds 6, 7, and 10 after 48 h of incubation displayed networks of fibrils (Fig. 6C).
Thus, the most plausible mechanism of action for compounds 6, 7, and 10 is an interaction with Aβ42 fibrils that did not lead to their disaggregation. In addition, compound 7 caused the formation of Aβ42 fibrils with lower β-sheet content.
Investigating the Inhibition Mechanism of Compounds 5, 6, 7, and 10 by FCS
Another ThT-independent method for the assessment of inhibition of Aβ42 aggregation properties of small molecules is FCS. FCS allows the determination of the diffusion time of a fluorescent molecule through a small volume, i.e. the confocal volume of a laser beam of about one femtoliter. Protein aggregation causes slower diffusion times, and highly labeled large aggregates cause large fluorescence bursts when they pass through the focus (36).
In the case of self-associating molecules, aggregation can be characterized by the number of fluorescence bursts (Fig. 7A) or by the product of number and intensity of fluorescence bursts (Fig. 7B). Unlike ThT-based methods that require the presence of cross-β-sheet structures in the protein aggregates to give rise to a signal, FCS detects any aggregates containing a fluorescent molecule passing through the detection volume. Very large aggregates, however, will not be detected as they will deposit below the confocal volume.
FIGURE 7.
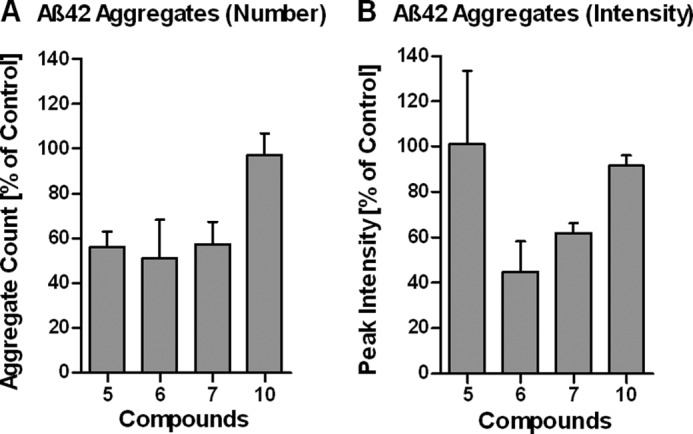
Fluorescence correlation spectroscopy of Aβ42 incubated with compounds 5, 6, 7, and 10. The aggregation of 5 nm Aβ42, N-terminally labeled with Oregon green in PBS and 3% DMSO, was monitored by FCS with or without 200 nm concentrations of compounds 5, 6, 7, and 10. Aggregate formation was quantified by counting the frequency of intensity spikes caused by Aβ42 aggregates passing the detection volume (A) and the height of the intensity spikes (B). Results were normalized to the control aggregation, and the data are expressed as the mean ± S.D.
Compounds 5, 6, and 7 reduced the number of small Aβ42 aggregates passing the laser beam by ∼50% when compared with conditions with DMSO alone. In contrast, compound 10 did not reduce the number of small Aβ42 aggregates (Fig. 7A). Taking into account not only the number of Aβ42 aggregates but also their size (product of peak number and intensity), it was revealed that compounds 6 and 7 were more potent than compound 5 in both reducing the number of Aβ42 aggregates as well as their size (Fig. 7B). Overall, compound 6 was most efficient in inhibiting Aβ42 aggregation monitored by FCS (Fig. 7, A and B).
Investigating the Interaction of Compounds 6, 7, and 10 with Aβ42 by Nuclear Magnetic Resonance Spectroscopy
NMR spectroscopy was used to follow the effects of compounds 6, 7, and 10 on Aβ42. As revealed by the intensity of 1H signals in one-dimensional NMR spectra, the addition of compound 6 resulted in a considerable loss of signal intensity. The NMR signal intensity loss showed a clear dependence on the concentration of the compound, resulting in a decrease of up to ∼30% at a compound/Aβ42 ratio of ∼30:1 (Fig. 8). The decrease in intensity was due to the presence of the compound, as the addition of a reference solution (pure DMSO) did not cause any significant changes in NMR signal intensities. Very similar changes in NMR spectra were also observed for compounds 7 (supplemental Fig. S2A) and 10 (supplemental Fig. S2B). Taken together the observed changes suggest that compounds 6, 7, and 10 induce formation of large Aβ42 oligomers that are broadened beyond the detection limit of liquid-state NMR due to slow tumbling. In addition, separate analysis of backbone and side chain signals of Aβ42 in the presence of compound 7 showed that the intensity decrease of the methyl groups is slower, indicating that they remain partially mobile in the oligomeric state (supplemental Fig. S2A). In contrast, almost identical signal decays for methyl and amide signals were observed for compounds 6 (Fig. 8) and 10 (supplemental Fig. S2B). Thus, in case of compounds 6 and 10, the formed Aβ42 oligomers had either less flexible side chains or had a larger molecular weight.
FIGURE 8.

Ligand-dependent conversion of Aβ42 into oligomers. Intensity changes of proton signals in one-dimensional 1H NMR spectra of Aβ42 upon the addition of compound 6 at various ratios are shown. Changes in the methyl and backbone amide signals are shown separately. Reference values were obtained after the addition of DMSO (without compound 6) at corresponding volumes.
To obtain residue-specific information about the interaction of compounds 6, 7, and 10 with Aβ42, two-dimensional [1H,15N]heteronuclear single quantum coherence spectra (49) of Aβ42 were measured in the absence and presence of the compounds. The variation of chemical shifts of amide 1H and 15N spins of Aβ42 upon the addition of the compounds represented alterations in their chemical environment after direct binding and/or induced conformational changes. In addition, the intensity of 1H,15N correlation peaks was affected by the exchange between the free and bound forms of the peptide and their intrinsic transverse relaxation rates. Therefore, a combined monitoring of chemical shifts and peak intensities can provide insights into the thermodynamic and kinetic aspects of the binding of small molecules to proteins (50). In line with the observation from one-dimensional 1H NMR spectra, the addition of compounds 6, 7, and 10 resulted in a decrease in the intensity of [1H,15N] correlation peaks (supplemental Fig. S3). The intensity decrease was quite uniform along the sequence with the exception of residues 25–29, in particular for compounds 7 (supplemental Fig. S3B) and 10 (supplemental Fig. S3C).
Besides the intensity decrease, a slight perturbation of backbone amide chemical shifts was observed (Fig. 9A). In case of compounds 7 and 10, it was similar both in magnitude and profile to the deviations observed upon the addition of identical volumes of pure DMSO. More pronounced chemical shift perturbations, however, were observed in the presence of compound 6, especially for residues 10–19 and for many residues from Ile-31 to Ala-42. With the exception of the basic His-13, His-14, and Lys-16 residues, all other amino acid residues are hydrophobic. Notably, these residues are located in the regions of the Aβ42 peptide sequence that constitute the N- and C-terminal β-strands of Aβ fibrils (Fig. 9B) (51).
FIGURE 9.

A, shown is the interaction of compounds 6, 7, and 10 with monomeric Aβ42. Shown is the average backbone amide proton and nitrogen chemical shift deviation obtained from two-dimensional 1H,15N heteronuclear single quantum coherence spectra of 15N-labeled Aβ42 in the absence and presence of the compounds 6, 7, and 10. The compounds 6 and 10 were present at a compound/peptide ratio of 30:1, whereas the corresponding value for compound 7 was 24:1. Reference data were obtained after the addition of DMSO (without compounds) at the corresponding volume. B, shown is an illustration of the main interactions of compound 6 with hydrophobic amino acids of monomeric Aβ42 using the NMR structure of Aβ42 fibrils (51) consisting of five peptides (PDB entry 2BEG).
The observed change of chemical shifts in dependence of the compound 6/Aβ42 ratio was nearly linear, i.e. far from the saturation regime (supplemental Fig. S4). This indicated that the interaction between monomeric, NMR-observable Aβ42 and compound 6 was weak with a Kd in the mm range. The NMR STD technique is a method that allows the detection of interactions between small molecules and proteins in a wide range of affinities (KD ∼ 10−8–10−3 m) (52). In these experiments, an 1H NMR signal from the peptide or protein was irradiated and kept saturated for a relatively long time. Then saturation transferred to all the other protons of the peptide as well as any ligand that was in contact with the peptide during the saturation period. A decrease in the 1H NMR signal intensities of the ligand, constituting the STD signal, represented the binding event. The part of the small molecule that had the strongest contact/binding with Aβ42 displays the most intense STD signal. For compound 6 the two peaks at ∼5.8 and 6.0 ppm were well separated from the Aβ42 peaks and selected for STD analysis (Fig. 10). The two peaks belong to the protons directly attached to the pyrazole rings. After the addition of an Aβ42 preparation enriched in oligomers, the peak at 5.8 ppm was already broadened in a conventional one-dimensional 1H NMR spectrum, suggesting that the corresponding proton of compound 6 was involved in the interaction with Aβ42 (Fig. 10A, upper signals). In line with its involvement in the interaction with Aβ42, a clear STD signal was observed at both 5.8 and 6.0 ppm after irradiation at 0.6 ppm that saturates the methyl signals of Aβ42 (Fig. 10A, lower signals). The aromatic protons from the 4-F-phenyl moieties (∼7.0–7.7 ppm) were not separated from the Aβ42 peaks but also showed clear STD signals after irradiation at 0.6 ppm.
FIGURE 10.

Preferential binding of compound 6 to oligomeric Aβ42. STD spectra of compound 6 in the presence of Aβ42. A, the positive spectra are one-dimensional (1D) 1H NMR spectra of compound 6 in the absence and presence of the peptide. The negative spectra are STD spectra obtained at irradiation frequencies of 0.6, −1, −2, −3, and −5 ppm. B, shown is the STD profile, observed for the ligand peaks at 5.8 and 6.0 ppm as a function of irradiation frequency. Note that the STD intensities obtained with the oligomer-enriched preparation of Aβ42 are larger and extend up to −5 ppm.
Next we tested the impact of the irradiation frequency on the STD effect. When the irradiation frequency was moved to −1, −2, −3, and −5 ppm, a continuous decrease of the STD effect was observed (Fig. 10B). Because no “visible” peptide signal is located at these up-field frequencies, the STD effects there should be caused by the “hidden” signals of the oligomeric peptide, which are broadened enough to be “NMR-invisible” but reach up to frequencies of −5 ppm due to severe line broadening. Therefore, the STD profile confirmed a binding event between compound 6 and Aβ42 oligomers (Fig. 10B). In line with a preferential interaction of compound 6 with Aβ42 oligomers, the normally solubilized Aβ42 preparation showed lower STD intensities (gray dots of Fig. 10B). In addition, the normally solubilized Aβ42 preparation that contains a smaller amount and potentially different types of oligomers did not show a STD effect at −5 ppm.
Rescue of Aβ42-induced Toxicity and Prevention of Aβ42-Uptake in Cultured Cells
To study the toxic effect of Aβ42 on SH-SY5Y cells, we selected a crude Aβ42 preparation containing mixtures of heterogeneous Aβ42 oligomers and abundant monomers (20, 39). This preparation was chosen to mimic the pathological situation in vivo where both monomeric and protofibrillar Aβ species populate the diseased AD brain. Furthermore, we reported that crude Aβ42 preparations, when compared with purified monomers and protofibrils, were much more toxic to cultured cells including neurons and fibrillized extensively (20, 45). Compounds 1, 5, 6, 7, and 10 were thus tested in a cell-based in vitro assay to determine their ability to increase the viability of SH-SY5Y cells treated with crude Aβ42 as measured by a MTT reduction assay (20). For this assay the ratio of compound to Aβ42 was 1:1 (10 μm) with a 1-h preincubation of the compound with crude Aβ42 followed by a 24-h treatment of the cells (20). The cell viability assay showed that compounds 5, 6, 7, and 10 but not compound 1 were able to reduce the toxicity of crude Aβ42, resulting in increased cell viability (Fig. 11A).
FIGURE 11.

Inhibition of crude Aβ42 induced toxicity and inhibition of internalization of crude Aβ42 by compounds 1, 5, 6, 7, and 10 monitored with SH-SY5Y neuroblastoma cells. A, assessment of crude Aβ42 toxicity in the presence of compounds 1, 5, 6, 7, and 10 by employing a MTT reduction assay is shown. Crude Aβ42 was preincubated for 1 h with the compounds a 1:1 molar ratio (10 μm) before the cells were treated with the mixture for 24 h. B, shown is an assessment of the efficacy of compounds 1, 5, 6, 7, and 10 to prevent crude Aβ42 internalization into SH-SY5Y cells. Crude Aβ42 was preincubated for 1 h with the compounds at a 1:1 molar ratio (3 μm) before the cells were treated with the mixture for 2 h. C, shown is correlation between inhibition of internalization of crude Aβ42 and inhibition of crude Aβ42-induced toxicity by compounds 1, 5, 6, 7, and 10. The data are expressed as the mean ± S.D. of three independent experiments.
To better understand the underlying mechanism by which compounds 5, 6, 7, and 10 increased the cell viability of SH-SY5Y cells treated with crude Aβ42, we developed an Aβ42 uptake assay (Fig. 11B). The assay is based on the fact that extracellular Aβ42 aggregates have been shown to interact with cellular membranes followed by internalization by endocytosis resulting in the accumulation of intra-neuronal Aβ42 (53). For this assay the ratio of compound to Aβ42 was 1:1 (3 μm) with a 1-h preincubation of the compound with crude Aβ42 followed by a 2-h treatment of the SH-SY5Y cells. Internalized Aβ42 was determined using an ELISA for human Aβ42. Compound 1 had little effect on crude Aβ42 internalization (<10% reduction) when compared with conditions without compounds (defined as 0% reduction), whereas compounds 5, 6, 7, and 10 interfered with crude Aβ42 internalization (∼30–60% reduction). Thus, the improved cell viability of Aβ42-treated SH-SY5Y cells observed in the presence of compounds 6, 7, and 10 correlated well with their capability to decrease the cellular uptake of crude Aβ42 (Fig. 11C).
DISCUSSION
The goal of this study was to rationally design small molecules capable of preventing the formation of toxic Aβ42 species. In recent years a number of small molecule inhibitors of Aβ42 aggregation have been studied. The majority of compounds, mainly natural products, contained phenolic moieties. Selected examples are apomorphine (18), curcumin (25), (−)-epigallocatechine gallate (32, 33), resveratrol (31), bi- and mono-flavonoids (54), tannic acid (34), nordihydroguaiaretic acid (34), tolcapone (45), RS-0466, RS-0406 (55), and O4 (56). Although all of these compounds have very different chemical structures, they share common features required for activity (25, 57). These features are hydroxyl substituents on the aromatic moiety, a second terminal aromatic moiety, and a linker unit to which the aromatic moieties are attached. The optimal linker length appeared to be between 8 and 16 Å, and the linker should not contain more than two rotating bonds (25, 57).
For our compounds we applied a rational design based on the 3-aminopyrazole moiety (35, 36) to retain entropically favorable multipoint hydrogen bond interactions with β-sheets (36). The distance between donor and acceptor should be in the range of 3.5–4.0 Å, and the distance between acceptor and donor should be 2.6–2.9 Å (35, 58). In this respect, the Ampox compound (N1,N2-bis(5-methyl-1H-pyrazol-3-yl)oxalamide; Refs. 35 and 36 and Fig. 1A) represented a suitable lead for compound optimization whereby the following criteria required to permeate biological membranes were targeted: molecular weight ≤ 450, polar surface area ≤90 Å2, number of hydrogen-bond acceptors ≤ 7, number of hydrogen-bond donors ≤ 4, and lipophilicity (logarithm of n-octanyl alcohol/water partition coefficient) ≤5 (59). Thus, we retained the 3-aminopyrazole ring, incorporated linker units with not more than two rotating bonds, and added aromatic moieties at each of the 3-aminopyrazole rings to enable π-stacking and/or hydrophobic interactions with the residues of Aβ42 (Fig. 1).
The use of ThT-based screening assays (Fig. 2) allowed the rapid screening of our compounds and resulted in the identification of compounds 1, 2, 4, 8, 9, 11, 12, and 14 with undesired structural features leading to inferior inhibition of Aβ42 fibrillization, i.e. aromatic substituents at the 4-position of the 3-aminopyrazole ring and rigid-linkers. The data from different in vitro assays (Fig. 3–10) suggested that the improved activity in inhibiting Aβ42 fibrillization by compounds 3, 5, 6, 7, 10, and 13 was linked to their ability to adopt a conformation where additional π-stacking and/or hydrophobic interactions with Aβ42 were possible. The weak mm binding of compound 6 to monomeric Aβ42 (supplemental Fig. S4) and the preference of compound 6 to bind to oligomeric Aβ42 (Fig. 10B) suggested that the structurally related compounds 3, 5, 7, 10, and 13 bind preferably to oligomeric Aβ42 species as well.
The preferred interaction of compound 6 with hydrophobic residues of the N- and C-terminal β-strands (Leu-17, Val-18, Phe-19, Ile-31, Ile-32, Val-36, Val-40, Ile-41, and Ala-42) of monomeric Aβ42 (Fig. 9) can be directly linked to the presence of the 3-aminopyrazole ring and the attached aromatic substituents at the 5-position. The STD spectra (Fig. 10) showed that at all irradiation frequencies there is an energy transfer from Aβ42 to compound 6 (Fig. 10A, lower signals), clearly indicating that both the 3-aminopyrazole ring and the 4-F-phenyl substituent have direct binding contacts with these Aβ42 residues. A similar mode of interaction was proposed earlier for the recognition of the hexapeptide sequence (KKLVFF) of residues 15–20 of Aβ42 by a trimeric 3-aminopyrazole ligand (36). The polyphenolic compound O4 preferably targets the hydrophobic residues 17–20 and 31–37 of Aβ40 as well (56). The importance of π-stacking and/or hydrophobic interactions was also observed in the preferred binding of phthalocyanine tetrasulfate to the heptapeptide sequence (EGVLYVG) of residues 35–41 of α-synuclein (50).
The suppression of the rise of the ThT-signal in kinetic experiments using Aβ42 monomers (Fig. 4A) and Aβ42 protofibrils (Fig. 5A) provide further evidence that compounds 5, 6, 7, and 10 interact with the β-sheets already present in oligomeric Aβ42. Infrared spectroscopy and x-ray diffraction have demonstrated the presence of significant amounts of β-sheet structure in Aβ42 protofibrils and fibrillar oligomers (60–62) that appear as the earliest fibrillar aggregates within the Aβ amyloidogenic pathway (47) and are putatively a major cytotoxic Aβ species (63). It was already demonstrated that ThT recognizes prefibrillar Aβ aggregates (64) and that the ThT fluorescence was only modestly increased (1.5-fold) when compared with fibrils (>100-fold). The lower ThT fluorescence of prefibrillar or oligomeric Aβ aggregates can be explained by the lower β-sheet content of oligomers (∼48–57%) when compared with fibrils (61).
Previous studies with small molecule inhibitors of Aβ42 aggregation allowed the differentiation of active compounds into three subsets (34, 46). Class I (e.g. Congo red, curcumin, nordihydroguaiaretic acid) inhibited Aβ42 oligomerization but not Aβ42 fibril formation. Class II (e.g. methylene blue, rhodamine B, phenol red) inhibited both Aβ42 oligomerization and Aβ42 fibril formation. Class III (e.g. orange G, piceid, tannic acid) inhibited Aβ42 fibril formation but not Aβ42 oligomerization (34, 46). Regarding the fibrillization of Aβ42 monomers and protofibrils, compounds 5, 6, 7, and 10 seemed to display the behavior of class III compounds and acted by kinetic stabilization of Aβ42 oligomeric intermediates (Figs. 4 and 5). In addition, compound 7 also increased the amount of soluble Aβ42 aggregates similar to SDS sensitive Aβ42 protofibrils (Fig. 5B). Unlike class III compounds reported in literature (34), compounds 5, 6, 7, and 10 were not able to disaggregate preformed Aβ42 fibrils (Fig. 6) into low molecular weight species. Again compound 7 displayed a somewhat different behavior by inducing the formation of Aβ42 fibrils with reduced ThT binding capacity (Fig. 6D).
FCS data revealed a reduction of the number of small Aβ42 aggregates for compounds 5, 6, and 7 but not compound 10 (Fig. 7A). The FCS data for larger Aβ42 aggregates (Fig. 7B) matched compound 6 when Aβ42 monomers were used, leading to an increased amount of soluble Aβ42 (Fig. 4B). For compound 7, however, the use of Aβ42 protofibrils resulted in the formation of Aβ42 aggregates that were indistinguishable from SDS-sensitive Aβ42 protofibrils (Fig. 5B). Thus, we speculate that the Aβ42 aggregates detected by FCS are different in size and conformation.
Compounds 6, 7, and 10 most efficiently increased the viability of cells treated with toxic Aβ42 (Fig. 11A) and decreased the cellular uptake of Aβ42 (Fig. 11B). NMR experiments showed an interaction between compound 6 and the two adjacent histidines at positions 13 and 14 of monomeric Aβ42 (Fig. 9A). Both residues are important for Aβ42 cell membrane binding and uptake (65). One possible mechanism for Aβ42 oligomer toxicity is related to their interaction with lipid bilayers in which they might cause perturbation and/or permeabilization (18, 66). An earlier study using human neuroblastoma cells showed that oligomeric Aβ42 was internalized more efficiently than fibrillar Aβ42 (67). We speculate that a similar interaction of compounds 6, 7, and 10 with oligomeric Aβ42 may in part explain their rescuing effect. Despite being a potent compound in the ThT IC50 and kinetic of Aβ42 monomer and protofibril fibrillization assays, compound 5 displayed a weaker rescuing capacity. This may be related to the relatively high degree of Aβ42 internalization found in the presence of compound 5 (Fig. 11B). This indicates that subtle differences between small molecule inhibitors of Aβ42 fibrillization (electron-deficient 4-F-phenyl substituent for compound 6; electron-rich substituent (4-(CH3)2N-phenyl for compound 7; electron-rich substituents 4-tolyl/2-thiophenyl for compound 10; electron-deficient 4-Cl-phenyl substituent for compound 5) can lead to different mechanistic interactions with Aβ42 species, resulting in a different reduction of Aβ42 mediated toxicity. A reduction of Aβ42 toxicity in vitro was also observed for curcumin (68), (−)-epigallocatechine gallate (32), resveratrol (69), RS-0406 (55, 70), and O4 (56), which have different interactions with Aβ42 as well. Furthermore, attenuation of Aβ42 toxicity was also observed when the kinetic stabilization of Aβ42 protofibrils was enhanced by adding Aβ40 monomers (23), and interestingly, small molecules stabilizing Aβ42 protofibrils in vitro improved behavioral performance in APP-transgenic mice (71).
In summary, compounds 6, 7, and 10 showed the validity of our inhibitor design (flexible (-CH2-CH2-) linker unit and aromatic substituents at the 5-position of 3-aminopyrazole) to target hydrophobic residues of Aβ42. Compounds 6, 7, and 10 efficiently prevented pathological self-assembly of Aβ42 monomers and Aβ42 protofibrils by binding to oligomeric Aβ42, broke down neurotoxic Aβ42 oligomers, increased the viability of SH-SY5Y neuroblastoma cells when challenged with toxic crude Aβ42 mixture, and decreased cellular uptake of Aβ42. Thus, compounds 6, 7, and 10 have potential as novel drug candidates for the treatment of neurodegeneration in AD and related amyloid diseases.
This work was supported by a Heisenberg scholarship (ZW71/2-2 and 3-2; to M. Z.). Heiko Kroth, Yvan Varisco, Nampally Sreenivasachary, Valérie Giriens, Sophie Lohmann, María Pilar López-Deber, Oskar Adolfsson, Maria Pihlgren, Paolo Paganetti, Wolfgang Froestl, Andrea Pfeifer, and Andreas Muhs are all full-time employees of AC Immune SA.

This article contains supplemental Figs. S1–S10.
- AD
- Alzheimer disease
- Aβ
- β-amyloid protein
- Bis-tris
- bis(2-hydroxyethyl)-amino-tris(hydroxymethyl)-methane
- FCS
- fluorescence correlation spectroscopy
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- O4
- 2,8-bis-(2,4-dihydroxyphenyl)-7-hydroxyphenoxacin-3-one
- RS-0406
- N′-bis(3-hydroxyphenyl)pyridazine-3,6-diamine
- RS-0466
- 6-ethyl-N,N′-bis(3-hydroxyphenyl)[1,3,5]triazine-2,4-diamine
- SH-SY5Y
- human-derived neuroblastoma cell line
- STD
- saturation transfer difference spectroscopy
- TEM
- transmission electron microscopy
- ThT
- thioflavin T.
REFERENCES
- 1. LaFerla F. M., Green K. N., Oddo S. (2007) Intracellular amyloid-β in Alzheimer disease. Nat. Rev. Neurosci. 8, 499–509 [DOI] [PubMed] [Google Scholar]
- 2. Smith M. A. (1998) Alzheimer disease. Int. Rev. Neurobiol. 42, 1–54 [DOI] [PubMed] [Google Scholar]
- 3. Clark C. M., Karlawish J. H. (2003) Alzheimer disease. Current concepts and emerging diagnostic and therapeutic strategies. Ann. Intern. Med. 138, 400–410 [DOI] [PubMed] [Google Scholar]
- 4. Cummings J. L. (2004) Alzheimer disease. N. Engl. J. Med. 351, 56–67 [DOI] [PubMed] [Google Scholar]
- 5. Grundman M., Thal L. J. (2000) Treatment of Alzheimer disease. Rationale and strategies. Neurol. Clin. 18, 807–828 [DOI] [PubMed] [Google Scholar]
- 6. Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 82, 4245–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Abnormal phosphorylation of the microtubule-associated protein Tau in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hardy J. (2009) The amyloid hypothesis for Alzheimer disease. A critical reappraisal. J. Neurochem. 110, 1129–1134 [DOI] [PubMed] [Google Scholar]
- 9. Oddo S., Caccamo A., Cheng D., Jouleh B., Torp R., LaFerla F. M. (2007) Genetically augmenting Tau levels does not modulate the onset or progression of Aβ pathology in transgenic mice. J. Neurochem. 102, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 10. Samura E., Shoji M., Kawarabayashi T., Sasaki A., Matsubara E., Murakami T., Wuhua X., Tamura S., Ikeda M., Ishiguro K., Saido T. C., Westaway D., St George Hyslop P., Harigaya Y., Abe K. (2006) Enhanced accumulation of Tau in doubly transgenic mice expressing mutant βAPP and presenilin-1. Brain Res. 1094, 192–199 [DOI] [PubMed] [Google Scholar]
- 11. Selkoe D. J. (1991) The molecular pathology of Alzheimer disease. Neuron 6, 487–498 [DOI] [PubMed] [Google Scholar]
- 12. Cohen F. E., Kelly J. W. (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426, 905–909 [DOI] [PubMed] [Google Scholar]
- 13. Dahlgren K. N., Manelli A. M., Stine W. B., Jr., Baker L. K., Krafft G. A., LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 14. McGowan E., Pickford F., Kim J., Onstead L., Eriksen J., Yu C., Skipper L., Murphy M. P., Beard J., Das P., Jansen K., Delucia M., Lin W. L., Dolios G., Wang R., Eckman C. B., Dickson D. W., Hutton M., Hardy J., Golde T. (2005) Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47, 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jarrett J. T., Berger E. P., Lansbury P. T., Jr. (1993) The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation. Implications for the pathogenesis of Alzheimer disease. Biochemistry 32, 4693–4697 [DOI] [PubMed] [Google Scholar]
- 16. Glabe C. C. (2005) Amyloid accumulation and pathogensis of Alzheimer disease. Significance of monomeric, oligomeric and fibrillar Aβ. Subcell. Biochem. 38, 167–177 [DOI] [PubMed] [Google Scholar]
- 17. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer disease. Progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 18. Lashuel H. A., Hartley D., Petre B. M., Walz T., Lansbury P. T., Jr. (2002) Neurodegenerative disease. Amyloid pores from pathogenic mutations. Nature 418, 291. [DOI] [PubMed] [Google Scholar]
- 19. Walsh D. M., Klyubin I., Fadeeva J. V., Rowan M. J., Selkoe D. J. (2002) Amyloid-β oligomers. Their production, toxicity, and therapeutic inhibition. Biochem. Soc. Trans. 30, 552–557 [DOI] [PubMed] [Google Scholar]
- 20. Jan A., Adolfsson O., Allaman I., Buccarello A. L., Magistretti P. J., Pfeifer A., Muhs A., Lashuel H. A. (2011) Aβ42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Aβ42 species. J. Biol. Chem. 286, 8585–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 22. Walsh D. M., Lomakin A., Benedek G. B., Condron M. M., Teplow D. B. (1997) Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372 [DOI] [PubMed] [Google Scholar]
- 23. Jan A., Gokce O., Luthi-Carter R., Lashuel H. A. (2008) The ratio of monomeric to aggregated forms of Aβ40 and Aβ42 is an important determinant of amyloid-β aggregation, fibrillogenesis, and toxicity. J. Biol. Chem. 283, 28176–28189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bandiera T., Lansen J., Post C., Varasi M. (1997) Inhibitors of Aβ peptide aggregation as potential anti-Alzheimer agents. Curr. Med. Chem. 4, 159–170 [Google Scholar]
- 25. Hawkes C. A., Ng V., McLaurin J. A. (2009) Small molecule inhibitors of Aβ-aggregation and neurotoxicity. Drug Dev. Res. 70, 111–124 [Google Scholar]
- 26. Adlard P. A., James S. A., Bush A. I., Masters C. L. (2009) β-Amyloid as a molecular therapeutic target in Alzheimer disease. Drugs Today 45, 293–304 [DOI] [PubMed] [Google Scholar]
- 27. Bush A. I. (2003) The metallobiology of Alzheimer disease. Trends Neurosci. 26, 207–214 [DOI] [PubMed] [Google Scholar]
- 28. Lorenzo A., Yankner B. A. (1994) β-Amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. U.S.A. 91, 12243–12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klunk W. E., Debnath M. L., Koros A. M., Pettegrew J. W. (1998) Chrysamine-G, a lipophilic analogue of congo red, inhibits Aβ-induced toxicity in PC12 cells. Life Sci. 63, 1807–1814 [DOI] [PubMed] [Google Scholar]
- 30. Ramassamy C. (2006) Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases. A review of their intracellular targets. Eur. J. Pharmacol. 545, 51–64 [DOI] [PubMed] [Google Scholar]
- 31. Ladiwala A. R., Lin J. C., Bale S. S., Marcelino-Cruz A. M., Bhattacharya M., Dordick J. S., Tessier P. M. (2010) Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 285, 24228–24237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ehrnhoefer D. E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E. E. (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566 [DOI] [PubMed] [Google Scholar]
- 33. Bieschke J., Russ J., Friedrich R. P., Ehrnhoefer D. E., Wobst H., Neugebauer K., Wanker E. E. (2010) EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ladiwala A. R., Dordick J. S., Tessier P. M. (2011) Aromatic small molecules remodel toxic soluble oligomers of amyloid β through three independent pathways. J. Biol. Chem. 286, 3209–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rzepecki P., Wehner M., Molt O., Zadmard R., Harms K., Schrader T. (2003) Aminopyrazole oligomers for β-sheet stabilization of peptides. Synthesis 12, 1815–1826 [Google Scholar]
- 36. Rzepecki P., Nagel-Steger L., Feuerstein S., Linne U., Molt O., Zadmard R., Aschermann K., Wehner M., Schrader T., Riesner D. (2004) Prevention of Alzheimer disease-associated Aβ aggregation by rationally designed nonpeptidic β-sheet ligands. J. Biol. Chem. 279, 47497–47505 [DOI] [PubMed] [Google Scholar]
- 37. Aggeli A., Bell M., Boden N., Keen J. N., Knowles P. F., McLeish T. C., Pitkeathly M., Radford S. E. (1997) Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature 386, 259–262 [DOI] [PubMed] [Google Scholar]
- 38. Gazit E. (2002) A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 16, 77–83 [DOI] [PubMed] [Google Scholar]
- 39. Jan A., Hartley D. M., Lashuel H. A. (2010) Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer disease research. Nat. Protoc. 5, 1186–1209 [DOI] [PubMed] [Google Scholar]
- 40. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 42. Widengren J., Chmyrov A., Eggeling C., Löfdahl P. A., Seidel C. A. (2007) Strategies to improve photostabilities in ultrasensitive fluorescence spectroscopy. J. Phys. Chem. A 111, 429–440 [DOI] [PubMed] [Google Scholar]
- 43. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer disease β-amyloid peptides. Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hudson S. A., Ecroyd H., Kee T. W., Carver J. A. (2009) The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 276, 5960–5972 [DOI] [PubMed] [Google Scholar]
- 45. Di Giovanni S., Eleuteri S., Paleologou K. E., Yin G., Zweckstetter M., Carrupt P. A., Lashuel H. A. (2010) Entacapone and tolcapone, two catechol O-methyltransferase inhibitors, block fibril formation of α-synuclein and β-amyloid and protect against amyloid-induced toxicity. J. Biol. Chem. 285, 14941–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Necula M., Kayed R., Milton S., Glabe C. G. (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324 [DOI] [PubMed] [Google Scholar]
- 47. Harper J. D., Wong S. S., Lieber C. M., Lansbury P. T. (1997) Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 4, 119–125 [DOI] [PubMed] [Google Scholar]
- 48. Goldsbury C. S., Wirtz S., Müller S. A., Sunderji S., Wicki P., Aebi U., Frey P. (2000) Studies on the in vitro assembly of a β1–40. Implications for the search for a β fibril formation inhibitors. J. Struct. Biol. 130, 217–231 [DOI] [PubMed] [Google Scholar]
- 49. Bodenhausen G., Ruben D. J. (1980) Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189 [Google Scholar]
- 50. Lamberto G. R., Binolfi A., Orcellet M. L., Bertoncini C. W., Zweckstetter M., Griesinger C., Fernández C. O. (2009) Structural and mechanistic basis behind the inhibitory interaction of PcTS on α-synuclein amyloid fibril formation. Proc. Natl. Acad. Sci. U.S.A. 106, 21057–21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., Riek R. (2005) Three-dimensional structure of Alzheimer amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mayer M., Mayer B. (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 53. Lai A. Y., McLaurin J. (2010) Mechanisms of amyloid-β peptide uptake by neurons. The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimer Dis. 2011:548380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thapa A., Woo E. R., Chi E. Y., Sharoar M. G., Jin H. G., Shin S. Y., Park I. S. (2011) Biflavonoids are superior to monoflavonoids in inhibiting amyloid-β toxicity and fibrillogenesis via accumulation of nontoxic oligomer-like structures. Biochemistry 50, 2445–2455 [DOI] [PubMed] [Google Scholar]
- 55. Walsh D. M., Townsend M., Podlisny M. B., Shankar G. M., Fadeeva J. V., El Agnaf O., Hartley D. M., Selkoe D. J. (2005) Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerization of natural Aβ and thereby rescue long term potentiation. J. Neurosci. 25, 2455–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bieschke J., Herbst M., Wiglenda T., Friedrich R. P., Boeddrich A., Schiele F., Kleckers D., Lopez del Amo J. M., Grüning B. A., Wang Q., Schmidt M. R., Lurz R., Anwyl R., Schnoegl S., Fändrich M., Frank R. F., Reif B., Günther S., Walsh D. M., Wanker E. E. (2012) Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101 [DOI] [PubMed] [Google Scholar]
- 57. Reinke A. A., Gestwicki J. E. (2007) Structure-activity relationships of amyloid β-aggregation inhibitors based on curcumin. Influence of linker length and flexibility. Chem. Biol. Drug. Des. 70, 206–215 [DOI] [PubMed] [Google Scholar]
- 58. Jorgensen W. L., Pranata J. (1990) Importance of secondary interactions in triply hydrogen bonded complexes. Guanine-cytosine vs. uracil-2,6-diaminopyridine. J. Am. Chem. Soc. 112, 2008–2010 [Google Scholar]
- 59. Pajouhesh H., Lenz G. R. (2005) Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2, 541–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Habicht G., Haupt C., Friedrich R. P., Hortschansky P., Sachse C., Meinhardt J., Wieligmann K., Gellermann G. P., Brodhun M., Götz J., Halbhuber K. J., Röcken C., Horn U., Fändrich M. (2007) Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing Aβ protofibrils. Proc. Natl. Acad. Sci. U.S.A. 104, 19232–19237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cerf E., Sarroukh R., Tamamizu-Kato S., Breydo L., Derclaye S., Dufrêne Y. F., Narayanaswami V., Goormaghtigh E., Ruysschaert J. M., Raussens V. (2009) Antiparallel β-sheet: a signature structure of the oligomeric amyloid β-peptide. Biochem. J. 421, 415–423 [DOI] [PubMed] [Google Scholar]
- 62. Stroud J. C., Liu C., Teng P. K., Eisenberg D. (2012) Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. U.S.A. 109, 7717–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lashuel H. A., Lansbury P. T., Jr. (2006) Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 39, 167–201 [DOI] [PubMed] [Google Scholar]
- 64. Reinke A. A., Gestwicki J. E. (2011) Insight into amyloid structure using chemical probes. Chem. Biol. Drug Des. 77, 399–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Poduslo J. F., Gilles E. J., Ramakrishnan M., Howell K. G., Wengenack T. M., Curran G. L., Kandimalla K. K. (2010) HH Domain of Alzheimer disease Aβ provides structural basis for neuronal binding in PC12 and mouse cortical/hippocampal neurons. PLoS One 5, e8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kayed R., Sokolov Y., Edmonds B., McIntire T. M., Milton S. C., Hall J. E., Glabe C. G. (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279, 46363–46366 [DOI] [PubMed] [Google Scholar]
- 67. Chafekar S. M., Baas F., Scheper W. (2008) Oligomer-specific Aβ toxicity in cell models is mediated by selective uptake. Biochim. Biophys. Acta 1782, 523–531 [DOI] [PubMed] [Google Scholar]
- 68. Yang F., Lim G. P., Begum A. N., Ubeda O. J., Simmons M. R., Ambegaokar S. S., Chen P. P., Kayed R., Glabe C. G., Frautschy S. A., Cole G. M. (2005) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 280, 5892–5901 [DOI] [PubMed] [Google Scholar]
- 69. Han Y. S., Zheng W. H., Bastianetto S., Chabot J. G., Quirion R. (2004) Neuroprotective effects of resveratrol against β-amyloid-induced neurotoxicity in rat hippocampal neurons. Involvement of protein kinase C. Br. J. Pharmacol. 141, 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nakagami Y., Nishimura S., Murasugi T., Kaneko I., Meguro M., Marumoto S., Kogen H., Koyama K., Oda T. (2002) A novel β-sheet breaker, RS-0406, reverses amyloid β-induced cytotoxicity and impairment of long term potentiation in vitro. Br. J. Pharmacol. 137, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hawkes C. A., Deng L. H., Shaw J. E., Nitz M., McLaurin J. (2010) Small molecule β-amyloid inhibitors that stabilize protofibrillar structures in vitro improve cognition and pathology in a mouse model of Alzheimer disease. Eur. J. Neurosci. 31, 203–213 [DOI] [PubMed] [Google Scholar]